Metal-free oxidative coupling of aminopyridines with nitroolefins to imidazo[1,2-a]pyridine in the presence of I2–TBHP–pyridine
-
Litao An
 ,
Xiaojun Sun
,
Xiaojun Sun

Abstract
A facile metal-free approach to the synthesis of imidazo[1,2-a]pyridine was developed through a tandem reaction of Michael addition and oxidative coupling. Iodine–t-butyl hydroperoxide–pyridine was found to be a green and efficient catalytic system for this approach.
1 Introduction
Metal-catalyzed coupling reactions have provided powerful tools for the formation of carbon–carbon bonds and carbon–heteroatom bonds to a wide range of compounds in many areas [1–4]. Coupling reactions have been extensively studied to develop innovative and greener approaches. Metal-free coupling is an important and increasingly trendy highlight that has attracted much attention in recent years [5–7]. Although traditional metal-catalyzed coupling reactions have been proven effective in many industrial applications, they are also generating harmful metal waste and much other organic waste. In addition, some metal catalysts are moisture and/or air sensitive. Owing to mild conditions, low cost, and good efficiency, metal-free catalysts could be candidates for many green processes in both academic and industrial research.
Imidazo[1,2-a]pyrimidines are important heterocyclic compounds [8]. They are prominent among nitrogen-containing fused heterocycles and are known for a plethora of biological and pharmacological properties [9], such as antidepressant [10], anticancer, antiviral, antiparasitic, and anti–human immunodeficiency virus activities. Furthermore, they are also the core structure of some currently marketed drugs, such as zolimidine, zolpidem, loprinone, and alpidem. Therefore, much interest has been drawn to develop the imidazo[1,2-a]pyridine preparation.
Traditionally, coupling of 2-aminopyridines with α-halocarbonyl compounds provides a practical method to imidazo[1,2-a]pyridine [11–13]. This protocol was also extended to similar α-functionalized ketones [14, 15]. Other methods included (i) the condensation of 2-aminopyridines and aromatic ketones [16–18]; (ii) one-pot condensations of 2-aminopyridines, aldehydes, and isonitriles [19–22]; (iii) three-component reactions of 2-aminopyridines, aldehydes, and alkynes [13–25], or oxidative cross-coupling/cyclization between 2-aminopyridines and terminal alkynes [26]; (iv) condensation of 2-aminopyridines with 1,3-dicarbonyl compounds [27, 28]; and (v) condensation of 2-aminopyridines with chalcones [29, 30]. (vi) Moreover, recently, Yan and co-workers have developed a CuBr-catalyzed method for the preparation of imidazo[1,2-a]pyridines from aminopyridines and nitroolefins [31]. Subsequently, FeCl2 [32] and FeCl3 [33, 34] were developed to promote the construction of imidazo[1,2-a]pyridine from in situ–generated or as-prepared nitroolefins. However, there are very few reports on metal-free catalysts for this route [35, 36]. In the former example, the authors found that Bu4NI–TBHP (t-butyl hydroperoxide) was an efficient catalytic system [35]. In the more recent example, a new protocol was presented in which the cycloaddition of N-aminopyridines and β-nitroolefins to substituted pyrazolo[1,5-a]pyridines had been developed [36]. Thus, a mild, green protocol for the synthesis of imidazo[1,2-a]pyridine is still desirable. Encouraged by the previous reports and as a part of our continuing study on metal-free/catalyst-free reactions in green chemistry [37, 38], we wish to report herein a metal-free I2–TBHP–pyridine combination mediated by the formation of imidazo[1,2-a]pyridine via Michael addition and oxidative coupling.
2 Results and discussion
To begin with our study, the reaction of 2-aminopyridine and β-nitrostyrene was chosen as a model reaction to screen the conditions (Table 1). The reaction occurred at room temperature, resulting in poor yield (Table 1, entry 1). Even at an elevated temperature, the material could not be consumed completely. Inspired by I2–TBHP in the C–N oxidative coupling [29, 39], the catalytic system was also tested for the model reaction. Fortunately, a good yield was achieved (Table 1, entry 2). When a catalytic amount of pyridine was added to the reaction mixture, the material was consumed in shorter time with comparative yield (Table 1, entry 3). When the loading of I2 was increased to 0.2 equiv, a better yield was given (Table 1, entry 4). Similarly, less TBHP gave poor yield (Table 1, entry 5). It is shown that it may be due to the synergistic effect in the catalytic system of I2–TBHP–Py [40]. However, the model reaction under the optimal condition without any solvent gave a negative result (Table 1, entry 6). Besides these trials, other candidates were also investigated, but they could not lead to better results (Table 1, entries 7–12).
