Activation of P4 by Li[SitBu3]: generation of lithium bis(supersilyl)heptaphosphanortricyclanide Li[P7(SitBu3)2]
-


Abstract
Treatment of P4 with one equivalent of Li[SitBu3] leads to the formation of a number of oligo-phosphanes and -phosphides, e.g. the bicyclo[1.1.0]tetraphosphane P4(SitBu3)2, the heptaphosphanortricyclane P7(SitBu3)3, the tetraphosphides Li3[P(PSitBu3)3] (Li3[2a]), and the pentaphosphacyclopentadienide Li[P5]. From this reaction we could isolate single crystals of Li3[2a]. However, this reaction took another course in the presence of Li[OSitBu3]. When P4 was treated with one equivalent of Li[SitBu3] in the presence of Li[OSitBu3], the heptaphosphanortricyclanide Li[P7(SitBu3)2] (Li[8a]) was formed. Single crystals of the cluster {Li4(C6H6)(OSitBu3)[8a]3}·C6H6 (orthorhombic, space group Pca21) were isolated from the reaction mixture at ambient temperature. This cluster compound consists of three chiral Li[P7(SitBu3)2] units, one silanolate Li[OSitBu3], and one benzene molecule. We further investigated the degradation reaction of the bicyclo[1.1.0]tetraphosphane P4(SitBu3)2. After heating a benzene solution to 60 °C for 24 h, we found 100 % conversion of P4(SitBu3)2, and P7(SitBu3)3 (monoclinic, space group P21/c) and tBu3SiPH2 were formed.
1 Introduction
In the past few decades, the reactivity of P4 towards nucleophilic agents has been extensively studied [1–6]. Previously we have reported that the products of the reaction between P4 and the silanides M[SitBu3] (M = Li, Na, K) [7–9] depend strongly on the stoichiometry and the solvent [9–17].
Using the reactants in molar ratios from 1:2 to 1:4, different phosphides were formed: (i) the tetraphosphenediides M2[tBu3SiPPPPSitBu3] (M2[1a]; M = Li, Na, K) and Na2[tBu2PhSiPPPPSiPhtBu2] (Na2[1b]) were obtained from the reaction of P4 with M[SitBu3] (M = Li, Na, K) [7–9] and Na[SiPhtBu2] [18, 19] in a molar ratio of 1:2 in thf [9–12, 20] (the supersilylated octaphosphides M4[P8(SitBu3)4] (M = Na, K) were also synthesized in a 1:2 stoichiometry but in weakly polar solvents (heptane, tBuOMe, etc.); see [9–11, 20]); (ii) the synthesis of the tetraphosphides M3[P(PSitBu3)3] (M3[2a]; M = Li, Na) and Na3[P(PSiPhtBu2)3] (Na3[2b]) was achieved by the reaction of P4 with the silanides M[SitBu3] (M = Li, Na) and Na[SiPhtBu2] in a 1:3 stoichiometry in benzene. However, in thf, M3[2a] (M = Li, Na, K) and Na3[2b] are unstable and thereby (iii) M[tBu3SiPPPSitBu3] (M[3a]; M = Li, Na, K) and Na[tBu2PhSiPPPSiPhtBu2] (Na[3b]) were formed [11–13]; (iv) the pentaphosphide Na2[P5(SitBu3)3] (Na2[4a]) could be synthesized by treating P4 with four equivalents of Na(thf)2[SitBu3] in benzene [12, 14, 15]. Recently, we have discovered that white phosphorus reacts with three molar equivalents of Li[Mes] in benzene forming the phosphide Li3[P(PMes)3] [21] that has been produced in analogous 1:3 reactions of P4 with the silanides M[SitBu3] (M = Li, Na) and Na[SiPhtBu2]. In this paper, we present the reaction of P4 with one equivalent of supersilyllithium Li[SitBu3] in the presence of Li[OSitBu3] by which the lithium bis(supersilyl)heptaphosphanortricyclanide Li[P7(SitBu3)2] (Li[8a]) has been obtained.
