Regioselective C-3 arylation of coumarins with arylhydrazines via radical oxidation by potassium permanganate
-
Jin-Wei Yuan
 ,
Wei-Jie Li
,
Wei-Jie Li

Abstract
An efficient protocol for oxidative C-3 arylation of coumarins with arylhydrazine has been developed using potassium permanganate as an oxidant. The arylated coumarins with different electronic properties were obtained in moderate to good yields. The developed protocol for direct C-3 arylation of coumarins could be extended to quinolinones.
1 Introduction
Coumarins are significant natural products, which display wide and interesting pharmacological properties such as anti-breast cancer, anti-HIV, anti-Alzheimer, vasorelaxant, and anti-aggregatory activities [1], [2], [3], [4]. Coumarin derivatives have proven to be useful skeletons in organic synthesis as valuable building blocks and in pharmaceuticals. In particular, 3-arylcoumarins have important applications in dyes [5], [6] and pharmaceutical industries [7], [8]. There are several drugs constituting functionalized 3-arylcoumarin as the basic core in them, which possess wide-ranging activities [9], [10], [11], [12], [13], [14], [15].
Despite pervasiveness of 3-arylcoumarins in several fields of science, they can be obtained by a few general methods such as using a cycloaddition ring construction approach [16], [17], [18], and by transition metal-catalyzed coupling reactions through C–H bond activation [19], [20], [21], [22], [23], [24], [25]. Of these pathways, a direct arylation approach to form 3-arylcoumarins has been realized via transition metal-mediated cross-coupling reactions [26], [27], [28], [29]. The common limitations of most of these methods are inevitability of transition metals which possess high cost and toxicity, necessity of prefunctionalization of the C–H bond at 3-position of the starting coumarins with a halogen or carboxyl group, and use of expensive arylating counterparts such as boronic acid, elevated temperature, and longer reaction time. Because a radical arylation reaction can basically offer a much more straightforward access to reaction substrates, mainly due to the fact that such arylations are comparable to C–H activation reactions and less functionalized starting materials are therefore required, there has recently been increasing interest in improving the reaction type [30], [31], [32]. Based on the traditional aryl radical sources for arylation reactions, which are arenediazonium salts, remarkable advances have recently been achieved by employing photocatalysts and optimized Gomberg–Bachmann conditions [33], [34], [35]. But arenediazonium salts are difficult to synthesize, unstable, known as explosive under certain circumstances, and give very poor yield with formation of azoproducts. Therefore, with the background of these inadequacies, developing an adaptable method for 3-arylation of coumarins, which has short reaction time under mild reaction conditions, is still a significant challenge.
Arylhydrazines are known to undergo oxidation to form aryl radicals in the presence of oxygen [36], [37], catalysts [38], [39], and stoichiometric amounts of high valent metal oxidants such as lead(IV) acetate [40], manganese(III) acetate [36], [41], ferrate salt [42], [43], or hypervalent iodine(V) reagent [44], [45]. Another aspect that will facilitate future applications is that arylhydrazines are simple and cheap starting materials. Recently, the oxidative radical 3-arylation of coumarins and quinolin-4-ones using arylhydrazines as aryl radical sources has been reported in the presence of oxygen from air [46], [47]. But the oxidative radical coupling reaction has longer time, and the scope of substrates is also narrow. Guided by the recent studies on using arylhydrazines as aryl radical sources, we decided to investigate the modular synthesis for 3-arylcoumarins by this method. Herein, we disclose an oxidant radical regioselective arylation of coumarins and quinolinones with arylhydrazines using KMnO4 as oxidant to produce 3-arylcoumarin and 3-arylquinolinone derivatives in moderate to good yields under mild conditions (Scheme 1).
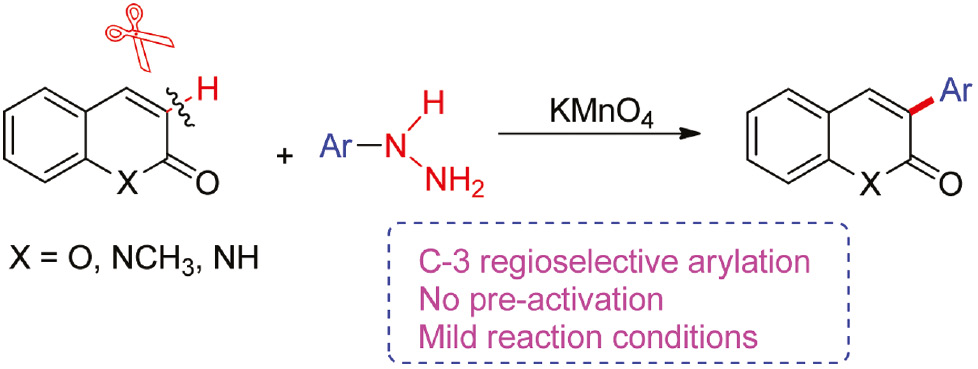
Synthesis of 3-arylcoumarins and 3-arylquinolinones.
2 Results and discussion
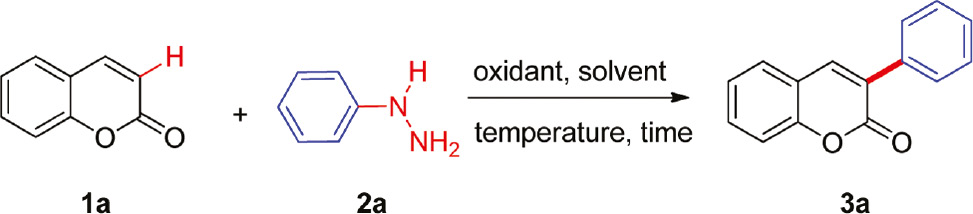
We began our study with the selection of coumarin (1a) and phenylhydrazine (2a) as model substrates to optimize the reaction condition (Table 1). Initially in the absence of the oxidant, the desired product 3-arylcoumarin (3a) was not obtained when the reaction was carried out in CH3CN at 80°C for 4.0 h under air atmosphere (entry 1). Fe(NO3)3, MnO2, Mn(OAc)2, Mn(OAc)3, KMnO4, H2O2, and t-butyl hydroperoxide (TBHP) were investigated as oxidants (Table 1, entries 2–8), but only Mn(OAc)3 and KMnO4 gave the desired product in decent yields, and KMnO4 proved to be the most effective. The amount of KMnO4 was also examined (Table 1, entries 6 and 9–12). The best yield was 65% when 3.0 eq KMnO4 was used, which indicated that the oxidant KMnO4 and the quantity of oxidant played important roles in this reaction. Subsequently, different solvents such as CH3CN, H2O, dioxane, CH3OH, dimethylsulfoxide (DMSO), 1,2-dichloroethane (DCE), and tetrahydrofuran (THF) were screened based on the solubility of the substrates (Table 1, entries 6 and 13–18) and CH3CN was found to be the best solvent for this transformation. The ratio of substrates coumarin and phenylhydrazine was investigated, and the ratio 1:2 of coumarin and phenylhydrazine proved to be the best result, providing 80% yield of 3a (Table S1, Supplementary Information available online). With increasing amount of phenylhydrazine, the yield of 3a decreased as the side reaction occurred to lead to byproducts. When the reaction temperature was improved from 40°C to 100°C, the yield of 3a was enhanced from 40% to 85% (Table S1, entry 4, and Table 1, entries 19–22). However, the product yields dramatically dropped if the reaction temperature was higher than 80°C, and the optimal reaction temperature was 80°C. Finally, reaction times were also examined, and 3.0 h gave good yield (Table 1, entries 23–26). After screening the reaction conditions, the optimal result was identified: the reaction of coumarin (1.0 equiv.) and phenylhydrazine (2.0 equiv.) was carried out in the presence of KMnO4 (3.0 equiv.) in CH3CN at 80°C for 3.0 h (Table 1, entry 26), which afforded 3-arylcoumarin in 85% yield.