Optimization of reaction conditions.a
 | |||||
|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | Temp. (°C) | Reaction time (h) | Yieldb (%) |
| 1 | I2 | CH3CN | r. t. | 24 | 36c |
| 2 | I2–TBHP | CH3CN | 80 | 20 | 80d |
| 3 | I2–TBHP–Py | CH3CN | 80 | 12 | 81e |
| 4 | I2–TBHP–Py | CH3CN | 80 | 12 | 90f |
| 5 | I2–TBHP–Py | CH3CN | 80 | 12 | 66g,c |
| 6 | I2–TBHP–Py | Solventless | 80 | 24 | 16f,c |
| 7 | TBHP | CH3CN | r. t. | 24 | – h |
| 8 | TBHP | CH3CN | 80 | 24 | – h |
| 9 | I2–Py | CH3CN | 80 | 12 | 71i |
| 10 | TBHP–Py | CH3CN | 80 | 24 | 80j |
| 11 | I2–30 % H2O2 | CH3CN | 80 | 12 | 52k |
| 12 | KI–TBHP | CH3CN | 80 | 17 | 65l |
aReaction condition: 2-aminopyridine (1.0 mmol), nitroolefin (1.05 mmol), solvent (10 mL), temperature means the oil bath temperature; bisolated yields; cI2 (0.2 mmol) was used, and the materials were not consumed completely; dI2 (0.1 mmol) and TBHP (2.0mmol) were added; eI2 (0.1 mmol), pyridine (0.62 mmol), and TBHP (2.0 mmol) were added; fI2 (0.2 mmol), pyridine (0.62 mmol), and TBHP (2.0 mmol) were added; gI2 (0.2 mmol), pyridine (0.62 mmol), and TBHP (1.0 mmol) were added; hno reaction, and TBHP (2.0 mmol) was used; iI2 (0.2 mmol) and pyridine (0.62 mmol) were added; jTBHP (2.0 mmol) and pyridine (0.62 mmol) were added; k30 % H2O2 (1 mL) was used; lKI (0.2 mmol) and TBHP (2.0 mmol) were added.
The substrate scope then was investigated under the optimized reaction conditions (Table 2). In most cases, the reaction proceeded smoothly in moderate to good yield. It was observed that some functional groups, such as amino, nitro, halo, and methoxy, were tolerable in the catalytic system. Generally speaking, 2-aminopyridine resulted in better yields than 3-methyl 2-aminopyridine. It was reported that ortho-substituted nitroolefins did not give the desired imidazo[1,2-a]pyridines in the presence of Bu4NI–TBHP [35]. It is noteworthy that fairly moderate yields were achieved for ortho-substituted nitroolefins under our catalytic system (Table 2, 3e, 3m). It was interesting that the catalytic system was effective even at room temperature in some cases (Table 2, 3d–i, 3k, 3m, 3o). Based on TLC analysis, a reaction at room temperature gave fewer spots than those at elevated temperature in these cases. Finally, the scope was extended to other substrates (Table 3). As for a heterocycle-substituted nitroolefin, it produced a complex mixture (Table 3, entry 1). It was the same when (E)-(2-nitroprop-1-en-1-yl)benzene was used instead of β-nitrostyrene (Table 3, entry 2). The reaction of β-nitrostyrene with acetylated 2-aminopyridine did not occur (Table 3, entry 3). It might be due to reduced activity of an amino in the presence of an acetyl group. If the amino was replaced with a benzyl group, only a poor yield could be obtained. It was more interesting that the benzyl was removed in the reaction process (Table 3, entry 4).
Synthesis of imidazo[1,2-a]pyridines.a
 | |||
|---|---|---|---|
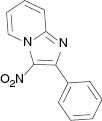 | 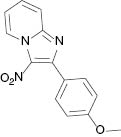 | 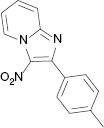 | 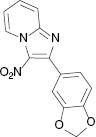 |
| 3a, 80 °C, 90 %b | 3b, 80 °C, 85 % | 3c, 80 °C, 87 % | 3d, r. t., 69 % |
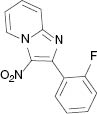 |  |  |  |
| 3e, r. t., 40 % | 3f, r. t., 71 % | 3g, r. t., 70 % | 3h, r. t., 78 % |
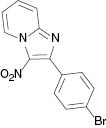
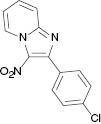
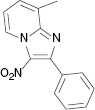 | 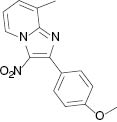 | 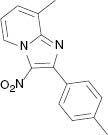 | 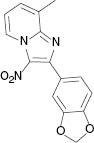 |
| 3i, r. t., 48 % | 3j, 100 °C, 46 % | 3k, r. t., 58 % | 3l, 80 °C, 71 % |
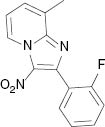 |  | 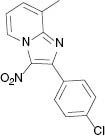 | |
| 3m, r. t., 47 % | 3n, 100 °C, 46 % | 3o, r. t., 65 % | |
aReaction conditions: 2-aminopyridine (1.0 mmol), nitroolefin (1.05 mmol), I2 (0.2 mmol), pyridine (0.62 mmol), and TBHP (2.0 mmol) were added in CH3CN (10 mL). The reaction time was 12 h. Temperature means the oil bath temperature. bIsolated yields.
