A dihydrochalcone derivative and further steroidal saponins from Sansevieria trifasciata Prain
-


Abstract
Phytochemical investigation of the aerial parts of Sansevieria trifasciata, one of the most common Dracaenaceae plants, has resulted in the isolation of a new dihydrochalcone derivative named trifasciatine C (1), four previously unreported steroidal saponins as two pairs of inseparable regioisomers: trifasciatosides K/L (2/3), M/N (4/5), together with the known 1,2-(dipalmitoyl)-3-O-β-D-galactopyranosylglycerol (6), aconitic acid (7), and 1-methyl aconitic acid (8). Their structures were elucidated mainly by extensive spectroscopic analysis (1D and 2D nuclear magnetic resonance) and high-resolution electronspray ionization-mass spectrometry, as well as chemical methods and comparison of their spectral data with those of related compounds. Compounds 2/3 and 4/5 were evaluated for their antiproliferative activity on Hela cells, and no significant effect was observed.
1 Introduction
Sansevieria trifasciata Prain belongs to the Draceanaceae family which includes more than 60 species distributed in tropical and subtropical dry climate regions throughout the world [1, 2]. In South Africa and tropical America, S. trifasciata has been used in traditional medicine for the treatment of inflammatory conditions and sold as a crude drug in the market to treat victims of snake bites [3]. Previous phytochemical investigation of the EtOAc soluble fraction of the MeOH extract of this plant led to the isolation of two new minor homoisoflavonoids (trifasciatines A and B) [4], while the n-butanol fraction yielded steroidal saponins [5, 6]. In our continuous search for bioactive secondary metabolites from Cameroonian medicinal plants, we undertook extensive phytochemical investigation of the EtOAc soluble fraction of the MeOH extract of this plant. The present paper describes the isolation and structure elucidation of one new dihydrochalcone derivative trivially named trifasciatine C (1) and two pairs of previously unreported steroidal saponins named trifasciatosides K and L (2, 3) and trifasciatosides M and N (4, 5) obtained as inseparable regioisomers.
2 Results and discussion
The EtOAc soluble fraction of the methanol extract of S. trifasciata was subjected to column chromatography on silica gel and Sephadex LH-20 to afford one previously undescribed dihydrochalcone derivative named trifasciatine C (1) and two pairs of new steroidal saponins named trifasciatosides K and L (2, 3) and trifasciatosides M and N (4, 5) obtained as inseparable regioisomers (Figure 1), together with the known 1,2-(dipalmitoyl)-3-O-β-D-galactopyranosylglycerol (6) [7], aconitic acid (7) [8] and 1-methyl aconitic acid (8) [8].
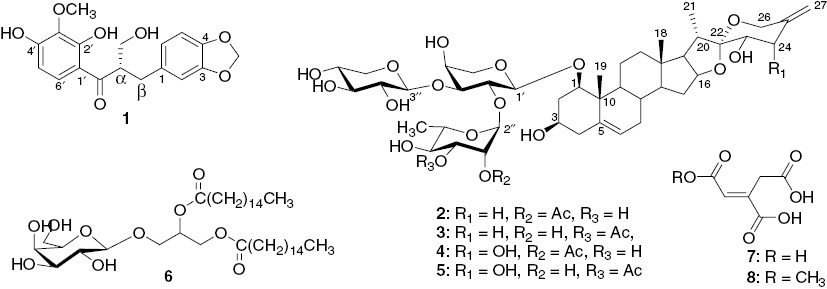
Structures of compounds 1–8 isolated from S. trifasciata.