Optimization of reaction conditionsa.
| Entry | Oxidant (eq) | Solvent | Temp. (°C) | Time (h) | Yieldb,c (%) |
|---|---|---|---|---|---|
| 1 | – | CH3CN | 80 | 4.0 | nr |
| 2 | Fe(NO3)3 (3.0) | CH3CN | 80 | 4.0 | 8 |
| 3 | MnO2 (3.0) | CH3CN | 80 | 4.0 | <5 |
| 4 | Mn(OAc)2 (3.0) | CH3CN | 80 | 4.0 | nr |
| 5 | Mn(OAc)3 (3.0) | CH3CN | 80 | 4.0 | 45 |
| 6 | KMnO4 (3.0) | CH3CN | 80 | 4.0 | 65 |
| 7 | H2O2 (3.0) | CH3CN | 80 | 4.0 | 10 |
| 8 | TBHP (3.0) | CH3CN | 80 | 4.0 | nr |
| 9 | KMnO4 (0.2) | CH3CN | 80 | 4.0 | 10 |
| 10 | KMnO4 (1.0) | CH3CN | 80 | 4.0 | 45 |
| 11 | KMnO4 (2.0) | CH3CN | 80 | 4.0 | 60 |
| 12 | KMnO4 (4.0) | CH3CN | 80 | 4.0 | 30 |
| 13 | KMnO4 (3.0) | H2O | 80 | 4.0 | 30 |
| 14 | KMnO4 (3.0) | Dioxane | 80 | 4.0 | 40 |
| 15 | KMnO4 (3.0) | CH3OH | 80 | 4.0 | 50 |
| 16 | KMnO4 (3.0) | DMSO | 80 | 4.0 | 63 |
| 17 | KMnO4 (3.0) | DCE | 80 | 4.0 | 60 |
| 18 | KMnO4 (3.0) | THF | 80 | 4.0 | 32 |
| 19 | KMnO4 (3.0) | CH3CN | 40 | 4.0 | nr |
| 20 | KMnO4 (3.0) | CH3CN | 60 | 4.0 | 40 |
| 21 | KMnO4 (3.0) | CH3CN | 90 | 4.0 | 80 |
| 22 | KMnO4 (3.0) | CH3CN | 100 | 4.0 | 75 |
| 23 | KMnO4 (3.0) | CH3CN | 80 | 0.5 | 40 |
| 24 | KMnO4 (3.0) | CH3CN | 80 | 1.0 | 60 |
| 25 | KMnO4 (3.0) | CH3CN | 80 | 2.0 | 80 |
| 26 | KMnO4 (3.0) | CH3CN | 80 | 3.0 | 85 |
aReaction conditions: coumarin 1a (0.5 mmol, 73 mg), phenylhydrazine 2a (1.0 mmol, 108 mg), oxidant in solvent (10 mL).
bIsolated yields.
cnr = no reaction.
With optimal reaction conditions in hand, the scope and generality of this methodology was further investigated (Table 2). First, a variety of substituted arylhydrazines were allowed to react with coumarin (Table 2, 3a–3o). Initially, the direct use of commercially available arylhydrazine hydrochlorides as the substrates did not lead to the desired products. Pretreatment of these hydrochloride salts with NaOH is required. Reactions of arylhydrazines having electron-donating groups such as –CH3, –C2H5, and –OCH3 with coumarin afforded 3-arylcoumarins 3a–3f in good to excellent yields of 78%–89%. Also the reaction of arylhydrazines bearing electron-withdrawing groups such as –F, –Cl, –CN, and –OCF3 with coumarin proceeded smoothly and afforded 3g–3j, 3m, and 3n in moderate yields of 40%–70%. Especially, arylhydrazines bearing the strong electron-withdrawing –CN and –OCF3 groups could also provide corresponding 3-arylated products in moderate yield (3i and 3j). It was noteworthy that arylhydrazine halides were well tolerated, providing handles for further functionalization (e.g. with a Suzuki reaction). However, 4-bromine function in the phenylhydrazine afforded only 20% yield (3l); the reason remained unknown. On the other hand, no expected product was observed in the case of ortho-bromophenylhydrazine (3k). It is not suitable for this transformation likely because of steric hindrance of ortho-bromophenylhydrazine. It is worth mentioning that an alkylhydrazine, tert-butylhydrazine, was also applied to the reaction, and the expected product was obtained (3o) in 35% yield. The fact demonstrated that alkylhydrazines are suitable precursors for alkyl radicals in this process.
Synthesis of 3-arylcoumarins from coumarins and arylhydrazinesa.
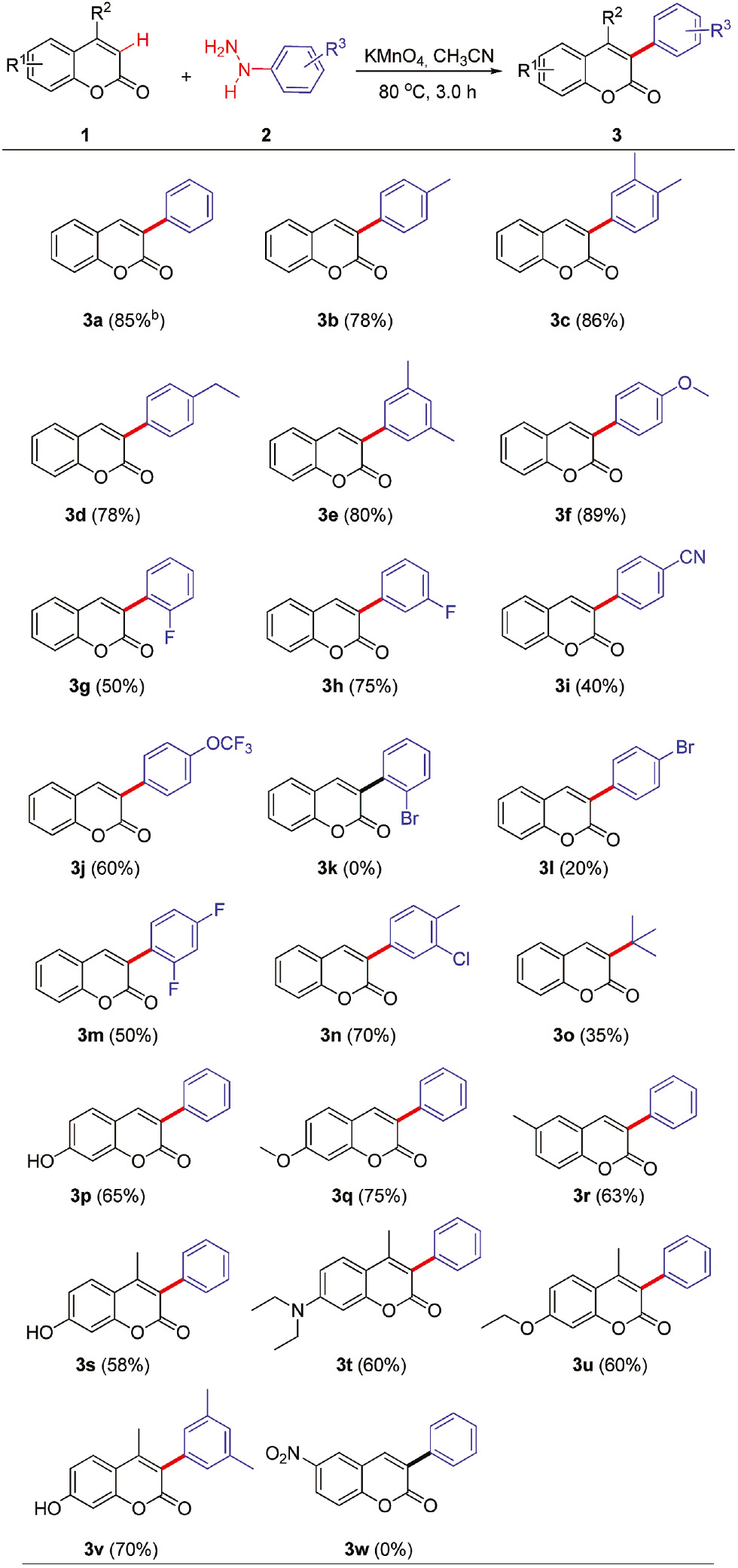
aReaction conditions: coumarins 1 (0.5 mmol), arylhydrazines 2 (1.0 mmol), KMnO4 (1.5 mmol), CH3CN (10 mL) at 80°C for 3.0 h.
bIsolated yields in parentheses.
Further, the scope of this methodology was also investigated with respect to coumarins (Table 2, 3p–3w). Various substituted coumarins were allowed to react with phenylhydrazine. It could be seen that the substituted coumarins bearing electron-rich groups such as –OH, –CH3, –OCH3, –OC2H5, and –N(C2H5)2 were transformed into the corresponding 3-arylcoumarins in good yields (3p–3v). Notably, the standard reaction conditions could also be applied to 4-substituted coumarins, affording the corresponding products (3s–3v) in 58%–70% yields, which indicated that the steric hindrance of coumarins played a weak role in this reaction. Pleasingly, even a sensitive functionality such as the hydroxyl group was also tolerated and the coupling reactions proceeded with no requisite for protection of this group (3p, 3s, and 3v). This feature is ubiquitous in hydroxycoumarin-based biologically active products, which eliminates the requirement of protection and deprotection of hydroxyl groups. Unfortunately, coumarin possessing an electron-withdrawing –NO2 group at the C6 position did not furnish the desired product 3w.
To further illustrate the generality and efficacy of this coupling reaction, a series of quinolinone substrates were also briefly surveyed (Table 3). Pleasingly, the C3 position of quinolinone derivatives was exclusively arylated, affording 3-arylquinolinones in 30%–83% yields. It was noteworthy that 2-quinolinone derivatives with a sensitive amide group were also tolerated and the coupling reaction proceeded with no requisite for protection of this group in good yield (5a and 5b). Notably, quinolinone derivatives bearing electron-rich (–OCH3) and electron-poor (–F, –Br) groups could proceed smoothly under the current condition. Although quinolinones with an electron-poor –F or –Br substituent had lower reactivity than that with an electron-rich group, the corresponding 3-arylated quinolinones (5band 5f) with intact halo groups could serve as good precursors for further functionalization. The reaction proved to be fairly general for other electron-poor and electron-rich heterocycles, such as quinolinone derivatives.