The trials on extension of scope under the present catalytic system.
| Entry | 1 | 2 | Product (yield) | Temp. (°C) |
|---|---|---|---|---|
| 1 | 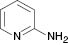 | 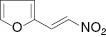 | Complex mixture | 80 |
| 2 | 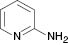 | 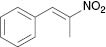 | Complex mixture | 100 |
| 3 | 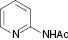 | 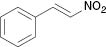 | No reaction | 100 |
| 4 | 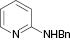 | 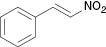 | 3a (20 %) | 100 |
| 5 | 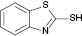 | 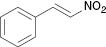 | No reaction | 100 |
| 6 | 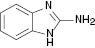 |  | No reaction | 100 |
| 7 | 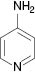 | 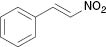 | No reaction | 100 |
| 8 | 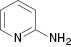 | 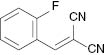 | No reaction | 100 |
aReaction conditions: 2-aminopyridine (1.0 mmol), nitroolefin (1.05 mmol), I2 (0.2 mmol), pyridine (0.62 mmol), TBHP (2.0 mmol), and CH3CN (10 mL). Temperature means the oil bath temperature. The reaction time was 12 h. bIsolated yields.
We have also attempted to construct new fused thiazoles and imidazoles by this protocol. Structures similar to 2-aminopyridine, such as 2-aminobenzimidazole and 2-aminobenzothiazole, were chosen as candidates (Table 3, entries 5 and 6). However, neither of them took part in the reaction. It was also observed that 4-aminopyridine does not undergo the Michael reaction with β-nitrostyrene under the catalytic condition (Table 3, entry 7). Activated alkene, 2-(2-fluorobenzylidene) malononitrile, which was suggested as a Michael addition acceptor, did not proceed as well in the reaction (Table 3, entry 8). Thus, this method showed good selectivity.
Based on the above results and previous reports [31, 35, 40], a plausible mechanism is put forward (Scheme 1). On the one hand, “outer complex” 4 was formed from pyridine and iodine. In the presence of TBHP, intermediate 5 was generated, which split into two kinds of radicals, 6 and 7. The radicals 6 or 7 resulted in PyHI 8 or TBHP after activating the intermediate 12, which would be illustrated in the following step. PyHI 8 and HI were oxidized by TBHP to “outer complex” 4. Thus, the catalytic cycle was achieved.

Proposed mechanism.
On the other hand, intermediate 9 was produced from β-nitrostyrene and 2-aminopyridine via Michael addition. The intermediate 9 was oxidized to imine 10, from which enamine 11 equilibrated. The intermediate 12, generated by one-electron oxidation, was abstracted of hydrogen by radicals 6 or 7 to intermediate 13. After intramolecular nucleophilic addition occurred, the final product was obtained by proton elimination of intermediate 14.
3 Conclusions
In summary, we have successfully developed a metal-free catalytic system for the synthesis of 3-nitro-2-arylimidazo[1,2-a]pyridine derivatives. The component in this system shows a synergistic effect for catalysis. It is simple and environmentally benign.
4 Experimental section
All materials were obtained from commercial suppliers and were used without further purification unless otherwise noted. The nitroolefins were prepared by the Henry reaction. All solvents were reagent grade. Flash chromatography was performed with silica gel (200–300 mesh). The nuclear magnetic resonance (NMR) spectra were measured on an AVANCE 400 spectrometer at 400 MHz for 1H and 100 MHz for 13C in CDCl3 solution with trimethylsilyl as an internal standard. Chemical shifts (δ) are given in parts per million and coupling constants (J) in Hertz. High-resolution mass spectroscopy (HRMS) spectra were recorded on an LTQ-Orbitrap mass spectrometer [(+)-ESI].
4.1 General procedure for the synthesis of imidazo[1,2-a]pyridine derivatives
In a 50-mL round-bottomed flask, a mixture of 2-aminopyridine (1 mmol) and nitroolefin (1.05 mmol) was stirred in 10 mL acetonitrile. Then, I2 (0.2 mmol), TBHP (2 mmol), and pyridine (0.05 mL) were added to the above mixture, and the resulting mixture was allowed to stir under reflux or at room temperature for the specified time (Table 2). The progress of the reaction was monitored by thin-layer chromatography (TLC). After the completion of the reaction, the reaction mixture was cooled to room temperature and poured into saturated sodium bisulfite solution (15 mL). After sonication for 10 min, the mixture was extracted with ethyl acetate (3 × 15 mL), followed by drying with anhydrous MgSO4. After evaporation of the solvent, the residue was purified by flash chromatography to afford the desired product.