Trifasciatine C (1) was isolated as a yellowish gum from hexane-EtOAc (1-1). Its high-resolution electronspray ionization-mass spectrometry (HRESIMS) exhibited a pseudomolecular ion peak ([M+Na]+) at m/z 369.0960, corresponding to the molecular formula C18H18O7 (calc. for C18H18O7Na: 369.0945). The IR spectrum of 1 showed characteristic absorption bands for hydroxy (3440 cm−1) and carbonyl (1643 cm−1) groups. In the proton nuclear magnetic resonance (1H NMR) spectrum, the signals at δH 7.49 (d, J=9.2, H-6′) and 6.38 (d, J=9.2, H-5′) (Table 1) suggested a pair of ortho-coupled protons in ring A [4, 9]. The substitution pattern of ring B was shown to be an ABX system with signals at δH 6.66 (d, J=7.9, H-5), 6.69 (d, J=1.4, H-2), and 6.63 (dd, J=7.9, 1.4, H-6). The 1H NMR spectrum also showed a signal integrating for two protons at δH 5.90 suggesting a methylenedioxyl (OCH2O) moiety [9, 10] and a singlet of three protons at δH 3.83 suggesting the presence of a methoxyl group in 1. In addition, the presence of two couples of geminal protons resonating at δH [3.70 (m, CH2O) and 3.90 (m, CH2O)] and [2.83 (dd, J=13.7, 5.3, Ha-β) and 2.91 (dd, J=13.9, 8.1, Hb-β)], as well as an aliphatic proton signal at δH 3.88 (H-α), revealed that compound 1 was related to 3-benzylchroman-4-one type homoisoflavonoids [4, 9, 10]. The 13C NMR spectrum exhibited 17 signals including those of 12 aromatic carbons, three methylenes, one sp3 methine, and one methyl signal. Extensive analysis of 1H-1H correlation spectroscopy (COSY), heteronuclear single quantum coherence (HSQC) and heteronuclear multiple-bond correlation spectroscopy (HMBC) spectra allowed us to assign 1H and 13C resonances (Figure 2). However, the chemical shift of the oxymethylene carbon resonating at δC 62.8 in 1, in comparison with the downfield shift observed for the related carbon in trifasciatine A (δC 71.3) previously isolated from this plant [4], suggested that 1 was an opened C-ring homoisoflavonoid derivative. This was also confirmed by the lack of HMBC correlations between the geminal protons resonating at δH 3.70 (CH2O) and 3.90 (CH2O) and the carbon at δC 158.0 (C-2′) as well as the presence of one additional oxygen atom in 1 compared to trifasciatine A depicted from the mass data. The location of the methylenedioxyl group was confirmed by the HMBC correlations observed between its protons at δH 5.90 with the O-bearing aromatic carbons at δC 148.0 (C-3) and 148.4 (C-4) (Figure 2). Furthermore, the cross peak correlation observed between the methoxy proton signal at δH 3.83 (OMe) and the carbon at δC 136.5 (C-3′) showed that it was linked at C-3′. Biogenetically, compound 1 could have derived from trifasciatine A previously isolated from the same plant due to the enzymatic cleavage of its C-ring since it was shown that etherase enzymes produced by some bacteria are able to break ether bonds [11]. The configuration at C-α was proposed to be S based on the positive sign of its optical rotation by opposition to that of C-3 in trifasciatine A whose configuration was recently determined by some of us using electronic circular dichroism [4]. Consequently, the structure of 1 was elucidated as (αS) α-hydroxymethyl-2′,4′-dihydroxy-3′-methoxy-3,4-methylenedioxydihydrochalcone to which we gave the trivial name trifasciatine C.
1H and 13C NMR data (700 and 175 MHz, respectively, CD3OD) of compound 1.
| Position | δH | δC |
|---|---|---|
| C=O | – | nd |
| α | 3.88 (m) | 50.1 |
| β | 2.83 (dd, J=13.7, 5.3); 2.91 (dd, J=13.7, 8.1) | 34.8 |
| 1 | – | 132.9 |
| 2 | 6.69 (d, J=1.4) | 108.3 |
| 3 | – | 148.0 |
| 4 | – | 148.4 |
| 5 | 6.66 (d, J=7.9) | 107.4 |
| 6 | 6.63 (dd, J=7.9, 1.4) | 121.6 |
| 1′ | – | 114.2 |
| 2′ | – | 158.0 |
| 3′ | – | 136.5 |
| 4′ | – | 161.6 |
| 5′ | 6.38 (d, J=9.2) | 107.6 |
| 6′ | 7.49 (d, J=9.2) | 127.0 |
| –CH2OH | 3.90 (m); 3.70 (m) | 62.8 |
| –OCH2O– | 5.90 (dd, J=4.8, 1.2) | 100.6 |
| –OMe | 3.83 (s) | 59.1 |
nd, Not determined.
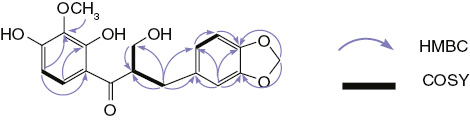
Selected HMBC and 1H-1H COSY correlations for compound 1.