2 Results and discussion
When P4 in benzene was treated with one molar equivalent of Li[SitBu3], several phosphorus-containing products were formed, as monitored by 31P NMR spectroscopy: e.g. the bicyclo[1.1.0]tetraphosphane P4(SitBu3)2 [10], the heptaphosphanortricyclane P7(SitBu3)3 [22], the tetraphosphide Li3[2a] [13], and the pentaphosphacyclopentadienide Li[P5] [23]. Furthermore, we could isolate single crystals of the tetraphosphide Li3[2a] (structural details: CCDC 1426534) from the reaction mixture. However, the reaction of P4 with Li[SitBu3] took another course in the presence of Li[OSitBu3] [24, 25]. When P4 was treated with one equivalent of Li[SitBu3] in the presence of Li[OSitBu3] [24, 25], the heptaphosphanortricyclanide Li[P7(SitBu3)2] (Li[8a]) was formed. Single crystals of the cluster {Li4(C6H6)(OSitBu3)[8a]3}·C6H6 were isolated from this reaction.
This result suggested a mechanism which is shown in Scheme 1: (i) At first, due to the poor solubility of P4 in benzene, and as consequence thereof, an excess of Li[SitBu3] being present in solution, Li3[2a] was formed. In a second step (ii) Li3[2a] releases Li2[PSitBu3] to give Li[3a], and finally, (iii) Li[3a] reacts with P4 to form the heptaphosphanortricyclanide Li[8a]. It is worth mentioning that the donor-supported degradation of M3[2a] (M = Li, Na, K) generally leads to the formation of the triphosphallyl compounds M[3a]. As alluded to above, in this case, Li[OSitBu3] was suggested to act as a donor for the degradation of Li3[2a].
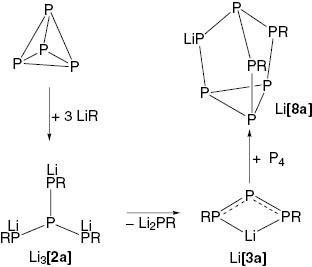
Reaction of P4 with supersilyllithium LiR (R = SitBu3).
Interestingly, cluster build-up also takes place when the bicyclo[1.1.0]tetraphosphane P4(SitBu3)2 is thermolized. After heating a benzene solution to 60 °C for 24 h, we found 100 % conversion of P4(SitBu3)2, and the heptaphosphanortricyclane P7(SitBu3)3 (structural details: CCDC 983799) and the supersilylphosphane tBu3SiPH2 were formed. As shown in Scheme 2, the supersilyl phosphirene 9a is apparently the key intermediate of this reaction.
![Scheme 2: Degradation of the bicyclo[1.1.0]tetraphosphane P4R2 (R = SitBu3).](/document/doi/10.1515/znb-2015-0156/asset/graphic/j_znb-2015-0156_scheme_002.jpg)
Degradation of the bicyclo[1.1.0]tetraphosphane P4R2 (R = SitBu3).
The crystals which were obtained from the reaction of P4 with one equivalent of Li[SitBu3] in the presence of Li[OSitBu3] are composed of three molecules of the heptaphosphanortricyclanide Li[8a], one molecule of Li[OSitBu3], and two molecules of benzene. While many examples of heptaphosphanortricyclanes of types I [26, 27] and IV [22, 27, 28] are known, types II [29] and III [30] are more elusive (Fig. 1). [PPh4][P7(PhCH2)2] is the only example of a type III cluster which has been reported as yet in the literature [30].
![Fig. 1: Heptaphosphanortricyclanides M3[P7] (I), M2[RP7] (II), M[R2P7] (III), and heptaphosphanortricyclanes [R3P7] IV and definition of the height h.](/document/doi/10.1515/znb-2015-0156/asset/graphic/j_znb-2015-0156_fig_001.jpg)
Heptaphosphanortricyclanides M3[P7] (I), M2[RP7] (II), M[R2P7] (III), and heptaphosphanortricyclanes [R3P7] IV and definition of the height h.