Synthesis of 3-arylquinolines from quinolinones and arylhydrazinesa.
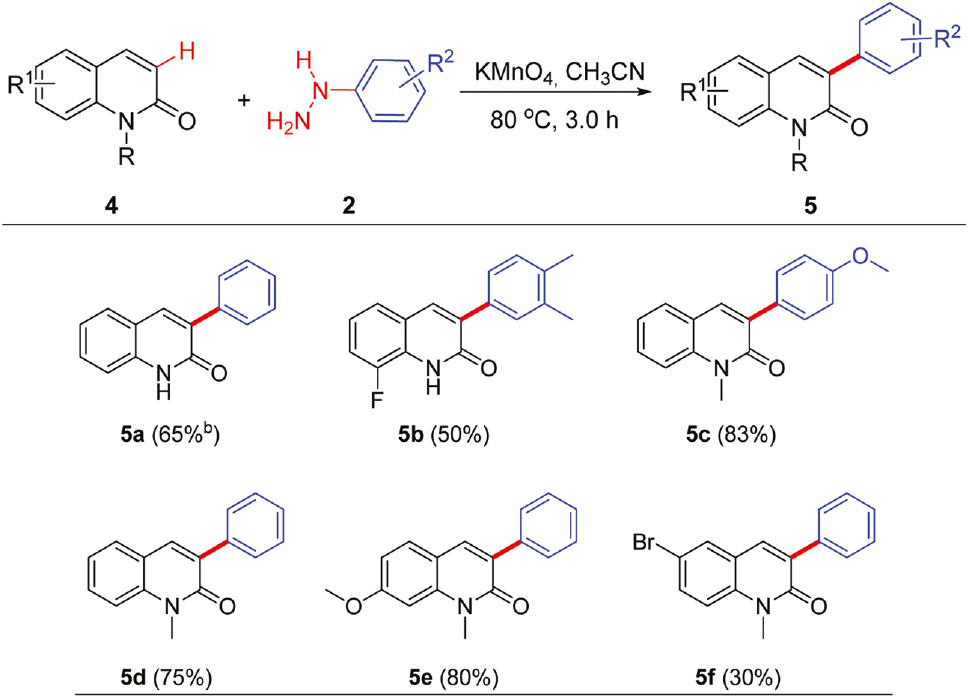
aReaction conditions: quinolinones 1 (0.5 mmol), arylhydrazines 2 (1.0 mmol), KMnO4 (1.5 mmol), CH3CN (10 mL) at 80°C for 3.0 h.
bIsolated yields in parentheses.
On the basis of the previous studies [36], [39], a plausible reaction mechanism for all arylations described above is proposed in Scheme 2. The phenylhydrazine A can be oxidized by KMnO4 to form a phenyldiazene B. The diazene B is then rapidly converted to a phenyl radical C by the release of N2 in the presence of KMnO4. The phenyl radical C can selectively add to the C3 position of coumarin to form a radical intermediate D, allowing the carbocation intermediate E to be formed by a single-electron transfer. Finally, the intermediate Eloses a proton to produce the C3 arylated coumarin F.
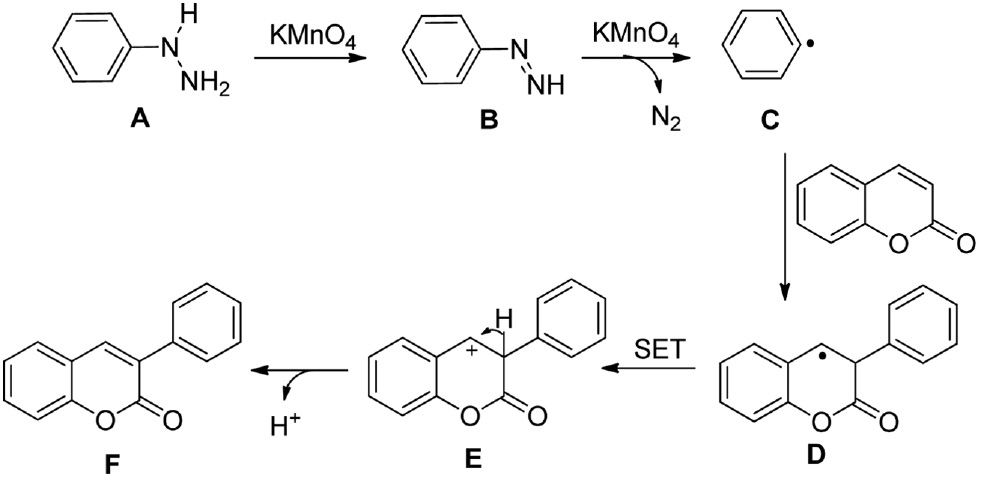
Plausible reaction mechanism for the formation of 3-arylcoumarin.
3 Conclusions
In summary, we have successfully developed an improved and regioselective C-3 arylation on coumarins and quinolinones with arylhydrazines using KMnO4 as an oxidant. This protocol provides a valuable approach to synthesize biologically interesting 3-arylcoumarins and 3-arylquinolinones. The advantages of this reaction are high efficiency, moderate to good yield, a broad functional group tolerance, and the use of cheap and readily available starting materials.
4 Experimental section
4.1 General
All substrates were purchased from J & K Scientific Ltd. without further purification. Nuclear magnetic resonance spectra were recorded on a Bruker Avance 400 MHz spectrometer. Chemical shifts for 1H NMR spectra are recorded in parts per million from tetramethylsilane. Data were reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, m = multiplet, and br = broad), coupling constant in Hz, and integration. Chemical shifts for 13C NMR spectra were recorded in parts per million from tetramethylsilane. High-resolution mass spectra (HR MS) were obtained on a Q-TOF instrument using the electrospray ionization (ESI) technique. IR spectra were recorded on a Shimadazu IR-408 Fourier transform infrared spectrophotometer using a thin film supported on KBr pellets. Melting points were measured on an XT4A microscopic apparatus and are uncorrected.
4.2 General experimental procedure for the synthesis of 3-arylcoumarins (3) and 3-arylquinolinone derivatives (5)
In a 50 mL Schlenk tube, a solution of coumarins 1 (or quinolinone derivatives 4) (0.5 mmol), arylhydrazines 2 (1.0 mmol), and KMnO4 (1.5 mmol, 237 mg) in CH3CN (10 mL) was stirred at 80°C for 3.0 h. After the reaction was finished, the mixture was diluted with saturated NaCl solution (50 mL). Then the aqueous layer was extracted with EtOAc (3 × 15 mL). The organic phase was dried over anhydrous Na2SO4 and concentrated under vacuum. The crude product was purified by silica gel column chromatography using ethyl acetate/petroleum ether (1:5 to 2:1) as eluant to obtain the desired product 3 (or 5).
4.2.1 3-Phenyl-2H-chromen-2-one (3a)
Colorless solid; m.p. 138–139°C (from EtOAc; lit. [18]: 136–137°C). − 1H NMR (400 MHz, CDCl3): δ = 7.81 (s, 1H), 7.71–7.69 (m, 2H), 7.53 (t, JH-H = 8.0 Hz, 2H), 7.47–7.40 (m, 3H), 7.36 (d, JH-H = 8.0 Hz, 1H), 7.29 (td, JH-H = 7.5 Hz, JH-H = 1.0 Hz, 1H). − 13C NMR (100 MHz, CDCl3): δ = 160.6, 153.5, 139.9 (CH), 134.7, 131.4 (CH), 128.8 (CH), 128.6 (CH), 128.5 (CH), 128.3, 127.9 (CH), 124.5 (CH), 119.7, 116.4 (CH). − IR (KBr): ν = 1716 (C=O), 1601, 1454, 1117 cm−1. − MS ((+)-ESI): m/z = 223.2 (calcd. 223.0 for C15H11O2, [M+H]+).
4.2.2 3-(p-Tolyl)-2H-chromen-2-one (3b)
Colorless solid; m.p. 160–161°C (from EtOAc; lit. [18]: 158–159°C). − 1H NMR (400 MHz, [D6]DMSO): δ = 8.19 (s, 1H), 7.75 (d, JH-H = 7.6 Hz, 1H), 7.63–7.58 (m, 3H), 7.41 (d, JH-H = 8.2 Hz, 1H), 7.36 (t, JH-H = 7.5 Hz, 1H), 7.25 (d, JH-H = 8.0 Hz, 2H), 2.34 (s, 3H). − 13C NMR (100 MHz, [D6]DMSO): δ = 160.2, 153.2, 140.3 (CH), 138.5, 132.1, 131.9 (CH), 129.2 (CH), 128.9 (CH), 128.7 (CH), 127.1, 125.0 (CH), 119.9, 116.2 (CH), 21.2 (CH3). − IR (KBr): ν = 1716 (C=O), 1610, 1452, 1113 cm−1. − MS ((+)-ESI): m/z = 237.3 (calcd. 237.0 for C16H13O2, [M+H]+).
