Reaction pathways and textural aspects of the replacement of anhydrite by calcite at 25 °C
-
Teresa Roncal-Herrero
 ,
José Manuel Astilleros
,
José Manuel Astilleros

Abstract
The replacement of sulfate minerals by calcium carbonate polymorphs (carbonation) has important implications in various geological processes occurring in Earth surface environments. In this paper we report the results of an experimental study of the interaction between anhydrite (100), (010), and (001) surfaces and Na2CO3 aqueous solutions under ambient conditions. Carbonation progress was monitored by glancing incidence X-ray diffraction (GIXRD) and scanning electron microscopy (SEM). We show that the reaction progresses through the dissolution of anhydrite and the simultaneous growth of calcite. The growth of calcite occurs oriented on the three anhydrite cleavage surfaces and its formation is accompanied by minor vaterite. The progress of the carbonation always occurs from the outer-ward to the inner-ward surfaces and its rate depends on the anhydrite surface considered, with the (001) surface being much more reactive than the (010) and (100) surfaces. The thickness of the formed carbonate layer grows linearly with time. The original external shape of the anhydrite crystals and their surface details (e.g., cleavage steps) are preserved during the carbonation reaction. Textural characteristics of the transformed regions, such as the gradation in the size of calcite crystals, from ∼2 µm in the outer region to ∼17 µm at the calcite-anhydrite interface, the local preservation of calcite crystalographic orientation with respect to anhydrite and the distribution of the microporosity mainly within the carbonate layer without development of any significant gap at the calcite-anhydrite interface. Finally, we compare these results on anhydrite carbonation with those on gypsum carbonation and can explain the differences on the basis of four parameters: (1) the molar volume change involved in the replacement process in each case, (2) the lack/existence of epitactic growth between parent and product phases, (3) the kinetics of dissolution of the different surfaces, and (4) the chemical composition (amount of structural water) of the parent phases.
Introduction
Mineral replacement reactions are common phenomena in a wide variety of environments and physicochemical conditions within the Earth. Such reactions often involve the dissolution of the parent phase as a result of its interaction with an aqueous fluid and the simultaneous precipitation of the product (Putnis 2002). The coupling between the kinetics of both processes, dissolution and crystallization, determines the preservation of the shape of the reactant to a scale that can reach the accurate reproduction of delicate details of its surface as well as its internal microstructure (Xia et al. 2009a). The scale of the pseudomorphism is indeed the result of how perfect this coupling is (Pöml et al. 2007; Fernández-Díaz et al. 2009).
The progress of mineral replacement reactions requires the continuous contact between the fluid phase and the interface between the primary and secondary phases where the reactions occur. Such contact is guaranteed when the mineral replacement involves the generation of significant microporosity (Putnis 2002). Such porosity is frequently a consequence of differences in solubility and molar volumes between the primary dissolving phase and the secondary precipitating phase. However, other factors such as the starting fluid composition and the solid-liquid volume ratio can also play an important role in the kinetics of mineral replacement reactions (Putnis 2002, 2009; Putnis et al. 2007; Ruiz-Agudo et al. 2014). When the reactant solid phase is more soluble and/or has a significantly higher molar volume than the product, the mineral replacement reaction can be accompanied by a volume reduction. This volume reduction is balanced by the generation of porosity, which allows the preservation of the original external shape (Pollok et al. 2011; Putnis 2009; Ruiz-Agudo et al. 2014, 2016). The specific contribution of the differences in solubility, molar volume, starting fluid composition, and solid-liquid volume ratio to porosity generation needs to be evaluated in each particular case.
Additionally, the existence of crystallographic relationships between the primary and secondary phases can promote the epitactic growth of the product on the surface of the reactant (Xia et al. 2009a, 2009b; Qian et al. 2011). This can lead to the partial armoring of the primary phase from the fluid and the subsequent stoppage of the replacement process (Prieto et al. 2003, 2013; Rodríguez-Blanco et al. 2007; Rodríguez et al. 2008; Pérez-Garrido et al. 2007) after a “partial equilibrium” end point is reached (Astilleros et al. 2006).
Gypsum (CaSO4⋅2H2O) and anhydrite (CaSO4) are the most common sulfate minerals in Earth’s surface environments (Anthony et al. 2003). Both sulfate minerals can be replaced by other phases as a result of their interaction with fluids of variable compositions and this happens particularly often under Earth surface conditions (e.g., Freyer and Voigt 2003). Different studies have highlighted the relevance of replacement phenomena involving sulfate minerals in the context of a range of geological problems. For example, Hanor (2004) proposed the interaction of gypsum and/or anhydrite with fluids that bear Ba and Sr as a key process in the formation of large sedimentary deposits of celestine (SrSO4). Many other authors have reported that calcium sulfate minerals can be replaced by calcium carbonate polymorphs in sedimentary basins where a steady aqueous carbonate supply is warranted (Pierre and Rouchy 1988; Rouchy et al. 1998; Dejonghe et al. 1998; Sanz-Rubio et al. 2001; Azam 2007; Stafford et al. 2008). Nevertheless, the actual kinetics and mechanisms of the calcium sulfate to calcium carbonate replacement reactions are still poorly understood.
Although gypsum is thermodynamically the most stable calcium sulfate phase below ∼60 °C, the solubility of anhydrite and gypsum are within the same order of magnitude (Ksp,gypsum = 10−4.58, Ksp,anhydrite = 10−4.36; Freyer and Voigt 2003). Both minerals have been reported to transform into CaCO3 when in contact with a carbonate-bearing aqueous solution (Flörke and Florke 1961; Freyer and Voigt 2003). For both polymorphs this transformation involves dissolution-crystallization reactions and commonly results in the preservation of the external shape of the initial sulfate crystals (Freyer and Voigt 2003). Experiments following the interaction between acidic,
To fill this gap in our knowledge, we report here on the results of an experimental study of the kinetics and mechanism of anhydrite carbonation under conditions that are relevant for industrial processes and can be extrapolated to those commonly found in natural environments. We compare our observations on anhydrite to results previously obtained on gypsum carbonation. Our goal was to evaluate to what extent (1) differences in the molar volume change involved in the replacement and (2) the lack/existence of epitactic growth between primary and secondary phases influences carbonation reaction pathways. We will show how the combination of both factors determines the development of specific textural features and affects the progress of the replacement reaction.
Experimental design
Experiments were carried out by putting freshly cleaved anhydrite fragments in glass vessels containing 1 mL of a commercial 0.5 M Na2CO3 aqueous solution (pH ≈ 11.4; Fluka). The vessels were immediately closed with a polypropylene cap to avoid exchange with CO2 in the atmosphere and evaporation. All experiments were performed using natural slightly blue, highly pure anhydrite crystals from Naica, Chihuahua (Mexico). Prior to the experiments, the crystals were cleaved parallel to (100), (010), and (001) faces with a knife edge to obtain fragments of approximately 2 × 2 × 1.5 mm. Anhydrite fragments were bounded by surfaces parallel to the main cleavage planes (100), (010), and (001), all of which are F-faces (Aquilano et al. 1992; Redfern and Parker 1998) and, therefore, very stable surfaces. Moreover, (100) together with the (010) are the most common surfaces in natural anhydrite crystals (Aquilano et al. 1992). All the experiments were run in triplicate at 25 ± 0.5 °C and atmospheric pressure. The samples were recovered from the solutions after times that ranged between 1 min and 1 month, followed by rapid drying using absorbent paper, and subsequently placing the resulting solids into a desiccator at room temperature prior to further analyses.