The cluster {Li4(C6H6)(OSitBu3)[8a]3} (shown in Figs. 2 and 3; for selected bond lengths and angles see the caption to Fig. 2) crystallizes in the orthorhombic space group Pca21 with an additional benzene molecule in the asymmetric unit (Fig. 2). The cluster consists of three chiral, crystallographically independent, and configurationally identical heptaphosphanortricyclanide Li[8a] units, one silanolate Li[OSitBu3], and one benzene molecule. Within the error of measurement, all three heptaphosphanortricyclanides show identical structural parameters. Therefore, we discuss only the structure of one of those three units. The compound features a Li triangle capped by one [OSitBu3]– anion. Each Li cation of this triangle is bonded to two different P atoms of one P7 cluster and to one P atom of another cluster. Two of the seven P atoms are substituted by a supersilyl residue pointing away from the center of the molecule. The three P7 clusters coordinate a fourth Li cation. The coordination sphere of this Li cation is completed by η6-coordination of a benzene molecule.
![Fig. 2: Molecular structure of {Li4(C6H6)(OSitBu3)[8a]3} in the solid state. Hydrogen atoms, carbon atoms on silicon, and cocrystallized C6H6 are omitted for clarity. Selected bond lengths (Å) and bond angles (deg): Li(1)–P(5) 2.568(8), Li(1)–COG(C6H6) 2.258, Li(2)–O(1) 1.922(8), Li(3)–O(1) 1.906(8), Li(4)–O(1) 1.912(9), Li(2)–P(2) 2.656(8), Li(4)–P(2) 2.691(8), Li(4)–P(3) 2.775(8), P(1)–P(2) 2.1598(16), P(1)–P(3) 2.1995(16), P(1)–P(4) 2.1910(16), P(2)–P(5) 2.1434(17), P(3)–P(6) 2.2015(16), P(4)–P(7) 2.1701(16), P(5)–P(6) 2.2188(17), P(5)–P(7) 2.2264(16), P(6)–P(7) 2.2405(16), P(3)–Si(1) 2.3455(17), P(4)–Si(2) 2.3046(16); P(5)–Li(1)–P(5B) 94.0(3), P(2)–Li(2)–P(3B) 102.3(2), P(2)–Li(2)–Li(4) 59.6(2), Li(3)–Li(2)–Li(4) 59.4(3), P(2)–P(1)–P(3) 95.46(6), P(2)–P(1)–P(4) 106.15(6), P(1)–P(3)–P(6) 102.58(6), P(6)–P(5)–P(7) 60.53(5).](/document/doi/10.1515/znb-2015-0156/asset/graphic/j_znb-2015-0156_fig_002.jpg)
Molecular structure of {Li4(C6H6)(OSitBu3)[8a]3} in the solid state. Hydrogen atoms, carbon atoms on silicon, and cocrystallized C6H6 are omitted for clarity. Selected bond lengths (Å) and bond angles (deg): Li(1)–P(5) 2.568(8), Li(1)–COG(C6H6) 2.258, Li(2)–O(1) 1.922(8), Li(3)–O(1) 1.906(8), Li(4)–O(1) 1.912(9), Li(2)–P(2) 2.656(8), Li(4)–P(2) 2.691(8), Li(4)–P(3) 2.775(8), P(1)–P(2) 2.1598(16), P(1)–P(3) 2.1995(16), P(1)–P(4) 2.1910(16), P(2)–P(5) 2.1434(17), P(3)–P(6) 2.2015(16), P(4)–P(7) 2.1701(16), P(5)–P(6) 2.2188(17), P(5)–P(7) 2.2264(16), P(6)–P(7) 2.2405(16), P(3)–Si(1) 2.3455(17), P(4)–Si(2) 2.3046(16); P(5)–Li(1)–P(5B) 94.0(3), P(2)–Li(2)–P(3B) 102.3(2), P(2)–Li(2)–Li(4) 59.6(2), Li(3)–Li(2)–Li(4) 59.4(3), P(2)–P(1)–P(3) 95.46(6), P(2)–P(1)–P(4) 106.15(6), P(1)–P(3)–P(6) 102.58(6), P(6)–P(5)–P(7) 60.53(5).
