Heteroleptic ruthenium(II) 2,2′-bipyridine complexes incorporating substituted pyrazol-1-yl-pyridazine ancillaries
-
Li-Hua Tang


Abstract
Condensation of 3,6-dichloropyridazine or 3,6-dichloro-4,5-dimethyl- pyridazine with 3-methyl-1H-pyrazole or 4-methyl-1H-pyrazole with the assistance of sodium metal in tetrahydrofuran at reflux afforded three 3,6-bis(pyrazolyl)- pyridazine-type ligands: 3,6-bis(3-methylpyrazolyl)pyridazine (L1), 3,6-bis(4-methyl- pyrazolyl)pyridazine (L2) and 4,5-dimethyl-3,6-bis(4-methylpyrazolyl)pyridazine (L3). Reactions of cis-[RuCl2(bpy)2] · 2H2O (bpy = 2,2′-bipyridine) and L1, L2 or L3 in the presence of NH4PF6 produced the heteroleptic cationic ruthenium(II) complexes [Ru(L1)(bpy)2](PF6)2 (1), [Ru(L2)(bpy)2](PF6)2 (2) and [Ru(L3)(bpy)2](PF6)2 (3), respectively. The three complexes have been characterized by UV/Vis and luminescence spectroscopy. The crystal structures of 1 · EtOH, 2 · EtOH and 3 have been determined by single-crystal X-ray diffraction.
1 Introduction
In recent years, ruthenium(II) complexes of the [Ru(bpy)3]2+ (bpy=2,2′-bipyridine) family have attracted great attention due to their possible applications in bioinorganic chemistry and photo-induced electron transfer processes [1], [2], [3], [4], [5]. In comparison to the polypyridyl systems, research based on ruthenium-bpy complexes with the other N-containing heterocycles, e.g. imidazole, pyrazole and pyridazine, has been limited [6], [7], [8], [9]. As a matter of fact, coordination of such nitrogen-containing multi-dentate heterocyclic ligands and their derivatives to the ruthenium center alters the electron transfer properties of the obtained ruthenium complexes due to the different electron donor/acceptor properties of the coordinating nitrogen atoms [10], [11]. For example, ruthenium(II) complexes incorporating dianionic bipyrazolate ancillaries are suited for high-performance dye-sensitized solar cells [12]. On the other hand, (1-pyrazolyl)pyridazines and their derivatives are able to act as bridges between transition metal centers, such as nickel, cobalt, copper, silver and ruthenium [13]. According to searches in the Cambridge Structural Database (CSD) [14], tetradentate (1-pyrazolyl)pyridazine ligands may adopt five coordination modes when coordinated to transition metals, as shown in Chart 1 (see modes a–e), among which the μ2,η4- (a) and η2- (b) fashions are more common [15], [16], [17], [18], [19]. A few ruthenium complexes bearing η2- and μ2,η4-(1-pyrazolyl)pyridazine ligands have been reported, such as [(η5-Cp*)Ru(η2-L)(PPh3)](PF6) (L=3,6-bis(pyrazolyl)pyridazine) [20], [(η6-p-cymene)Ru(η2-L1)Cl](ClO4), [(η6-C6H6)Ru(η2-L′)Cl](PF6) (L′=3,6-bis(3,5-dimethylpyrazolyl)pyridazine) [21], [(η6-p-cymene)Ru(η2-L′)Cl](BPh4) [22], and an oxo-centered triruthenium-acetate complex [Ru3O(OAc)5(py)-(μ2,η4-L)](PF6) (py=pyridine) [23]. Recently, we have reported the syntheses and phosphorescent properties of several heteroleptic ruthenium(II)/(III) polypyridine complexes with a series of substituted 2,2′-bipyridines l, Schiff base N,O-ligands and N,C-phenylphthalazines [24], [25], [26]. In this paper, we report the synthesis, structure and spectroscopic properties of three heteroleptic ruthenium(II)-bpy complexes with (1-pyrazolyl)pyridazine ligands (Scheme 2).
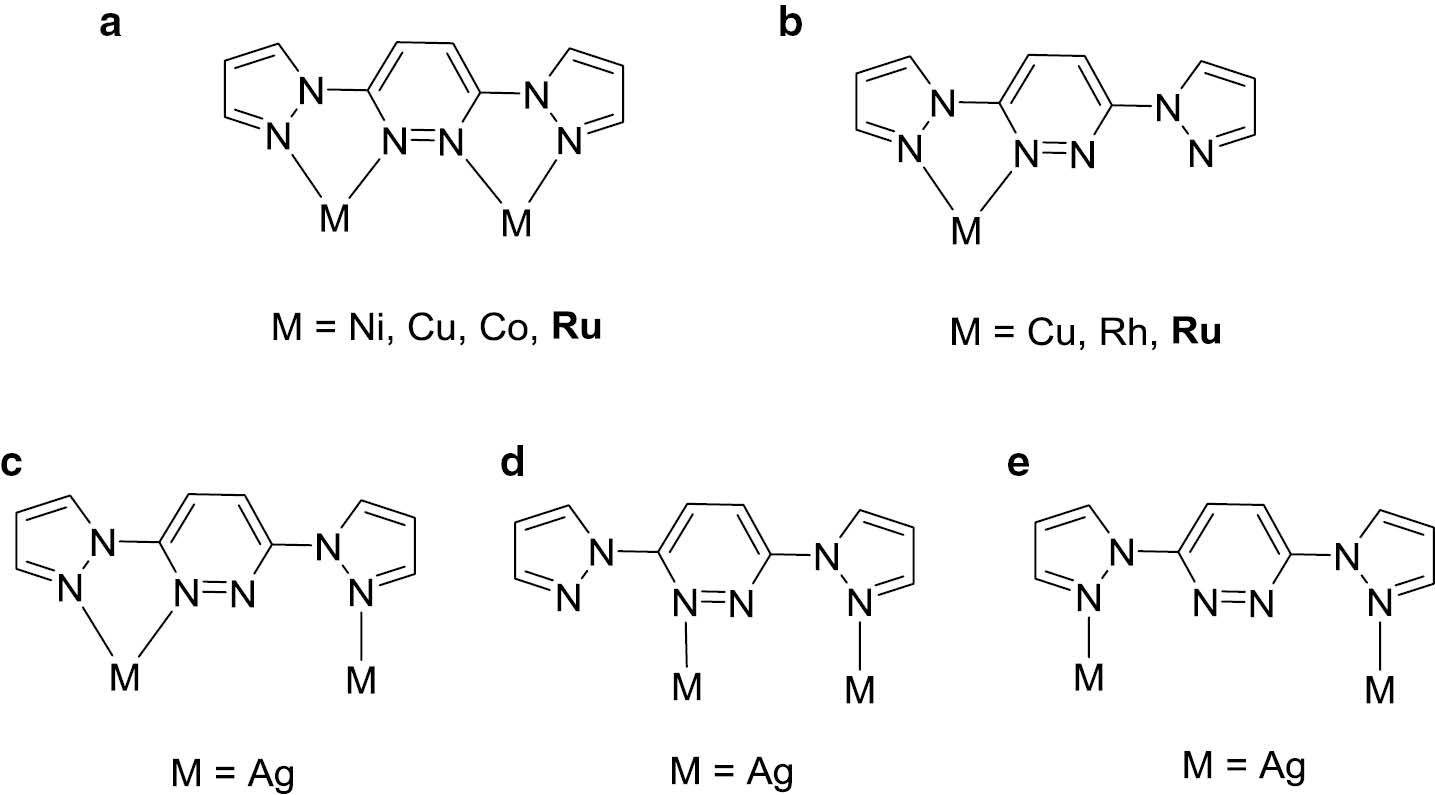
Coordination modes of the (1-pyrazolyl)pyridazine ligands.