Trifasciatosides K/L (2/3), isolated as a white amorphous powder from MeOH, had the molecular formula C45H68O18, as determined from its HRESIMS which showed the pseudomolecular ion peak at m/z 919.4387 ([M+Na]+) (calcd for C45H68O19Na: 919.4298). The IR spectrum exhibited a broad absorption band due to hydroxyl groups at 3300 cm−1, as well as a strong absorption due to carbonyl groups at 1724 cm−1. It was shown to be a mixture of two monoacetylated spirostanol saponins 2 and 3 from the interpretation of the NMR data. The 1H NMR spectrum showed two tertiary methyl signals at δH 1.12/1.13 (H-18) and 1.09/1.10 (H-19) (each s), two secondary methyl signals at δH 1.27/1.30 (d, J=6.2, H-6″), exomethylene proton signals at δH 4.79 ( H-27a) and 4.83 (H-27b), the olefinic proton signal at δH 5.58 (brd, J=5.1, H-6), and three anomeric proton signals at δH 4.35/4.37 (d, J=7.3, H-1′), 4.40/4.41 (d, J=6.0, H-1″′), and 5.32/5.40 (brs, H-1″). The 13C NMR spectrum exhibited signals characteristic of 1,3,23-trihydroxyspirosta-5,25(27)-diene [5, 6] at δC 83.4/83.6 (C-1), 68.5/68.7 (C-3), 138.1(C-5), 124.5 (C-6), 82.5/82.4 (C-16), 68.7/68.9 (C-23), 143.1 (C-25), and 108.5 (C-27) (Table 2). Assignment of the other proton and carbon signals of the aglycone part was achieved by careful examination of the HSQC, HMBC, and 1H-1H COSY spectra. Evaluation of spin-spin couplings and chemical shifts in the sugar part allowed the identification of one 2,3-disubstituted arabinopyranosyl unit, one terminal rhamnopyranosyl residue, and one xylopyranosyl unit in each compound. In addition, 2/3 afforded arabinose, xylose, and rhamnose on acid hydrolysis. Their absolute configurations were determined by gas chromatography (GC) analysis of the corresponding trimethylsilated L-cysteine adducts [12]. The presence of acetyl groups was indicated by methyl singlets at δH 2.12/2.13 (s, MeCO), whereas in the 13C NMR spectrum, the signals of ester carbonyls at δC 171.3/171.4 (MeCO) and methyl carbons at δC 19.7/20.1 (MeCO) were observed. The 1H-1H COSY spectrum allowed the resonances observed at a lower field at δH 5.14 (dd, J=3.6; 1.6) and 4.94 (dd, J=10.0; 3.2) to be assigned to H-2″ and H-3″ of the two rhamnopyranosyl units, respectively, which suggested that they were acetylated. This was confirmed by HMBC correlations between H-2″ and H-3″ and the acetyl carbonyl carbons at δC 171.3 and 171.4, respectively. The HMBC correlations between the anomeric proton signals at δH 4.40/4.41 (d, J=6.0, H-1″′) and the carbon at δC 84.2/84.6 (C-3′), 5.40 (brs, H-1″) and 74.1 (C-2′), 5.32 (d, J=1.8, H-1″) and 74.3 (C-2′), and finally between the protons at δH 4.37/4.35 (d, J=7.3, H-1′) and the carbons at δC 83.4/83.6 (C-1) proved the sequence of the sugar chain at C-1 to be α-L-rhamnopyranosyl-(1→2)-[β-D-xylopyranosyl-(1→3)]-α-L-arabinopyranosyl in both compounds. Based on the above data, 2/3 was elucidated as a mixture of (23S)-3β,23-dihydroxyspirosta-5,25(27)-dien-1β-yl O-(2-O-acetyl-α-L-rhamnopyranosyl)-(1→2)-O-[β-D-xylopyranosyl-(1→3)]-α-L-arabinopyranoside (Trifasciatoside K) and (23S)-3β,23-dihydroxyspirosta-5,25(27)-dien-1β-yl O-(3-O-acetyl- α-L-rhamnopyranosyl)-(1→2)-O-[β-D-xylopyranosyl-(1→3)]-α-L-arabinopyranoside (Trifasciatoside L).