![Fig. 3: The structure of the heptaphosphanortricyclanide Li[8a] in {Li4(C6H6)(OSitBu3)[8a]3}. Only one of the three crystallographically independent and configurationally identical P7 clusters is shown.](/document/doi/10.1515/znb-2015-0156/asset/graphic/j_znb-2015-0156_fig_003.jpg)
The structure of the heptaphosphanortricyclanide Li[8a] in {Li4(C6H6)(OSitBu3)[8a]3}. Only one of the three crystallographically independent and configurationally identical P7 clusters is shown.
The structural parameters of Li[8a] are closer to those of type IV clusters (P7(SitBu3)3, structural details: CCDC 983799) than to those in [Li(tmeda)]3P7 [28] (type I). The mean length of the P–P bonds in the P3 ring in Li[8a] is 2.228 Å. This is comparable to the value of P7(SitBu3)3 (2.224 Å), whereas the length of the related P–P bonds in [Li(tmeda)]3P7 is 2.255 Å. The distances between the bridgehead P atom and the bridging P atoms of two of these three bonds in Li[8a] are comparable to the other two types, but the bond P(1)–P(2) is 0.04 Å shorter than those in type I and IV clusters.
The distance P(2)–P(5) (2.1434(17) Å) is related to those found in [Li(tmeda)]3P7 (2.150 Å), whereas the bond lengths P(3)–P(6) and P(4)–P(7) (mean value: 2.186 Å) are like those in P7(SitBu3)3 (2.185 Å). A comparison of the heights h reveals that the height of Li[8a] (h = 3.112 Å) is in between the h values for [Li(tmeda)]3P7 (3.02 Å) and P7(SitBu3)3 (3.158 Å).
3 Experimental section
The solvents thf, heptane, benzene, and C6D6 were stored over sodium/benzophenone and distilled prior to use. Li[SitBu3] [7] and P4(SitBu3)2 [10] were prepared according to the published procedures. All other starting materials were purchased from commercial sources and used without further purification. The NMR spectra were recorded on Bruker AM 250, DPX 250, Avance 400, and Avance 500 spectrometers. NMR chemical shifts (δ) are reported in ppm.
3.1 Reaction of P4 with Li[SitBu3]
A solution of Li[SitBu3] [7] (554 mg, 2.7 mmol) in heptane (10 mL) was added to a mixture of P4 (300 mg, 2.4 mmol) in benzene (10 mL). In the 31P NMR spectrum of the reaction, mixture signals were observed which are attributable to, e.g. the bicyclo[1.1.0]tetraphosphane P4(SitBu3)2 [10], the tetraphosphides Li3[2a] [13], the heptaphosphanortricyclane P7(SitBu3)3 [22], and the pentaphosphacyclopentadienide Li[P5] [23]. Furthermore, we could isolate single crystals of the tetraphosphides Li3[2a] (structural details: CCDC 1426534) from the reaction mixture at ambient temperature.
3.2 Reaction of P4 with Li[SitBu3] in the presence of Li[OSitBu3]
A solution of Li[SitBu3] [7] (75 mg, 0.36 mmol) and Li[OSitBu3] [25] (8 mg, 0.04 mmol) in benzene (0.8 mL) was added to a mixture of P4 (43 mg, 0.35 mmol) in benzene (3 mL). Cocrystals {Li4(C6H6)(OSitBu3)[8a]3}·C6H6 were obtained from the reaction mixture at ambient temperature (yield: 31 mg, 36 %). {Li4(C6H6)(OSitBu3)[8a]3}: – 1H NMR (300.1 MHz, C6D6): δ = 1.35 (br, SitBu3), 1.23 ppm (s, OSitBu3). – 31P NMR (101.25 MHz, C6D6): δ = –177.4 ppm (due to fluxionality arising from Cope rearrangement of the cluster cage at room temperature, only one signal was observed in the 31P NMR spectrum; cf. [31]). – Anal. for C96H201Li4OP21Si7 (2246.43) calcd. C 51.33, H 9.02; found C 51.75, H 9.25.
3.3 Thermolysis of P4(SitBu3)2
A solution of P4(SitBu3)2 (52 mg, 0.1 mmol) in benzene (1 mL) was heated to 60 °C for 24 h. By cooling to room temperature single crystals of P7(SitBu3)3 (structural details: CCDC 983799) were grown from the solution. The 31P NMR spectrum of the mother liquor revealed solely signals which are attributable to the heptaphosphanortricyclane P7(SitBu3)3 and the supersilylphosphane tBu3SiPH2.