4.2.3 3-(3,4-Dimethylphenyl)-2H-chromen-2-one (3c)
Light yellow solid; m.p. 130–131°C (from EtOAc; lit. [47]: 110–112°C). − 1H NMR (400 MHz, CDCl3): δ = 7.75 (s, 1H), 7.51–7.46 (m, 3H), 7.42 (dd, JH-H = 7.8 Hz, JH-H = 1.4 Hz, 1H), 7.33 (d, JH-H = 8.1 Hz, 1H), 7.26 (t, JH-H = 7.5 Hz, 1H), 7.18 (d, JH-H = 7.8 Hz, 1H), 2.30 (s, 3H), 2.28 (s, 3H). − 13C NMR (100 MHz, CDCl3): δ = 160.7, 153.4, 139.1 (CH), 137.6, 136.6, 132.2, 131.1 (CH), 129.7 (CH), 129.6 (CH), 128.4, 127.8 (CH), 125.9 (CH), 124.4 (CH), 119.8, 116.4 (CH), 19.9 (CH3), 19.6 (CH3). − IR (KBr): ν = 2922, 1718 (C=O), 1605, 1454, 1101 cm−1. − MS ((+)-ESI): m/z = 251.3 (calcd. 251.1 for C17H15O2, [M+H]+).
4.2.4 3-(4-Ethylphenyl)-2H-chromen-2-one (3d)
Colorless solid [29]; m.p. 118–119°C (from EtOAc). − 1H NMR (400 MHz, CDCl3): δ = 7.79 (s, 1H), 7.63 (d, JH-H = 8.2 Hz, 2H), 7.54–7.49 (m, 2H), 7.36 (d, JH-H = 8.2 Hz, 1H), 7.31–7.27 (m, 3H), 2.69 (q, JH-H = 7.6Hz, 2H), 1.26 (t, JH-H = 7.6 Hz, 3H). − 13C NMR (100 MHz, CDCl3): δ = 160.7, 153.4, 145.2, 139.2 (CH), 132.0, 131.2 (CH), 128.5 (CH), 128.3, 128.0, 127.8 (CH), 124.4 (CH), 119.8, 116.4 (CH), 28.7 (CH2), 15.5 (CH3). − IR (KBr): ν = 3012, 2948, 1716 (C=O), 1558, 1452, 1115 cm−1. − MS ((+)-ESI): m/z = 251.0 (calcd. 251.1 for C17H15O2, [M+H]+).
4.2.5 3-(3,5-Dimethylphenyl)-2H-chromen-2-one (3e)
Yellow solid; m.p. 126–127°C (from EtOAc). − 1H NMR (400 MHz, CDCl3): δ = 7.78 (s, 1H), 7.52 (td, JH-H = 7.9 Hz, JH-H = 1.9 Hz, 2H), 7.36 (d, JH-H = 8.1 Hz, 1H), 7.30–7.26 (m, 3H), 7.05 (s, 1H), 2.37 (s, 6H). − 13C NMR (100 MHz, CDCl3): δ = 160.6, 153.4, 139.6 (CH), 138.0, 134.6, 131.2 (CH), 130.6 (CH), 128.7, 127.8 (CH), 126.3 (CH), 124.4 (CH), 119.7, 116.4 (CH), 21.3 (CH3). − IR (KBr): ν = 2922, 2852, 1724 (C=O), 1608, 1456, 1117 cm−1. − HR MS ((+)-ESI): m/z = 251.1064 (calcd. 251.1067 for C17H15O2, [M+H]+).
4.2.6 3-(4-Methoxyphenyl)-2H-chromen-2-one (3f)
Colorless solid; m.p. 138–139°C (from EtOAc; lit. [18]: 140–141°C). − 1H NMR (400 MHz, CDCl3): δ = 7.74 (s, 1H), 7.66 (d, JH-H = 8.9 Hz, 2H), 7.52–7.46 (m, 2H), 7.33 (d, JH-H = 8.2 Hz, 1H), 7.27 (td, JH-H = 7.5 Hz, JH-H = 1.1 Hz, 1H), 6.96 (d, JH-H = 8.9 Hz, 2H), 2.83 (s, 3H). − 13C NMR (100 MHz, CDCl3): δ = 160.8, 160.1, 153.2, 138.5 (CH), 131.0 (CH), 129.8 (CH), 127.8, 127.7 (CH), 127.1, 124.4 (CH), 119.8, 116.3 (CH), 113.9 (CH), 55.3 (CH3). − IR (KBr): ν = 2918, 1716 (C=O), 1608, 1514, 1452, 1252, 1128 cm−1. − MS ((+)-ESI): m/z = 253.4 (calcd. 253.0 for C16H13O3, [M+H]+).
4.2.7 3-(2-Fluorophenyl)-2H-chromen-2-one (3g)
Yellow solid; m.p. 165–166°C (from EtOAc). − 1H NMR (400 MHz, CDCl3): δ = 7.84 (s, 1H), 7.56–7.53 (m, 3H), 7.39–7.37 (m, 2H), 7.30 (t, JH-H = 7.3 Hz, 1H), 7.24–7.14 (m, 2H). − 13C NMR (100 MHz, CDCl3): δ = 161.1 (d, JF-C = 247.7 Hz), 159.9, 153.7, 142.6 (d, JF-C = 3.2 Hz) (CH), 131.6 (d, JF-C = 50.2 Hz) (CH), 131.3 (CH), 130.6 (d, JF-C = 8.2 Hz) (CH), 128.1 (CH), 124.5 (CH), 124.1 (d, JF-C = 3.5 Hz) (CH), 123.2, 122.4 (d, JF-C = 13.7 Hz), 119.2, 116.6 (CH), 116.0 (d, JF-C = 21.9 Hz) (CH). − 19F NMR (376 MHz, CDCl3): δ = –114.5. − IR (KBr): ν = 1728 (C=O), 1610, 1452, 1120 cm−1. − HR MS ((+)-ESI): m/z = 241.0663 (calcd. 241.0659 for C15H10FO2, [M+H]+).
4.2.8 3-(3-Fluorophenyl)-2H-chromen-2-one (3h)
Colorless solid; m.p. 173–174°C (from EtOAc; lit. [47]: 152–154°C). − 1H NMR (400 MHz, CDCl3): δ = 7.83 (s, 1H), 7.54 (t, JH-H = 7.4 Hz, 2H), 7.47 (t, JH-H = 7.5 Hz, 2H), 7.43–7.39 (m, 1H), 7.36 (d, JH-H = 8.6 Hz, 1H), 7.30 (d, JH-H = 7.9 Hz, 1H), 7.09 (td, JH-H = 8.6 Hz, JH-H = 1.8 Hz, 1H). − 13C NMR (100 MHz, CDCl3): δ = 162.6 (d, JF-C = 244.4 Hz), 160.2, 153.5, 140.4 (CH), 136.6 (d, JF-C = 8.2 Hz), 131.8 (CH), 130.0 (d, JF-C = 8.2 Hz, CH), 128.1 (CH), 127.0 (d, JF-C = 2.2 Hz, CH), 124.6 (CH), 124.1 (d, JF-C = 2.9 Hz, CH), 119.4, 116.5 (CH), 115.8 (d, JF-C = 8.8 Hz, CH), 115.6 (d, JF-C = 10.7 Hz, CH). − 19F NMR (376 MHz, CDCl3): δ = –112.6. − IR (KBr): ν = 1709 (C=O), 1610, 1588, 1456, 1180 cm−1. − MS ((+)-ESI): m/z = 241.3 (calcd. 241.0 for C15H10FO2, [M+H]+).
4.2.9 4-(2-Oxo-2H-chromen-3-yl)benzonitrile (3i)
Light yellow solid; m.p. 239–240°C (from EtOAc; lit. [47]: 152–154°C). − 1H NMR (400 MHz, CDCl3): δ = 7.90 (s, 1H), 7.85 (d, JH-H = 8.4 Hz, 2H), 7.74 (d, JH-H = 8.4 Hz, 2H), 7.59 (d, JH-H = 7.5 Hz, 2H), 7.39 (d, JH-H = 8.4 Hz, 1H), 7.34 (td, JH-H = 8.4 Hz, JH-H = 0.7 Hz, 1H). − 13C NMR (100 MHz, CDCl3): δ = 159.9, 153.7, 141.3 (CH), 139.1 (CH), 132.4 (CH), 132.2 (CH), 129.2 (CH), 128.3 (CH), 126.4, 124.8 (CH), 119.2, 118.5, 116.6 (CH), 112.4. − IR (KBr): ν = 1709 (C=O), 1614, 1446, 1115 cm−1. − MS ((+)-ESI): m/z = 248.2 (calcd. 248.0 for C16H10NO2, [M+H]+).
