Metal carbonyl complexes of potentially ambidentate 2,1,3-benzothiadiazole and 2,1,3-benzoselenadiazole acceptors
-
Sebastian Plebst


Abstract
The compounds [W(CO)5(btd)], [W(CO)5(bsd] and [Re(CO)3(bpy)(bsd)](BF4), btd=2,1,3-benzothiadiazole and bsd=2,1,3-benzoselenadiazole were isolated and characterized experimentally (crystal structure, spectroscopy, spectroelectrochemistry) and by density functional theory calculations. The results confirm single N-coordination in all cases, binding to Se was calculated to be less favorable. Studies of one-electron reduced forms indicate that the N-coordination is maintained during electron transfer.
1 Introduction
The 2,1,3-benzochalcogenadiazoles (bcd) btd and bsd (Fig. 1) have become widely used as electron acceptor components in electronically functional organic (low band-gap) polymers [1], [2], [3], [4], [5], [6], [7], [8]. Other physical properties such as optical and electrochemical activity have also been well studied [1], [2], [3], [4], [5], [6], [7], [8]. However, little is known about the interaction of such molecules with metals [9], [10], [11], although the chalcogen, the two N-donor atoms, and the conjugated π system are potential metal binding sites (Fig. 2). It was shown in previous EPR reports that the reduced forms, the radical anions btd˙− or bsd˙−, can even serve as bridging ligands in symmetrical dinuclear complexes (μ-btd˙−)[MLn]2 and (μ-bsd˙−)[MLn]2 [12], [13], [14], [15]. EPR Spectra suggested high symmetry and thus the possibility of chalcogen coordination in certain mononuclear compounds [12].

The 2,1,3-benzochalcogenadiazole (bcd) ligands.
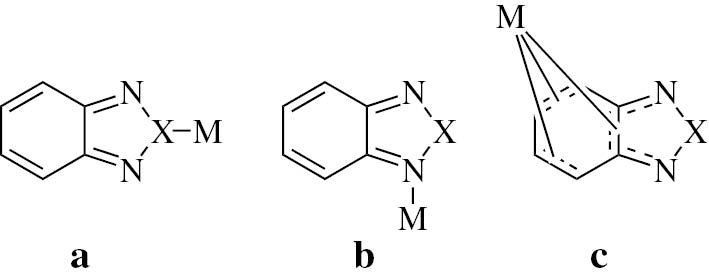
Different metal coordination alternatives for mononuclear metal complexes of the 2,1,3-benzochalcogenadiazoles.
In this work we describe the experimental and computational investigation of the coordination potential of btd and bsd acceptors versus the electron-rich metal carbonyl complex fragments W(CO)5 and [Re(CO)3(bpy)]+. Whereas the former has been used in earlier studies [12], [13], [14], [15], the latter proved to be an excellent binding site for weakly basic acceptor ligands such as TCNQ or N-heterocyclic cations [16], [17], [18].
2 Experimental section
2.1 Instrumentation
EPR spectra in the X band were recorded with a Bruker System EMX. 1H NMR spectra were taken on a Bruker AC 250 spectrometer. IR spectra were obtained using a Nicolet 6700 FT-IR instrument. UV/Vis/NIR absorption spectra were recorded on a J&M TIDAS spectrophotometer. Cyclic voltammetry was carried out in 0.1 M Bu4NPF6 solutions using a three-electrode configuration (glassy carbon working electrode, Pt counter electrode, Ag/AgCl reference) and a PAR 273 potentiostat and function generator. The ferrocene/ferrocenium (Fc/Fc+) couple served as internal reference. Spectroelectrochemistry was performed using an optically transparent thin-layer electrode (OTTLE) cell [19]. A two-electrode capillary served to generate intermediates for X band EPR studies [20].
2.2 Syntheses
2.2.1 [W(CO)5(bcd)] (1 and 2)
The synthesis from bcd and photogenerated W(CO)5(THF) has been described earlier [12], [13], [14], [15].