3.4 Crystal structure determination
Data of {Li4(C6H6)(OSitBu3)[8a]3} · C6H6 were collected on a Stoe IPDS II two-circle diffractometer with a Genix Microfocus tube with mirror optics using MoKα radiation (λ = 0.71073 Å) and were scaled using the frame scaling procedure in the X-area program system [32]. The structure was solved by Direct Methods using the program Shelxs [33, 34] and refined against F2 with full-matrix least-squares techniques using the program Shelxl-97 [33, 34]. Details of the crystal structure analysis are summarized in Table 1.
Crystal data and numbers pertinent to data collection and structure refinement of {Li4(C6H6)(OSitBu3)[8a]3} · C6H6.
| ({Li4(C6H6)(OSitBu3)[8a]3} · C6H6 | |
|---|---|
| Empirical formula | C96H201Li4OP21Si7 |
| Mr | 2246.32 |
| Crystal size, mm3 | 0.42 × 0.38 × 0.23 |
| Crystal system | Orthorhombic |
| Space group | Pca21 |
| a, Å | 29.2281(6) |
| b, Å | 15.0606(3) |
| c, Å | 29.3704(5) |
| V, Å3 | 12928.6(4) |
| Z | 4 |
| Dcalcd., g cm–3 | 1.154 |
| μ(MoKα), mm–1 | 0.373 |
| F(000), e | 4840 |
| hkl range | ±35, –18/17, ±35 |
| ((sinθ)/λ)max, Å–1 | 0.6171 |
| Refl. measured | 124 037 |
| Refl. unique | 24 556 |
| Rint | 0.0510 |
| Param. refined | 1163 |
| R(F)a/wR(F2)b (all refls.) | 0.0430/0.0916 |
| GoF (F2)c | 1.063 |
| a/bb | 0.035/11.9894 |
| x(Flack) | 0.37(8) |
| Δρfin (max/min), e Å–3 | 0.674/–0.282 |
aR1 = Σ||Fo| – |Fc||/Σ|Fo|.
bwR2 = [Σw(Fo2 – Fc2)2/Σw(Fo2)2]1/2, w = [σ2(Fo2) + (aP)2 + bP]–1, where P = [Max(Fo2, 0) + 2Fc2]/3.
cGoF = S = [Σw(Fo2 – Fc2)2/(nobs – nparam)]1/2.
CCDC 983160 ({Li4(C6H6)(OSitBu3)[8a]3} · C6H6) contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre viawww.ccdc.cam.ac.uk/data_request/cif.
References
[1] M. Scheer, G. Balazs, A. Seitz, Chem. Rev.2010, 110, 4236.10.1021/cr100010eSearch in Google Scholar
[2] B. M. Cossairt, N. A. Piro, C. C. Cummins, Chem. Rev.2010, 110, 4164.10.1021/cr9003709Search in Google Scholar
[3] A. R. Fox, R. J. Wright, E. Rivard, P. P. Power, Angew. Chem. Int. Ed.2005, 44, 7729.10.1002/anie.200502865Search in Google Scholar
[4] J. D. Masuda, W. W. Schoeller, B. Donnadieu, G. Bertrand, Angew. Chem. Int. Ed.2007, 46, 7052.Search in Google Scholar
[5] J. D. Masuda, W. W. Schoeller, B. Donnadieu, G. Bertrand, J. Am. Chem. Soc.2007, 129, 14180.10.1021/ja077296uSearch in Google Scholar
[6] J. J. Weigand, M. Holthausen, R. Fröhlich, Angew. Chem. Int. Ed.2009, 48, 295, and references cited therein.Search in Google Scholar
[7] N. Wiberg, K. Amelunxen, H.-W. Lerner, H. Schuster, H. Nöth, I. Krossing, M. Schmidt-Amelunxen, T. Seifert, J. Organomet. Chem.1997, 542, 1.10.1016/S0022-328X(97)00306-9Search in Google Scholar
[8] H.-W. Lerner, I. Sänger, F. Schödel, K. Polborn, M. Bolte, M. Wagner, Z. Naturforsch.2007, 62b, 1285.Search in Google Scholar
[9] H.-W. Lerner, Coord. Chem. Rev.2005, 249, 781.10.1016/j.ccr.2004.08.020Search in Google Scholar