(a–e) Represents the five coordination modes.
2 Experimental section
2.1 General
All synthetic manipulations were carried out under dry nitrogen by standard Schlenk techniques. RuCl3·3H2O was used as purchased from Pressure Chemical Co. Ltd. cis-[RuCl2(bpy)2]2·H2O [27] and ligands L1‒L3 [21] were prepared according to the literature methods. NMR spectra were recorded on a Bruker ALX 400 spectrometer operating at 400 MHz for 1H, 376 MHz for 19F and 162 MHz for 31P. Electronic absorption spectra were obtained on a Shimadzu UV-3000 spectrophotometer. Photoluminescence (PL) spectra were measured with a Shimadzu RF-5301PC fluorescence spectrophotometer. Elemental analyses were carried out using a Perkin-Elmer 2400 CHN analyzer.
2.2 Synthesis of 3,6-di(3-methyl-pyrazol-1-yl)pyridazine (L1)
A mixture of 3-methylpyrazole (464 mg, 5.65 mmol) and sodium metal (130 mg, 5.65 mmol) was stirred in 15 mL THF (absolute) at room temperature for 20 min; then 3,6-dichloropyridazine (500 mg, 2.82 mmol) was added, and the mixture was heated at 70°C for 2 h. After cooling to room temperature, the mixture was poured into ice water. A pink solid was precipitated, washed with water and dried in vacuo. Yield: 386 mg, 57%. – 1H NMR (400 MHz, CDCl3): δ=8.70–8.55 (m, 2H), 8.29 (s, 1H), 8.26 (s, 1H), 6.37–6.23 (m, 2H), 2.39 (s, 6H, CH3).
2.3 Synthesis of 3,6-di(4-methyl-pyrazol-1-yl)pyridazine (L2)
L2 was prepared similarly to the synthesis of L1 using 4-methylpyrazole instead of 3-methylpyrazole. A light yellow solid of L2 was obtained. Yield: 400 mg, 59%. – 1H NMR (400 MHz, CDCl3): δ=8.75 (d, J=2.6 Hz, 1H), 8.46 (d, J=2.4 Hz, 1H), 8.21 (d, J=0.8 Hz, 1H), 7.82 (dd, J=4.0, 1.4 Hz, 2H), 6.55 (dd, J=2.6, 1.7 Hz, 1H), 6.52 (dd, J=2.5, 1.8 Hz, 1H), 2.73 (s, 6H, CH3).
2.4 Synthesis of 4,5-dimethyl-3,6-di(4-methyl-pyrazol-1-yl)pyridazine (L3)
The synthetic method was similar to that used for L2, employing 3,6-dichloro-4,5-dimethylpyridazine instead of 3,6-dichloropyridazine. The resulting precipitate was filtered off, washed with water and dried in vacuo. Yield: 514 mg, 68%. – 1H NMR (400 MHz, CDCl3): δ=8.09–8.03 (m, 2H), 7.63 (d, J=7.0 Hz, 2H), 2.54 (s, 6H, CH3), 2.22 (s, 6H, CH3).
2.5 Synthesis of [Ru(L1)(bpy)2](PF6)2 (1)
A mixture of L1 (30 mg, 0.12 mmol) and cis-[Ru(bpy)2Cl2]·2H2O (52 mg, 0.10 mmol) was stirred in 20 mL ethanol at reflux for 3 h, during which the color of the solution turned into orange-red. After filtering the solution, an excess of an ammonium hexafluorophosphate solution in ethanol was added, and the mixture was further stirred at room temperature for 10 min. After removing the solvent by rotary evaporation, the residue was washed with n-hexane (3×7 mL) to give the desired red product. Recrystallization with dichloromethane–n-hexane (v:v=1:4) gave red crystals of 1·EtOH after 6 days. Yield: 36 mg, 47% (based on Ru). – IR (KBr, cm−1): ν(C=N)1630, ν(N–N)1448. – 1H NMR (400 MHz, DMSO-d6): δ=9.24 (d, J=3.1 Hz, 1H), 8.86 (d, J=7.8 Hz, 2H), 8.78 (d, J=9.6 Hz, 3H), 8.43 (d, J=9.5 Hz, 1H), 8.19 (dt, J=20.5, 6.7 Hz, 5H), 8.06 (d, J=5.6 Hz, 1H), 7.92 (d, J=5.5 Hz, 1H), 7.70–7.61 (m, 2H), 7.56 (dd, J=8.9, 4.6 Hz, 3H), 7.16 (d, J=2.6 Hz, 1H), 6.86 (d, J=3.1 Hz, 1H), 6.41 (d, J=2.6 Hz, 1H), 2.26 (s, 3H, CH3), 2.10 (s, 3H, CH3). – 19F NMR (376 MHz, DMSO-d6): δ=‒70.20 (d, J=680 Hz, PF6). – 31P NMR (162 MHz, DMSO-d6): δ=(ppm): ‒144.20 (sept, J=680 Hz, PF6). – Analysis for C34H34F12N10OP2Ru: calcd. C 41.26, H 3.46, N 14.15; found C 41.20, H 3.45, N 14.19%.
