Assessment of R18, COG1410, and APP96-110 in excitotoxicity and traumatic brain injury
-
Li Shan Chiu


Abstract
Cationic arginine-rich and poly-arginine peptides (referred to as CARPs) have potent neuroprotective properties in in vitro excitotoxicity and in vivo models of stroke. Traumatic brain injury (TBI) shares many pathophysiological processes as stroke, including excitotoxicity. Therefore, we evaluated our lead peptide, poly-arginine R18, with the COG1410 and APP96-110 peptides, which have neuroprotective actions following TBI. In an in vitro cortical neuronal glutamic acid excitotoxicity injury model, R18 was highly neuroprotective and reduced neuronal calcium influx, while COG1410 and APP96-110 displayed modest neuroprotection and were less effective at reducing calcium influx. In an impact-acceleration closed-head injury model (Marmarou model), R18, COG1410, and APP96-110 were administered intravenously (300 nmol/kg) at 30 minutes after injury in male Sprague-Dawley rats. When compared to vehicle, no peptide significantly improved functional outcomes, however the R18 and COG1410 treatment groups displayed positive trends in the adhesive tape test and rotarod assessments. Similarly, no peptide had a significant effect on hippocampal neuronal loss, however a significant reduction in axonal injury was observed for R18 and COG1410. In conclusion, this study has demonstrated that R18 is significantly more effective than COG1410 and APP96-110 at reducing neuronal injury and calcium influx following excitotoxicity, and that both R18 and COG1410 reduce axonal injury following TBI. Additional dose response and treatment time course studies are required to further assess the efficacy of R18 in TBI.
Introduction
Neuroprotective pharmacological agents aimed at minimising harm to the brain and improving patient outcomes after a traumatic brain injury (TBI) are currently lacking. As such, TBI places a massive burden on society and the economy, a situation further compounded by its rising incidence [1]. Current surgical and rehabilitative interventions are limited in their ability to improve outcomes caused by TBI, and a large proportion of survivors endure debilitating neurological deficits as a consequence [2]. With limited efficacious treatment options, the development of an effective neuroprotective agent would be of great clinical significance in terms of reducing the impact of TBI on patients and the wider community.
Our laboratory has demonstrated that cationic arginine-rich peptides including polyarginine peptides (hereafter referred to as CARPs) have potent neuroprotective properties in an in vitro glutamic acid excitotoxicity neuronal injury model [3, 4, 5, 6], as well as in vivo after both permanent and transient middle cerebral artery occlusion (MCAO) stroke in rats [4, 7, 8, 9, 10]. Furthermore, increasing arginine content and positive charge are critical for peptide neuroprotective potency (Meloni et al., 2015a), with poly-arginine peptide R18 identified as our lead peptide (18-mer of arginine; Table 1). The COG1410 and APP96-110 peptides are derived from the apolipoprotein E and amyloid precursor proteins, respectively, and have been demonstrated to improve outcomes in several acute brain injury models, including TBI [11, 12, 13, 14, 15, 16, 17, 18]. Interestingly, both COG1410 and APP96-110 are also cationic and arginine-rich (Table 1).
Summary of peptides used in study.
| Peptide | Sequence[*] | Net charge at pH 7 | Purity |
|---|---|---|---|
| R18 | H-RRRRRRRRRRRRRRRRRR-OH | +18 | 98% |
| APP96-110 | Ac-NWCKRGRKQCKTHPH-NH2 | +5 | 98% |
| COG1410 | Ac-AS-Aib-LRKL-Aib-KRLL-NH2 | +3 | 98% |
In view of the involvement of excitotoxicity in secondary central nervous system injury cascades, and the pathophysiological parallels between stroke and TBI, it is likely that CARPs could also have beneficial effects following TBI [19, 20]. Therefore, we assessed our lead peptide, R18, along with the COG1410 and APP96-110 peptides in an in vitro glutamic acid neuronal excitotoxicity injury model and an in vivo impact-acceleration TBI model.
Methods
Peptides used in this study
Details of the peptides used in this study are provided in Table 1. Peptides for this study were synthesised by Mimotopes (Australia) and purified by high performance liquid chromatography. For both in vitro and in vivo studies, peptides were respectively prepared as a 500 μM stock solution in water for irrigation (Baxter, Australia), and in 0.9% sodium chloride for injection (Pfizer, Australia). Reconstituted peptides were stored at -20°C until use.