1:1H NMR ([D6]benzene): δ (ppm)=6.67 (br ddd, 1H(d)), 6.79 (br ddd, 1H(a)), 7.27 (d, 1H, (b), 3JHH=8.75 Hz), 7.47 (d, 1H(c), 3JHH=8.75 Hz). – Analysis for C11H4N2O5SW (460.07): calcd. C 28.72, N 6.09, H 0.88; found C 27.95, N 5.56, H 0.64.
2:1H NMR ([D8]toluene): δ [ppm]=6.59 (ddd, 1H(d), 3JHH=9 Hz, 4JHH=6 Hz, 5JHH=1 Hz), 6.72 (br ddd, 1H(a), 3JHH=9 Hz, 4JHH=6 Hz, 5JHH=1.25 Hz), 7.20 (d, 1H(b), 3JHH=9 Hz), 7.31 (d, 1H(c), 3JHH=9.25 Hz). – Analysis for C11H4N2O5SeW (506.97): calcd. C 26.06, N 5.53, H 0.80; found C 25.95, N 5.56, H 0.74.
2.2.2 [Re(CO)3(bpy)(bsd)](BF4) ([3](BF4))

Pentacarbonylchloridorhenium (150 mg, 0.41 mmol) and 65 mg (0.41 mmol) of 2,2′-bipyridine were heated in 40 mL of toluene for 5 h. Cooling yielded bipyridinetricarbonylchloridorhenium as a yellow solid, of which 100 mg (0.22 mmol) was dissolved in 35 mL of dichloromethane and treated with 42 mg (0.22 mmol) AgBF4. The mixture was stirred overnight, and the precipitated AgCl was removed. 40 mg (0.22 mmol) of bsd was added and the orange-yellow product was obtained in 58% yield (100 mg, 0.13 mmol) after cooling to −8°C. – 1H NMR ([D6]acetone): δ [ppm]=7.64 (br m, 1H), 7.88–7.79 (br m, 2H), 8.00 (br m, 2H), 8.14 (br d, t, 1H), 8.51 (br t, d, 2H), 8.82 (br d, 2H), 9.56 (br d, t, 2H). – Analysis for C19H14BCl3F4N4O2ReSe (781.22): calcd. C 28.94, N 7.10, 1.79; found C 27.55, N 6.56, H 1.88.
2.3 Crystal structure analyses
Single crystals were obtained by cooling concentrated solutions in dichloromethane (1, 3(BF4)) or hexane (2) to about –10°C. An Apex II Duo diffractometer was used at −173°C measuring temperature with MoKα radiation (λ=71.073 pm). After absorption correction the structures were solved by Patterson methods and refined by full-matrix least-squares on F2 [21], [22], [23]. The hydrogen atoms were included via the riding model. Ortep representations depict 50% probability regions.
Despite repeated attempts the poor crystal quality of 1 precluded a refinement to a comparable standard. Due to the isotropic refinement and the large number of necessary restraints the detailed structural data will thus not be discussed.
CCDC 1557781 and 1557782 contain the supplementary crystallographic data for 2 and 3(BF4) which can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
2.4 Calculations
The electronic structure calculations on all compounds were performed by density functional theory (DFT) method using the Gaussian 09 [24] program package. DFT calculations employed the Perdew-Burke-Ernzerhof [25], [26] (PBE0) hybrid functional. The geometry of the open shell form was calculated by a UKS approach. For H, C, N, O, S and Se atoms polarized triple-ζ basis sets 6-311G(d) [27], together with quasirelativistic effective core pseudopotentials and a corresponding optimized set of basis functions for Re and W [28], [29] were used. Geometry optimizations were followed by vibrational analysis. The solvent was described by the polarizable conductor model (PCM) [30].
3 Results and discussion
In the non-reduced neutral forms the ligands btd and bsd form only mononuclear isolable complexes with the carbonyl-tungsten and -rhenium complex fragments employed here. This was observed both for the solids (single crystal XRD) and for solutions (1H NMR). The EPR-detected N,N′-coordinated dinuclear systems of reduced states are thus due to a strongly enhanced charge at the partially quinonoid nitrogen centers [12], [13], [14], [15], leading to an ECEC mechanism as apparent from cyclic voltammetry (charge transfer assisted polynucleation) [15].