Samples were analyzed using glancing incident X-ray diffraction (GIXRD) analyses with a PANalytical X’Pert PRO MRD diffractometer equipped with a CuKα X-ray source, an X-ray parabolic mirror in the incident beam and a parallel plate collimator with flat graphite monochromator in the diffracted beam (Xe proportional detector). Scans were recorded between 2 and 80 °θ at 2 °/s scan rate and a step size of 0.05°. The angle of incidence was 1°, which allowed obtaining information corresponding to the shallow layers, minimizing the presence of peaks coming from the bulk of the anhydrite crystal. Diffraction patterns were compared to standard powder diffraction files from the ICDD-PDF2 database (release 2007): 00-037-1496 for anhydrite, 01-071-3699 for calcite, and 00-024-0030 for vaterite. A semi-quantitative estimate of the percentage of the different crystalline phases forming on the outer part of the carbonated layer was performed using the reference intensity ratio (RIR) method (Snyder 1992). Reacted samples were placed on an aluminum stub covered by a sticky carbon pad or mounted on glass slide and embed in a synthetic epoxy resin and polished using diamond laps to investigate the contact between the anhydrite and the replaced layer. Then samples were imaged using a JEOL JSM6400 Scanning Electron Microscope (SEM) (20 kV; working distance of 15 nm) equipped with a LINK Ex1 energy dispersive spectrometer. Both GIXRD analyses and SEM observations were conducted on specific anhydrite surfaces after set interaction times. In those cases the orientation of the anhydrite surface of interest was established prior to the experiments by observing the interference figures in a polarized light microscope. The progress of the replacement and its thickness were determined by measuring the width of the transformed layer on SEM images of sections of post-reaction samples using the Image J software (Rasband 2016).
Results
Reaction pathways in anhydrite carbonation
The carbonation reaction begins on anhydrite surfaces immediately after the crystals are immersed into the 0.5 M Na2CO3 aqueous solution. GXRD data (Fig. 1) confirm calcite as the main component forming on the outer layer of the reacted anhydrite samples from the very early stages of the carbonation process. This was irrespective the anhydrite surface considered. This reaction seems to progress via the dissolution of anhydrite and the simultaneous formation of calcite crystals. Evidences of both processes can be clearly detected on the images of anhydrite surfaces that even after 1 min are covered by small calcite crystals (Fig. 2a).

GXRD patterns obtained after the interaction of anhydrite (001) cleavage surfaces with 0.5 M Na2CO3 aqueous solution during (a) 1 h, (b) 3 h, and (c) 1 day. The main (hkl) reflections for (+) anhydrite (ANH) and (*) calcite (cc) are indicated. All other minor peaks correspond to either calcite or anhydrite.

SEM photomicrographs of an anhydrite (100) surface after reaction with a 0.5 M Na2CO3 aqueous solutions for (a) 1 min, (b) 30 min, (c) 5 h, and (d) 7 days. Note the progressive increase in both, the degree of coverage of the anhydrite surface and the size of the growing calcite crystals.
Although at the very beginning calcite nuclei are small and scarce (Fig. 2a), their density and sizes rapidly increase on all considered anhydrite surfaces during the first 30 min of interaction (Fig. 2b). The newly formed calcite crystals were not randomly distributed on the anhydrite surface but they were spatially related to etch pits or step edges (Fig. 2b) and their size increases with increasing interaction time: after 1 min the size of calcite crystals varied between ∼0.2–0.5 µm and it reached ∼12 µm after 1 week (Fig. 2d). The progress of the interaction leads to a complete coverage of the anhydrite surface after a few hours (∼5 h) (Fig. 2c). Calcite crystals show the typical rhombohedral habit, with flat (104) faces and sharp edges and no modification in the general characteristics of the rhombohedral habit of calcite were observed.
Significant differences in the extent of coverage by CaCO3 phases and the pathway of carbonation were detected at the early stages of the reaction on different anhydrite surfaces. For example, after 1 h of reaction, the coverage was almost complete on the anhydrite (001) surface (Fig. 3a), while on the (100) surface it reached ∼33% (Fig. 3b) and on the (010) surface ∼47% only (Fig. 3c). In addition, both calcite and vaterite were present on the anhydrite surfaces. However, the amount of vaterite observed coexisting with calcite at the early stages of reaction also varies depending on the anhydrite surface considered. Numerous vaterite aggregates could be distinguished on anhydrite (010) and (001) surfaces, while these were scarce on the (100) surface (Figs. 3d, 3e, and 3f). GXRD patterns collected on reacted samples 2 h after the beginning of experiments evidence that irrespective the anhydrite surface studied calcite was the most abundant CaCO3 polymorph. Weak reflections that could be assigned to vaterite were distinguishable in the GXRD pattern collected from (001) anhydrite surfaces (Fig. 4a). These reflections are extremely weak in the GXRD pattern collected on the (010) surface and absent on the patterns obtained from the (100) anhydrite surface (Figs. 4b and 4c). These results were also confirmed by the scarcity observed of vaterite aggregates in SEM images of each surface (Figs. 3a and 3d). Semi-quatitative estimates derived from GXDR patterns using the Reference Intensity Ratio (RIR) method (Snyder 1992) yielded vaterite percentages in the carbonate layer after 2 h reaction below 1% on the (010) and ∼1.5% on the (001) surface, respectively. After 15 h of reaction the GXRD patterns revealed the presence of only reflections that could be assigned to calcite and this was irrespective the anhydrite cleavage surface considered (e.g., Fig. 1c).

SEM photomicrographs showing anhydrite (001), (100), and (010) cleaved surfaces partially covered by calcite and vaterite crystals after 1 h of interaction with Na2CO3-aqueous solutions (a–f). White arrows in photomicrograph d are pointing out some vaterite aggregates.