Primary cortical neuronal cultures
The isolation and culturing of primary cortical neurons was undertaken as previously described [3]. Briefly, cortical tissue extracted from E18-19 Sprague-Dawley rats were dissociated in Dulbecco’s modified Eagle medium (DMEM; Life Technologies, Australia) supplemented with 1.3 mM L-cysteine, 0.9 mM NaHCO3, 10 U/mL papain (Sigma Aldrich, USA), 50 U/mL DNase (Sigma Aldrich), and washed in cold DMEM/10% horse serum. Neurons were resuspended in Neurobasal media (Life Technologies, Australia) containing 2% B27 supplement (B27; Life Technologies) and glutamine (0.5 mM), penicillin G (0.1 mg/ mL) and streptomycin (0.06 mg/mL). Neurons were seeded into 96-well plastic plates (Nunc, Australia) or 96-well glass wells (7 mm diameter, ProTech, Australia) pre-coated with poly-D-lysine (70 – 150 kDa; Sigma-Aldrich) and maintained in a CO2 incubator (5% CO2, 95% air balance, 98% humidity) at 37°C until use on days in vitro 10 to 14. On day in vitro four the mitotic inhibitor cytosine β-D-arabinofuranoside (1 μM; Sigma-Aldrich) was added to the cultures to inhibit the proliferation of non-neuronal cells. Under these conditions, cultures routinely consist of > 95% neurons and 1 - 3% astrocytes.
Glutamic acid excitotoxicity model and peptide treatments
The glutamic acid model is routinely performed on cortical neurons maintained in 96-well plastic culture plates [3, 4]. Peptides were added to culture wells 10 minutes prior to glutamic acid (L-glutamic acid; Sigma Aldrich) exposure by removing media and adding 50 μL of MEM (Minimal Essential Medium; Life Technologies; Cat. No.: 11090. Supplemented with glutamine, penicillin G and streptomycin as above)/2% B27 containing the specific peptide To induce excitotoxicity, 50 μL of MEM/2% B27 containing glutamate (200 μM; final concentration 100 μM) was added to the culture wells and incubated at 37°C in the CO2 incubator for 5 minutes (note: peptide concentration reduced by half during this step). Media in wells was then replaced with 100 μL of MEM/2% B27 and cultures incubated for a further 20 - 24 hours at 37°C in the CO2 incubator. For all experiments, untreated controls with or without glutamic acid treatment underwent the same incubation steps and media additions. In this model, neuronal survival following glutamic acid treatment typically ranges from 2 - 10% (i.e. 90 - 98% cell death).
Neuronal viability assessment
Cell viability was examined with light microscopy to qualitatively assess morphological cell death at 30 - 40 minutes and 24 hours after glutamic acid exposure. The MTS colorimetric viability assay (Promega, Australia) was used to quantitatively assess cell viability 24 hours after glutamic acid exposure. The MTS assay quantifies the ability of cytosolic enzymes in viable cells to reduce the tetrazolium salt (MTS; 4,5-dimethyliazol-2-yl)-5-(3-carboxymethoxy-phenyl)-2-(4- sulfophenyl)-2H-tetrazolium salt) to the water-soluble brown formazan salt, which is detected spectrophotometrically at 490 nm.
Intracellular calcium kinetics
Intracellular calcium influx was monitored in neuronal culture wells (glass wells) using Fura-2 AM (5 μM; Sigma Aldrich, Australia) in real time using a fluorescent plate reader, as previously described [5]. The aim of these experiments was to determine the relative change in intracellular calcium before and after glutamic acid exposure. Briefly, cells were loaded with the fluorescent calcium ion indicator Fura-2 AM in 50 μL MEM/2% B27, 0.1% pluronic F-127 (Sigma-Aldrich), for 20 minutes at 37°C in the CO2 incubator. Fura-2 AM solution was removed from wells, replaced with 50 μL MEM/2% B27 containing peptide (1 or 5 μM) or NMDA/AMPA receptor blockers (MK801/CNQX; 5 μM/5 μM; Tocris Bioscience, UK) and incubated for 10 minutes at 37°C in the CO2 incubator. Control cultures received 50 μL of MEM/2% B27 only. After the 10-minute incubation period, media in wells was replaced with 50 μL of balanced salt solution (mM: 116 NaCl, 5.4 KCl, 1.8 CaCl2, 0.8 MgSO4, 1 NaH2PO4; pH 7.2) and wells were transferred to a spectrophotometer (CLARIOstar, BMG Labtec, Australia) while maintaining temperature at 37°C. Starting at 30 seconds prior to glutamic acid addition (50 μL of 200 μM in MEM/2% B27; final concentration 100 μM) to wells, spectrophotometer measurements (excitation 355 nm/emission 495 nm) were recorded every 5 seconds until 2 minutes after glutamic acid addition. Control wells did not receive glutamic acid treatment. Experiments were performed in triplicate. For fluorescent kinetic tracers, data was converted to reflect proportional neuronal calcium influx relative to both control (no glutamic acid) and glutamic acid treated control, with no glutamic acid control taken as 0% calcium influx (baseline) and glutamic acid control treated as 100% calcium influx.