2.6 Synthesis of [Ru(L2)(bpy)2](PF6)2 (2)
A mixture of L2 (28.2 mg, 0.11 mmol) and cis-[Ru(bpy)2Cl2]·2H2O (52 mg, 0.10 mmol) was stirred in 20 mL ethanol at reflux for 3 h, during which the color of the solution turned into orange-red. After filtering the solution, an excess of an ammonium hexafluorophosphate solution in ethanol was added, and the mixture was further stirred at room temperature for 10 min. After removing the solvent by rotary evaporation, the residue was washed with diethyl ether (3×7 mL) to give the desired orange-red product. Recrystallization with dichloromethane–n-hexane (v:v=1:4) gave red crystals of 2·EtOH after 1 week. Yield: 32 mg, 46% (based on Ru). – IR (KBr, cm−1): ν(C=N)1637, ν(N–N)1445. – 1H NMR (400 MHz, DMSO-d6): δ=9.13 (s, 1H), 8.92–8.66 (m, 6H), 8.32 (d, J=9.3 Hz, 1H), 8.28–8.07 (m, 6H), 7.91 (d, J=6.2 Hz, 1H), 7.79–7.75 (m, 1H), 7.67–7.47 (m, 6H), 2.33 (s, 3H, CH3), 2.08 (s, 3H, CH3). – 19F NMR (376 MHz, DMSO-d6): δ=‒70.24 (d, J=673 Hz, PF6). – 31P NMR (162 MHz, DMSO-d6): δ=‒144.22 (sept, J=673 Hz, PF6). – Analysis for C34H34F12N10OP2Ru: calcd. C 41.26, H 3.46, N 14.15; found C 41.21, H 3.44, N 14.19%.
2.7 Synthesis of [Ru(L3)(bpy)2](PF6)2 (3)
A mixture of L3 (29 mg, 0.11 mmol) and cis-[Ru(bpy)2Cl2]·2H2O (52 mg, 0.10 mmol) was stirred in 20 mL ethanol at reflux for 2.5 h, during which the color of the solution turned into red. After filtering the solution, an excess of an ammonium hexafluorophosphate solution in ethanol was added, and the mixture was further stirred at room temperature for 10 min. After removing the solvent by rotary evaporation, the residue was washed with diethyl ether (3×5 mL) to give an orange-red product. Recrystallization with dichloromethane–n-hexane (v:v=1:4) gave red crystals of 3 after 5 days. Yield: 41 mg, 57% (based on Ru). – IR (KBr, cm−1): ν(C=N)1604, ν(N–N)1443. – 1H NMR (400 MHz, DMSO-d6): δ=9.05 (s, 1H), 8.84 (d, J=7.7 Hz, 2Hy), 8.75 (d, J=8.1 Hz, 2H), 8.17 (dd, J=15.2, 7.8 Hz, 4H), 8.00 (dd, J=16.5, 6.4 Hz, 2Hy), 7.85–7.80 (m, 1H), 7.64 (d, J=28.5 Hz, 4H), 7.54 (s, 3H), 7.06 (s, 1H), 2.78 (s, 3H, CH3), 2.56 (s, 3H, CH3), 2.10 (s, 3H, CH3), 1.99 (s, 3H, CH3). – 19F NMR (376 MHz, DMSO-d6): δ=‒70.19 (d, J=670 Hz, PF6). – 31P NMR (162 MHz, DMSO-d6): δ=‒144.20 (sept, J=670 Hz, PF6). – Analysis for C34H32F12N10P2Ru: calcd. C 42.03, H 3.32, N 14.41; found C 42.08, H 3.30, N 14.44%.
2.8 X-ray crystallography
The crystallographic data and experimental details for [Ru(L1)(bpy)2](PF6)2·EtOH (1·EtOH), [Ru(L2)(bpy)2](PF6)2·EtOH (2·EtOH) and [Ru(L3)(bpy)2](PF6)2 (3) are summarized in Table 1. Selected bond lengths and bond angles are given in Table 2. Selected hydrogen bond systems are listed in Table 3. Intensity data were collected on a Bruker SMART APEX 2000 CCD diffractometer using graphite-monochromatized MoKα radiation (λ=0.71073 Å) at T=296(2) K. The collected frames were processed with the software Saint [28]. The data were corrected for absorption using the program Sadabs [29]. Structures were solved by Direct Methods and refined by full-matrix least squares on F2 using the Shelxtl software package [30]. All non-hydrogen atoms were refined anisotropically. The positions of all hydrogen atoms were generated geometrically (Csp3–H=0.97 and Csp2–H=0.93 Å), assigned isotropic displacement parameters, and allowed to ride on their respective parent carbon atoms before the final cycle of least-squares refinement. The low ratios of observed to unique reflections for complexes 2 and 3 may be due to relatively poor crystal quality. OMIT commands were used to delete the most disagreeable reflections. DELU and ISOR restraints were used to model the disorder of some carbon and nitrogen atoms in complex 3.