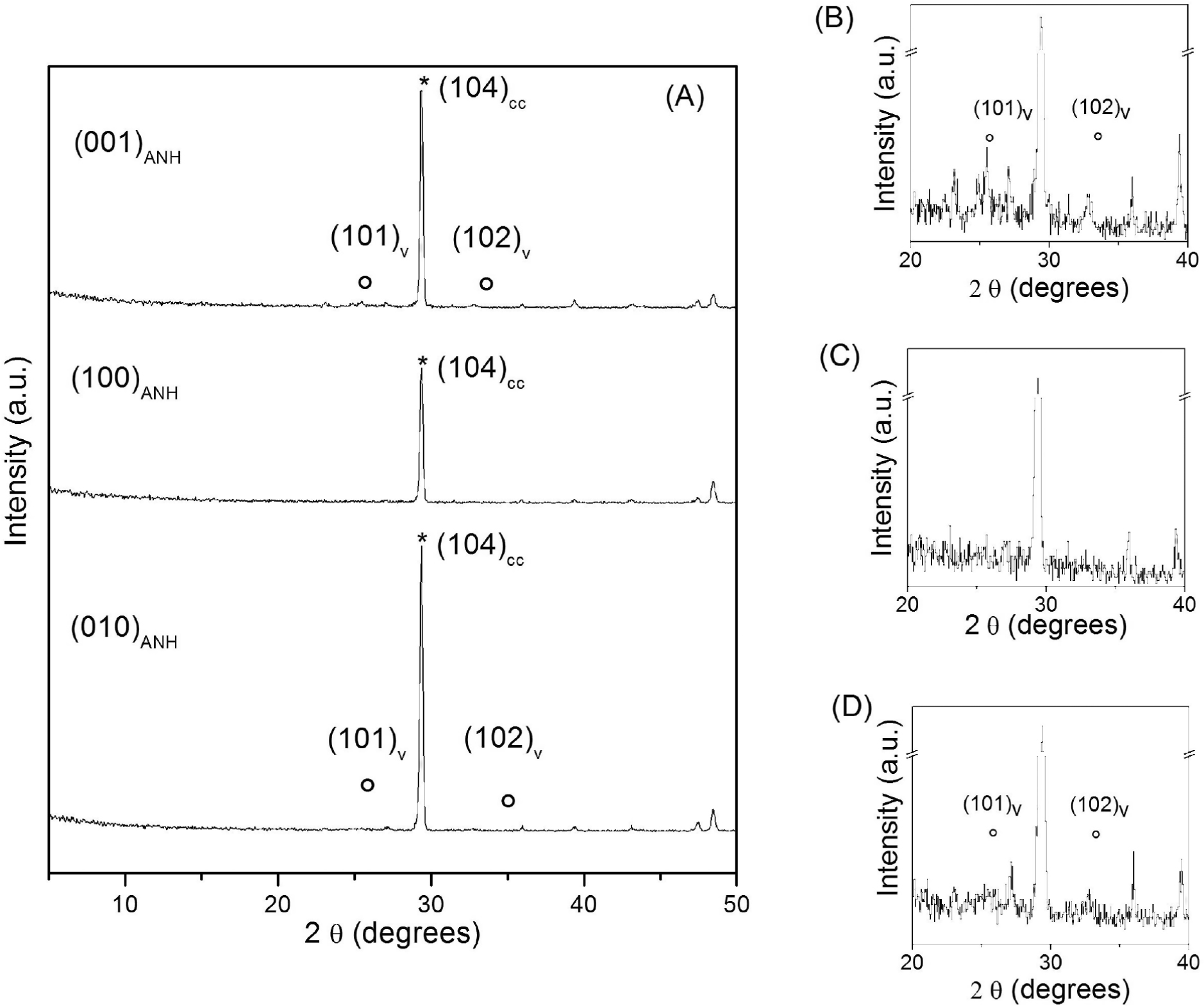
(a) GXRD patterns taken on anhydrite (001), (100), and (010) cleaved surfaces after 1 h of interaction with Na2CO3 aqueous solutions. The main (hkl) reflections for (*) calcite and (o) vaterite are indicated. In plots in b, c, and d, the vertical scale has been enlarged to more clearly show vaterite reflections in the diagrams taken on (001), (100), and (010) surfaces, respectively. SEM photomicrographs of these surfaces are shown in Figure 3.
Textural characteristics of calcite pseudomorphs after anhydrite
SEM photomicrographs reveal that calcite crystals appear to share a mutual orientation with the initial anhydrite, suggesting epitactic growth on each anhydrite surface (Fig. 2). We focus from hereon on the observations on the anhydrite (100) surface. Similar features are also observed on (010) and (001). On (100) this epitactic relationship becomes apparent after 1 min of interaction (Fig. 2a) and, at least locally, remains well defined after prolonged interaction times (7 days; Fig. 2d).
The original external shape of the anhydrite crystals as well as fine details of their surfaces including their cleavage steps are preserved during the carbonation reaction (Fig. 5a). Calcite pseudomorphs after anhydrite are bounded by well-defined faces and sharp edges (Fig. 5a). The backscatter electron (BSE) micrograph in Figure 5b shows a section of an anhydrite crystal partially transformed into CaCO3 after 3 days of reaction. Differences in contrast between the CaSO4 and CaCO3 phase evidence the extent of the transformation. It is apparent that the carbonation reaction progresses from outward to inward of the anhydrite crystal and a well-defined reaction front developed. Eventually, the reaction progresses and finishes with the formation of pseudomorphs exclusively consisting of calcite (Fig. 5a). The replaced layer consists of calcite crystals, which at least locally were oriented with respect to the original anhydrite surface (Figs. 5c and 5d). A gradual increase in the size of the formed calcite crystals is also apparent, with those closer to the interface with the retreating anhydrite surface being significantly larger (mean size 17 ± 2.5 µm) than those on the outer rim (mean size 2.2 ± 0.8 µm). The contact between the replaced layer and the retreating anhydrite surface is sharp and no significant gap was observed at the interface. The replaced layer armoring the original anhydrite shows a homogeneous aspect, yet the carbonate layer also shows an evenly distributed porosity consisting of small pores located at the contact between slightly misoriented calcite crystals or between calcite crystals that show different orientations with respect to anhydrite (Fig. 5d).

(a) SEM photomicrograph of an anhydrite surface fully replaced by calcite after 24 h interaction with a 0.5 M Na2CO3 aqueous solution. (b) BSE photomicrograph of a section of an anhydrite (ANH) crystal partially replaced by calcite (CC) after 3 days in contact with a 0.5 M Na2CO3 aqueous solution; the rectangle indicates the same area shown in c. (c) BSE photomicrograph of calcite (cc) at the carbonate-anhydrite (ANH) interface. The edge of several calcite crystals have been outlined to highlight their common orientation. (d) SEM photomicrograph of the carbonate-anhydrite interface revealing the porosity associated with the replacement process. The image was acquired from a polished section of a partially replaced anhydrite crystal reacted for three days with a 0.5 M Na2CO3 aqueous solution
Reaction kinetics of anhydrite carbonation
The change in the width of the transformed region over time as obtained from evaluating SEM images is show in Figure 6. The thickness of the carbonate layer increase linearly with time, indicating no change in the transformation rate as the thickness of the carbonate layer (coating the anhydrite core) increases. It takes approximately 14 days for the whole replacement of an anhydrite single crystal to occur (crystal volume ≈ 6 mm3).