4.1.1 3-Nitro-2-phenylimidazo[1,2-a]pyridine (3a) [35]
1H NMR (400 MHz, CDCl3): δ = 9.53 (d, J = 6.9 Hz, 1H), 7.91–7.92 (m, 2H), 7.86 (d, J = 8 Hz, 1H), 7.67 (t, J = 7.9 Hz, 1H), 7.52–7.53 (m, 3H), 7.28–7.32 (m, 1H) ppm. – 13C NMR (100 MHz, CDCl3): δ = 150.3, 145.2, 131.9, 130.8, 130.2, 130.1, 128.2, 128.2, 118.3, 116.5. – HRMS [(+)-ESI]: m/z = 240.0776 (calcd. 240.0773 for C13H10N3O2, [M+H]+).
4.1.2 2-(4-Methoxyphenyl)-3-nitroimidazo[1,2-a]pyridine (3b) [31]
1H NMR (400 MHz, CDCl3): δ = 9.50 (d, J = 6.8 Hz, 1H), 7.95 (d, J = 8.5 Hz, 2H), 7.81 (d, J = 8.8 Hz, 1H), 7.64 (t, J = 8 Hz, 1H), 7.24 (d, J = 6.8 Hz, 1H), 7.02 (d, J = 8.5 Hz, 2H), 3.89 (s, 3H) ppm. – HRMS [(+)-ESI]: m/z = 270.0871 (calcd. 270.0879 for C14H12N3O3, [M+H]+).
4.1.3 3-Nitro-2-p-tolylimidazo[1,2-a]pyridine (3c) [35]
1H NMR (400 MHz, CDCl3): δ = 9.51 (d, J = 6.9 Hz, 1H), 7.83 (d, J = 8.0 Hz, 3H), 7.64 (t, J = 7.9 Hz, 1H), 7.31 (d, J = 8 Hz, 2H), 7.22–7.27 (m, 1H), 2.44 (s, 3H) ppm. – 13C NMR (100 MHz, CDCl3): δ = 150.4, 145.2, 140.5, 130.7, 130.0, 129.0, 128.9, 128.2, 118.2, 116.3, 21.5 ppm. – HRMS [(+)-ESI]: m/z = 276.0740 (calcd. 276.0749 for C14H11N3O2Na, [M+Na]+).
4.1.4 2-(Benzo[d][1, 3]dioxol-5-yl)-3-nitroimidazo[1,2-a]pyridine (3d) [41]
1H NMR (400 MHz, CDCl3): δ = 9.50 (d, J = 6.8 Hz, 1H), 7.80 (d, J = 8.9 Hz, 1H), 7.64 (t, J = 7.9 Hz, 1H), 7.52 (dd, J = 8.1, 1.4 Hz, 1H), 7.43 (d, J = 1.2 Hz, 1H), 7.24–7.27 (m, 1H), 6.93 (d, J = 8.1 Hz, 1H), 6.05 (s, 1H) ppm. – 13C NMR (100 MHz, CDCl3): δ = 150.0, 149.5, 147.5, 145.1, 131.0, 128.3, 125.5, 125.1, 118.2, 116.3, 110.5, 108.2, 101.5 ppm. – HRMS [(+)-ESI]: m/z = 284.0670 (calcd. 284.0671 for C14H10N3O4, [M+H]+).
4.1.5 2-(2-Fluorophenyl)-3-nitroimidazo[1,2-a]pyridine (3e)
1H NMR (400 MHz, CDCl3): δ = 9.47 (d, J = 7.0 Hz, 1H), 7.85 (d, J = 8.9 Hz, 1H), 7.63–7.70 (m, 2H), 7.48–7.53 (m, 1H), 7.28–7.32 (m, 2H), 7.21 (t, J = 9.2 Hz, 1H) ppm. – 13C NMR (100 MHz, CDCl3): δ = 161.8, 159.3, 145.2, 144.3, 131.9, 131.8, 131.1, 131.1, 130.7, 127.8, 124.2, 124.2, 121.0, 120.8, 118.4, 116.7, 115.9, 115.7 ppm. – HRMS [(+)-ESI]: m/z = 280.0496 (calcd. 280.0498 for C13H8FN3O2Na, [M+Na]+).