Crystallographic data and experimental details for complexes 1·EtOH, 2·EtOH and 3.
| Complex | 1·EtOH | 2·EtOH | 3 |
|---|---|---|---|
| Empirical formula | C34H34F12N10OP2Ru | C34H34F12N10OP2Ru | C34H32F12N10P2Ru |
| Formula weight | 989.72 | 989.72 | 971.71 |
| Crystal system | Monoclinic | Triclinic | Monoclinic |
| Space group | P21/n | P1̅ | P21/c |
| a, Å | 10.3891(18) | 10.650(3) | 19.585(13) |
| b, Å | 16.569(3) | 12.051(3) | 11.414(9) |
| c, Å | 23.720(4) | 16.298(4) | 19.251(15) |
| α, deg | 90 | 96.360(4) | 90 |
| β, deg | 90.940(3) | 105.262(4) | 97.937(13) |
| γ, deg | 90 | 91.047(4) | 90 |
| Volume, Å3 | 4082.5(12) | 2003.0(8) | 4263(5) |
| Z | 4 | 2 | 4 |
| Dcalcd, Mg m−3 | 1.610 | 1.641 | 1.514 |
| T, K | 296(2) | 296(2) | 296(2) |
| F(000), e | 1992 | 996 | 1952 |
| μ(MoKα), mm−1 | 0.6 | 0.6 | 0.5 |
| Refl. total/unique | 25 458/9324 | 12 680/8831 | 12 279/8919 |
| Rint | 0.0731 | 0.0526 | 0.1037 |
| R1a/wR2b (I>2σ(I)) | 0.0690/0.1734 | 0.0764/0.1734 | 0.1030/0.2425 |
| R1/wR2 (all data) | 0.1338/0.2126 | 0.1750/0.2311 | 0.2494/0.3230 |
| GoFc | 0.979 | 0.917 | 0.950 |
| Final max/min diff. peaks, e Å−3 | +0.75/‒0.73 | +0.69/‒0.61 | +0.94/‒0.83 |
aR1=Σ||Fo|–|Fc||/Σ|Fo|; bwR2= [Σw(Fo2 – Fc2)2/Σw(Fo2)2]1/2, w= [σ2(Fo2)+(AP)2+BP]−1, where P= (Max(Fo2, 0)+2Fc2)/3; cGoF=S=[Σw(Fo2 – Fc2)2/(nobs – nparam)]1/2.
Selected bond lengths (Å) and angles (deg) for the heteroleptic ruthenium(II) complexes 1·EtOH, 2·EtOH and 3.
| Complex | Ru‒N(bpy) | Ru‒N(L) | N(bpy)‒Ru‒N(bpy) | N(L)‒Ru‒N(L) |
|---|---|---|---|---|
| 1·EtOH | 2.064(4) 2.047(5) 2.051(5) 2.057(5) | 2.025(4) 2.065(4) | 78.47(19) 79.3(2) | 78.23(17) |
| 2·EtOH | 2.051(6) 2.053(6) 2.055(6) 2.054(6) | 2.026(6) 2.055(6) | 78.6(3) 79.5(2) | 78.1(2) |
| 3 | 2.021(12) 2.024(11) 2.086(10) 2.089(11) | 2.048(9) 2.051(8) | 78.8(5) 79.0(5) | 77.5(4) |
Selected weak hydrogen bonds for complexes 1·EtOH, 2·EtOH and 3a.
| Complex | D–H⋯A | d(D–H) (Å) | d(HA) (Å) | d(DA) (Å) | ∠(DHA) (deg) |
|---|---|---|---|---|---|
| 1·EtOH | C(2S)–H(2S2)⋯F(2)i | 0.97 | 2.38 | 3.320(15) | 164.2 |
| C(1)–H(1)⋯N(5) C(24)–H(24A)⋯N(3)ii | 0.93 0.96 | 2.61 2.45 | 3.136(9) 3.335(8) | 116.4 153.5 | |
| 2·EtOH | C(26)–H(26)⋯F(4)iii C(2S)–H(2S2)⋯F(3) C(1)–H(1)⋯N(3) | 0.93 0.96 0.93 | 2.35 1.93 2.66 | 3.246(9) 2.839(18) 3.201(11) | 161.8 157.5 118.2 |
| 3 | C(31)–H(31)⋯F(6)iv C(11)–H(11)⋯N(10) | 0.93 0.93 | 2.37 2.62 | 3.25(2) 3.21(2) | 157.9 121.8 |
aSymmetry codes: (i) –x+½, y+½, –z+3/2; (ii) –x+1, –y+1, –z+1; (iii) –x+2, –y+2, –z+1; (iv) –x+2, y – ½, –z+½.
CCDC 1585065 (for [Ru(L1)(bpy)2](PF6)2·EtOH (1·EtOH)), 1585066 (for [Ru(L2)(bpy)2](PF6)2·EtOH (2·EtOH)), and 1585068 (for [Ru(L3)(bpy)2](PF6)2 (3)) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre viawww.ccdc.cam.ac.uk/data_request/cif.
3 Results and discussion
Previously, Thompson has reported an effective procedure for the synthesis of 3,6-bis(pyrazolyl)pyridazine by the treatment of pyrazole with potassium metal to produce the potassium pyrazolide, which was then reacted with 3,6-dichloropyridazine to afford the target compounds [15]. As shown in Scheme 1, condensation of 3,6-dichloropyridazine or 3,6-dichloro-4,5-dimethylpyridazine with 3-methyl-1H-pyrazole or 4-methyl-1H-pyrazole with the assistance of sodium metal afforded three substituted pyrazol-1-yl-pyridazine ligands L1‒L3, which were easily purified with good yields. As displayed in Scheme 2, interactions of cis-[Ru(bpy)2Cl2]·2H2O with an equivalent of L1‒L3 in the presence of NH4PF6 led to the isolation of the corresponding heteroleptic ruthenium(II) complexes [Ru(L1)(bpy)2](PF6) (1), [Ru(L2)(bpy)2](PF6) (2) and [Ru(L3)(bpy)2](PF6) (3), respectively. The separate singlets for the two methyl groups in 1 and 2 (four methyl signals for 3) in their 1H NMR spectra show that the structures are rigid in solution (no fluxionality of the bis(pyrazolyl)piperazine ligand). Moreover, the 31P and 19F NMR spectra of complexes 1–3 exhibit the fluorine signal as a doublet at about δ=–70 ppm and the phosphorus signal as a septet at about δ=–144 ppm, which are typical for (PF6)‒ anions.