The complexes {W(CO)5(btd)] (1), [W(CO)5(bsd)] (2) and [Re(CO)3(bpy)(bsd)](BF4) (3(BF4)), bpy=2,2′-bipyridine, were characterized analytically and, in part, by X-ray diffraction of single crystals (Figs. 3–5 and Tables 1 and 2). Due to poor crystal and X-ray data quality the individual bond parameters of 1 will not be discussed in detail.

Molecular structure of 2 in the crystal. H atoms are omitted for clarity.
![Fig. 4: Molecular structure of [3+] in the crystal of [3] (BF4)×CH2Cl2.](/document/doi/10.1515/znb-2017-0100/asset/graphic/j_znb-2017-0100_fig_004.jpg)
Molecular structure of [3+] in the crystal of [3] (BF4)×CH2Cl2.

SeN/SeN Dimer formation of 2 (left) and π/π interaction of 2 in the crystal (right).
Selected crystallographic data of 1, 2 and [3](BF4)×CH2Cl2.
| 1 | 2 | [3](BF4)×CH2Cl2 | |
|---|---|---|---|
| Empirical formula | C11H4N2O5SW | C11H4N2O5SeW | C19H12N4O3ReSe, CH2Cl2, BF4 |
| Formula mass Mr | 460.07 | 506.97 | 781.23 |
| Crystal system | Monoclinic | Triclinic | Monoclinic |
| Space group | P21/c | P1̅ | P21/n |
| a, pm | 787.04(13) | 700.56(4) | 1014.33(6) |
| b, pm | 1338.6(2) | 823.86(5) | 1879.60(9) |
| c, pm | 2478.3(4) | 1201.57(7) | 1374.06(8) |
| α, deg | 90 | 97.668(3) | 90 |
| β, deg | 95.753(5) | 92.709(3) | 111.554(3) |
| γ, deg | 90 | 108.070(2) | 90 |
| V, pm3 | 259 770(70) | 650 500(70) | 243 650(20) |
| Z | 8 | 2 | 4 |
| μ, mm−1 | 9.1 | 11.17 | 6.8 |
| T, K | 100(2) | 100(2) | 100(2) |
| ρcalcd, g cm−3 | 2.35 | 2.55 | 2.13 |
| F(000), e | 1712.0 | 464.0 | 1480 |
| 2θ range, deg | 3.04–50.10 | 3.44–61.29 | 3.85–61.38 |
| Data/restraints/parameters | 4606/532/362 | 3964/0/182 | 7529/0/325 |
| R1/wR2 [I>2 σ(I)] | 0.0896/0.2628 | 0.0122/0.0266 | 0.0207/0.0469 |
| R1/wR2 (all data) | 0.0995/0.2754 | 0.0140/0.0270 | 0.0269/0.0489 |
| GOF on F2 | 1.131 | 1.049 | 1.114 |
| Δρfin (max/min), e×106 pm−3 | 5.68/–3.87 | 8.39/–7.08 | 1.67/–1.42 |
Selected experimental and DFT calculated bond lengths of 2.
| Bindung | pm (exp) | pm (DFT) |
|---|---|---|
| W1–N2 | 224.2(1) | 226.1 |
| W1–C1 | 206.1(2) | 205.4 |
| W1–C2 | 205.8(2) | 205.4 |
| W1–C3 | 203.5(2) | 205.0 |
| W1–C4 | 204.7(2) | 205.0 |
| W1–C5 | 198.1(2) | 199.1 |
| Se–N1 | 178.7(1) | 175.9 |
| Se–N2 | 181.1(1) | 179.2 |
| N1–C10 | 133.0(2) | 132.2 |
| N2–C15 | 133.7(2) | 132.2 |
| C10–C11 | 142.8(2) | 142.1 |
| C10–C15 | 144.6(2) | 144.9 |
| C11–C12 | 136.1(2) | 135.7 |
| C12–C13 | 143.0(2) | 142.5 |
| C13–C14 | 136.4(2) | 136.0 |
| C14–C15 | 142.8(2) | 141.7 |
The structural studies reveal the expected [31] o-quinonediimine-type C–C and C–N bond alternancy and the metal coordination to one of the two equivalent N atoms, with bond parameters that are typical and which can be well reproduced by DFT calculations (Table 2, Fig. 6). The complex ion [Re(CO)3(bpy)(bsd)]+ in 3+ exhibits the common [16], [17], [18] fac arrangement of the carbonyl ligands (Fig. 4). Three kinds of intermolecular interactions were observed: The tungsten compound 2 reveals intermolecular SeN/SeN interactions (Fig. 5) at 282.3 pm – a previously observed feature [32], [33]. The tungsten-containing compounds 1 and 2 show stacking through π/π interactions [34] of the btd (380 pm) or bsd (350 pm) ligands with anti arrangement (Fig. 5). The BF4− anion in the crystal of [3](BF4)×CH2Cl2 exhibits F···H hydrogen bonding to the solvent and to the bsd and bpy ligands.