4.1.6 2-(3-Bromophenyl)-3-nitroimidazo[1,2-a]pyridine (3f) [31]
1H NMR (400 MHz, CDCl3): δ = 9.42 (d, J = 7.2 Hz, 1H), 8.01 (s, 1H), 7.81 (t, J = 8.4 Hz, 2H), 7.57–7.66 (m, 2H), 7.24–7.35(m, 2H) ppm. – 13C NMR (100 MHz, CDCl3): δ = 148.2, 145.0, 133.9, 133.0, 132.8, 131.1, 129.6, 128.8, 128.0, 122.0, 118.3, 116.8 ppm. – HRMS [(+)-ESI]: m/z = 339.9702 (calcd. 339.9698 for C13H8BrN3O2Na, [M+Na]+).
4.1.7 2-(4-Bromophenyl)-3-nitroimidazo[1,2-a]pyridine (3g) [41]
1H NMR (400 MHz, CDCl3): δ = 9.5 (d, J = 7.2 Hz, 1H), 7.80–7.85 (m, 3H), 7.63–7.69 (m, 3H), 7.27–7.32 (m, 1H) ppm. – 13C NMR (100 MHz, CDCl3): δ = 149.1, 145.2, 131.7, 131.4, 131.1, 130.8, 128.2, 124.9, 118.4, 116.7 ppm. – HRMS [(+)-ESI]: m/z = 339.9695 (calcd. 339.9698 for C13H8BrN3O2Na, [M+Na]+).
4.1.8 2-(4-Chlorophenyl)-3-nitroimidazo[1,2-a]pyridine (3h) [31]
1H NMR (400 MHz, DMSO): δ = 9.42 (d, J = 6.8 Hz, 1H), 7.97 (d, J = 8.8, 1H), 7.82–7.88 (m, 3H), 7.59 (d, J = 8 Hz, 2H), 7.49 (t, J = 7.2 Hz, 1H). – 13C NMR (100 MHz, DMSO): δ = 148.3, 145.2, 135.1, 132.4, 132.2, 131.6, 128.9, 128.5, 118.3, 117.7. – HRMS [(+)-ESI]: m/z = 274.0387 (calcd. 274.0383 for C13H9ClN3O2, [M+H]+).
4.1.9 8-Methyl-3-nitro-2-phenylimidazo[1,2-a]pyridine (3i) [31]
1H NMR (400 MHz, CDCl3): δ = 9.35 (d, J = 6.8 Hz, 1H), 7.91 (d, J = 3.6 Hz, 2H), 7.48–7.50 (m, 3H), 7.43 (d, J = 6.8 Hz, 1H),7.16 (t, J = 8 Hz, 1H), 2.73 (s, 3H) ppm. – 13C NMR (100 MHz, CDCl3): δ = 149.7, 145.2, 132.3, 130.1, 130.0, 129.9, 128.7, 128.1, 125.9, 116.5, 77.4, 77.1, 76.7, 16.6 ppm. – HRMS [(+)-ESI]: m/z = 254.0933 (calcd. 254.0930 for C14H12N3O2, [M+H]+).
4.1.10 2-(4-Methoxyphenyl)-8-methyl-3-nitroimidazo[1,2-a]pyridine (3j)
1H NMR (400 MHz, CDCl3): δ = 9.32 (d, J = 6.0 Hz, 1H), 7.94 (d, J = 8.6 Hz, 2H), 7.40 (d, J = 6.9 Hz, 1H), 7.11 (t, J = 6.9 Hz, 1H), 7.00 (d, J = 8.4 Hz, 2H), 3.87 (s, 3H), 2.69 (s, 3H) ppm. – 13C NMR (100 MHz, CDCl3): δ = 161.22, 149.59, 145.21, 131.89, 129.85, 128.32, 125.97, 124.48, 116.11, 113.59, 55.37, 16.52 ppm. – HRMS [(+)-ESI]: m/z = 306.0857 (calcd. 306.0855 for C15H13N3O3Na, [M+Na]+).
4.1.11 8-Methyl-3-nitro-2-p-tolylimidazo[1,2-a]pyridine (3k) [31]
1H NMR (400 MHz, CDCl3): δ = 9.33 (d, J = 6.9 Hz, 1H), 7.83 (d, J = 7.9 Hz, 2H), 7.40 (d, J = 7.0 Hz, 1H), 7.30 (d, J = 7.8 Hz, 2H), 7.13 (t, J = 7.0 Hz, 1H), 2.70 (s, 3H), 2.43 (s, 3H) ppm. – 13C NMR (100 MHz, CDCl3): δ = 149.8, 145.2, 140.3, 130.1, 129.8, 129.3, 128.8, 128.5, 125.9, 116.3, 21.5, 16.6 ppm. – HRMS [(+)-ESI]: m/z = 268.1082 (calcd. 268.1086 for C15H14N3O2, [M+H]+).