4.2.10 3-(4-(Trifluoromethoxy)phenyl)-2H-chromen- 2-one (3j)
Colorless solid; m.p. 160–161°C (from EtOAc). − 1H NMR (400 MHz, CDCl3): δ = 7.83 (s, 1H), 7.75 (dd, JH-H = 6.8 Hz, JH-H = 2.0 Hz, 2H), 7.57–7.54 (m, 2H), 7.38 (d, JH-H = 8.6 Hz, 1H), 7.34–7.28 (m, 3H). − 13C NMR (100 MHz, CDCl3): δ = 160.4, 153.6, 149.5, 140.2 (CH), 133.2, 131.7 (CH), 130.1 (CH), 128.0 (CH), 127.0, 124.6 (CH), 121.7, 120.8 (CH), 119.4, 116.5 (CH). − 19F NMR (376 MHz, CDCl3): δ = –57.7. − IR (KBr): ν = 1709 (C=O), 1610, 1516, 1456, 1140 cm−1. − HR MS ((+)-ESI): m/z = 301.0578 (calcd. 307.0577 for C16H10F3O3, [M+H]+).
4.2.11 3-(4-Bromophenyl)-2H-chromen-2-one (3l)
Colorless solid; m.p. 189–190°C (from EtOAc; lit. [18]:188–189°C). − 1H NMR (400 MHz, CDCl3): δ = 7.81 (s, 1H), 7.60–7.52 (m, 6H), 7.35 (d, JH-H = 8.7 Hz, 1H), 7.30 (td, JH-H = 8.6 Hz, JH-H = 1.0 Hz, 1H). − 13C NMR (100 MHz, CDCl3): δ = 160.2, 153.5, 139.9 (CH), 133.5, 131.7 (CH), 131.6 (CH), 130.1 (CH), 128.0 (CH), 127.1, 124.6 (CH), 123.1, 119.5, 116.5 (CH). − IR (KBr): ν = 1712 (C=O), 1610, 1487, 1450, 1012 cm−1. − MS ((+)-ESI): m/z = 301.1 (calcd. 301.0 for C15H10BrO2, [M+H]+).
4.2.12 3-(2,4-Difluorophenyl)-2H-chromen-2-one (3m)
Light yellow solid; m.p. 172–173°C (from EtOAc). − 1H NMR (400 MHz, CDCl3): δ = 7.83 (s, 1H), 7.61–7.53 (m, 3H), 7.39 (d, JH-H = 8.2 Hz, 1H), 7.32 (td, JH-H = 7.8 Hz, JH-H = 0.8 Hz, 1H), 7.00–6.91 (m, 2H). − 13C NMR (100 MHz, CDCl3): δ = 164.5, 162.0, 160.0, 153.8, 142.7 (d, JF-C = 3.8 Hz) (CH), 132.3 (dd, JF-C = 9.6 Hz, JF-C = 9.4 Hz) (CH), 132.0 (CH), 128.1 (CH), 124.6 (CH), 122.3, 119.1, 116.6 (CH), 111.6 (dd, JF-C = 3.5 Hz, JF-C = 3.6 Hz) (CH), 104.4 (t, JF-C = 25.4 Hz) (CH). − 19F NMR (376 MHz, CDCl3): δ = –108.3, –110.1. − IR (KBr): ν = 1710 (C=O), 1611, 1584, 1450, 1141 cm−1. − HR MS ((+)-ESI): m/z = 259.0568 (calcd. 259.0565 for C15H9F2O2, [M+H]+).
4.2.13 3-(3-Chloro-4-methylphenyl)-2H-chromen- 2-one (3n)
Yellow solid; m.p. 193–194°C (from EtOAc). − 1H NMR (400 MHz, CDCl3): δ = 7.79 (s, 1H), 7.70 (d, JH-H = 1.6 Hz, 1H), 7.55–7.51 (m, 3H), 7.35 (d, JH-H = 8.6 Hz, 1H), 7.29 (t, JH-H = 7.8 Hz, 2H), 2.40 (s, 3H). − 13C NMR (100 MHz, CDCl3): δ = 160.3, 153.5, 139.8 (CH), 136.8, 134.4, 133.7, 131.6 (CH), 130.9 (CH), 128.8 (CH), 127.9 (CH), 126.9, 126.7 (CH), 124.6 (CH), 119.5, 116.5 (CH), 19.9 (CH3). − IR (KBr): ν = 1718 (C=O), 1454, 1356, 1113 cm−1. − HR MS ((+)-ESI): m/z = 271.7179 (calcd. 271.7177 for C16H12ClO2, [M+H]+).
4.2.14 3-(tert-Butyl)-2H-chromen-2-one (3o)
Fait yellow solid; m.p. 81–82°C (from EtOAc; lit. [48]:82–83°C). − 1H NMR (400 MHz, CDCl3): δ = 7.54 (s, 1H), 7.47–7.43 (m, 2H), 7.29 (d, JH-H = 8.7 Hz, 1H), 7.23 (t, JH-H = 6.7 Hz, 1H), 1.40 (s, 9H). − 13C NMR (100 MHz, CDCl3): δ = 159.9, 153.2, 137.1, 136.6 (CH), 130.5 (CH), 127.5 (CH), 123.9 (CH), 119.4, 116.0, 35.0, 28.6 (CH3). − IR (KBr): ν = 2965, 1720 (C=O), 1455, 1354 cm−1. − MS ((+)-ESI): m/z = 203.2 (calcd. 203.1 for C13H15O2, [M+H]+).
4.2.15 7-Hydroxy-3-phenyl-2H-chromen-2-one (3p)
White solid; m.p. 201–202°C (from EtOAc; lit. [28]:203–204°C. − 1H NMR (400 MHz, [D6]DMSO): δ = 10.66 (s, 1H), 8.15 (s, 1H), 7.69 (d, JH-H = 7.3 Hz, 2H), 7.60 (d, JH-H = 8.5 Hz, 1H), 7.43 (d, JH-H = 7.6 Hz, 2H), 7.37 (t, JH-H = 7.3 Hz, 1H), 6.82 (dd, JH-H = 8.5 Hz, JH-H = 2.2 Hz, 1H), 6.76 (d, JH-H = 2.0 Hz, 1H). − 13C NMR (100 MHz, [D6]DMSO): δ = 161.7, 160.6, 155.3, 141.6 (CH), 135.6, 130.4 (CH), 128.7 (CH), 128.6 (CH), 128.5 (CH), 122.6, 113.8 (CH), 112.4 (CH), 102.2 (CH). − IR (KBr): ν = 3228, 1680 (C=O), 1615, 1595, 1570, 1445, 1080 cm−1. − MS ((+)-ESI): m/z = 239.1 (calcd. 239.0 for C15H11O3, [M+H]+).
4.2.16 7-Methoxy-3-phenyl-2H-chromen-2-one (3q)
Light yellow solid; m.p. 119–120°C (from EtOAc; lit. [18]: 118–119°C). − 1H NMR (400 MHz, CDCl3): δ = 7.75 (s, 1H), 7.68 (d, JH-H = 8.4 Hz, 2H), 7.45–7.41 (m, 3H), 7.39–7.35 (m, 1H), 6.87–6.84 (m, 2H), 3.87 (s, 3H). − 13C NMR (100 MHz, CDCl3): δ = 162.6, 160.8, 155.3, 139.9 (CH), 135.0, 128.8 (CH), 128.4 (CH), 124.8, 113.3, 112.7 (CH), 100.4 (CH), 55.7 (CH3). − IR (KBr): ν = 3053, 2970, 1716 (C=O), 1614, 1506, 1464, 1439, 1120 cm−1. − MS ((+)-ESI): m/z = 253.3 (calcd. 253.1 for C16H13O3, [M+H]+).
4.2.17 6-Methyl-3-phenyl-2H-chromen-2-one (3r)
Colorless solid; m.p. 148–149°C (from EtOAc; lit. [27]:149–150°C). − 1H NMR (400 MHz, CDCl3): δ = 7.74 (s, 1H), 7.70–7.67 (m, 2H), 7.46–7.37 (m, 3H), 7.33–7.31 (m, 2H), 7.24 (dd, JH-H = 7.4 Hz, JH-H = 1.6 Hz, 1H), 2.41 (s, 3H). − 13C NMR (100 MHz, CDCl3): δ = 160.8, 151.6, 139.9 (CH), 134.8, 134.1, 132.4 (CH), 128.7 (CH), 128.5 (CH), 128.4 (CH), 128.2, 127.7 (CH), 119.4, 116.1 (CH), 20.8 (CH3). − IR (KBr): ν = 2916, 2848, 1720 (C=O), 1616, 1577, 1448, 1111 cm−1. − MS ((+)-ESI): m/z = 237.2 (calcd. 237.1 for C16H13O2, [M+H]+).