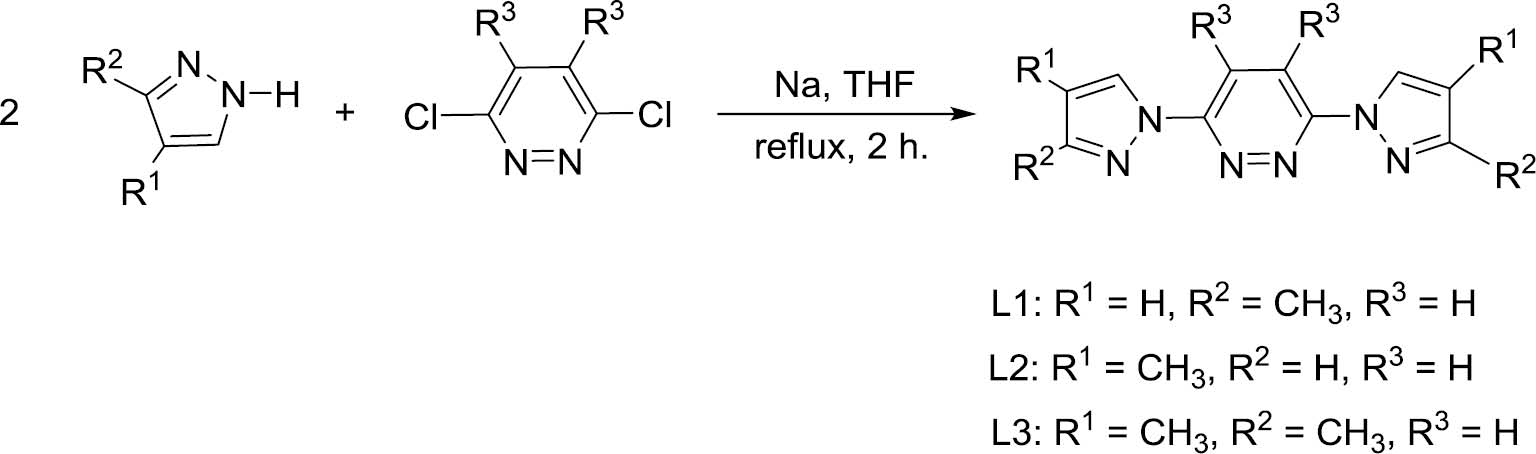
Syntheses of ligands L1, L2 and L3.
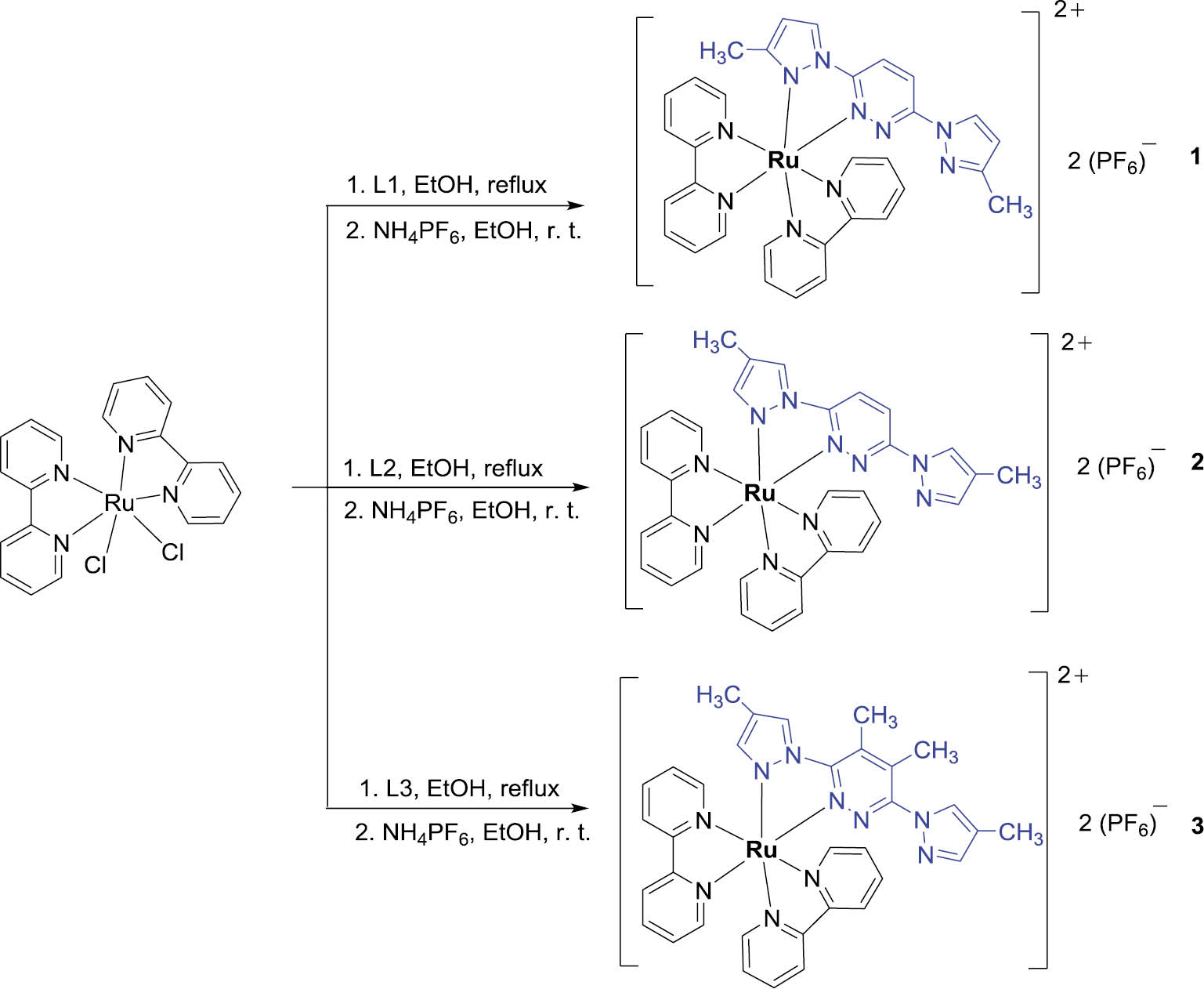
Syntheses of heteroleptic ruthenium(II) complexes 1, 2 and 3.