![Fig. 6: DFT (PBE0 in vacuo) calculated energy minima of the isomers 2a and 2b from [W(CO)5(bsd)].](/document/doi/10.1515/znb-2017-0100/asset/graphic/j_znb-2017-0100_fig_006.jpg)
DFT (PBE0 in vacuo) calculated energy minima of the isomers 2a and 2b from [W(CO)5(bsd)].
According to DFT calculations both N and Se coordination of W(CO)5 result in local energy minima for 2 (Fig. 6), with the N-bonded isomer being more stable by 0.42 eV. This value is diminished to 0.34 eV for the reduced forms 2a− and 2b−; the rhenium complex ion 3+ is calculated with an even larger preference (0.51 eV) for N vs. Se coordination.
The reversible reduction of complexes [12], [13], [14], [15], including the E1/2=–1.25 V vs. Fc+/0 in CH2Cl2/0.1 M Bu4NPF6, invited studies of the monoanionic forms by EPR, IR and UV/VIS spectroelectrochemistry [35].
In addition to the previously reported pentacarbonyltungsten radical complexes [12], [13], [14], [15] we have now investigated by EPR the electrochemically generated neutral rhenium compound [Re(CO)3(bpy)(bsd)]˙ (3˙) (Fig. 7). The partially resolved spectrum (g=2.0057) reveals hyperfine coupling from 185,187Re (0.94 mT), 14N (0.63 mT) and 1H (0.21 mT); the 185,187Re isotopes (37.4 and 62.6% nat. abundance) have I=5/2 and very similar nuclear magnetic moment. These results are fully compatible with previously reported data of related carbonylrhenium radical complexes [16], [17], [18], [36] and with DFT calculated spin densities as depicted in Fig. 8. The spin is confined to the bsd ligand in all cases. The contribution from the chalcogen is supported by experiment, a coupling (6.8 mT) of the unpaired electron with 77Se (I=½, 7.6% nat. abundance) is observed in an amplified spectrum. The spin density on Re was calculated at only –0.020, and there is negligible spin density on bpy although the corresponding π* acceptor MO is only 0.3 eV higher than that of bsd.

ESR spectrum of electrogenerated 3˙ in 0.1 M CH2Cl2/NBu4PF6 (black, bottom) with simulation (red, top).

Spin distribution of 2˙− (left) and 3˙ (right) (PBE0, PCM/CH2Cl2).
The calculated bond length changes on reduction of 2 or 3+ (Table 3) are rather small, in agreement with the excellent acceptor capacity of the bsd ligand.
Selected experimental and DFT optimized bond lengths (pm) of 2, 2− and 3+.