Traumatic brain injury model and peptide administration
This study was approved by the Animal Ethics Committee of the University of Western Australia and follows the guidelines outlined in the “Australian Code for the Care and Use of Animals for Scientific Purposes”. Male Sprague- Dawley rats weighing 360 – 400 g were housed under controlled conditions with a 12-hour light-dark cycle and free access to food and water ad libitum before and after surgery. A total of 36 animals underwent the procedure, but only 27 survived to the four-day end-point (25% mortality). Nine peptide-treated animals either died unexpectedly, or were euthanased for welfare reasons as per the animal ethics guidelines such as excessive weight-loss (>15%), or persistent respiratory distress (see Table 2 for details). Peptide-treatment groups consisted of 4 – 5 animals, while sham and vehicle treatment groups consisted of 5 and 8 animals, respectively.
Summary of animal deaths following TBI.
| Treatment | Reason for death |
|---|---|
| APP96-110 | Died 2 days post-TBI: autopsy revealed subdural haematoma |
| APP96-110 | Died 2 days post-TBI: animal displayed signs of respiratory distress prior to death |
| APP96-110 | Died shortly after extubation following surgery: autopsy revealed possible lung pathology (pneumonia) |
| APP96-110 | Euthanased 2 days post-TBI due to excessive weight loss >15% |
| COG1410 | Died at conclusion of surgery: animal did not regain spontaneous breathing |
| COG1410 | Euthanased 2 days post-TBI due to respiratory distress |
| COG1410 | Died 3 days post-TBI: autopsy revealed subarachnoid haemorrhage |
| R18 | Euthanased 2 days post-TBI due to excessive weight loss >15% |
| R18 | Euthanased 1 day post-TBI due to blood in urine: autopsy revealed testicular neoplasia |
The Marmarou weight-drop impact-acceleration injury model [21] was adapted for this study. Briefly, rats underwent anaesthesia induction with 5% halothane (mix 30% O2/70% N2O gas), intubated, and maintained using 1 - 2% halothane. To produce the injury, a 435 g brass weight was dropped from a height of 180 cm, onto a steel disc (1 cm diameter and 2 mm thick) adhered to the skull of the rat with cyanoacrylate. Just prior to the induction of injury, the rat was placed prone and secured on a foam bed, with masking tape across the upper back and pelvic region on the dorsal surface. The rat was then temporarily disconnected from anaesthetic (< 1 min) to induce the injury. Sham animals underwent the same surgical and TBI procedures, but were placed adjacent to the apparatus as the weight was released.
At 30 minutes post-impact, treatments were administered intravenously (600 μL over 6 min) through the right internal jugular vein using an infusion pump. Treatments consisted of the vehicle control (0.9% NaCl for injection) and R18, COG1410, and APP96-110 peptides at 300 nmol/kg. Animals were randomised into treatment groups, and all personnel carrying-out animal and histological procedures were blinded to treatments.
Post-surgical animal care and monitoring
At the conclusion of surgery, pethidine (IM: 1 mg in 0.2 mL saline) and bupivacaine were administered (SC: 0.1 mg in 0.2 mL saline per site) to the head surgical wound. A 2 mL volume of injectable saline was also subcutaneously administered to aid hydration. To avoid hypothermia, rat cages were placed on a heating mat during post-surgical monitoring and housed in a holding room maintained at 26 - 28°C. Post-surgery, animals were monitored twice a day. Pethidine (SC: 1 mg in 0.2 mL saline) was administered if the animal’s behaviour was indicative of discomfort, and saline (SC: 2 mL) if animal weight continued to decline. Rats were also provided with sweetened nourishments to encourage food intake.
Functional assessments
Due to the exploratory nature of the study, three behavioural tests (Barnes maze, adhesive tape removal, and rotarod) were assessed to measure potential injury deficits. These assessments were carried out daily, starting at 24 hours after surgery and continued until study end-point (4 days post-TBI). Each animal was given three attempts at each test, and the first attempt was used for statistical analyses.
The Barnes maze is a well-characterised spatial learning and memory test [22] that utilises a rat’s innate need to escape brightly-lit areas. Animals are required to find the location of an escape hole from 20 other holes set in a round table top within 180 seconds. During the test, a bright light is placed above the table to serve as a mild aversion stimulus. The adhesive tape test assesses sensorimotor function [23], and has been previously used to detect deficits in a similar model of TBI [24]. Alternating between right and left forepaws, a 10 mm x 10 mm piece of adhesive tape (Diversified Biotech, USA) is placed on the palmer surface of the forepaw, and the time taken to remove the tape recorded; animals were allowed 180 seconds to remove the tape. The rotarod test is commonly used in experimental TBI [25], and assesses vestibulomotor deficits. The test involves placing the animal on a rotating rod (Model number: MK-630B; Muromachi, Japan) at a speed of four revolutions per minute (rpm), increasing incrementally to 40 rpm; the time that the animal remained on the rod is recorded (maximum time allowable was 180 s).