13C NMR data (175 MHz, CD3OD) of compounds 2–5 (δ in ppm).
| Position | 2 | 3 | 4 | 5 |
|---|---|---|---|---|
| 1 | 83.4 | 83.6 | 83.4 | 83.5 |
| 2 | 35.2 | 35.5 | 35.8 | 36.3 |
| 3 | 68.5 | 68.7 | 68.4 | 68.6 |
| 4 | 42.2 | 42.3 | 42.0 | 42.1 |
| 5 | 138.1 | 138.1 | 138.0 | 138.1 |
| 6 | 124.5 | 124.5 | 124.6 | 124.5 |
| 7 | 32.2 | 32.5 | 31.4 | 31.3 |
| 8 | 33.4 | 33.6 | 32.5 | 32.5 |
| 9 | 50.0 | 50.1 | 49.7 | 49.9 |
| 10 | 42.0 | 42.1 | 42.0 | 42.0 |
| 11 | 23.3 | 23.3 | 23.3 | 23.3 |
| 12 | 40.1 | 40.1 | 40.0 | 40.1 |
| 13 | 40.5 | 40.3 | 40.5 | 41.1 |
| 14 | 56.5 | 56.7 | 56.2 | 56.4 |
| 15 | 32.5 | 32.6 | 32.5 | 31.8 |
| 16 | 82.5 | 82.4 | 83.0 | 82.9 |
| 17 | 61.8 | 62.0 | 61.8 | 62.5 |
| 18 | 15.7 | 15.8 | 15.7 | 15.8 |
| 19 | 15.1 | 15.0 | 16.0 | 16.8 |
| 20 | 35.9 | 36.9 | 35.4 | 35.7 |
| 21 | 13.9 | 13.9 | 13.0 | 13.9 |
| 22 | 110.6 | 110.6 | 111.6 | 112.1 |
| 23 | 68.7 | 68.9 | 69.7 | 69.7 |
| 24 | 38.5 | 38.7 | 74.2 | 74.2 |
| 25 | 143.1 | 143.1 | 144.6 | 144.5 |
| 26 | 63.4 | 63.4 | 60.0 | 59.7 |
| 27 | 108.5 | 108.5 | 112.0 | 112.0 |
| Ara | ||||
| 1′ | 99.5 | 99.6 | 99.6 | 99.5 |
| 2′ | 74.1 | 74.3 | 73.3 | 74.0 |
| 3′ | 84.6 | 84.2 | 84.0 | 84.2 |
| 4′ | 69.3 | 69.6 | 69.2 | 69.5 |
| 5′ | 67.1 | 67.4 | 67.8 | 67.7 |
| Rha | ||||
| 1″ | 97.0 | 100.2 | 97.1 | 100.2 |
| 2″ | 72.5 | 68.6 | 72.5 | 68.7 |
| 3″ | 69.1 | 74.2 | 69.1 | 74.5 |
| 4″ | 73.8 | 69.0 | 73.5 | 69.7 |
| 5″ | 68.4 | 69.7 | 68.0 | 67.6 |
| 6″ | 16.4 | 17.1 | 17.0 | 17.1 |
| Xyl | ||||
| 1″′ | 104.3 | 104.9 | 104.9 | 104.3 |
| 2″′ | 74.4 | 74.6 | 74.2 | 74.1 |
| 3″′ | 76.4 | 76.6 | 76.0 | 76.2 |
| 4″′ | 71.0 | 71.2 | 69.9 | 70.1 |
| 5″′ | 66.9 | 66.5 | 67.0 | 66.4 |
| Ac | 171.3 | 171.4 | 171.3 | 171.4 |
| 19.7 | 20.1 | 19.7 | 19.7 |
The positive HRESIMS of compounds 4/5 exhibited a pseudomolecular ion peak at m/z 935.4335 ([M+Na]+) corresponding to the molecular formula C45H68O19 with 16 mass units more than 2/3. The 1H NMR spectrum exhibited one signal with regard to H-21 at δH 0.95 (d, J=7), two signals for H-18 at δH 1.12 (s) and 1.11 (s), one signal due to H-19 at δH 0.90 (s) together with the olefinic proton signal at δH 5.55 (brs, H-6), and the exomethylene proton signals H-27 at δH 4.98 (brs) and 5.06 (brs). The 13C NMR spectrum showed signals characteristic of 1,3,23,24-tetrahydroxyspirosta-5,25(27)-diene [5] at δC 83.4/83.6 (C-1), 68.4/68.6 (C-3), 138.0/138.1 (C-5), 124.6/124.5 (C-6), 83.0/82.9 (C-16), 69.7 (C-23), 74.2 (C-24), 144.6/144.5 (C-25), and 112.0 (C-27) (Table 2). GC analysis of chiral derivatives of sugars from acid hydrolysate of 4/5 showed the presence of L-rhamnose, D-xylose, and L-arabinose [12]. Comparison of the 1H and 13C NMR spectra of 4/5 with those of 2/3 revealed that the structures of the rings A–E portion and sugar moiety (including the acetylation patterns) attached at C-1 of the aglycone were identical. However, significant differences were recognized in the signals from the ring F portion (C-22 to C-27) (Table 2). This indicated that the only difference between the above compounds is the presence of a hydroxyl group on ring F in 4/5. The location of this OH group was determined by using a combination of 1H, 13C NMR, 1H-1H COSY, HSQC, and HMBC spectra. HMBC correlations were depicted between the proton signal at δH 3.76 (d, J=3.2, H-23) and the carbon signal at δC 111.6 (C-22) and between the proton signal at δH 4.22 (d, J=3.2, H-24) and the carbons at δC 144.6 (C-25) and 112.0 (C-27), respectively. The structures of 4/5 were elucidated as (23S,24S)-3β,23,24-trihydroxyspirosta-5,25(27)-dien-1β-yl O-(2-O-acetyl-α-L-rhamnopyranosyl)-(1→2)-O-[β-D-xylopyranosyl-(1→3)]-α-L-arabinopyranoside and (23S,24S)-3β,23,24-trihydroxyspirosta-5,25(27)-dien-1β-yl O-(3-O-acetyl-α-L-rhamnopyranosyl)-(1→2)-O-[β-D-xylopyranosyl-(1→3)]-α-L-arabinopyranoside to which we gave the trivial named trifasciatosides M and N, respectively.