The crystal structures of complexes 1·EtOH, 2·EtOH and 3 have been established by X-ray crystallography. The molecular structures of the heteroleptic ruthenium(II)-bpy complex cations are shown in Figs. 1–3. The central ruthenium atoms of the three complexes are in an octahedral coordination environment, with two bpy units and one L ligand. The pyrazol-1-yl-pyridazine (L) ligands in the three complexes all adopt the η2-coordination mode (Chart 1b), forming a stable five-membered metallocycle of RuCN3, similar to those in other related half-sandwich ruthenium(II) complexes [20], [21], [22]. The Ru–N(bpy) bond lengths in 1·EtOH, 2·EtOH and 3 range from 2.021(12) to 2.089(11) Å. The average Ru–N(L) bond length of 2.051(8) Å in complex 3 is similar to those in complexes 1·EtOH (2.065(4) Å) and 2·EtOH (2.055(6) Å). The chelate bond angles of N(L)–Ru–N(L) in 1·EtOH, 2·EtOH and 3 are 78.23(17)°, 78.1(2)° and 77.5(4)°, respectively, which are slightly larger than those in the reported half-sandwich ruthenium(II) complexes (75.43°‒75.90°) [20], [21], [22]. A tendency for increased ruthenium–nitrogen bond lengths in the trans position with respect to the Ru–N(pz) bond is observed in 1·EtOH (2.064(4) Å vs 2.047(5) Å) and 2·EtOH (2.053(6) Å vs 2.047(6) Å), which is identical with that in related [Ru(bpy)2(C^N)]+ complexes [31]. The three complexes have small N(bpy)–Ru–N(bpy) bond angles (78.47(19)°‒79.5(2)°), due to the rigidity of the bpy units. Views of the packing of the complexes 1·EtOH, 2·EtOH and 3 in the crystals are shown in Figs. 1b, 2b, and 3b, respectively. The organization of the components of 1·EtOH, 2·EtOH and 3 seems to be influenced by very weak interionic and intermolecular C–H⋯F and C–H⋯N hydrogen-bonding interactions (see Table 3). The separations H⋯N in the three compounds are in the range 2.45–2.66 Å, and the H⋯F distances in 1·EtOH, 2·EtOH and 3 are between 1.97 and 2.38 Å. The hydrogen bond angles C–H⋯N are 116.4° and 153.5° for complex 1·EtOH, 118.2° for complex 2·EtOH and 121.8° for complex 3. The C–H⋯F bond angles are in the range 153.5°–164.4° for complexes 1·EtOH, 2·EtOH and 3. Hydrogen bonding with the ethanol molecules certainly contributes to the stabilization of the crystal structures of complexes 1·EtOH and 2·EtOH.
![Fig. 1: (a) Molecular structure of the cation [Ru(L1)(bpy)2]2+ of the complex 1·EtOH. The ethanol solvent molecule and the counter anions of PF6‒ are omitted for clarity. (b) Packing diagram of 1·EtOH in a unit cell, viewed along the crystallographic bc plane. C‒H···F and C‒H···N hydrogen bonds are shown as dashed lines.](/document/doi/10.1515/znb-2019-0233/asset/graphic/j_znb-2019-0233_fig_001.jpg)
(a) Molecular structure of the cation [Ru(L1)(bpy)2]2+ of the complex 1·EtOH. The ethanol solvent molecule and the counter anions of PF6‒ are omitted for clarity. (b) Packing diagram of 1·EtOH in a unit cell, viewed along the crystallographic bc plane. C‒H···F and C‒H···N hydrogen bonds are shown as dashed lines.
![Fig. 2: (a) Molecular structure of the cation [Ru(L2)(bpy)2]2+ of the complex 2·EtOH. The ethanol solvent molecule and the counter anions of PF6‒ are omitted for clarity. (b) Packing diagram of 2·EtOH in a unit cell, viewed along the crystallographic ab plane. C‒H···F and C‒H···N hydrogen bonds are shown as dashed lines.](/document/doi/10.1515/znb-2019-0233/asset/graphic/j_znb-2019-0233_fig_002.jpg)
(a) Molecular structure of the cation [Ru(L2)(bpy)2]2+ of the complex 2·EtOH. The ethanol solvent molecule and the counter anions of PF6‒ are omitted for clarity. (b) Packing diagram of 2·EtOH in a unit cell, viewed along the crystallographic ab plane. C‒H···F and C‒H···N hydrogen bonds are shown as dashed lines.
![Fig. 3: (a) Molecular structure of the cation [Ru(L3)(bpy)2]2+ of complex 3. The ethanol solvent molecule and the counter anions of PF6‒ are omitted for clarity. (b) Packing diagram of 3 in a unit cell, viewed along the crystallographic ab plane. C‒H···F and C‒H···N hydrogen bonds are shown as dashed lines.](/document/doi/10.1515/znb-2019-0233/asset/graphic/j_znb-2019-0233_fig_003.jpg)
(a) Molecular structure of the cation [Ru(L3)(bpy)2]2+ of complex 3. The ethanol solvent molecule and the counter anions of PF6‒ are omitted for clarity. (b) Packing diagram of 3 in a unit cell, viewed along the crystallographic ab plane. C‒H···F and C‒H···N hydrogen bonds are shown as dashed lines.
The UV/Vis absorption spectra of the new heteroleptic ruthenium(II) complexes 1–3, together with that of the parent complex [Ru(bpy)3](PF6)2, in CH2Cl2 at room temperature are shown in Fig. 4. At a first glance, the spectra are grossly similar and reveal three intense transitions located at 230–260, 270–330 and 410–470 nm. The strong absorption bands around 250 and 300 nm are assigned to typical spin-allowed π‒π* transitions of the ligands, and the comparatively less intense broad bands around 450 nm are ascribed to the metal-to-ligand charge transfer. These spectra are similar to that of [Ru(bpy)3](PF6)2 and related ruthenium(II)-bpy complexes with additional N,N-donors [12], [24]. It is interesting to note that there is a red-shift of about 60 nm observed for the low-lying transition depending on the presence of coordination of the L ligands. This considerable shift suggests that the low-energy transition is the main signature of the charge transfer arising between the ruthenium atom and bpy ligand. The room temperature PL spectra of the heteroleptic ruthenium(II) complexes 1–3 and [Ru(bpy)3](PF6)2 in the CH2Cl2 solution are illustrated in Fig. 5. The emission maximum for the complexes 1–3 appears at about 595 nm but with weaker intensity compared to the parent complex [Ru(bpy)3](PF6)2, possibly due to a reduced intracationic π-electron conjugation system.