Histological assessment for axonal injury and hippocampal neuronal loss
Rats were euthanased 4 days post-TBI with pentobarbital (IP; 100 mg/kg) and transcardially perfused with 200 mL of saline followed by 200 mL of 10% neutral buffered formalin. Brains were removed and post-fixed in 4% formalin for 1 week before embedding in paraffin. Brains were sectioned in 10 µm coronal sections at approximately bregma -4.5 for Bielschowsky’s silver staining, and bregma -3.8 for Nissl staining. Stained sections were imaged using light microscopy to assess axonal injury within the corpus callosum and neuronal loss in the cornu ammonis (CA) of the hippocampus. The severity of axonal injury was semi-quantitatively assessed in three consecutive sections to obtain an overall grade, ranging from 0 (indicating absence of injury) to 4, with increasing grade indicating increasing levels of axons displaying disorganised architecture, undulation and varicosities, and disordered orientation of oligodendrocyte nuclei. To determine the number of surviving hippocampal neurons, CA1/CA2 and CA3 pyramidal neurons with an intact cell body and the presence of an obvious nucleus and/or nucleolus at 400X magnification were counted in the entire length of both hippocampi using Image J counter (v1.51j8; National Institutes of Health, USA). Light microscopic images were captured using an Olympus DP-70 digital camera fitted to an Olympus IX70 inverted microscope.
Statistical analysis
All statistical analyses were carried out using SPSS (v.24; IBM, USA) and presented as mean ± standard error of the mean (SEM). Cell viability, calcium kinetics measurements, and CA counts were analysed by an analysis of variance (ANOVA), followed by a Fisher post-hoc test. Area under the curve (AUC) for calcium kinetics data was calculated by trapezoidal approximation of the AUC using fluorescent kinetic data obtained from 5 to 85 seconds after the addition of glutamic acid to wells. Animal mortality was analysed using Chi-square test. All functional and axonal injury grading data were analysed by a Kruskal-Wallis non-parametric test. For statistical purposes, peptide treatment groups were compared to the vehicle treatment group. A value of P < 0.05 was considered statistically significant.
Results
Effect of peptides on cultured neurons exposed to glutamic acid excitotoxicity
The neuroprotective dose response efficacy of R18, COG1410, and APP96-110 peptides was compared in cortical neuronal cultures exposed to glutamic acid excitotoxicity (Figure 1). As previously reported [5], R18 was highly neuroprotective following glutamic acid excitotoxicity, achieving a 98% protective effect at the 1 μM dose. In contrast, COG1410 and APP96-110 provided only modest neuroprotection following glutamic acid excitotoxicity, achieving approximately 10-15% protection at the 1 and 10 μM dose, respectively.

Glutamic acid neuronal excitotoxicity model; peptide dose response experiments. Peptides present in neuronal cultures for 10 minutes before and during the 5-min glutamic acid exposure. Neuronal viability measured 24 h following glutamic acid exposure. MTS data are expressed as percentage of neuronal viability, with no insult and glutamic acid (Glut.) controls at 100% and 5%, respectively. Concentration of peptides are in μM. Data are presented as mean ± SEM; N = 4; *P < 0.05 when compared to glutamic acid control.
Effect of peptides on neuronal intracellular calcium kinetics following glutamic acid excitotoxicity
The ability of R18, COG1410, and APP96-110 peptides at concentrations of 1 and 5 μM to reduce neuronal intracellular calcium influx following exposure to glutamic acid was assessed using a Fura-2 AM calcium kinetics assay (Figure 2a - d). At 1 and 5 μM, R18 significantly reduced neuronal intracellular calcium influx (Figure 2a, d). Comparatively, APP96-110 significantly reduced neuronal calcium influx at 5 μM, but not at 1 μM, while COG1410 displayed a non-significant reduction in calcium influx at 5 μM.

Intracellular calcium assessment using Fura-2 AM after glutamic acid exposure in cortical neuronal cultures. a – c: Fluorescent tracers; fluorescence intensity (FI) of neuronal cultures 30 s before and after the addition (arrow) of glutamic acid (100 μM final concentration) expressed as a percentage of intracellular neuronal calcium influx. Peptides (1 and 5 μM) and NMDA/AMPA receptor blockers (MK801/CNQX; 5 μM/5 μM) were added to neuronal cultures for 10 min and removed (time = 0) before glutamic acid addition. Glutamic acid control received glutamic acid exposure only. Control did not receive peptide or glutamic acid treatment. d: Neuronal calcium influx expressed as trapezoidal area under the curve (AUC). Data are presented as mean ± SEM; N = 3. * P < 0.05 when compared to glutamic acid control.