Since some steroidal saponins were shown to exhibit potent cytotoxicity [5, 13, 14], compounds 2/3 and 4/5 were evaluated for their antiproliferative activity on Hela cells, and no considerable effect was observed.
In conclusion, phytochemical investigation of the EtOAc extract of S. trifasciata has resulted in the isolation of a new dihydrochalcone derivative named trifasciatine C and four previously unreported monoacetylated steroidal saponins obtained as two pairs of inseparable regioisomers: trifasciatosides K/L (2/3) and M/N (4/5). It has been established that acetyl saponins are typical unstable compounds extensively distributed in many plant species, and this structural stability depends on different solvents and separation materials used for their purification [15, 16]. Therefore, it could be assumed that compound 3 (or 5) could have resulted from compound 2 (or 4) and vice versa from the migration of the acetyl group from 2 to 3 positions (vice versa) of the rhamnose moiety as shown by Zeng et al. [16], thus resulting to the two inseparable mixtures 2/3 and 4/5. Although the isolated saponins exhibited no significant antiproliferative activity, the present work indicated that S. trifasciata represents a potential source of new steroidal saponins and phenolic compounds. Given the biological importance and the various uses of this medicinal plant, the isolated compounds will be screened for other biological activities in our future investigation.
3 Experimental section
3.1 General experimental procedures
Optical rotations were measured on a Perkin-Elmer 241 MC polarimeter (Perkin-Elmer, Waltham, MA, USA). IR spectra were recorded on a Shimadzu infrared spectrometer (Shimadzu, Kyoto, Japan). UV spectra were registered on a Photolab 6600 UV-VIS spectrophotometer (Orbit Technologies Pvt. Ltd., India), and HRESIMS were recorded on an Agilent 6210 ESI-TOF mass spectrometer (Agilent Technologies, Santa Clara, CA, USA). GC-MS analysis was performed on a GCMS-QP2010SE (Shimadzu, Japan) with inert cap 5MS/Sil i.d.0.25×30 m (GL Sciences Inc., Japan) [Column temperature: 100–280°C, rate of temperature increase: 10°C/min]. The following sugar samples and reagents were commercially obtained: L-rhamnose, D-rhamnose (Funakoshi Co., Ltd., Japan), D-xylose, L-xylose (Wako Pure Chemical Industries, Ltd., Japan), D-arabinose, L-arabinose (Kishida Chemical Co., Ltd., Japan), L-cysteine methyl ester hydrochloride (Kanto Chemical Co., Inc., Japan), and N-trimethylsilylimidazole (TMS-imidazole) (Tokyo Kasei Kogyo Co., Ltd., Japan). 1H and 13C NMR spectra were recorded on a Bruker AVANCE 700 spectrometer (Bruker BioSpin, Billerica, MA, USA) (700 MHz for 1H and 175 MHz for 13C) in deuterated MeOH. All chemical shifts (δ) are given in ppm with reference to the residual solvent signal and coupling constants (J) are in Hz. Thin-layer chromatography was carried out on precoated silica gel 60 F254 (Merck, Darmstadt, Germany) plates developed with hexane:EtOAc, EtOAc:MeOH, and EtOAc:MeOH-H2O. They were visualized under UV light (254 and 365 nm) and by spraying with 10% aqueous H2SO4 followed by heating.