4.2.18 7-Hydroxy-4-methyl-3-phenyl-2H-chromen- 2-one (3s)
White solid; m.p. 231–232°C (from EtOAc; lit. [49]:225–227°C). − 1H NMR (400 MHz, [D6]DMSO): δ = 10.5 (s, -OH), 7.61 (d, JH-H = 8.8 Hz, 1H), 7.43 (t, JH-H = 7.5 Hz, 2H), 7.36 (t, JH-H = 7.2 Hz, 1H), 7.28 (d, JH-H = 6.9 Hz, 2H), 6.83 (dd, JH-H = 8.8 Hz, JH-H = 2.2 Hz, 1H), 6.74 (d, JH-H = 2.2 Hz, 1H), 2.17 (s, 3H). − 13C NMR (100 MHz, [D6]DMSO): δ = 161.3, 160.7, 154.3, 148.7, 135.3, 130.7 (CH), 128.4 (CH), 128.0 (CH), 127.5 (CH), 122.7, 113.4 (CH), 112.8, 102.3 (CH), 16.7 (CH3). − IR (KBr): ν = 3228, 1682 (C=O), 1614, 1593, 1577, 1446, 1078 cm−1. − MS ((+)-ESI): m/z = 253.2 (calcd. 253.1 for C16H13O3, [M+H]+).
4.2.19 7-Hydroxy-4-methyl-3-phenyl-2H-chromen- 2-one (3t)
Deep yellow solid; m.p. 102–103°C (from EtOAc). − 1H NMR (400 MHz, DMSO): δ = 7.46–7.40 (m, 3H), 7.34 (t, JH-H = 7.3 Hz, 1H), 7.29 (d, JH-H = 7.0 Hz, 2H), 6.61(dd, JH-H = 9.0 Hz, JH-H = 2.5 Hz, 1H), 6.54 (d, JH-H = 2.5 Hz, 1H), 3.41 (q, JH-H = 7.1 Hz, 4H), 2.22 (s, 3H), 1.21 (t, JH-H = 7.1 Hz, 6H). − 13C NMR (100 MHz, DMSO): δ = 162.1, 155.1, 150.2, 148.3, 135.3, 130.5 (CH), 128.2 (CH), 127.5 (CH), 126.1 (CH), 121.0, 109.5, 108.6 (CH), 97.4 (CH), 44.7 (CH2), 16.3 (CH3), 12.4 (CH3). − IR (KBr): ν = 2987, 2856, 1707 (C=O), 1618, 1587, 1522, 1074 cm−1. − HR MS ((+)-ESI): m/z = 308.1648 (calcd. 308.1645 for C20H22NO2, [M+H]+).
4.2.20 7-Ethoxy-4-methyl-3-phenyl-2H-chromen- 2-one (3u)
Yellow solid; m.p. 117–118°C (from EtOAc). − 1H NMR (400 MHz, CDCl3): δ = 7.52 (d, JH-H = 8.8 Hz, 1H), 7.41 (d, JH-H = 7.0 Hz, 2H), 7.35 (d, JH-H = 7.3 Hz, 1H), 7.29–7.25 (m, 2H), 6.85 (dd, JH-H = 8.8 Hz, JH-H = 2.5 Hz, 1H), 6.79 (d, JH-H = 2.2 Hz, 1H), 4.06 (q, JH-H = 7.0 Hz, 2H), 2.23 (s, 3H), 1.43 (t, JH-H = 7.0 Hz, 3H). − 13C NMR (100 MHz, CDCl3): δ = 161.7, 161.3, 154.3, 148.0, 134.7, 130.2 (CH), 128.3 (CH), 127.9 (CH), 126.1 (CH), 124.0, 113.9, 112.6 (CH), 101.0 (CH), 64.1 (CH2), 16.5 (CH3), 14.6 (CH3). − IR (KBr): ν = 3062, 2979, 2947, 1712 (C=O), 1604, 1508, 1385, 1361, 1074 cm−1. − HR MS ((+)-ESI): m/z = 281.1175 (calcd. 281.1172 for C18H17O3, [M+H]+).
4.2.21 3-(3,5-Dimethylphenyl)-7-hydroxy-4-methyl- 2H-chromen-2-one (3v)
Yellow solid; m.p. 228–230°C (from EtOAc). − 1H NMR (400 MHz, CDCl3): δ = 8.01 (s, 1H), 7.51 (d, JH-H = 8.7 Hz, 1H), 7.01–7.00 (m, 2H), 6.89 (bs, 2H), 6.85 (dd, JH-H = 8.7 Hz, JH-H = 2.3 Hz, 1H), 2.33 (s, 6H), 2.26 (s, 3H). − 13C NMR (100 MHz, CDCl3): δ = 162.7, 159.9, 153.9, 149.2, 137.9, 134.3, 129.8 (CH), 127.7 (CH), 126.3 (CH), 123.8, 113.7, 113.6 (CH), 103.1 (CH), 21.3 (CH3), 16.6 (CH3). − IR (KBr): ν = 3062, 2979, 1712 (C=O), 1604, 1508, 1385, 1361, 1074 cm−1. − HR MS ((+)-ESI): m/z = 281.1176 (calcd. 281.1172 for C18H17O3, [M+H]+).
4.2.22 3-Phenylquinolin-2(1H)-one (5a)
White solid; m.p. 226–227°C (from EtOAc; lit. [50]:228–230°C). − 1H NMR (400 MHz, [D6]DMSO): δ = 11.96 (s, 1H), 8.10 (s, 1H), 7.77–7.72 (m, 3H), 7.50 (td, JH-H = 8.3 Hz, JH-H = 1.2 Hz, 1H), 7.45–7.41 (m, 2H), 7.40 (d, JH-H = 7.3 Hz, 1H), 7.34 (d, JH-H = 8.1 Hz, 1H), 7.20 (td, JH-H = 7.9 Hz, JH-H = 0.8 Hz, 1H). − 13C NMR (100 MHz, [D6]DMSO): δ = 161.5, 138.8, 138.0 (CH), 136.7, 131.9, 130.6 (CH), 129.1 (CH), 128.6 (CH), 128.4 (CH), 128.3 (CH), 122.3 (CH), 120.0, 115.1 (CH). − IR (KBr): ν = 3428, 1635 (C=O), 1610, 1464, 1454, 1120, 1028 cm−1. − MS ((+)-ESI): m/z = 222.3 (calcd. 222.1 for C15H12NO, [M+H]+).
4.2.23 3-(3,4-Dimethylphenyl)-8-fluoroquinolin- 2(1H)-one (5b)
Light yellow solid; m.p. 238–239°C (from EtOAc). − 1H NMR (400 MHz, CDCl3): δ = 9.82 (s, 1H), 7.84 (s, 1H), 7.55 (s, 1H), 7.49 (d, JH-H = 7.2 Hz, 1H), 7.37 (d, JH-H = 7.8 Hz, 1H), 7.21 (d, JH-H = 7.9 Hz, 2H), 7.15–7.10 (m, 1H), 2.33 (s, 3H), 2.30 (s, 3H). − 13C NMR (100 MHz, CDCl3): δ = 150.1, 147.6, 137.2, 136.7 (CH), 136.4, 133.1, 129.9 (CH), 129.6 (CH), 126.6, 126.5, 126.2 (CH), 123.2 (d, JF-C = 7.9 Hz) (CH), 122.1 (d, JF-C = 7.9 Hz) (CH), 114.8 (d, JF-C = 7.9 Hz) (CH), 19.9, 19.6. − 19F NMR (376 MHz, CDCl3): δ = –135.3. − IR (KBr): ν = 2944, 2848, 1645 (C=O), 1614, 1454, 1259 cm−1. − HR MS ((+)-ESI): m/z = 268.1130 (calcd. 268.1132 for C17H15FNO, [M+H]+).
4.2.24 3-(4-Methoxyphenyl)-1-methylquinolin- 2(1H)-one (5c)
Pale yellow solid; m.p. 113–114°C (from EtOAc; lit. [50]:114–116°C). − 1H NMR (400 MHz, CDCl3): δ = 7.73 (s, 1H), 7.67 (dd, JH-H = 7.0 Hz, JH-H = 1.9 Hz, 2H), 7.56 (dd, JH-H = 7.8 Hz, JH-H = 1.0 Hz, 1H), 7.51 (td, JH-H = 7.2 Hz, JH-H = 1.4 Hz, 1H), 7.32 (d, JH-H = 8.5 Hz, 1H), 7.21 (td, JH-H = 7.2 Hz, JH-H = 0.6 Hz, 1H), 6.95 (d, JH-H = 8.8 Hz, 2H), 3.83 (s, 3H), 3.76 (s, 3H). − 13C NMR (100 MHz, CDCl3): δ = 161.6, 159.5, 139.3, 135.7 (CH), 131.9, 130.2 (CH), 129.9 (CH), 129.2, 128.6 (CH), 122.1 (CH), 120.8, 113.9 (CH), 113.6 (CH), 55.3 (CH3), 29.9 (CH3). − IR (KBr): ν = 3032, 2952 (–CH3), 1637 (C=O), 1604, 1598, 1510, 1458 (Ar–), 1246, 1178, 1030 cm−1. − MS ((+)-ESI): m/z = 266.2 (calcd. 266.1 for C17H16NO2, [M+H]+).