Animal survival rate and weight loss following TBI
All sham and vehicle-treated animals survived to end of experiment day 4. In contrast, 2 of 7, 3 of 9, and 4 of 9 animals in the R18 (P = 0.83), COG1410 (P = 0.28) and APP96-110 (P = 0.18) treatment groups respectively, did not survive to day 4. Details regarding animal deaths are summarised in Table 2. No significant differences in weight loss were observed between peptide and vehicle treatment groups (data not shown).
Effect of peptides on functional outcomes following TBI
When administered 30 minutes after injury, R18, COG1410, and APP96-110 peptide treatments did not result in any significant improvements in functional outcomes when compared to vehicle (Figures 3 - 5). Although not statistically significant, R18 treatment did improve functional measurements in the adhesive tape (days 1 – 4; Figure 3a - d) and rotarod (days 3 and 4; Figure 4c, d) tests. Similarly, the COG1410 treatment group displayed an improvement in the rotarod test (day 4), but not to a statistically significant level. In contrast, APP96-110 treatment appeared to worsen Barnes maze performance (days 2 - 4; Figure 5b - d).

Adhesive tape test measurements at days 1 - 4 (a - d) post-TBI. Peptide dose 300 nmol/kg. Sham (N = 5), vehicle (N = 8), APP96-110 (N = 5), COG1410 (N = 4), and R18 (N = 5). Data are presented as mean ± SEM. There were no significant differences between vehicle and peptide treatment groups.

Rotarod measurements at days 1 - 4 (a - d) post-TBI. Peptide dose 300 nmol/kg. Sham (N = 5), vehicle (N = 8), APP96-110 (N = 5), COG1410 (N = 4), and R18 (N = 5). Data are presented as mean ± SEM. There were no significant differences between vehicle and peptide treatment groups.

Barnes maze measurements at days 1 - 4 (a - d) post-TBI. Peptide dose 300 nmol/kg. Sham (N = 5), vehicle (N = 8), APP96-110 (N = 5), COG1410 (N = 4), and R18 (N = 5). Data are presented as mean ± SEM. There were no significant differences between vehicle and peptide treatment groups.
Effect of peptides on histological outcomes following TBI
Severity of axonal injury was significantly reduced in the R18 (P = 0.02) and COG1410 (P = 0.05) treatment groups, while APP96-110 had no effect on axonal injury (Figure 6). In the absence of injury, axons within the corpus callosum appeared smooth and well-organised, with oligodendrocyte nuclei arranged parallel to axons (Figure 7a). Following TBI, the architecture of callosal axons showed varying degrees of disorganisation and undulation and at times displayed varicosities, with loss of the normal longitudinal orientation of oligodendrocyte nuclei (Figure 7b - e). Hippocampal CA neuronal loss was increased in vehicle-treated animals compared to sham animals, but not to a statistically significant level (Figure 8). Similarly, no significant differences were observed in CA1/CA2 and CA3 hippocampal counts between peptide and vehicle treatment groups (Figure 8a, b).