| Bond | 2 | 2 (DFT) | 2˙− (DFT) | Bond | [3+ ] | [3+ ] (DFT) |
|---|---|---|---|---|---|---|
| W(1)–N(2) | 242.2(1) | 226.1 | 223.2 | Re(1)–N(3) | 218.7(2) | 221.5 |
| Se(1)–N(1) | 178.7(1) | 175.9 | 182.9 | Se(1)–N(3) | 180.6(2) | 181.3 |
| Se(1)–N(2) | 181.1(1) | 179.2 | 179.4 | Se(1)–N(4) | 176.9(2) | 176.1 |
| N(1)–C(10) | 133.0(2) | 132.2 | 134.4 | N(3)–C(1) | 133.9(3) | 133.5 |
| N(2)–C(15) | 133.7(2) | 132.2 | 135.4 | N(4)–C(6) | 133.2(3) | 132.4 |
| C(10)–C(15) | 144.6(2) | 144.9 | 144.4 | C(1)–C(2) | 142.4(3) | 142.0 |
| C(1)–C(6) | 144.4(3) | 145.3 | ||||
| C(2)–C(3) | 136.1(3) | 136.4 | ||||
| C(3)–C(4) | 143.0(4) | 142.8 | ||||
| C(4)–C(5) | 134.8(4) | 136.0 | ||||
| C(5)–C(6) | 143.4(3) | 142.5 | ||||
| C–O(cis)a | 114.6(3) | 115.0 | ||||
| 115.0 | ||||||
| C–O(trans)a | 114.3(3) | 114.8 |
acis/trans relative to the bsd ligand.
Infrared spectroelectrochemistry in the carbonyl stretching region (Fig. 9, Table 4) exhibits only minor low-frequency shifts (10–60 cm−1) on reduction, confirming the EPR-supported notion of a btd- or bsd-centered electron transfer. The larger spread of the bands in the reduced states of 1˙− and 2˙− agrees with the diminished π acceptor capacity of bcd˙− relative to unreduced bcd. The decreased low frequency split (“E”) for 3˙ relative to 3+ signifies similar ligand effects of bsd˙− and unreduced bpy.
 (bottom) in the CO stretching region in 0.1 M CH2Cl2/NBu4PF6.](/document/doi/10.1515/znb-2017-0100/asset/graphic/j_znb-2017-0100_fig_009.jpg)
IR spectroelectrochemical measurements of the reduction of 1 (left, top) and 2 (right, top) and of [3](BF4) (bottom) in the CO stretching region in 0.1 M CH2Cl2/NBu4PF6.
IR vibrational bands of 1n and 2n.
| Complex | Absorption maximum (cm–1) |
|---|---|
| 1 | 2075, 1975, 1935, 1920 (sh) |
| 1˙− | 2060, 1970, 1915, 1865 |
| 2 | 2075, 1975, 1935, 1915 (sh) |
| 2˙− | 2060, 1965, 1915, 1860 |
| [3+] | 2040, 1945, 1935 |
| 3˙ | 2020, 2010 (sh), 1915 |
The long-wavelength absorptions of the compounds 1, 2 and 3(BF4) are due to metal-to-ligand charge transfer (MLCT) processes [12], [13], [14], [15]. In each case the LUMO is located at the btd or bsd acceptor, even in the presence of the secondary acceptor bpy for 3+. Reduction causes the MLCT bands at about λmax=500 nm to disappear in 1°/− and 2°/− from the visible region due to single occupancy of the acceptor MO. On the other hand, a new long-wavelength band emerges at 485 nm on reduction of 3+ which reflects the presence of a close lying second acceptor MO π*(bpy) (Fig. 10, Table 5).
 (bottom) in 0.1 M CH2Cl2/NBu4PF6.](/document/doi/10.1515/znb-2017-0100/asset/graphic/j_znb-2017-0100_fig_010.jpg)
UV/Vis/NIR spectroelectrochemical response on reduction of 2 (top) and of [3](BF4) (bottom) in 0.1 M CH2Cl2/NBu4PF6.
UV/Vis/NIR spectroelectrochemical data of 1n and 2n in 0.1 M NBu4PF6/CH2Cl2 in an OTTLE cell.
| Complex | λ (in nm) (ε in M–1 cm–1) |
|---|---|
| 1 | 315 (10 575), 395 (2645), 480 (5465) |
| 1˙− | 320 (6300), 350 (5620), 395 (2630) |
| 2 | 340 (11 100), 405 (2570), 510 (3410) |
| 2˙− | 330 (5520), 370 (6410), 405 (2570) |
| [3+ ] | 325, 340, 390 (sh) |
| 3˙ | 295, 380, 485br |
4 Conclusion
This study has shown that the neutral btd and bsd ligands are indeed good π acceptors but poor σ bases. Coordination in the unreduced state is thus restricted to mononuclear N-coordinated complexes even with electron rich carbonylmetal fragments. Reduction occurs exclusively on the bcd ligands with almost vanishing spin distribution to the metal or co-ligands such as bpy. It is not surprising, therefore, that the well used bcd acceptor components have not been used more in coordination chemistry despite the presence of several potential coordination sites. Less conventional metal species may be required to take advantage of this potential, extending the application of such components beyond pure organic systems [1], [2].