Linear increase in thickness of the transformed layer as a function of time. Thickness values correspond to the average of a minimum of 20 measurements on 3 sections of partially replaced anhydrite crystals at each time step. The error bars corresponds to the error of measurement.
Discussion
The interaction between carbonate-bearing solution with anhydrite leads to the generation of pseudomorphs consisting of aggregates of calcite crystals. This interaction shows the typical characteristics of a mineral replacement process (Putnis 2002; Putnis and Putnis 2007). Our data reveal that the reaction progresses via the dissolution of the primary phase (anhydrite) and the growth of CaCO3 at the anhydrite-solution interface. The reaction involves the formation of a sharp, inward moving, reaction front separating the primary and secondary phases. Although anhydrite carbonation shares major characteristics with gypsum carbonation (Fernández-Díaz et al. 2009), it also shows specific differences regarding (1) the reaction pathways, (2) the textural characteristics of the calcite crystal aggregates forming the pseudomorphs after the specific calcium sulfate mineral, and (3) the reaction kinetics. The factors responsible for these differences between anhydrite and gypsum carbonation are discussed below.
Reaction pathways in anhydrite carbonation vs. gypsum carbonation
The interaction between anhydrite (100) surface and a 0.5 M Na2CO3 aqueous solution involves the formation of the CaCO3 polymorph calcite since the very early stages of the reaction, with vaterite appearing only as a very minor phase that readily transforms into calcite after a few hours. The vaterite to calcite transformation is well known to proceed via dissolution and reprecipitation specially in the presence of sulfate (Fernández-Díaz et al. 2010; Rodriguez-Blanco et al. 2011; Bots et al. 2012). This relatively simple reaction pathway contrast with the more complex sequence of dissolution-crystallization reactions observed during the carbonation of gypsum under identical experimental conditions (Fernández-Díaz et al. 2009). In the case of gypsum carbonation the reaction started with the formation of a thin layer of amorphous calcium carbonate (ACC) within the first minutes of the replacement process. This early ACC layer readily transformed to a mixture of vaterite and calcite. Furthermore, both crystalline carbonate polymorphs were still present after the carbonation reaction had finished, although the proportion of vaterite continued to decrease as a result of its solvent mediated transformation into calcite and after 72 h of reaction only calcite was detected.
Two main factors can explain the different reaction pathways observed in the carbonation of both calcium sulfate phases. The first factor and, probably, the most important one is the existence or absence of epitactic relationships between the calcite overgrowth and the calcium sulfate substrate. Whereas the overgrowth clearly is epitactic on anhydrite (100) surface (Figs. 2 and 3), no evident epitactic relationship were observed when calcite crystals initially form on gypsum (010) surfaces (Fernandez-Diaz et al. 2009). Indeed, during the carbonation of gypsum calcite crystals first nucleate randomly oriented within the thin ACC layer that carpeted the initial gypsum (010) surface after the first minutes of reaction. Only later did the calcite crystals become oriented with their 𝒸 axis perpendicular to the retreating (010) gypsum surface as their growth progressed. The formation of metastable phases like ACC and vaterite at the early stages of the interaction in the case of gypsum carbonation was the consequence of the predominance of kinetic factors over thermodynamics. This predominance is associated with the energy barrier that needs to be overcome for the heterogeneous nucleation of calcite on the surface of a calcium sulfate mineral to occur. This energy barrier significantly decreases when heterogeneous nucleation has an epitactic character, as it is the case for calcite forming on anhydrite (100) surface (Chernov 1984). The difference in complexity of the reaction pathways operating during the interaction between a carbonate-bearing aqueous solution and anhydrite or gypsum highlights the importance of crystallographic relationships in defining mineralogical evolutions during mineral replacement processes. A significant influence of crystallographic relationships in the development of mineral replacement processes has been found in several systems. For example, Xia et al. (2009a) showed that the similarity of the crystal structures of pentlandite, (Fe, Ni)9S8, and violarite, (NiFe)3S4, which facilitates the epitactic nucleation, promotes the replacement of petlandite by violarite under conditions at which other sulfides ([e.g., vaesite (NiS2) and polydymite (Ni3S4)] are thermodynamically more stable than violarite. Similarly, Qian et al. (2011) concluded that the replacement of pyrrhotite by marcasite instead of pyrite under low-temperature hydrothermal conditions is facilitated by the higher crystallographic similarity between the S lattices in pyrrhotite and marcasite structures.
The second factor that might influence the different reaction pathways observed during the carbonation reaction is the differences in dissolution kinetics of gypsum vs. anhydrite. Bulk experiments revealed that anhydrite dissolves around two orders of magnitude slower than gypsum (Jeschke et al. 2001; Jeschke and Draybrodt 2002). Similarly, macroscopic dissolution measurements conducted on anhydrite (010) and gypsum (010) surfaces yielded dissolution rate constants (k) of 2.6 and 46 ± 6 µmol m−2s−1, respectively (Pachon-Rodriguez and Colombani 2013). Based on atomic force microscopic (AFM) studies much faster dissolution kinetics for gypsum than for anhydrite were also confirmed (Bosbach et al. 1995; Shindo et al. 1996). Consequently, supersaturation of the aqueous solution at the interface with respect to CaCO3 phases increases much more rapidly during interaction with gypsum (010) surfaces than with anhydrite (100) surfaces, thereby explaining the formation of metastable ACC as well as higher amounts of vaterite during the early stages of the carbonation of gypsum compared to the total lack of ACC and only minor vaterite for anhydrite carbonation.
The influence of the different dissolution kinetics for each face could also explain the slightly different mineralogical pathways observed on anhydrite (010) and (001) surfaces in comparison to observed on anhydrite (100) surface. While only calcite is detected on (100) anhydrite surface by GXRD at any reaction step, SEM observations evidence the formation of small amounts of vaterite together to calcite at early stages of the interaction on both (010) and (001) surfaces, being these amounts larger in the later. AFM studies by Shindo et al. (2010) showed that anhydrite (001) dissolves at much faster rate than (100) and (010), which are similarly reactive. This means that Ca2+ ions are released to the solution at a faster rate from (001), leading to larger departures from equilibrium in the layer of aqueous solution close to this surface. This can explain both the formation of higher percentages of vaterite and its preservation during longer times as a main component of the carbonate layer on (001) anhydrite cleavage surfaces.
Textural characteristics of calcite pseudomorphs after anhydrite vs. calcite pseudomorphs after gypsum
Calcite pseudomorphs after gypsum are characterized by a thin, fine-grained rim (15–20 µm) of randomly oriented calcite crystals and a core of parallel columns of calcite crystals in a stockade-like arrangement (Fernández-Díaz et al. 2009). This textural feature is the consequence of an initial nucleation of randomly oriented calcite crystals on gypsum (010). Due to the faster calcite growth along the [001] direction, differently oriented crystals compete for space as they grow. Only those calcite crystals whose c-axis is perpendicular to the original gypsum surface are able to grow without interfering with other crystals. As a result, the progress of the replacement leads to the development of parallel column-like calcite crystals elongated along [001], which grow perpendicular to the retreating gypsum surface.