4.2.25 1-Methyl-3-phenylquinolin-2(1H)-one (5d)
White solid; m.p. 134–135°C (from EtOAc; lit. [50]:135–137°C). − 1H NMR (400 MHz, CDCl3): δ = 7.77 (s, 1H), 7.70 (dd, JH-H = 7.0 Hz, JH-H = 1.4 Hz, 2H), 7.59–7.25 (m, 2H), 7.42 (t, JH-H = 7.5 Hz, 2H), 7.35 (t, JH-H = 8.4 Hz, 2H), 7.22 (t, JH-H = 7.3 Hz, 1H), 3.77 (s, 3H). − 13C NMR (100 MHz, CDCl3): δ = 161.5, 139.6, 136.8 (CH), 132.4, 130.3 (CH), 128.9 (CH), 128.8 (CH), 128.1 (CH), 128.0 (CH), 122.2 (CH), 120.7, 114.0 (CH), 29.9 (CH3). − IR (KBr): ν = 3049, 3032, 1645 (C=O), 1591, 1454 cm−1. − MS ((+)-ESI): m/z = 236.2 (calcd. 236.1 for C16H14NO, [M+H]+).
4.2.26 7-Methoxy-1-methyl-3-phenylquinolin- 2(1H)-one (5e)
Light yellow solid; m.p. 134–135°C (from EtOAc). − 1H NMR (400 MHz, CDCl3): δ = 7.70–7.67 (m, 3H), 7.48 (d, JH-H = 8.6 Hz, 1H), 7.40 (td, JH-H = 7.2 Hz, JH-H = 1.2 Hz, 2H), 7.35–7.31 (m, 1H), 6.81 (dd, JH-H = 8.6 Hz, JH-H = 2.2 Hz, 1H), 6.76 (d, JH-H = 2.2 Hz, 1H), 3.90 (s, 3H), 3.72 (s, 3H). − 13C NMR (100 MHz, CDCl3): δ = 161.8, 161.6, 141.2, 137.0, 136.7 (CH), 130.2 (CH), 129.2, 128.8 (CH), 128.1 (CH), 127.7 (CH), 114.9, 109.8 (CH), 98.4 (CH), 55.6 (CH3), 29.9 (CH3). − IR (KBr): ν = 2979, 2927, 1645 (C=O), 1616, 1595, 1508, 1454, 1248, 1211, 1036 cm−1. − HR MS ((+)-ESI): m/z = 266.1179 (calcd. 266.1176 for C17H16NO2, [M+H]+).
4.2.27 6-Bromo-1-methyl-3-phenylquinolin- 2(1H)-one (5f)
Pale yellow solid, m.p. 175–176°C (from EtOAc; lit. [50]:176–178°C). − 1H NMR (400 MHz, CDCl3): δ = 7.73 (d, JH-H = 2.2 Hz, 1H), 7.70–7.67 (m, 3H), 7.63 (dd, JH-H = 8.9 Hz, JH-H = 2.2 Hz, 1H), 7.44 (t, JH-H = 7.6 Hz, 2H), 7.40–7.37 (m, 1H), 7.25 (d, JH-H = 6.7 Hz, 1H), 3.77 (s, 3H). − 13C NMR (100 MHz, CDCl3): δ = 161.1, 138.5, 136.3, 135.3 (CH), 133.7, 132.9 (CH), 130.8 (CH), 128.9 (CH), 128.4 (CH), 128.2 (CH), 122.2, 115.7 (CH), 114.9, 30.1 (CH3). − IR (KBr): ν = 2954, 2924, 1647 (C=O), 1579, 1487, 1444, 1415, 1228, 1209, 1118 cm−1. − MS ((+)-ESI): m/z = 314.1 (calcd. 314.0 for C16H13BrNO, [M+H]+).
5 Supplementary information
Further experimental details and NMR spectra of compounds 3a–3v and 5a–5fare given as Supplementary Information available online (http://dx.doi.org/10.1515/znb-2016-0109).
Funding source: National Natural Science Foundation of China
Award Identifier / Grant number: 21172055
Award Identifier / Grant number: 21302042
Funding statement: The work was supported by the National Natural Science Foundation of China (Nos. 21172055 and 21302042), Natural Science Foundation of Henan scientific committee (No. 142102210410), Natural Science Foundation of Henan Educational Committee (No. 14B150053), and the Program for Innovative Research Team from Zhengzhou (No. 131PCXTD605), and Scientific Fund Project of Zhengzhou Science and Technology Bureau (No. 20130883).
Acknowledgments
The work was supported by the National Natural Science Foundation of China (Nos. 21172055 and 21302042), Natural Science Foundation of Henan scientific committee (No. 142102210410), Natural Science Foundation of Henan Educational Committee (No. 14B150053), and the Program for Innovative Research Team from Zhengzhou (No. 131PCXTD605), and Scientific Fund Project of Zhengzhou Science and Technology Bureau (No. 20130883).
References
[1] M. A. Musa, J. S. Cooperwood, M. Khan, F. Omar, Curr. Med. Chem. 2008, 15, 2664.10.2174/092986708786242877Search in Google Scholar PubMed PubMed Central
[2] I. Kempen, D. Papapostolou, N. Thierry, L. Pochet, S. Counerotte, B. Masereel, J. M. Foidart, M. J. Reboud-Ravaux, A. Noel, B. Pirotte, Br. J. Cancer2003, 88, 1111.10.1038/sj.bjc.6600856Search in Google Scholar PubMed PubMed Central
[3] C. Spino, M. Dodier, S. Sotheeswaran, Bioorg. Med. Chem. Lett. 1998, 8, 3475.10.1016/S0960-894X(98)00628-3Search in Google Scholar PubMed
[4] P. Anand, B. Singh, N. Singh, Bioorg. Med. Chem. 2012, 20, 1175.10.1016/j.bmc.2011.12.042Search in Google Scholar PubMed
[5] C. Wang, C. Wu, J. Zhu, R. H. Miller, Y. Wang, J. Med. Chem. 2011, 54, 2331.10.1021/jm101489wSearch in Google Scholar PubMed PubMed Central
[6] K. G. Reddie, W. H. Humphries, C. P. Bain, C. K. Payne, M. L. Kemp, N. Murthy, Org. Lett. 2012, 14, 680.10.1021/ol203105cSearch in Google Scholar PubMed PubMed Central
[7] L. You, R. An, X. Wang, Y. Li, Bioorg. Med. Chem. Lett. 2010, 20, 7426.10.1016/j.bmcl.2010.10.027Search in Google Scholar PubMed
[8] L. Piazzi, A. Cavalli, F. Belluti, A. Bisi, S. Gobbi, S. Rizzo, M. Bartolini, V. Andrisano, M. Recanatini, A. Rampa, J. Med. Chem. 2007, 50, 4250.10.1021/jm070100gSearch in Google Scholar PubMed
[9] M. J. Matos, D. Viña, E. Quezada, C. Piciau, G. Delogu, F. Orallo, L. Santana, E. Uriarte, Bioorg. Med. Chem. Lett.2009, 19, 3268.10.1016/j.bmcl.2009.04.085Search in Google Scholar PubMed
[10] M. J. Matos, C. Terán, Y. Pérez-Castillo, E. Uriarte, L. Santana, D. Viña, J. Med. Chem. 2011, 54, 7127.10.1021/jm200716ySearch in Google Scholar PubMed
[11] L. M. Kabeya, A. A. Marchi, A. Kanashiro, N. P. Lopes, C. H. T. P. Silva, M. T. Pupo, Y. M. Lucisano-Valim, Bioorg. Med. Chem. 2007, 15, 1516.10.1016/j.bmc.2006.10.068Search in Google Scholar PubMed
[12] E. B. B. Ong, N. Watanabe, A. Saito, Y. Futamura, K. H. A. E. Galil, A. Koito, N. Najimudin, H. Osada, J. Biol. Chem. 2011, 286, 14049.10.1074/jbc.M110.185397Search in Google Scholar PubMed PubMed Central
[13] D. Olmedo, R. Sancho, L. M. Bedoya, J. L. Lopez-Perez, E. D. Olmo, E. Munoz, J. Alcami, M. P. Gupta, A. S. Feliciano, Molecules2012, 17, 9245.10.3390/molecules17089245Search in Google Scholar PubMed PubMed Central
[14] M. de Souza Santos, M. P. F. de Morais Del Lama, L. A. Deliberto, F. da Silva Emery, M. T. Pupo, R. M. Z. G. Naal, Arch. Pharmacal. Res. 2013, 36, 731.10.1007/s12272-013-0084-8Search in Google Scholar PubMed
[15] H. Zhao, B. Yan, L. B. Peterson, B. S. J. Blagg, ACS Med. Chem. Lett. 2012, 3, 327.10.1021/ml300018eSearch in Google Scholar PubMed PubMed Central
[16] J. M. Liu, X. Zhang, L. J. Shi, M. W. Liu, Y. Y. Yue, F. W. Li, K. L. Zhao, Chem. Commun. 2014, 50, 9887.10.1039/C4CC04377DSearch in Google Scholar
[17] K. V. Sashidhara, G. R. Palnati, S. R. Avula, A. Kumar, Synlett2012, 4, 611.10.1055/s-0031-1290344Search in Google Scholar
[18] H. Y. Zeng, C. J. Li, Angew. Chem., Int. Ed. 2014, 53, 13862.10.1002/anie.201407589Search in Google Scholar PubMed
[19] M. J. Matos, S. Vazquez-Rodriguez, F. Borges, L. Santana, E. Uriarte, Tetrahedron Lett.2011, 52, 1225.10.1016/j.tetlet.2011.01.048Search in Google Scholar