Dedicated to: Professor Dietrich Gudat on the occasion of his 60th birthday.
Acknowledgements
This work has been supported by the Land Baden-Württemberg and the DFG. We thank Dr. Wolfgang Frey for crystal structure determinations.
References
[1] B. A D. Neto, P. H. P. R. Carvalho, J. R. Correa, Acc. Chem. Res.2015, 48, 1560.10.1021/ar500468pSearch in Google Scholar
[2] S. Mondal, M. Konda, B. Kauffmann, M. K. Manna, A. K. Das, Cryst. Growth Des.2015, 15, 5548.10.1021/acs.cgd.5b01179Search in Google Scholar
[3] X. Song, M. Wang, L. Kong, J. Zhao, RSC Adv.2017, 7, 18189.10.1039/C7RA01449JSearch in Google Scholar
[4] E. Zhou, J. Cong, K. Hashimoto, K. Tajima, Macromolecules2013, 46, 763.10.1021/ma302596kSearch in Google Scholar
[5] J.-H. Kim, J. B. Park, S. A. Shin, M. H. Hyun, D.-H. Hwang, Polymer2014, 55, 3605.10.1016/j.polymer.2014.05.055Search in Google Scholar
[6] B. Shaik, J.-H. Han, D. J. Song, H.-M. Kang, S.-G. Lee, Can. J. Chem.2016, 94, 553.10.1139/cjc-2015-0542Search in Google Scholar
[7] S. Yao, B. Kim, X. Yue, M. Y. Colon Gomez, M. V. Bondar, K. D. Belfield, ACS Omega2016, 1, 1149.10.1021/acsomega.6b00289Search in Google Scholar
[8] W.-Q. Zhang, Q.-Y. Li, Q. Zhang, Y. Lu, H. Lu, W. Wang, X. Zhao, X.-J. Wang, Inorg. Chem.2016, 55, 1005.10.1021/acs.inorgchem.5b02626Search in Google Scholar
[9] R. H. Hanson, C. E. Meloan, Inorg. Nucl. Chem. Lett.1971, 7, 467.10.1016/0020-1650(71)80185-XSearch in Google Scholar
[10] M. Herberhold, A. F. Hill, J. Organomet. Chem.1989, 377, 151.10.1016/0022-328X(89)80061-0Search in Google Scholar
[11] D. A. Bashirov, T. S. Sukhikh, N. V. Kuratieva, D Y. Naumov, S. N. Konchenko, N. A. Semenov, A. V. Zibarev, Polyhedron2012, 42, 168.10.1016/j.poly.2012.05.015Search in Google Scholar
[12] W. Kaim, V. Kasack, Angew. Chem. Int. Ed. Engl.1982, 21, 700.10.1002/anie.198207002Search in Google Scholar
[13] W. Kaim, J. Organomet. Chem.1984, 264, 317.10.1016/0022-328X(84)85078-0Search in Google Scholar
[14] W. Kaim, S. Kohlmann, Inorg. Chim. Acta1985, 101, L21.10.1016/S0020-1693(00)85989-7Search in Google Scholar
[15] W. Kaim, S. Kohlmann, A. J. Lees, M. M. Zulu, Z. Anorg. Allg. Chem.1989, 575, 97.10.1002/zaac.19895750113Search in Google Scholar
[16] H. Hartmann, W. Kaim, I. Hartenbach, T. Schleid, M. Wanner, J. Fiedler, Angew. Chem. Int. Ed.2001, 40, 2842.10.1002/1521-3773(20010803)40:15<2842::AID-ANIE2842>3.0.CO;2-RSearch in Google Scholar
[17] H. Hartmann, W. Kaim, M. Wanner, A. Klein, S. Frantz, C. Duboc-Toia, J. Fiedler, S. Záliš, Inorg. Chem.2003, 42, 7018.10.1021/ic034232lSearch in Google Scholar
[18] S. Berger, A. Klein, W. Kaim, J. Fiedler, Inorg. Chem.1998, 37, 5664.10.1021/ic980268fSearch in Google Scholar
[19] F. Hartl, H. Luyten, H. A. Nieuwenhuis, G. Schoemaker, Appl. Spectrosc.1994, 48, 1522.10.1366/0003702944027787Search in Google Scholar
[20] W. Kaim, S. Ernst, V. Kasack, J. Am. Chem. Soc.1990, 112, 173.10.1021/ja00157a028Search in Google Scholar