4.1.12 2-(Benzo[d][1,3]dioxol-5-yl)-8-methyl-3-nitroimidazo[1,2-a]pyridine (3l)
1H NMR (400 MHz, CDCl3): δ = 9.35 (d, J = 6.9 Hz, 1H), 7.51–7.58 (dd, J = 6,4 Hz, 1.6 Hz, 1H), 7.43(d, J = 1.6 Hz, 2H), 7.15 (t, J = 7.1 Hz, 1H), 6.93 (d, J = 8.1 Hz, 1H), 6.04 (s, 1H), 2.71 (s, 3H) ppm. – 13C NMR (100 MHz, CDCl3): δ = 149.4, 149.3, 147.5, 145.1, 129.9, 128.4, 125.9, 125.1, 116.3, 110.6, 108.2, 101.5, 16.6 ppm. – HRMS [(+)-ESI]: m/z = 320.0626 (calcd. 320.0647 for C15H11N3O4Na, [M+Na]+).
4.1.13 2-(2-Fluorophenyl)-8-methyl-3-nitroimidazo[1,2-a]pyridine (3m)
1H NMR (400 MHz, CDCl3): δ = 9.36 (d, J = 6.8 Hz, 1H), 7.66–7.70 (m, 1H), 7.45–7.53 (m, 2H), 7.28–7.32 (m, 1H), 7.18–7.23 (m, 2H), 2.74 (s, 3H) ppm. – 13C NMR (100 MHz, CDCl3): δ = 161.9, 159.4, 145.3, 143.8, 131.8, 131.7, 131.2, 131.2, 129.7, 128.7, 125.5, 124.2, 124.2, 121.2, 121.1, 116.6, 115.9, 115.6, 16.7 ppm. – HRMS [(+)-ESI]: m/z = 294.0651 (calcd. 294.0655 for C14H10FN3O2Na, [M+Na]+).
4.1.14 2-(4-Bromophenyl)-8-methyl-3-nitroimidazo[1,2-a]pyridine (3n)
1H NMR (400 MHz, CDCl3): δ = 9.37 (d, J = 6.9 Hz, 1H), 7.82 (d, J = 8.4 Hz, 1H), 7.65 (d, J = 8.4 Hz, 1H), 7.47 (d, J = 7.0 Hz, 1H), 7.20 (t, J = 7.0 Hz, 1H), 2.73 (s, 2H) ppm. – 13C NMR (100 MHz, CDCl3): δ = 148.5, 145.2, 131.8, 131.4, 131.1, 130.1, 128.7, 125.9, 124.6, 116.7, 16.6 ppm. – HRMS [(+)-ESI]: m/z = 332.0016 (calcd. 332.0029 for C14H11BrN3O2, [M+H]+).
4.1.15 2-(4-Chlorophenyl)-8-methyl-3-nitroimidazo[1,2-a]pyridine (3o)
1H NMR (400 MHz, CDCl3): δ = 9.36 (d, J = 6.9 Hz, 1H), 7.88 (d, J = 8.4 Hz, 2H), 7.47 (t, J = 7.2 Hz, 3H), 7.19 (t, J = 7.0 Hz, 1H), 2.72 (s, 3H) ppm. – 13C NMR (100 MHz, CDCl3): δ = 148.5, 145.2, 136.2, 131.6, 130.7, 130.1, 128.7, 128.4, 125.9, 116.7, 16.6 ppm. – HRMS [(+)-ESI]: m/z = 288.0530 (calcd. 288.0540 for C14H11ClN3O2, [M+H]+).
5 Supporting Information
General experimental information, spectroscopic data, and spectra of compounds 3a–3o are summarized as Supporting Information available online (DOI: 10.1515/znb-2015-0171).
Acknowledgments
We are grateful to the National Natural Science Foundation of China (21375044), University Natural Science Foundation of Jiangsu Province (12KJD150002), and Administration of Science & Technology of Huaian City (HAG2013011) for financial support.