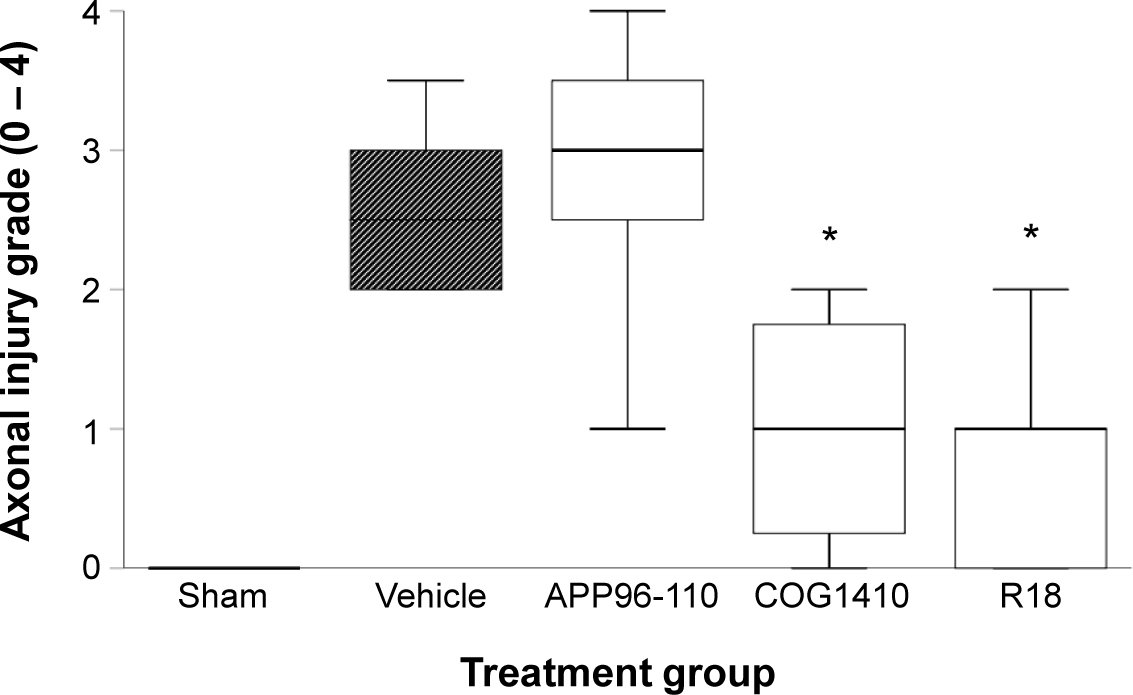
Axonal injury at day 4 post-TBI. Axonal injury grade of sham (N = 5), vehicle (N = 8), APP96-110 (N = 5), COG1410 (N = 4), and R18 (N = 5) treatment groups. Peptide dose 300 nmol/kg. Data are presented as box plots showing median and grade distribution; *P < .05 when compared to vehicle treatment group.

Representative images of axonal injury grading criteria from 0–4 (a – e) following Bielschowsky’s silver stain, as indicated in the key. Examples of axons exhibiting undulated appearance and varicosities are designated ‘V’. Areas showing disordered arrangement of oligodendrocyte nuclei are designated ‘N’. Scale bar represents 100 μM.
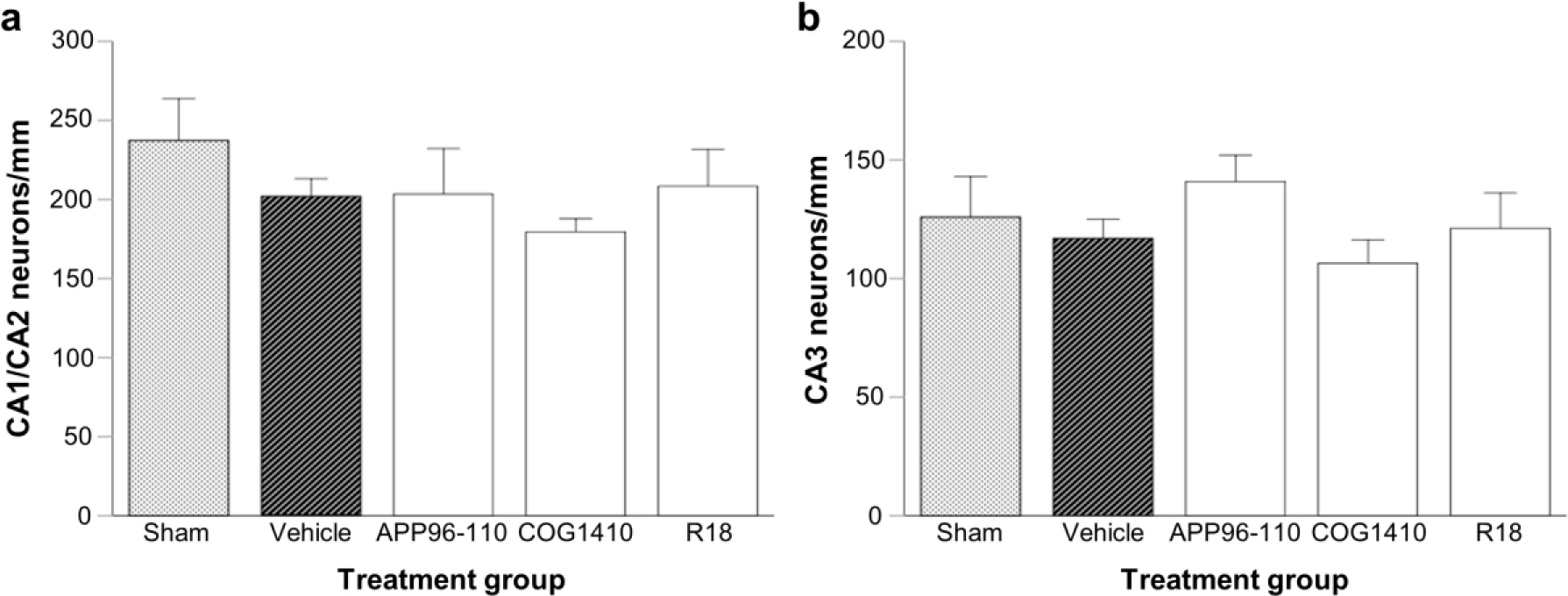
Number of normal appearing CA neurons per millimetere of hippocampus at day 4 post-TBI. a: CA1/CA2; and b: CA3 regions at approximately bregma -3.8. Peptide dose 300 nmol/kg. Data are presented as mean ± SEM. Sham (N = 5), vehicle (N = 8), APP96-110 (N = 5), COG1410 (N = 4), and R18 (N = 5). There were no significant differences between sham, vehicle, and peptide treatment groups.