[21] G. M. Sheldrick, Shelxl-97, Program for the Refinement of Crystal Structures, University of Göttingen, Göttingen (Germany) 1997.Search in Google Scholar
[22] G. M. Sheldrick, Shelxtl, Bruker AXS Inc., Madison, Wisconsin (USA) 1998.Search in Google Scholar
[23] G. M. Sheldrick, Acta Crystallogr.2008, A64, 112.10.1107/S0108767307043930Search in Google Scholar PubMed
[24] M. J. Frisch, G. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Blonio, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, D. J. Fox, Gaussian 09 (revision E.01), Gaussian, Inc, Wallingford CT (USA) 2009.Search in Google Scholar
[25] J. P. Perdew, K. Burke, M. Ernzerhof, Phys. Rev. Lett.1996, 77, 3865.10.1103/PhysRevLett.77.3865Search in Google Scholar PubMed
[26] C. Adamo, V. Barone, J. Chem. Phys.1999, 110, 6158.10.1063/1.478522Search in Google Scholar
[27] P. J. Hay, J. Chem. Phys.1977, 66, 4377.10.1063/1.433731Search in Google Scholar
[28] D. Andrae, U. Häussermann, M. Dolg, H. Stoll, H. Preuss, Theor. Chim. Acta1990, 77, 123.10.1007/BF01114537Search in Google Scholar
[29] J. M. L. Martin, A. Sundermann, J. Chem. Phys.2001, 114, 3408.10.1063/1.1337864Search in Google Scholar
[30] M. Cossi, N. Rega, G. Scalmani, V. Barone, J. Comput. Chem.2003, 24, 669.10.1002/jcc.10189Search in Google Scholar PubMed
[31] A. C. Gomes, G. Biswas, A. Banerjee, W. L. Duax, Acta Crystallogr.1989, C45, 73.10.1107/S0108270188008376Search in Google Scholar
[32] A.-J. Zhou, S.-L. Zheng, Y. Fang, M.-L. Tong, Inorg. Chem.2005, 44, 4457.10.1021/ic050472kSearch in Google Scholar PubMed
[33] C.-K. Tan, J. Wang, J.-D. Leng, L.-L. Zheng, M.-L. Tong, Eur. J. Inorg. Chem.2008, 2008, 771.10.1002/ejic.200700930Search in Google Scholar
[34] C. Janiak, J. Chem. Soc. Dalton Trans.2000, 29, 3885.10.1039/b003010oSearch in Google Scholar
[35] W. Kaim, J. Fiedler, Chem. Soc. Rev.2009, 38, 3373.10.1039/b504286kSearch in Google Scholar PubMed
[36] A. Grupp, M. Bubrin, F. Ehret, H. Kvapilová, S. Záliš, W. Kaim, J. Organomet. Chem.2014, 751, 678.10.1016/j.jorganchem.2013.08.030Search in Google Scholar
©2017 Walter de Gruyter GmbH, Berlin/Boston
Articles in the same Issue
- Frontmatter
- In this Issue
- Preface
- Congratulations to Dietrich Gudat
- On the dimorphism of Pr6Mo10O39
- Rhodium-rich silicides RERh6Si4 (RE=La, Nd, Tb, Dy, Er, Yb)
- Coordination of the ambiphilic phosphinoborane tBu2PCH2BPh2 to Cu(I)Cl
- N-Heterocyclic germylenes and stannylenes of the type [Fe{(η5-C5H4)NR}2E] with bulky alkyl substituents
- Die Europium(II)-Oxidhalogenide Eu2OBr2 und Eu2OI2
- Structure and spectroscopic properties of porphyrinato group 14 derivatives: Part I – Phenylacetylido ligands
- Synthesis, solid-state structures and reduction reactions of heteroleptic Ge(II) and Sn(II) β-ketoiminate complexes
- Reactions of Al/P, Ga/P and P–H functionalized frustrated Lewis pairs with azides and a diazomethane – formation of adducts and capture of nitrenes