Stockade-like textures are absent in calcite pseudomorphs after anhydrite. As explained above, calcite nucleation on anhydrite cleavage surfaces is epitactic. In all the cases, the first calcite crystals to form have one of their (104) surfaces laying on the corresponding anhydrite cleavage surface. As all these calcite crystals are equally oriented on anhydrite surface, competitive growth between them does not occur. On the contrary, the existence of epitactic relationships promotes their growth parallel to the anhydrite surface (Sunagawa 1987), which leads to their coalescence and the formation of a compact calcite layer that shields the anhydrite.
Further textural differences between gypsum and anhydrite result from the very different change in molar volume involved in the carbonation of these two calcium sulfate phases. Both gypsum and anhydrite have larger molar volumes than calcite. This means that in both cases carbonation is accompanied by a negative volume change. Consequently for a pseudomorphic process this volume change needs to be balanced by the generation of porosity. This is indeed the case in the replacement of anhydrite by calcite, which involves a volume change of ∼−9.2 cm3/mol; ∼20% (Smyth and McCormick 1995). The porosity generated to compensate this volume change is confined within the transformed layer and shows a basically homogeneous distribution during the early stages of the carbonation process. The difference between the molar volumes of gypsum and calcite is significantly larger (∼−37.6 cm3/mol; >50%) (Smyth and McCormick 1995), which determines that the volume change associated to the carbonation reaction can only be partially compensated by the generation of porosity. Part of this volume change is compensated by the formation of a gap between the replaced layer and the retreating surface of gypsum. The size of this gap increases as the transformation advances. The formation of similar gaps at the interface between primary and secondary phases has been observed during replacement processes, which either involve large volume changes, i.e., replacement of pyrochlore as a result of hydrothermal alteration (Pöml et al. 2007), or are kinetically limited by the precipitation of the secondary phase, i.e., pentlandite, (Fe, Ni)9S8, by violarite (NiFe)3S4 (Xia et al. 2009b). The development of such a gap during the carbonation of gypsum determines the loss of adhesion between the retreating gypsum surface and the replaced layer and eventually leads to the formation of a hollow core within calcite pseudomorphs after gypsum. This, together to the larger amount of porosity formed, confers these pseudomorphs with a much more fragile nature than calcite pseudomorphs formed after anhydrite. In nature the transformation of anhydrite to calcite generates reduction of volume that may cause the development of porosity or deformation in the sediment layers. For example: the nodules of sulfur of the Messinian of Cyprus (Rouchy et al. 1994), and the sulfate evaporates in the Middle Miocene (Pierre and Rouchy 1988) in the Gulf of Suez are associated with abnormally deformed secondary carbonates formation associated to sulfate evaporates in the Middle Miocene (Pierre and Rouchy 1988).
Reaction kinetics of anhydrite carbonation vs. gypsum carbonation
The carbonation of anhydrite occurs at much lower rate than the carbonation of gypsum. For example, a gypsum single crystal with volume ≈ 6 mm3 is completely replaced by CaCO3 after being in contact with a 0.5 M Na2CO3 aqueous solution during 1 day (Fernández-Díaz et al. 2009). In contrast, the whole replacement of an anhydrite single crystal of similar volume takes approximately 14 days. This much slower kinetics of anhydrite carbonation is a consequence of two factors. The first factor is the different dissolution kinetics of gypsum and anhydrite. Since the solution layer closer to the calcium sulfate mineral surface becomes rapidly supersaturated with respect to any CaCO3 polymorphs, CaCO3 precipitation occurs immediately. Therefore, dissolution of the calcium sulfate mineral is the rate determining step of the carbonation process. Consequently, the much slower dissolution of anhydrite in comparison to gypsum can explain the much slower anhydrite carbonation. The second factor that can contribute to make anhydrite carbonation a slower process is the significantly smaller volume of porosity that is generated during the former reaction. For the carbonation process to proceed, a permanent communication between the interface where the reaction occurs and the bulk solution is required. This communication through the formed porosity guarantees continuous supply of
It is interesting that, although anhydrite carbonation occurs at a much slower rate than that of gypsum, its rate remains constant until the process is complete and anhydrite single crystals are fully transformed into aggregates of small calcite crystals. On the contrary, gypsum carbonation rate is very high during the early stages of the process but it significantly slows down at later stages. Fernández-Díaz et al. 2009 explained this change in the gypsum carbonation kinetics as due to three main causes: (1) the formation of a thick layer coating the gypsum surface that prevent percolation of the fluid to the interface, (2) the depletion of
Implications for calcium sufate carbonation processes
The experimental results show that the carbonation of anhydrite involves the early nucleation of epitactic calcite on the three main anhydrite cleavage surfaces. Calcite can be accompanied by small amounts of vaterite at early stages of the carbonation processes in two of these surfaces, defining two different surface-related reaction pathways: anhydrite → calcite and anhydrite → calcite + vaterite → calcite. We interpret these reaction pathways as reflecting the differences in supersaturation with respect to CaCO3 phases at the fluid-anhydrite interface that result from the different dissolution rate of anhydrite cleavage surfaces. We interpret these pathways during anhydrite carbonation as resulting from two factors: (1) The two orders of magnitude slower dissolution kinetics of anhydrite in comparison to gypsum, which determines a much slower supersaturation rate for CaCO3 phases at the fluid-anhydrite interface. (2) The existence of epitatic relationships between calcite and all three main anhydrite cleavage surfaces. Anhydrite carbonation reaction involves a negative volume change, which is compensated by the generation of microporosity. For the reaction times considered in this work, this microporosity is homogeneously distributed within the carbonate layer and a sharp contact exists between the carbonate layer and the anhydrite core. This porosity provides a continuous communication between the carbonate-bearing fluid and the calcite-anhydrite interface, thereby guaranteeing the progress of anhydrite carbonation at a constant rate. Anhydrite carbonation progresses at a much slower rate than the carbonation of gypsum. The different carbonation kinetics of these two calcium sulfate phases can be explain as arising from (1) the slower dissolution kinetics of anhydrite compared to gypsum and (2) the much higher volume change and, thus, microporosity generated in gypsum carbonation.
Open access: Article available to all readers online. This is CC-BY.
Acknowledgments
This study was supported by the MINECO (Spain) under projects CGL2013-47988-C2-1-P and CGL2016-77138-C2-1-P, the Marie Curie EU-FP6 Mineral Nucleation and Growth Kinetics (MIN-GRO) Research and Training Network under contract MRTNCT-2006-035488, and by the UK Engineering and Physical Research Council through grant number EP/P015514/1.