Discussion
Following previous studies in our laboratory demonstrating the neuroprotective efficacy of CARPs, including our lead peptide R18 in in vivo models of stroke [3, 4, 5, 8, 9, 10, the present study set out to determine if R18 could exert beneficial effects in an in vivo model of TBI. Two other peptides, COG1410 and APP96-110, which have previously demonstrated positive effects in in vivo TBI models were also included for comparative purposes.
An initial in vitro glutamic acid neuronal excitotoxicity study demonstrated R18 to be more neuroprotective and reduced neuronal intracellular calcium influx to a greater degree than both COG1410 and APP96-110. While modest, the ability of COG1410 and APP96-110 to reduce excitotoxic neuronal calcium influx is of interest, as these two peptides were not designed based on this specific mechanism of action. The modest inhibitory action of COG1410 and APP96-110 on calcium influx is likely due to their low cationic charge and the presence of only two arginine residues. We have previously demonstrated that CARPs can reduce excitotoxic calcium influx [4, 7, 26], and that calcium influx inhibition is influenced by peptide cationic charge and arginine content [5, 7].
In the in vivo study, administration of R18, COG1410 and APP96-110 after TBI did not appear to have any significant effects in reducing CA neuronal loss, however CA injury was modest, with vehicle-treated and sham animals not recording significantly different CA neuronal cell counts. In contrast to the results obtained for hippocampal CA injury, R18 and COG1410 significantly reduced axonal injury following TBI, while APP96-110 was ineffective. Diffuse axonal injury is a feature common across all severities of traumatic brain injury [27] and is caused by both mechanical and chemical disturbances [28, 29, 30, 31]. The mechanical forces produce axonal stretching, while chemical disruptions can induce glutamate excitotoxicity, both of which lead to an increase in neuronal intracellular calcium influx.
Furthermore, the toxic increase in neuronal intracellular calcium can subsequently increase mitochondrial calcium levels with detrimental effects on the organelle, which ultimately lead to cell death [31, 32]. Consequently, given the capacity of R18 and COG1410 to reduce excitotoxicity-induced neuronal intracellular calcium, in addition to the known ability of CARPs to target and exert positive effects on mitochondria [20, 33, 34], this could explain the observed positive effects on axonal injury. Furthermore, the reduced axonal injury in R18 and COG1410 treatment groups could explain the trends in improved functional outcomes for these treatments.
The peptides did not have any significant effect on functional outcomes following TBI, although some positive trends were observed for R18 and COG1410 in the adhesive tape and/or rotarod tests. The lack of significant functional improvements most likely reflects the low number of animals used and/or the high variability associated with functional outcomes after TBI. While the small sample size is a limitation of the study, additional animals were not recruited to the study for several reasons: i) the study was considered largely exploratory to establish experimental procedures and uncover any initial evidence of functional and/or histological treatment effects; and ii) on animal welfare grounds due to the unexpectedly high mortality rate experienced in the peptide treatment groups, particularly COG1410 and APP96-110.
Both COG1410 and APP96-110 peptides have previously demonstrated the capacity to reduce axonal injury and functional outcomes following TBI [12, 14]. However, APP96-110 was given intracerebroventricularly rather than intravenously, which may account for its lack of efficacy in the present study, despite reducing in vitro excitotoxicity-induced neuronal intracellular calcium levels. Intracerebroventricular administration of APP96-110 represents a more direct route of delivery into the brain and overcomes many of the systemic pharmacokinetic interactions that would reduce uptake into the brain if administered intravenously such as the blood brain barrier, elimination by the kidney and liver, and degradation by vascular proteases. Similarly, successful outcomes with COG1410 have been achieved with higher doses [11, 14, 15, 35] than used in the present study. The 300 nmol/kg dose was chosen in the present study because R18 is neuroprotective at this dose following MCAO [10], and to increase the possibility of uncovering a differential treatment effect with R18, COG1410, and APP96-110.