3.2 Plant material
The aerial part of S. trifasciata was collected in Dschang in March 2014 and authentified at the Cameroon National Herbarium, Yaounde, where voucher specimens are deposited (No 43509/HCN).
3.3 Extraction and isolation
The powdered aerial parts of S. trifasciata (3.3 kg) were extracted three times with MeOH (3×10 L) at room temperature, and the filtrate obtained was evaporated under reduced pressure to yield the crude MeOH extract (497.3 g, 15% yield). Part of this extract (450 g) was triturated with EtOAc to yield 102 g of the EtOAc soluble fraction. About 97 g of the EtOAc soluble fraction was submitted to a silica gel column chromatography eluted with hexane:EtOAc and EtOAc:MeOH with increasing polarity yielding several fractions. The fraction eluted with hexane:EtOAc 6:4 (1.2 g) was subjected to Sephadex LH-20 (Merck, Darmstadt, Germany) column chromatography eluted with MeOH to give a mixture (30 mg) which was repeatedly chromatographed on silica gel (hexane:EtOAc 65:35) to afford 1 (2.1 mg). The fraction eluted with hexane:EtOAc 2:8 (3.0 g) was subjected to Sephadex LH-20 column chromatography eluted with MeOH to give a subfraction which was recrystallized to yield 6 (15 mg). The fraction eluted with EtOAc:MeOH 90:10 (3.8 g) was purified on a Sephadex LH-20 column eluted with MeOH and was repeatedly chromatographed on silica gel column (eluted with EtOAc:MeOH:H2O 90:10:5) to afford 2/3 (10 mg) and 4/5 (7 mg), respectively. Recrystallization of the fraction eluted with EtOAc:MeOH 85:15 (15 g) yielded 7 (110 mg) and 8 (25 mg).
Trifasciatine C (1): Yellowish gum. – [α]25D=+20 (c=0.3, MeOH). – UV (MeOH): λmax (log ε)=291 (3.17). – IR (NaCl): νmax=3400, 2920, 1643, 1245 cm−1. – 1H (CD3OD, 700 MHz) and 13C NMR (CD3OD, 175 MHz): see Table 1. – HRESIMS: m/z 369.0960 [M+Na]+ (calcd. for C18H18O7Na: 369.0945).
Trifasciatosides K/L (2/3): Amorphous powder. – [α]25D=−30 (c=0.8, MeOH). – IR (KBr): νmax=3300, 2972, 1724, 1446, 1373, 1253, 1137, 1048, 981 cm−1. – 1H NMR (CD3OD, 700 MHz): 5.58 (brd, J=5.1, H-6), 5.40 (brs, H-1″), 5.32 (d, J=1.8, H-1″), 5.14 (dd, J=3.6, 1.6, H-2″), 4.94 (o, H-3″), 4.83 (brs, H-27), 4.79 (brs, H-27), 4.40/4.41 (d, J=6.0, H-1″′), 4.35/4.37 (d, J=7.3, H-1′), 3.42 (m, H-3), 1.27/1.30 (d, J=6.2, H-6″), 1.12/1.13 (s, H-18), 1.09/1.10 (s, H-19), 0.95 (d, J=7.0, H-21). – 13C NMR (CD3OD, 175 MHz,): see Table 2. – HRESIMS: 919.4387 [M+Na]+ (calcd. for C45H68O18Na: 919.4303).
Trifasciatosides M/N (4/5): Amorphous powder. – [α]25D=−23.3 (c=0.3, MeOH). – IR (KBr): νmax – 3330, 2921, 1726, 1377, 1253, 1047 cm−1. – 1H NMR (CD3OD, 700 MHz): 5.55 (brs, H-6), 5.40 (d, J=1.0, H-1″), 5.32 (d, J=1.7, H-1″), 5.15 (dd, J=3.5, 1.5, H-2″), 5.06 (brs, H-27), 4.90 (o, H-3″), 4.98 (brs, H-27), 4.42/4.44 (d, J=6.3, H-1″′), 4.37/4.33 (d, J=7.3, H-1′), 3.34 (m, H-3), 1.27/1.24 (d, J=6.1, H-6″), 1.11/1.12 (s, H-18), 0.95/0.92 (d, J=7.0, H-21), 0.90/0.91 (s, H-19). – 13C NMR (CD3OD, 175 MHz): see Table 2. HRESIMS: 935.4335 [M+Na]+ (calcd. for C45H68O19Na: 935.4247).