[10] A. Lorbach, A. Nadj, S. Tüllmann, F. Dornhaus, F. Schödel, I. Sänger, G. Margraf, J. W. Bats, M. Bolte, M. C. Holthausen, M. Wagner, H.-W. Lerner, Inorg. Chem.2009, 48, 1005.10.1021/ic8016003Search in Google Scholar
[11] H.-W. Lerner, M. Bolte, K. Karaghiosoff, M. Wagner, Organometallics2004, 23, 6073.10.1021/om049348jSearch in Google Scholar
[12] A. Lorbach, S. Breitung, I. Sänger, F. Schödel, M. Bolte, M. Wagner, H.-W. Lerner, Inorg. Chim. Acta, 2011, 378, 1.10.1016/j.ica.2011.07.050Search in Google Scholar
[13] H.-W. Lerner, M. Wagner, M. Bolte, Chem. Commun.2003, 990.10.1039/b210984kSearch in Google Scholar PubMed
[14] H.-W. Lerner, G. Margraf, L. Kaufmann, J. W. Bats, M. Bolte, M. Wagner, Eur. J. Inorg. Chem.2005, 1932.Search in Google Scholar
[15] N. Wiberg, A. Wörner, H.-W. Lerner, K. Karaghiosoff, H. Nöth, Z. Naturforsch.1998, 53b, 1004.Search in Google Scholar
[16] H.-W. Lerner, G. Margraf, J. Bats, M. Wagner, Chem. Commun.2005, 4545.10.1039/b507833dSearch in Google Scholar PubMed
[17] N. Wiberg, A. Wörner, H.-W. Lerner, K. Karaghiosoff, D. Fenske, G. Baum, A. Dransfeld, P. V. Ragué Schleyer, Eur. J. Inorg. Chem.1998, 833.10.1002/(SICI)1099-0682(199806)1998:6<833::AID-EJIC833>3.3.CO;2-TSearch in Google Scholar
[18] H.-W. Lerner, S. Scholz, M. Bolte, Z. Anorg. Allg. Chem.2001, 627, 1638.10.1002/1521-3749(200107)627:7<1638::AID-ZAAC1638>3.0.CO;2-CSearch in Google Scholar
[19] H.-W. Lerner, S. Scholz, M. Bolte, M. Wagner, Z. Anorg. Allg. Chem.2004, 630, 443.10.1002/zaac.200300347Search in Google Scholar
[20] N. Wiberg, A. Wörner, K. Karaghiosoff, D. Fenske, Chem. Ber.1997, 130, 135.10.1002/cber.19971300123Search in Google Scholar
[21] A. Hübner, T. Bernert, I. Sänger, E. Alig, M. Bolte, L. Fink, M. Wagner, H.-W. Lerner, Dalton Trans.2010, 39, 7528.Search in Google Scholar
[22] I. Kovács, G. Baum, G. Fritz, D. Fenske, N. Wiberg, H. Schuster, K. Karaghiosoff, Z. Anorg. Allg. Chem.1993, 619, 453.10.1002/zaac.19936190305Search in Google Scholar
[23] M. Baudler, Angew. Chem. Int. Ed.1987, 26, 419.10.1002/anie.198704191Search in Google Scholar
[24] H.-W. Lerner, S. Scholz, M. Bolte, Organometallics2002, 21, 3827.10.1021/om020318vSearch in Google Scholar
[25] A. Budanow, H. Hashemi Haeri, I. Sänger, F. Schödel, M. Bolte, T. Prisner, M. Wagner, H.-W. Lerner, Chem. Eur. J.2014, 20, 10236.10.1002/chem.201403854Search in Google Scholar
[26] W. Hönle, H. G. von Schnering, A. Schmidpeter, G. Burget, Angew. Chem. Int. Ed.1984, 10, 817.10.1002/anie.198408171Search in Google Scholar
[27] W. Hönle, H. G. von Schnering, Z. Anorg. Allg. Chem.1978, 440, 171.Search in Google Scholar
[28] P. Noblet, V. Cappello, G. Tekautz, J. Baumgartner, K. Hassler, Eur. J. Inorg. Chem.2011, 101.10.1002/ejic.201000749Search in Google Scholar
[29] R. S. P. Turbervill, J. M. Goicoechea, Organometallics2012, 31, 2452.10.1021/om300072zSearch in Google Scholar
[30] S. P. Mattamana, K. Promprat, J. C. Fettinger, B. W. Eichhorn, Inorg. Chem.1998, 37, 6222.10.1021/ic9804770Search in Google Scholar