2 in the CH2Cl2 solution.](/document/doi/10.1515/znb-2019-0233/asset/graphic/j_znb-2019-0233_fig_004.jpg)
The UV/Vis absorption spectra of complexes 1–3 and [Ru(bpy)3](PF6)2 in the CH2Cl2 solution.
2 in the CH2Cl2 solution (λex=460 nm).](/document/doi/10.1515/znb-2019-0233/asset/graphic/j_znb-2019-0233_fig_005.jpg)
Luminescence emission spectra of complexes 1–3 and [Ru(bpy)3](PF6)2 in the CH2Cl2 solution (λex=460 nm).
In summary, three new heteroleptic ruthenium(II) complexes incorporating substituted pyrazol-1-yl-pyridazine ancillaries were synthesized by the reaction of cis-[RuCl2(bpy)2]·2H2O with 3,6-bis(pyrazolyl)pyridazine-type ligands in moderate yields. According to the CSD search results, only five ruthenium(II) complexes with 3,6-bis(pyrazolyl)pyridazine-type ligands have previously been reported [20], [21], [22], [23]. The structure determination of the three polypyridyl ruthenium(II) complexes confirmed that the 3,6-bis(pyrazolyl)pyridazine ligand coordinates to the ruthenium(II)-bpy moiety in the usual η2-coodination mode (Chart 1b) as an enrichment of the structural diversity of the limited series of this type of ruthenium complexes highlighting the specific features of the Ru–N(pz) and trans-Ru–N(py) bonds. The three crystal structures appear to be stabilized by C–H⋯N and C–H⋯F hydrogen-bonding interactions. The optical properties of the three dicationic complexes in solution show close similarities, but a significant difference from the parent [(bipy)3Ru]2+ dication both in absorption and emission. No significant effect of the methyl substituents has been observed.
Acknowledgment
This project was supported by the Natural Science Foundation of China (21372007).
References
[1] J. M. Van Raden, S. Louie, L. N. Zakharov, R. Jasti, J. Am. Chem. Soc.2017, 139, 2936.10.1021/jacs.7b00359Search in Google Scholar PubMed
[2] A. Stumper, T. D. Pilz, M. Schaub, H. Görls, D. Sorsche, K. Peuntinger, D. Guldi, S. Rau, Eur. J. Inorg. Chem.2017, 3799.10.1002/ejic.201700548Search in Google Scholar
[3] F. E. Poynton, S. S. Bright, S. Blasco, D. C. William, J. M. Kelly, T. Gunnlaugssson, Chem. Soc. Rev.2017, 46, 7706.10.1039/C7CS00680BSearch in Google Scholar
[4] M. S. Meijer, S. Bonnet, Inorg. Chem.2019, 58, 11689.10.1021/acs.inorgchem.9b01669Search in Google Scholar PubMed PubMed Central
[5] K. Qiu, Y. Wen, O. Cheng, X. Liao, C. Liu, T. W. Rees, Q. Zhang, L. Jib, H. Chao, Chem. Commun.2019, 55, 11235.10.1039/C9CC05962HSearch in Google Scholar PubMed
[6] D. Mulhern, S. Brooker, H. Görls, S. Rau, J. G. Vos, Dalton Trans.2006, 51.10.1039/B510751BSearch in Google Scholar
[7] W. Zhang, J.-H. Liu, J.-X. Pan, P. Li, L.-C. Sun, Polyhedron2008, 27, 1168.10.1016/j.poly.2007.12.023Search in Google Scholar
[8] T. Kundu, A. D. Chowdhury, D. De, S. M. Mobin, V. G. Puranik, A. Datta, G. K. Lahiri, Dalton Trans.2012, 41, 4484.10.1039/c2dt12126cSearch in Google Scholar PubMed
[9] K. Heussner, K. Peuntinger, N. Rockstroh, S. Rau, C. Streb, Dalton Trans.2015, 44, 330.10.1039/C4DT03017FSearch in Google Scholar
[10] G. S. Huff, W. K. C. Lo, R. Horvath, X.-Z. Sun, G. R. Weal, H. J. Davidson, A. D. W. Kenneddy, C. John McAAdam, J. D. Crowley, M. W. George, K. C. Gordon, Inorg. Chem.2016, 55, 12238.10.1021/acs.inorgchem.6b01959Search in Google Scholar PubMed
[11] S. Naskar, B. Pakhira, D. Mishra, P. Mitra, S. K. Chattopadhyay, S. Naskar, Polyhedron2015, 100, 170.10.1016/j.poly.2015.07.058Search in Google Scholar
[12] H.-H. Yeh, S.-T. Ho, Y. Chi, J. N. Clifford, E. Palomares, S.-H. Liu, P.-T. Chou, J. Mater. Chem. A2013, 1, 7681.10.1039/c3ta10988gSearch in Google Scholar
[13] Q. Yu, A.-S. Zhang, T.-L. Hu, X.-H. Bu, Solid State Sci.2010, 12, 1484.10.1016/j.solidstatesciences.2010.06.013Search in Google Scholar
[14] C. R. Groom, I. J. Bruno, M. P. Lightfoot, S. C. Ward, Acta Crystallogr.2016, B72, 171.10.1107/S2052520616003954Search in Google Scholar
[15] L. K. Thompson, T. C. Woon, D. B. Murphy, E. J. Gabe, F. L. Lee, Y. L. Page, Inorg. Chem.1985, 24, 4719.10.1021/ic00220a057Search in Google Scholar
[16] M. P. Gamasa, J. Gimeno, E. Lastra, J. M. R. Gonzalez, S. Garcia-Granda, Polyhedron1990, 9, 2603.10.1016/S0277-5387(00)86835-7Search in Google Scholar
[17] H. Gao, Z.-H. Zhang, P. Jiang, X.-R. Li, Transition Met. Chem.2006, 31, 1088.10.1007/s11243-006-0114-1Search in Google Scholar