3.4 Acid hydrolysis and GC-MS
Compounds 2/3 and 4/5 (ca. 1.0 mg) were individually heated in 1 M HCl (0.1 mL) at 90°C for 3 h. Each reaction mixture was dried under reduced pressure and dissolved in pyridine (0.2 mL). TMS-imidazole (50 μL) was added to part of the solution (0.1 mL) then heated at 50°C for 30 min. The reaction mixture was diluted with H2O (0.2 mL) and extracted with n-hexane (0.1 mL) then analyzed by GC-MS by comparison with standard samples. L-cysteine methyl ester hydrochloride (ca. 1.0 mg) was added to the remaining pyridine solution (0.1 mL) and heated at 60°C for 1 h, then the TMS derivative was prepared the same manner mentioned above and analyzed by GC-MS. Standard TMS-sugars (tR, min.) were the following: TMS-xylose, tR=12.08, 12.68; TMS-rhamnose, tR=11.06, 11.88; and TMS-arabinose, tR=10.87, 10.92, 11.29. Standard TMS-thiazolidine derivatives (tR, min.) were the following: D-xylose, tR=17.49; L-xylose, tR=17.74; D-rhamnose, tR=18.25; L-rhamnose, tR=18.13; D-arabinose, tR=17.80; and L-arabinose, tR=17.48.
3.5 Cell culture and cell proliferation assay
Human malignant epithelial cells (HeLa) were cultured in Eagle’s minimum essential medium supplemented with 10% fetal bovine serum (FBS) kept in an incubator at 37°C in a humidified air containing 5% CO2. FBS was purchased from Nichirei Bioscience Inc. (Tokyo, Japan). Cell viability was determined by a Cell-Titer 96 Aqueous Non-Radioactive Cell Proliferation (MTS) Assay (Promega, WI) according to the manufacturer’s protocol. HeLa cells (1×104 cells/well) were seeded in 96 well plates and incubated for 24 h, subsequently grown with compounds for additional 48 h, and then cell proliferation assay was performed.
4 Supplementary data
HRESIMS, 1H and 13C NMR, COSY, HSQC and HMBC spectra for new compounds are available.
Acknowledgments
The authors are grateful to the Alexander von Humboldt Foundation (AvH), Bonn, Germany, for the financial support of this work through the equipment grant to Prof Dr. Tapondjou A. Léon.
References
1. Mabberley DJ. Mabberley’s plant-book. Cambridge: Cambridge University Press, 2008.Suche in Google Scholar
2. Lu P-L, Morden CW. Phylogenetic relationships among dracaenoid genera (Asparagaceae: Nolinoideae) inferred from chloroplast DNA loci. Syst Bot 2014;39:90‒104.10.1600/036364414X678035Suche in Google Scholar
3. Antunes AD, Da Silva BP, Parente JP, Valente AP. A new bioactive steroidal saponin from Sansevieria cylindrica. Phytother Res 2003;17:179‒82.10.1002/ptr.1059Suche in Google Scholar
4. Tchegnitegni BT, Teponno RB, Tanaka C, Gabriel AF, Tapondjou AL, Miyamoto T. Sappanin-type homoisoflavonoids from Sansevieria trifasciata Prain. Phytochem Lett 2015;12:262–6.10.1016/j.phytol.2015.04.017Suche in Google Scholar
5. Mimaki Y, Inoue T, Kuroda M, Sashida Y. Steroidal saponins from Sansevieria trifasciata. Phytochemistry 1996;43:1325‒31.10.1016/S0031-9422(96)00397-4Suche in Google Scholar
6. Teponno RB, Tanaka C, Jie B, Tapondjou AL, Miyamoto T. Trifasciatosides A–J, steroidal saponins from Sansevieria trifasciata. Chem Pharm Bull 2016;64:1347–55.10.1248/cpb.c16-00337Suche in Google Scholar PubMed
7. Weil MJ, ZhangY, Nair MG. Tumor cell proliferation and cyclooxygenase inhibitory constituents in horseradish (Armoracia rusticana) and wasabi (Wasabia japonica). J Agric Food Chem 2005;3:1440–4.10.1021/jf048264iSuche in Google Scholar PubMed
8. Lee JI, Kong C-S, Baek SO, Seo Y. Isolation and antioxidant activity of methyl aconitates from Arctic Red Alga Polysiphonia stricta. Ocean Polar Res 2014;36:247–54.10.4217/OPR.2014.36.3.247Suche in Google Scholar