[20] L. Zhang, T. H. Meng, R. H. Fan, J. Wu, J. Org. Chem. 2007, 72, 7279.10.1021/jo071117+Search in Google Scholar PubMed
[21] J. B. Meng, M. G. Shen, D. C. Fu, Z. H. Gao, R. J. Wang, H. G. Wang, T. Matsuura, Synthesis1990, 8, 719.10.1055/s-1990-26993Search in Google Scholar
[22] F. Jafarpour, S. Zarei, M. B. A. Olia, N. Jalalimanesh, J. Org. Chem. 2013, 78, 2957.10.1021/jo302778dSearch in Google Scholar PubMed
[23] S. Messaoudi, J. D. Brion, M. Alami, Org. Lett. 2012, 14, 1496.10.1021/ol300235kSearch in Google Scholar PubMed
[24] A. Unsinn, S. H. Wunderlich, P. Knochel, Adv. Synth. Catal. 2013, 355, 989.10.1002/adsc.201300049Search in Google Scholar
[25] M. Mosrin, G. Monzon, T. Bresser, P. Knochel, Chem. Commun. 2009, 37, 5615.10.1039/b913693bSearch in Google Scholar PubMed
[26] F. Jafarpour, M. B. A. Olia, H. Hazrati, Adv. Synth. Catal.2013, 355, 3407.10.1002/adsc.201300707Search in Google Scholar
[27] Z. J. She, Y. Shi, Y. M. Huang, Y. Y. Cheng, F. J. Song, J. S. You, Chem. Commun. 2014, 50, 13914.10.1039/C4CC05827ESearch in Google Scholar PubMed
[28] F. Jafarpour, H. Hazrati, N. Mohasselyazdi, M. Khoobi, A. Shafiee, Chem. Commun. 2013, 49, 10935.10.1039/c3cc46959jSearch in Google Scholar PubMed
[29] S. Martins, P. S. Branco, M. C. de la Torre, M. A. Sierra, A. Pereira, Synlett2010, 19, 2918.10.1055/s-0030-1259014Search in Google Scholar
[30] R. Bolton, G. H. Williams, Chem. Soc. Rev. 1986, 15, 261.10.1039/cs9861500261Search in Google Scholar
[31] W. R. Bowman, J. M. D. Storey, Chem. Soc. Rev. 2007, 36, 1803.10.1039/b605183aSearch in Google Scholar PubMed
[32] C. L. Ciana, R. J. Phipps, J. R. Brandt, F. M. Meyer, M. J. Gaunt, Angew. Chem., Int. Ed. 2011, 50, 458.10.1002/anie.201004703Search in Google Scholar PubMed
[33] C. Galli, Chem. Rev. 1988, 88, 765.10.1021/cr00087a004Search in Google Scholar
[34] G. Pratsch, T. Wallaschkowski, M. R. Heinrich, Chem. Eur. J. 2012, 18, 11555.10.1002/chem.201200430Search in Google Scholar PubMed
[35] Y. H. Chen, M. Lee, Y. Z. Lin, D. S. Leow, Chem. Asian J. 2015, 10, 1618.10.1002/asia.201500497Search in Google Scholar PubMed
[36] J. Hofmann, H. Jasch, M. R. Heinrich, J. Org. Chem. 2014, 79, 2314.10.1021/jo500063rSearch in Google Scholar PubMed
[37] T. Jiang, S. Y. Chen, H. Zhuang, R. S. Zeng, J. P. Zou, Tetrahedron Lett. 2014, 55, 4549.10.1016/j.tetlet.2014.06.099Search in Google Scholar
[38] M. Y. Li, Y. Ye, ChemCatChem2015, 7, 4137.10.1002/cctc.201500575Search in Google Scholar
[39] T. Jiang, S. Y. Chen, G. Y. Zhang, R. S. Zeng, J. P. Zou, Org. Biomol. Chem. 2014, 12, 6922.10.1039/C4OB00798KSearch in Google Scholar
[40] J. B. Aylward, J. Chem. Soc. C1969, 12, 1663.10.1039/j39690001663Search in Google Scholar
[41] Z. X. Chen, G. W. Wang, J. Org. Chem. 2005, 70, 2380.10.1021/jo047894gSearch in Google Scholar PubMed
[42] H. Firouzabadi, D. Mohajer, M. Entezari-Moghadam, Bull. Chem. Soc. Jpn. 1988, 61, 2185.10.1246/bcsj.61.2185Search in Google Scholar
[43] T. Tangiguchi, H. Zaimoku, H. Ishibashi, Chem. Eur. J.2011, 17, 4307.10.1002/chem.201003060Search in Google Scholar PubMed
[44] P. Patil, A. Nimonkar, K. G. Akamanchi, J. Org. Chem. 2014, 79, 2331.10.1021/jo500131hSearch in Google Scholar PubMed
[45] R. R. Jadhav, S. N. Huddar, K. G. Akamanchi, Eur. J. Org. Chem. 2013, 30, 6779.10.1002/ejoc.201300917Search in Google Scholar
[46] M. Ravi, P. Chauhan, R. Kant, S. K. Shukla, P. P. Yadav, J. Org. Chem. 2015, 80, 5369.10.1021/acs.joc.5b00739Search in Google Scholar PubMed
[47] P. Chauhan, M. Ravi, S. Singh, P. Prajapati, P. P. Yadav, RSC Adv. 2016, 6, 109.10.1039/C5RA20954DSearch in Google Scholar
[48] G. A. Russell, B. Z. Shi, W. Jiang, S. S. Hu, B. H. Kim, W. Baik, J. Am. Chem. Soc. 1995, 117, 3952.10.1021/ja00119a009Search in Google Scholar
[49] S. Kim, D. J. Kang, C. H. Lee, P. H. Lee, J. Org. Chem. 2012, 77, 6530.10.1021/jo301086kSearch in Google Scholar PubMed
[50] L. Liu, H. Lu, H. Wang, C. Yang, X. Zhang, D. Zhang-Negrerie, Y. F. Du, K. Zhao, Org. Lett.2013, 15, 2906.10.1021/ol400743rSearch in Google Scholar PubMed
Supplemental Material:
The online version of this article (DOI: 10.1515/znb-2016-0109) offers supplementary material, available to authorized users.
©2016 Walter de Gruyter GmbH, Berlin/Boston
Articles in the same Issue
- Frontmatter
- In this Issue
- Li2Pt3Se4: a new lithium platinum selenide with jaguéite-type crystal structure by multianvil high-pressure/high-temperature synthesis
- RE4B4O11F2 (RE = Sm, Tb, Ho, Er): four new rare earth fluoride borates isotypic to Gd4B4O11F2
- Regioselective C-3 arylation of coumarins with arylhydrazines via radical oxidation by potassium permanganate
- Two new POM-based compounds containing a linear tri-nuclear copper(II) cluster and an infinite copper(II) chain, respectively
- Environmentally benign synthesis of methyl 6-amino-5-cyano-4-aryl-2,4-dihydropyrano[2,3-c]pyrazole-3-carboxylates using CeO2 nanoparticles as a reusable and robust catalyst
- Synthesis and structural characterization of Ca12Ge17B8O58
- Synthesis of some new hydrazide-hydrazones related to isatin and its Mannich and Schiff bases
- Purpureone, an antileishmanial ergochrome from the endophytic fungus Purpureocillium lilacinum
- Syntheses and crystal structures of two new silver–organic frameworks based on N-pyrazinesulfonyl-glycine: weak Ag···O/N interaction affecting the coordination geometry
Articles in the same Issue
- Frontmatter
- In this Issue
- Li2Pt3Se4: a new lithium platinum selenide with jaguéite-type crystal structure by multianvil high-pressure/high-temperature synthesis
- RE4B4O11F2 (RE = Sm, Tb, Ho, Er): four new rare earth fluoride borates isotypic to Gd4B4O11F2
- Regioselective C-3 arylation of coumarins with arylhydrazines via radical oxidation by potassium permanganate
- Two new POM-based compounds containing a linear tri-nuclear copper(II) cluster and an infinite copper(II) chain, respectively
- Environmentally benign synthesis of methyl 6-amino-5-cyano-4-aryl-2,4-dihydropyrano[2,3-c]pyrazole-3-carboxylates using CeO2 nanoparticles as a reusable and robust catalyst
- Synthesis and structural characterization of Ca12Ge17B8O58
- Synthesis of some new hydrazide-hydrazones related to isatin and its Mannich and Schiff bases
- Purpureone, an antileishmanial ergochrome from the endophytic fungus Purpureocillium lilacinum
- Syntheses and crystal structures of two new silver–organic frameworks based on N-pyrazinesulfonyl-glycine: weak Ag···O/N interaction affecting the coordination geometry