- Metal carbonyl complexes of potentially ambidentate 2,1,3-benzothiadiazole and 2,1,3-benzoselenadiazole acceptors
- Lithium alkaline earth tetrelides of the type Li2AeTt (Ae=Ca, Ba, Tt=Si, Ge, Sn, Pb): synthesis, crystal structures and physical properties
- Magnetic properties of the germanides RE3Pt4Ge6 (RE=Y, Pr, Nd, Sm, Gd–Dy)
- Overcrowded aminophospanitrenes: a case study
- PCl bond length depression upon coordination of a diazaphosphasiletidine to a group 13 element Lewis acid or a transition metal carbonyl fragment – Synthesis and structural characterization of diazaphosphasiletidine adducts with P-coordination
- Iminopyridine ligand complexes of group 14 dihalides and ditriflates – neutral chelates and ion pair formation
- On the structure of the P-iodo-, bromo- and chloro-bis(imino)phosphoranes: A DFT study
- (Dicyclohexyl(2-(dimesitylboryl)phenyl)phosphine: en route to stable frustrated Lewis pairs-hydrogen adducts in water
- Insertion of phenyl isocyanate into mono- and diaminosilanes
Articles in the same Issue
- Frontmatter
- In this Issue
- Preface
- Congratulations to Dietrich Gudat
- On the dimorphism of Pr6Mo10O39
- Rhodium-rich silicides RERh6Si4 (RE=La, Nd, Tb, Dy, Er, Yb)
- Coordination of the ambiphilic phosphinoborane tBu2PCH2BPh2 to Cu(I)Cl
- N-Heterocyclic germylenes and stannylenes of the type [Fe{(η5-C5H4)NR}2E] with bulky alkyl substituents
- Die Europium(II)-Oxidhalogenide Eu2OBr2 und Eu2OI2
- Structure and spectroscopic properties of porphyrinato group 14 derivatives: Part I – Phenylacetylido ligands
- Synthesis, solid-state structures and reduction reactions of heteroleptic Ge(II) and Sn(II) β-ketoiminate complexes
- Reactions of Al/P, Ga/P and P–H functionalized frustrated Lewis pairs with azides and a diazomethane – formation of adducts and capture of nitrenes
- Metal carbonyl complexes of potentially ambidentate 2,1,3-benzothiadiazole and 2,1,3-benzoselenadiazole acceptors
- Lithium alkaline earth tetrelides of the type Li2AeTt (Ae=Ca, Ba, Tt=Si, Ge, Sn, Pb): synthesis, crystal structures and physical properties
- Magnetic properties of the germanides RE3Pt4Ge6 (RE=Y, Pr, Nd, Sm, Gd–Dy)
- Overcrowded aminophospanitrenes: a case study
- PCl bond length depression upon coordination of a diazaphosphasiletidine to a group 13 element Lewis acid or a transition metal carbonyl fragment – Synthesis and structural characterization of diazaphosphasiletidine adducts with P-coordination
- Iminopyridine ligand complexes of group 14 dihalides and ditriflates – neutral chelates and ion pair formation
- On the structure of the P-iodo-, bromo- and chloro-bis(imino)phosphoranes: A DFT study
- (Dicyclohexyl(2-(dimesitylboryl)phenyl)phosphine: en route to stable frustrated Lewis pairs-hydrogen adducts in water
- Insertion of phenyl isocyanate into mono- and diaminosilanes