9. Teponno RB, Ponou KB, Fiorini D, Tapondjou AL, Barboni L. Chemical constituents from the roots of Furcraea bedinghausii Koch. Int Lett Chem Phys Astron 2013;11:9–19.10.18052/www.scipress.com/ILCPA.16.9Suche in Google Scholar
10. Said A, Aboutabl AE, Melek RF, Jaleel GA, Raslan M. Steroidal saponins and homoisoflavanone from the aerial parts of Sansevieria cylindrica Bojer ex Hook. Phytochem Lett 2015;12:113–8.10.1016/j.phytol.2015.03.006Suche in Google Scholar
11. White GF, Russel NJ, Tidswell EC. Bacterial scission of ether bonds. Microbiol Rev 1996;60:216–32.10.1128/mr.60.1.216-232.1996Suche in Google Scholar PubMed PubMed Central
12. Hara S, Okabe H, Mihashi K. Gas-liquid chromatographic separation of aldose enantiomers as trimethylsilyl ethers of methyl 2-(polyhydroxyalkyl)-thiazolidine-4 (R) –carboxylates. Chem Pharm Bull 1987;35:501–6.10.1248/cpb.35.501Suche in Google Scholar
13. Kougan GB, Miyamoto T, Tanaka C, Paululat T, Mirjolet JF, Duchamp O, et al. Steroidal saponins from two species of Dracaena. J Nat Prod 2010;73:1266–70.10.1021/np100153mSuche in Google Scholar PubMed
14. Fouedjou TR, Teponno RB, Quassinti L, Bramucci M, Petrelli D, Vitali LA, et al. Steroidal saponins from the leaves of Cordyline fruticosa (L.) A. Chev. and their cytotoxic and antimicrobial activity. Phytochem Lett 2014;7:62–8.10.1016/j.phytol.2013.10.001Suche in Google Scholar
15. Sparg SG, Light ME, Van Staden J. Biological activities and distribution of plant saponins. J Ethnopharmacol 2004;94:219–43.10.1016/j.jep.2004.05.016Suche in Google Scholar PubMed
16. Zeng L, Zhu M, Zhong J, Yan W. Structural stability of acetyl saponins in different solvents and separation materials. Phytochem Lett 2015;11:368–72.10.1016/j.phytol.2014.10.020Suche in Google Scholar
Supplemental Material:
The online version of this article offers supplementary material (DOI: https://doi.org/10.1515/znc-2017-0027)
©2017 Walter de Gruyter GmbH, Berlin/Boston
Artikel in diesem Heft
- Frontmatter
- Ginkgetin inhibits proliferation of human leukemia cells via the TNF-α signaling pathway
- Effects of a water extract of Lepidium meyenii root in different models of persistent pain in rats
- Interaction of tubulin and protein kinase CK2 in Trypanosoma equiperdum
- Synthesis, biological activity and molecular modeling study of new Schiff bases incorporated with indole moiety
- A dihydrochalcone derivative and further steroidal saponins from Sansevieria trifasciata Prain
- Chemical composition and biological activities of leaf and fruit essential oils from Eucalyptus camaldulensis
- Fungal production of the polysaccharide pullulan from a plant hydrolysate
- Acetogenins and alkaloids during the initial development of Annona muricata L. (Annonaceae)
- Corrigendum
- Corrigendum to: Ginkgetin inhibits proliferation of human leukemia cells via the TNF-α signaling pathway
Artikel in diesem Heft
- Frontmatter
- Ginkgetin inhibits proliferation of human leukemia cells via the TNF-α signaling pathway
- Effects of a water extract of Lepidium meyenii root in different models of persistent pain in rats
- Interaction of tubulin and protein kinase CK2 in Trypanosoma equiperdum
- Synthesis, biological activity and molecular modeling study of new Schiff bases incorporated with indole moiety
- A dihydrochalcone derivative and further steroidal saponins from Sansevieria trifasciata Prain
- Chemical composition and biological activities of leaf and fruit essential oils from Eucalyptus camaldulensis
- Fungal production of the polysaccharide pullulan from a plant hydrolysate
- Acetogenins and alkaloids during the initial development of Annona muricata L. (Annonaceae)
- Corrigendum
- Corrigendum to: Ginkgetin inhibits proliferation of human leukemia cells via the TNF-α signaling pathway