References cited
Alonso-Zarza, A.M., Sánchez Moya, Y., Bustillo Revuelta, M.A., Sopeña, A., and Delgado Huertas, A. (2002) Silicification and dolomitization of anhydrite nodules in argillaceous terrestrial deposits: an example of meteoric-dominated diagenesis from the Triassic of central Spain. Sedimentology, 49, 303–317.10.1046/j.1365-3091.2002.00442.xSuche in Google Scholar
Anthony, J.W., Bideaux, R.A., Bladh, K.W., and Nichols, M.C. (2003) Handbook of Mineralogy, vol. V. Borates, Carbonates, Sulfates. Mineral Data Publishing, Tucson, Arizona, Mineralogical Society of America, Chantilly, Virginia, http://www.handbookofmineralogy.org/.Suche in Google Scholar
Aquilano, D., Rubbo, M., Catti, M., Pavese, A., and Ugliengo, P. (1992) Theoretical equilibrium and growth morphology of anhydrite (CaSO4) crystals. Journal of Crystal Growth, 125, 519–532.10.1016/0022-0248(92)90292-QSuche in Google Scholar
Astilleros, J.M., Pina, C.M., Fernández-Díaz, L., Prieto, M., and Putnis, A. (2006) Nanoscale phenomena during the growth of solid solutions on calcite {1014} surfaces. Chemical Geology, 225, 322–335.10.1016/j.chemgeo.2005.08.025Suche in Google Scholar
Azam, S. (2007) Study on the geological and engineering aspects of anhydrite/gypsum transition in the Arabian Gulf coastal deposits. Bulletin of Engineering Geology and the Environment, 66, 177–185.10.1007/s10064-006-0053-2Suche in Google Scholar
Azdarpour, A., Asadullah, M., Mohammadian, E., Junin, R., Hamidi, H., Manan, M., and Daud, A.R.M. (2015) Mineral carbonation of red gypsum via pH-swing process: Effect of CO2 pressure on the efficiency and products characteristics. Chemical Engineering Journal, 264, 425–436.10.1016/j.cej.2014.11.125Suche in Google Scholar
Barna, P.B., and Adamik, M. (1998) Fundamental structure forming phenomena of polycrystalline films and the structure zone models. Thin Solid Films, 317, 27–33.10.1016/S0040-6090(97)00503-8Suche in Google Scholar
Booth, J., Hong, Qi, Compton, R.G., Prout, K., and Payne, R.M. (1997) Gypsum overgrowths passivate calcite to acid attack. Journal of Colloid and Interface Science, 192, 207–214.10.1006/jcis.1997.4978Suche in Google Scholar PubMed
Bosbach, D., Jordan, G., and Rammensee, W. (1995) Crystal growth and dissolution kinetics of gypsum and fluorite; an in situ scanning force microscopy study. European Journal of Mineralogy, 7, 267–276.10.1127/ejm/7/2/0267Suche in Google Scholar
Bots, P., Benning, L.G., Rodriguez-Blanco, J-D., Roncal-Herrero, T., and Shaw, S. (2012) Mechanistic Insight into the crystallization of Amorphous Calcium Carbonate (ACC). Crystal Growth and Design, 12, 3806–3841.10.1021/cg300676bSuche in Google Scholar
Chernov, A.A. (1984) Modern Crystallography III. Crystal Growth. Springer.10.1007/978-3-642-81835-6Suche in Google Scholar
Dejonghe, L., Demaiffe, D., and Weis, D. (1998) Strontium isotope geochemistry of anhydrites and calcite pseudomorphs after anhydrite from Paleozoic formations in Belgium. Chemical Geology, 144, 63–71.10.1016/S0009-2541(97)00110-1Suche in Google Scholar
Fernández-Díaz, L., Pina, C.M., Astilleros, J.M., and Sánchez-Pastor, N. (2009) The carbonatation of gypsum: Pathways and pseudomorph formation. American Mineralogist, 94, 1223–1234.10.2138/am.2009.3194Suche in Google Scholar
Fernández-Díaz, L., Fernández-González, A., and Prieto, M. (2010) The role of sulphate groups in the polymorphism of CaCO3. Geochimica et Cosmochimica Acta, 74, 6064–6076.10.1016/j.gca.2010.08.010Suche in Google Scholar
Flörke, W., and Flörke, O.W. (1961) Vateritbildung aus Gips in Sodalösung. Neues Jahrbuch für Mineralogie Monatschaften, 179–181.Suche in Google Scholar
Freyer, D., and Voigt, W. (2003) Crystallization and phase stability of CaSO4 and CaSO4–Based Salts. Monatshefte für Chemie, 134, 693–719.10.1007/s00706-003-0590-3Suche in Google Scholar
Hanor, J.S. (2004) A model for the origin of large carbonate-and evaporite-hosted celestine (SrSO4) deposits. Journal of Sedimentary Research, 74, 168–175.10.1306/092203740168Suche in Google Scholar
Hu, G., Dam-Johansen, K., and Wedel, S. (2008) Oriented nucleation and growth of anhydrite during direct sulfation of limestone. Crystal Growth & Design, 8, 1081–1085.10.1021/cg0704403Suche in Google Scholar
Jeschke, A.A., and Dreybrodt, W. (2002) Pitfalls in the determination of empirical dissolution rate equations of minerals from experimental data and a way out: an iterative procedure to find valid rate equations, applied to Ca-carbonates and -sulphates. Chemical Geology, 192, 183–194.10.1016/S0009-2541(02)00135-3Suche in Google Scholar
Jeschke, A.A., Vosbeck, K., and Dreybrodt, W. (2001) Surface controlled dissolution rates of gypsum in aqueous solutions exhibit nonlinear dissolution rates. Geochimica et Cosmochimica Acta, 65, 27–34.10.1016/S0016-7037(00)00510-XSuche in Google Scholar
Kendall, A.C. (2001) Late diagenetic calcitization of anhydrite from the Mississippian of Saskatchewan, western Canada. Sedimentology, 48, 29–55.10.1046/j.1365-3091.2001.00350.xSuche in Google Scholar
Offedu, F.G., Cama, J., Soler, J.M., and Putnis, C.V. (2014) Direct nanoscale observations of the coupled dissolution of calcite and dolomite and the precipitation of gypsum. Beilstein Journal of Nanotechnology, 4, 1245–1253.10.3762/bjnano.5.138Suche in Google Scholar PubMed PubMed Central
Pachon-Rodriguez, E.A., and Colombani, J. (2013) Pure dissolution kinetics of anhydrite and gypsum in inhibiting aqueous salts solutions. American Institute of Chemical Engineers Journal, 59, 1622–1626.10.1002/aic.13922Suche in Google Scholar
Pérez-Garrido, C., Fernández-Díaz, L., Pina, C.M., and Prieto, M. (2007) In situ AFM observations of the interaction between calcite {104} surfaces and Cd-bearing aqueous solutions, Surface Science, 601, 5499–5509.10.1016/j.susc.2007.09.021Suche in Google Scholar
Pérez-Moreno, S.M., Gázquez, M.J., and Bolívar, J.P. (2015) CO2 sequestration by indirect carbonation of artificial gypsum generated in the manufacture of titanium dioxide pigments. Chemical Engineering Journal, 262, 737–746.10.1016/j.cej.2014.10.023Suche in Google Scholar