References
[1] Z. H. Shao, H. B. Zhang, Chem. Soc. Rev.2012, 41, 560.Search in Google Scholar
[2] C. F. Lee, Y. C. Liu, S. S. Badsara, Chem.-Asian J.2014, 9, 706.Search in Google Scholar
[3] Q. Xiao, Y. Zhang, J. B. Wang, Acc. Chem. Res.2013, 46, 236.Search in Google Scholar
[4] A. Bhunia, S. R. Yetra, A. T. Biju, Chem. Soc. Rev.2012, 41, 3140.Search in Google Scholar
[5] J. J. Mousseau, A. B. Charette, Acc. Chem. Res.2013, 46, 412.Search in Google Scholar
[6] R. A. D. Arancon, C. S. K. Lin, C. Vargas, R. Luque, Org. Biomol. Chem.2014, 12, 10.Search in Google Scholar
[7] R. Samanta, K. Matcha, A. P. Antonchick, Eur. J. Org. Chem.2013, 2013, 5769.Search in Google Scholar
[8] F. Couty, G. Evano, in Comprehensive Heterocyclic Chemistry III (Eds. A. R. Katritzky, C. A. Ramsden, E. F. V. Scriven, R. J. K. Taylor), Elsevier, Oxford, UK, 2008, p. 409.Search in Google Scholar
[9] C. Enguehard-Gueiffier, A. Gueiffier, Mini-Rev. Med. Chem. Oxford, UK, 2007, 7, 888.10.2174/138955707781662645Search in Google Scholar PubMed
[10] M. H. Wiegand, Drugs2008, 68, 2411.10.2165/0003495-200868170-00001Search in Google Scholar PubMed
[11] Y. Vara, E. Aldaba, A. Arrieta, J. L. Pizarro, M. I. Arriortua, F. P. Cossio, Org. Biomol. Chem.2008, 6, 1763.Search in Google Scholar
[12] S. El Kazzouli, S. Berteina-Raboin, A. Mouaddib, G. Guillaumet, Lett. Org. Chem.2012, 9, 118.Search in Google Scholar
[13] M. Adib, A. Mohamadi, E. Sheikhi, S. Ansari, H. R. Bijanzadeh, Synlett2010, 1606.10.1055/s-0029-1219962Search in Google Scholar
[14] J. S. Yadav, B. V. S. Reddy, Y. G. Rao, M. Srinivas, A. V. Narsaiah, Tetrahedron Lett.2007, 48, 7717.Search in Google Scholar
[15] C. M. Yu, X. Chen, R. Wu, G. H. Yang, J. Y. Shi, L. L. Pan, Eur. J. Org. Chem.2014, 2014, 2037.Search in Google Scholar
[16] A. J. Stasyuk, M. Banasiewicz, M. K. Cyranski, D. T. Gryko, J. Org. Chem.2012, 77, 5552.Search in Google Scholar
[17] K. Pericherla, P. Kaswan, P. Khedar, B. Khungar, K. Parang, A. Kumar, RSC Adv.2013, 3, 18923.Search in Google Scholar
[18] D. C. Mohan, R. R. Donthiri, S. N. Rao, S. Adimurthy, Adv. Synth. Catal.2013, 355, 2217.Search in Google Scholar
[19] M. Adib, M. Mahdavi, M. A. Noghani, P. Mirzaei, Tetrahedron Lett.2007, 48, 7263.Search in Google Scholar
[20] B. Maiti, K. Chanda, M. Selvaraju, C. C. Tseng, C. M. Sun, ACS Comb. Sci.2013, 15, 291.Search in Google Scholar
[21] A. H. Shinde, M. Srilaxmi, B. Satpathi, D. S. Sharada, Tetrahedron Lett.2014, 55, 5915.Search in Google Scholar
[22] A. Maleki, S. Javanshir, M. Naimabadi, RSC Adv.2014, 4, 30229.Search in Google Scholar
[23] J. B. Bharate, S. K. Guru, S. K. Jain, S. Meena, P. P. Singh, S. Bhushan, B. Singh, S. B. Bharate, R. A. Vishwakarma, RSC Adv.2013, 3, 20869.Search in Google Scholar
[24] S. K. Guchhait, A. L. Chandgude, G. Priyadarshani, J. Org. Chem.2012, 77, 4438.Search in Google Scholar
[25] T. Palani, K. Park, M. R. Kumar, H. M. Jung, S. Lee, Eur. J. Org. Chem.2012, 5038.10.1002/ejoc.201200679Search in Google Scholar
[26] C. He, J. Hao, H. Xu, Y. Mo, H. Liu, J. Han, A. Lei, Chem. Commun.2012, 48, 11073.Search in Google Scholar
[27] L. Ma, X. Wang, W. Yu, B. Han, Chem. Commun.2011, 47, 11333.Search in Google Scholar
[28] X. Wang, L. Ma, W. Yu, Synthesis2011, 43, 2445.Search in Google Scholar
[29] Y. F. Zhang, Z. K. Chen, W. L. Wu, Y. H. Zhang, W. P. Su, J. Org. Chem.2013, 78, 12494.Search in Google Scholar
[30] K. Monir, A. K. Bagdi, S. Mishra, A. Majee, A. Hajra, Adv. Synth. Catal.2014, 356, 1105.Search in Google Scholar
[31] R. -L. Yan, H. Yan, C. Ma, Z. -Y. Ren, X. -A. Gao, G. -S. Huang, Y. -M. Liang, J. Org. Chem.2012, 77, 2024.Search in Google Scholar
[32] H. Yan, S. Yang, X. Gao, K. Zhou, C. Ma, R. Yan, G. Huang, Synlett2012, 23, 2961.10.1055/s-0032-1317685Search in Google Scholar