[18] B. R. Manzano, F. A. Jalon, I. M. Ortiz, M. L. Soriano, F. G. de la Torre, J. Elguero, M. A. Maestro, K. Mereiter, T. D. W. Claridge, Inorg. Chem.2008, 47, 413.10.1021/ic701117aSearch in Google Scholar PubMed
[19] P. Hubberstey, C. E. Russell, Chem. Commun.1995, 959.10.1039/c39950000959Search in Google Scholar
[20] G. Gupta, K. T. Prasad, A. V. Rao, S. J. Geib, B. Das, K. M. Rao, Inorg. Chim. Acta2010, 363, 2287.10.1016/j.ica.2010.03.052Search in Google Scholar
[21] G. Gupta, K. T. Prasad, B. Das, G. P. A. Yap, K. M. Rao, J. Organomet. Chem.2009, 694, 2618.10.1016/j.jorganchem.2009.03.043Search in Google Scholar
[22] M. Laura Soriano, F. A. Jalón, B. R. Manzano, M. Maestro, Inorg. Chim. Acta2009, 362, 4486.10.1016/j.ica.2009.04.011Search in Google Scholar
[23] F.-R. Dai, H.-Y. Ye, B. Li, L.-Y. Zhang, Z.-N. Chen, Dalton Trans.2009, 8696.10.1039/b908798bSearch in Google Scholar PubMed
[24] Z.-M. Wang, S.-M. Shen, X.-Y. Shen, Y.-Q. Xu, A.-Q. Jia, Q.-F. Zhang. J. Coord. Chem.2016, 69, 851.10.1080/00958972.2016.1156098Search in Google Scholar
[25] C.-J. Wang, W.-F. Xu, B.-H. Tong, A.-Q. Jia, Q.-F. Zhang. J. Coord. Chem.2017, 70, 1617.10.1080/00958972.2017.1317346Search in Google Scholar
[26] J. Ji, G.-Q. Li, Y.-Q. Xu, A.-Q. Jia, Q.-F. Zhang. Z. Naturforsch.2019, 74b, 267.10.1515/znb-2018-0211Search in Google Scholar
[27] B. P. Sullivan, D. J. Salmon, T. J. Meyer. Inorg. Chem. 1978, 17, 3334.10.1021/ic50190a006Search in Google Scholar
[28] Smart, Saint+ for Windows NT (version 6.02a), Area Detector Control and Integration Software, Bruker AXS Inc., Madison, WI (USA) 1998.Search in Google Scholar
[29] G. M. Sheldrick, Sadabs, University of Göttingen, Göttingen (Germany) 1996.Search in Google Scholar
[30] G. M. Sheldrick, Shelxtl (version 5.1), Software Reference Manual, Bruker AXS Inc., Madison, WI (USA) 1997.Search in Google Scholar
[31] B.-S. Chen, K. Chen, Y.-H. Hong, W.-H. Liu, T.-H. Li, C.-H. Lai, P.-T. Chou, Y. Chi, G.-H. Lee, Chem. Commun.2009, 5844.10.1039/b914197aSearch in Google Scholar PubMed
©2020 Walter de Gruyter GmbH, Berlin/Boston
Articles in the same Issue
- Frontmatter
- In this Issue
- The symmetry principle of antiaromaticity
- Assembly of C-propyl-pyrogallol[4]arene with bipyridine-based spacers and solvent molecules
- A new two-fold interpenetrating two-dimensional copper(II) coordination polymer constructed from 4,4′-bis(2-methylimidazol-1-yl)diphenyl ether
- Synthesis, structure, fluorescence, and electrochemical properties of a binuclear Ag(I) complex with 1,4-bis(benzo[d]oxazol-2-yl)butane as a ligand
- Design, synthesis, and characterization of 6-[(trimethyl)silylethynyl]naphthalene-2-ethene: a new precursor for the preparation of high-refractive-index organic materials
- A MOF based on a lead(II) 2-oxido-6-methylpyridine-4-carboxylate network
- Transition metal complexes of monodentate 2-oxazolines containing long chain alkyl groups. The crystal structure of trans-PdCl2(κ1-N-rac-2-heptadecyl-4,5-dihydro-5-methyl-2-oxazole)2
- A Zn(II) complex with a pyridyl- and carboxylate-containing ligand: synthesis and structural characterization
- Heteroleptic ruthenium(II) 2,2′-bipyridine complexes incorporating substituted pyrazol-1-yl-pyridazine ancillaries
- Crystal structure of mechanochemically synthesized Ag2CdSnS4
Articles in the same Issue
- Frontmatter
- In this Issue
- The symmetry principle of antiaromaticity
- Assembly of C-propyl-pyrogallol[4]arene with bipyridine-based spacers and solvent molecules
- A new two-fold interpenetrating two-dimensional copper(II) coordination polymer constructed from 4,4′-bis(2-methylimidazol-1-yl)diphenyl ether
- Synthesis, structure, fluorescence, and electrochemical properties of a binuclear Ag(I) complex with 1,4-bis(benzo[d]oxazol-2-yl)butane as a ligand
- Design, synthesis, and characterization of 6-[(trimethyl)silylethynyl]naphthalene-2-ethene: a new precursor for the preparation of high-refractive-index organic materials
- A MOF based on a lead(II) 2-oxido-6-methylpyridine-4-carboxylate network
- Transition metal complexes of monodentate 2-oxazolines containing long chain alkyl groups. The crystal structure of trans-PdCl2(κ1-N-rac-2-heptadecyl-4,5-dihydro-5-methyl-2-oxazole)2
- A Zn(II) complex with a pyridyl- and carboxylate-containing ligand: synthesis and structural characterization
- Heteroleptic ruthenium(II) 2,2′-bipyridine complexes incorporating substituted pyrazol-1-yl-pyridazine ancillaries
- Crystal structure of mechanochemically synthesized Ag2CdSnS4