Pierre, C., and Rouchy, J.M. (1988) Carbonate replacements after sulphate evaporites in the Middle Miocene of Egypt. Journal of Sedimentary Petrology, 58, 446–456.Suche in Google Scholar
Pollok, K., Putnis, C.V., and Putnis, A. (2011) Mineral replacement reactions in solid solution-aqueous solution systems: Volume changes, reactions paths and end-points using the example of model salt systems. American Journal of Science, 311, 211–223.10.2475/03.2011.02Suche in Google Scholar
Pöml, P., Menneken, M., Stephan, T., Niedermeie, D.R.D., Geisler, T., and Putnis, A. (2007) Mechanism of hydrothermal alteration of natural self-irradiated and synthetic crystalline titanate-based pyrochlore. Geochimica et Cosmochimica Acta, 71, 3311–3322.10.1016/j.gca.2007.03.031Suche in Google Scholar
Prieto, M., Cubillas, P., and Fernández-González, A. (2003) Uptake of dissolved Cd by biogenic and abiogenic aragonite: a comparison with sorption onto calcite. Geochimica et Cosmochimica Acta, 67, 3859–3869.10.1016/S0016-7037(03)00309-0Suche in Google Scholar
Prieto, M., Astilleros, J.M., and Fernández-Díaz, L. (2013) Environmental remediation by crystallization of solid solutions. Elements, 9, 195–201.10.2113/gselements.9.3.195Suche in Google Scholar
Putnis, A. (2002) Mineral replacement reactions: from macroscopic observations to microscopic mechanism. Mineralogical Magazine, 66, 689–708.10.1180/0026461026650056Suche in Google Scholar
Putnis, A. (2009) Mineral replacement reactions. In E.H. Oelkers and J. Schott, Eds., Thermodynamics and Kinetics of Water-Rock Interactions, 30, p. 87–124. Reviews in Mineralogy and Geochemistry, Mineralogical Society of America, Chantilly, Virginia.10.1515/9781501508462-005Suche in Google Scholar
Putnis, A., and Putnis, C.V. (2007) The mechanism of reequilibration of solids in the presence of a fluid phase. Journal of Solid State Chemistry, 180, 1783–1786.10.1016/j.jssc.2007.03.023Suche in Google Scholar
Putnis, C.V., Austrheim, H., and Putnis, A. (2007) A mechanism of fluid transport through minerals. Geochimica et Cosmochimica Acta, 71, A814.Suche in Google Scholar
Qian, G., Xia, F., Brugger, J., Skinner, W.M., Bel, J., Chen, G., and Pring, A. (2011) Replacement of pyrrhotite by pyrite and marcasite under hydrothermal conditions up to 220 °C: An experimental study of reaction textures and mechanisms. American Mineralogist, 96, 1878–1898.10.2138/am.2011.3691Suche in Google Scholar
Rasband, W.S (2016) ImageJ. National Institutes of Health, Bethesda, Maryland, https://imagej.nih.gov/ij/, 1997–2016.Suche in Google Scholar
Redfern, S.E., and Parker, S.C. (1998) Atomistic simulation of the effects of calcium and strontium defects on the surface structure and stability of BaSO4. Journal of the Chemical Society, Faraday Transactions, 94, 1947–1952.10.1039/a801533cSuche in Google Scholar
Rodríguez, J.D., Jiménez, A., Prieto, M., Torre, L., and García-Granda, S. (2008) Interaction of gypsum with As(V)-bearing aqueous solutions: Surface precipitation of guerinite, sainfeldite, and Ca2NaH(AsO4)2·6H2O, a synthetic arsenate. American Mineralogist, 93, 928–939.10.2138/am.2008.2750Suche in Google Scholar
Rodríguez-Blanco, J.D., Jiménez, A., and Prieto, M. (2007) Oriented overgrowth of pharmacolite (CaHAsO4·2H2O) on gypsum (CaSO4·2H2O). Crystal Growth & Design, 7, 2756–2763.10.1021/cg070222+Suche in Google Scholar
Rodriguez-Blanco, J-D., Shaw, S., and Benning, L.G. (2011) The kinetics and mechanisms of amorphous calcium carbonate (ACC) crystallization to calcite, via vaterite. Nanoscale, 2011, 265–271.10.1039/C0NR00589DSuche in Google Scholar
Rouchy, J.M., Bernet-Rollande, M.C., and Maurin, A.F. (1994) Descriptive petrography of evaporites: application in the field, subsurface, and laboratory. In M. Majithia, Ed., Evaporitic Sequences in the Petroleum Exploration, p. 70–123. Geological Methods, Technip edition.Suche in Google Scholar
Rouchy, J.M., Taberner, C., Blanc-Valleron, M.M., Sprovieri, R., Russell, M., Pierre, C., Di Stefano, E., Pueyo, J.J., Caruso, A., Dinarés, J., and others. (1998) Sedimentary and diagenetic markers of the restriction in a marine basin: the Lorca basin (SE Spain) during the Messinian. Sedimentary Geology, 121, 23–55.10.1016/S0037-0738(98)00071-2Suche in Google Scholar
Ruiz-Agudo, E., Putnis, C.V., and Putnis, A. (2014) Coupled dissolution and precipitation at mineral–fluid interfaces. Chemical Geology, 383, 132–146.10.1016/j.chemgeo.2014.06.007Suche in Google Scholar
Ruiz-Agudo, E., Putnis, C.V., Hövelmann, J., Àlvarez-Lloret, P., Ibañez-Velasco, A., and Putnis, A. (2015) Experimental study of the replacement of calcite by calcium sulphates. Geochimica et Cosmochimica Acta, 156, 75–93.10.1016/j.gca.2015.02.012Suche in Google Scholar
Ruiz-Agudo, E., Àlvarez-Lloret, P., Ibáñez-Velasco, A., and Ortega-Huertas, M. (2016) Crystallographic control in the replacement of calcite by calcium sulphates. Crystal Growth and Design, 16, 4950–4959.10.1021/acs.cgd.6b00522Suche in Google Scholar
Sanz-Rubio, E., Sánchez-Moral, S., Cañaveras, J.C., Calvo, J.P., and Rouchy, J.M. (2001) Calcitization of Mg-Ca carbonate and Ca sulphate deposits in a continental Tertiary basin (Calatayud Basin, NE Spain). Sedimentary Geolology, 140, 123–142.10.1016/S0037-0738(00)00175-5Suche in Google Scholar
Shindo, H., Seo, A., Itasaka, M., Odaki, T., and Tanaka, K. (1996) Stability of surface atomic structures of ionic crystals studied by atomic microscopy observation of various faces of CaSO4 crystal in solutions. Journal of Vacuum Science and Technology B, 14, 1365–1368.10.1116/1.589099Suche in Google Scholar
Shindo, H., Igarashi, T., Karino, W., Seo, A., Yamanobe-Hada, M., and Haga, M. (2010) Stabilities of crystal faces of anhydrite (CaSO4) compared by AFM observation of facet formation processes in aqueous solutions. Journal of Crystal Growth, 312, 573–579.10.1016/j.jcrysgro.2009.12.001Suche in Google Scholar
Smyth, J.R.S., and McCormick, T.C. (1995) Crystallographic Data for Minerals. In J. Thomas, Ed., Mineral Physics & Crystallography: A Handbook of Physic Contrasts, p. 1–17. American Geophysical Union, Washington D.C.10.1029/RF002p0001Suche in Google Scholar