[33] H. Yan, Y. L. Wang, C. M. Pan, H. Zhang, S. Z. Yang, X. Y. Ren, J. Li, G. S. Huang, Eur. J. Org. Chem.2014, 2014, 2754.Search in Google Scholar
[34] S. Santra, A. K. Bagdi, A. Majee, A. Hajra, Adv. Synth. Catal.2013, 355, 1065.Search in Google Scholar
[35] X. S. Xu, P. Z. Hu, W. B. Yu, G. Hong, Y. C. Tang, M. W. Fang, X. Q. Li, Synlett2014, 25, 718.10.1055/s-0033-1340485Search in Google Scholar
[36] C. Ravi, D. Chandra Mohan, N. Naresh Kumar Reddy, S. Adimurthy, RSC Adv.2015, 5, 42961.Search in Google Scholar
[37] D. P. Li, G. L. Zhang, L. T. An, J. P. Zou, W. Zhang, Green Chem.2011, 13, 594.Search in Google Scholar
[38] L. T. An, F. Q. Ding, J. P. Zou, Dyes Pigm.2008, 77, 478.Search in Google Scholar
[39] X. Liu, G. Q. Yu, J. H. Li, D. Wang, Y. X. Chen, K. Q. Shi, B. H. Chen, Synlett2013, 24, 1588.10.1055/s-0033-1338871Search in Google Scholar
[40] J. T. Zhang, Z. T. Wang, Y. Wang, C. F. Wan, X. Q. Zheng, Z. Y. Wang, Green Chem.2009, 11, 1973.Search in Google Scholar
[41] H. Yan, R. Yan, S. Yang, X. Gao, Y. Wang, G. Huang, Y. Liang, Chem.-Asian J.2012, 7, 2028.Search in Google Scholar
Supplemental Material
The online version of this article (DOI: 10.1515/znb-2015-0171) offers supplementary material, available to authorized users.
©2016 by De Gruyter
Articles in the same Issue
- Frontmatter
- In this Issue
- Salaterpene E, a eudesmane-type sesquiterpene from Salacia longipes var. camerunensis
- N-Acyl-N-(4-chlorophenyl)-4-nitrobenzenesulfonamides: highly selective and efficient reagents for acylation of amines in water
- Synthesis and characterization of Cu(II)-halide 1-methylimidazole complexes
- Highly selective naked-eye anion sensors based on thioureido or amido calix[4]arenes
- A tetranuclear ruthenium complex with bridging pyridine-2,4-dicarboxylato ligands forming a square metallamacrocycle
- Ab initio studies of the structural, electronic, and optical properties of quaternary BxAlyGa1–x–yN compounds
- Activation of P4 by Li[SitBu3]: generation of lithium bis(supersilyl)heptaphosphanortricyclanide Li[P7(SitBu3)2]
- Metal-free oxidative coupling of aminopyridines with nitroolefins to imidazo[1,2-a]pyridine in the presence of I2–TBHP–pyridine
- La3Cu4P4O2 and La5Cu4P4O4Cl2: synthesis, structure and 31P solid state NMR spectroscopy
- Notes
- Synthesis and single-crystal structure of the pseudo-ternary compounds LiA[N(CN)2]2 (A = K or Rb)
- The first pseudo-ternary thiocyanate containing two alkali metals – synthesis and single-crystal structure of LiK2[SCN]3
Articles in the same Issue
- Frontmatter
- In this Issue
- Salaterpene E, a eudesmane-type sesquiterpene from Salacia longipes var. camerunensis
- N-Acyl-N-(4-chlorophenyl)-4-nitrobenzenesulfonamides: highly selective and efficient reagents for acylation of amines in water
- Synthesis and characterization of Cu(II)-halide 1-methylimidazole complexes
- Highly selective naked-eye anion sensors based on thioureido or amido calix[4]arenes
- A tetranuclear ruthenium complex with bridging pyridine-2,4-dicarboxylato ligands forming a square metallamacrocycle
- Ab initio studies of the structural, electronic, and optical properties of quaternary BxAlyGa1–x–yN compounds
- Activation of P4 by Li[SitBu3]: generation of lithium bis(supersilyl)heptaphosphanortricyclanide Li[P7(SitBu3)2]
- Metal-free oxidative coupling of aminopyridines with nitroolefins to imidazo[1,2-a]pyridine in the presence of I2–TBHP–pyridine
- La3Cu4P4O2 and La5Cu4P4O4Cl2: synthesis, structure and 31P solid state NMR spectroscopy
- Notes
- Synthesis and single-crystal structure of the pseudo-ternary compounds LiA[N(CN)2]2 (A = K or Rb)
- The first pseudo-ternary thiocyanate containing two alkali metals – synthesis and single-crystal structure of LiK2[SCN]3