[31] R. S. P. Turbervill, J. M. Goicoechea, Chem. Commun.2012, 48, 1470–1472.10.1039/C1CC12089ASearch in Google Scholar
[32] X-area, Diffractometer control program system, STOE & Cie GmbH, Darmstadt (Germany) 2002.Search in Google Scholar
[33] G. M. Sheldrick, shelxs/l-97, Programs for Crystal Structure Determination, University of Göttingen, Göttingen (Germany) 1997.Search in Google Scholar
[34] G. M. Sheldrick, Acta Crystallogr.2008, A64, 112–122.10.1107/S0108767307043930Search in Google Scholar
©2016 by De Gruyter
Articles in the same Issue
- Frontmatter
- In this Issue
- Salaterpene E, a eudesmane-type sesquiterpene from Salacia longipes var. camerunensis
- N-Acyl-N-(4-chlorophenyl)-4-nitrobenzenesulfonamides: highly selective and efficient reagents for acylation of amines in water
- Synthesis and characterization of Cu(II)-halide 1-methylimidazole complexes
- Highly selective naked-eye anion sensors based on thioureido or amido calix[4]arenes
- A tetranuclear ruthenium complex with bridging pyridine-2,4-dicarboxylato ligands forming a square metallamacrocycle
- Ab initio studies of the structural, electronic, and optical properties of quaternary BxAlyGa1–x–yN compounds
- Activation of P4 by Li[SitBu3]: generation of lithium bis(supersilyl)heptaphosphanortricyclanide Li[P7(SitBu3)2]
- Metal-free oxidative coupling of aminopyridines with nitroolefins to imidazo[1,2-a]pyridine in the presence of I2–TBHP–pyridine
- La3Cu4P4O2 and La5Cu4P4O4Cl2: synthesis, structure and 31P solid state NMR spectroscopy
- Notes
- Synthesis and single-crystal structure of the pseudo-ternary compounds LiA[N(CN)2]2 (A = K or Rb)
- The first pseudo-ternary thiocyanate containing two alkali metals – synthesis and single-crystal structure of LiK2[SCN]3
Articles in the same Issue
- Frontmatter
- In this Issue
- Salaterpene E, a eudesmane-type sesquiterpene from Salacia longipes var. camerunensis
- N-Acyl-N-(4-chlorophenyl)-4-nitrobenzenesulfonamides: highly selective and efficient reagents for acylation of amines in water
- Synthesis and characterization of Cu(II)-halide 1-methylimidazole complexes
- Highly selective naked-eye anion sensors based on thioureido or amido calix[4]arenes
- A tetranuclear ruthenium complex with bridging pyridine-2,4-dicarboxylato ligands forming a square metallamacrocycle
- Ab initio studies of the structural, electronic, and optical properties of quaternary BxAlyGa1–x–yN compounds
- Activation of P4 by Li[SitBu3]: generation of lithium bis(supersilyl)heptaphosphanortricyclanide Li[P7(SitBu3)2]
- Metal-free oxidative coupling of aminopyridines with nitroolefins to imidazo[1,2-a]pyridine in the presence of I2–TBHP–pyridine
- La3Cu4P4O2 and La5Cu4P4O4Cl2: synthesis, structure and 31P solid state NMR spectroscopy
- Notes
- Synthesis and single-crystal structure of the pseudo-ternary compounds LiA[N(CN)2]2 (A = K or Rb)
- The first pseudo-ternary thiocyanate containing two alkali metals – synthesis and single-crystal structure of LiK2[SCN]3