Snyder, R.L. (1992) The use of reference intensity ratios in X-ray quantitative analysis. Powder Diffraction, 7, 186–193.10.1017/S0885715600018686Suche in Google Scholar
Stafford, K.W., Ulmer-Scholle, D., and Rosales-Lagarde, L. (2008) Hypogene calcitization: Evaporite diagenesis in the western Delaware Basin. Carbonates and Evaporites, 23, 89–103.10.1007/BF03176155Suche in Google Scholar
Sunagawa, I. (1987) Morphology of minerals. In I. Sunagawa, Ed., Morphology of Crystals, p. 509–588. Terra Scientific, Tokyo.Suche in Google Scholar
Xia, F., Brugger, J., Ngothai, Y., O’Neill, B., Chen, G., and Pring, A. (2009a) Three-dimensional ordered arrays of zeolite nanocrystals with uniform size and orientation by a pseudomorphic coupled dissolution-reprecipitation replacement route. Crystal Growth & Design, 9, 4902–4906.10.1021/cg900691aSuche in Google Scholar
Xia, F., Brugger, J., Chen, G., Ngothai, Y., O’Neill, B., Putnis, A., and Pring, A. (2009b) Mechanism and kinetics of pseudomorphic mineral replacement reactions: A case study of the replacement of pentlandite by violarite. Geochimica et Cosmochimica Acta, 73, 1945–1969.10.1016/j.gca.2009.01.007Suche in Google Scholar
© 2017 by Walter de Gruyter Berlin/Boston
This work is licensed under the Creative Commons Attribution-NonCommercial-NoDerivatives 3.0 License.
Artikel in diesem Heft
- Actinides in geology, energy, and the environment
- Thermodynamic investigation of uranyl vanadate minerals: Implications for structural stability
- Actinides in geology, energy, and the environment
- Uranium-bearing opals: Products of U-mobilization, diffusion, and transformation processes
- Special Collection: Olivine
- Quantifying and correcting the effects of anisotropy in XANES measurements of chromium valence in olivine: Implications for a new olivine oxybarometer
- Special collection: Dynamics of magmatic processes
- High-resolution geochemistry of volcanic ash highlights complex magma dynamics during the Eyjafjallajökull 2010 eruption
- Special collection: Water in nominally hydrous and anhydrous minerals
- Evidence for post-depositional diffusional loss of hydrogen in quartz phenocryst fragments within ignimbrites
- Special collection: Martian rocks and minerals: Perspectives from rovers, orbiters, and meteorites
- Visible to near-infrared MSL/Mastcam multispectral imaging: Initial results from select high-interest science targets within Gale Crater, Mars
- Multi-stage formation of REE minerals in the Palabora Carbonatite Complex, South Africa
- Spin orientation in solid solution hematite-ilmenite
- Constraints on aluminum and scandium substitution mechanisms in forsterite, periclase, and larnite: High-resolution NMR
- Shock-induced P-T conditions and formation mechanism of akimotoite-pyroxene glass assemblages in the Grove Mountains (GRV) 052082 (L6) meteorite
- The spin state of Fe3+ in lower mantle bridgmanite
- Reaction pathways and textural aspects of the replacement of anhydrite by calcite at 25 °C
- Majorite-olivine–high-Ca pyroxene assemblage in the shock-melt veins of Pervomaisky L6 chondrite
- Cu and Fe diffusion in rhyolitic melts during chalcocite “dissolution”: Implications for porphyry ore deposits and tektites
- Field-based accounting of CO2 sequestration in ultramafic mine wastes using portable X-ray diffraction
- NanoSIMS study of seismically deformed zircon: Evidence of Y, Yb, Ce, and P redistribution and resetting of radiogenic Pb
- Study on structure variations of incommensurately modulated labradorite feldspars with different cooling histories
- Carbocernaite from Bear Lodge, Wyoming: Crystal chemistry, paragenesis, and rare-earth fractionation on a microscale
- Magma mush chemistry at subduction zones, revealed by new melt major element inversion from calcic amphiboles
Artikel in diesem Heft
- Actinides in geology, energy, and the environment
- Thermodynamic investigation of uranyl vanadate minerals: Implications for structural stability
- Actinides in geology, energy, and the environment
- Uranium-bearing opals: Products of U-mobilization, diffusion, and transformation processes
- Special Collection: Olivine
- Quantifying and correcting the effects of anisotropy in XANES measurements of chromium valence in olivine: Implications for a new olivine oxybarometer
- Special collection: Dynamics of magmatic processes
- High-resolution geochemistry of volcanic ash highlights complex magma dynamics during the Eyjafjallajökull 2010 eruption
- Special collection: Water in nominally hydrous and anhydrous minerals
- Evidence for post-depositional diffusional loss of hydrogen in quartz phenocryst fragments within ignimbrites
- Special collection: Martian rocks and minerals: Perspectives from rovers, orbiters, and meteorites
- Visible to near-infrared MSL/Mastcam multispectral imaging: Initial results from select high-interest science targets within Gale Crater, Mars
- Multi-stage formation of REE minerals in the Palabora Carbonatite Complex, South Africa
- Spin orientation in solid solution hematite-ilmenite
- Constraints on aluminum and scandium substitution mechanisms in forsterite, periclase, and larnite: High-resolution NMR
- Shock-induced P-T conditions and formation mechanism of akimotoite-pyroxene glass assemblages in the Grove Mountains (GRV) 052082 (L6) meteorite
- The spin state of Fe3+ in lower mantle bridgmanite
- Reaction pathways and textural aspects of the replacement of anhydrite by calcite at 25 °C
- Majorite-olivine–high-Ca pyroxene assemblage in the shock-melt veins of Pervomaisky L6 chondrite
- Cu and Fe diffusion in rhyolitic melts during chalcocite “dissolution”: Implications for porphyry ore deposits and tektites
- Field-based accounting of CO2 sequestration in ultramafic mine wastes using portable X-ray diffraction
- NanoSIMS study of seismically deformed zircon: Evidence of Y, Yb, Ce, and P redistribution and resetting of radiogenic Pb
- Study on structure variations of incommensurately modulated labradorite feldspars with different cooling histories
- Carbocernaite from Bear Lodge, Wyoming: Crystal chemistry, paragenesis, and rare-earth fractionation on a microscale
- Magma mush chemistry at subduction zones, revealed by new melt major element inversion from calcic amphiboles