Neuroprotection by the APP96-110 peptide is purported to act mainly via its heparin-binding activity [12], however its exact mechanism has not been elucidated. A similar peptide also derived from the amyloid precursor protein suggests that APP96-110 could modulate calcium influx and activate NFκβ transcription factors [36]. Similarly, COG1410 may also affect calcium influx [37] through interactions with cell-surface heparan sulfate proteoglycans during internalisation into the cell [38], but it is generally argued that its neuroprotective action is mainly through interactions with the lipoprotein receptor-associated protein [39]. In light of these reports and the findings of the present study, we propose that the neuroprotective and/or calcium influx inhibitory activity of COG1410 and APP96-110 are mediated at least in part by the positive charge and arginine content of the peptides. In support of this, a more recent CARP derived from selected non-sequential amino acids from the same apolipoprotein E region as COG1410 (CN-105; Ac-VSRRR-NH2, charge +3) has demonstrated histological and functional benefits in a closed-head TBI model [40]. In addition, the CN-105 peptide has also demonstrated efficacy in in vivo models of stroke [41] and intracerebral haemorrhage [42].
While the in vivo study uncovered potential therapeutic effects with R18 and COG1410, several difficulties were encountered with the animal functional assessment procedures. With regards to the rotarod assessment, it was observed that with repeated exposure, rats had a tendency to leap off the rotating rod during the test, leading to a shorter recorded latency. This behaviour was observed in both sham and injured rats. It was also observed that in the Barnes maze test, the bright light became a less effective aversion stimulus with rats willing to explore the table rather than enter the escape hole. Considering this, we suggest additional functional tests and/or minimising the exposure of animals to these tests in future studies. For example, as an alternative learning and memory test the Morris water maze may be employed.
In conclusion, this study has demonstrated that the CARP R18 is more effective than either COG1410 or APP96-110 at reducing neuronal death and calcium influx following in vitro excitotoxicity. In addition, R18 showed greater efficacy than APP96-110 in reducing axonal injury and improving some functional outcomes after TBI. Additional dose response and therapeutic time window studies are required to further evaluate the potential of R18 as a neuroprotective therapy for TBI.
Acknowledgements
This study was supported by grants from the Neurotrauma Research Program of Western Australia and Brain Foundation (Australia), and financial support was also provided by the Perron Institute for Neurological and Translational Science and the Department of Neurosurgery at Sir Charles Gairdner Hospital. We also thank Jim Litis for providing a student PhD scholarship for Li Shan Chiu, Argeo Siliquini for constructing the weight-drop device, and Professor Frank Mastaglia for his assistance in the preparation of this manuscript.
Conflicts of interest: Bruno P. Meloni and Neville W. Knuckey are the holders of several patents regarding the use of arginine-rich peptides as neuroprotective treatments. The other authors declare no conflict of interest.
References
[1] Stein SC, Georgoff P, Meghan S, et al. 150 Years of Treating Severe Traumatic Brain Injury: a Systematic Review of Progress in Mortality. J. Neurotrauma [Internet]. 2010;27:1343–1353. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20392140.10.1089/neu.2009.1206Search in Google Scholar PubMed
[2] Dahdah MN, Barnes S, Buros A, et al. Variations in Inpatient Rehabilitation Functional Outcomes Across Centers in the Traumatic Brain Injury Model Systems Study and the Influence of Demographics and Injury Severity on Patient Outcomes. Arch. Phys. Med. Rehabil. [Internet]. 2016 [cited 2017 Apr 25];97:1821–1831. Available from: http://www.sciencedirect.com/science/article/pii/S0003999316301903.10.1016/j.apmr.2016.05.005Search in Google Scholar PubMed
[3] Meloni BP, Craig AJ, Milech N, et al. The neuroprotective efficacy of cell-penetrating peptides TAT, penetratin, Arg-9, and Pep-1 in glutamic acid, kainic acid, and in vitro ischemia injury models using primary cortical neuronal cultures. Cell. Mol. Neurobiol. 2014;34:173–181.10.1007/s10571-013-9999-3Search in Google Scholar PubMed
[4] Meloni BP, Milani D, Edwards AB, et al. Neuroprotective peptides fused to arginine-rich cell penetrating peptides: Neuroprotective mechanism likely mediated by peptide endocytic properties. Pharmacol. Ther. [Internet]. 2015;153:36–54. Available from: http://linkinghub.elsevier.com/retrieve/pii/S0163725815001175.10.1016/j.pharmthera.2015.06.002Search in Google Scholar PubMed
[5] Meloni BP, Brookes LM, Clark VW, et al. Poly-arginine and arginine-rich peptides are neuroprotective in stroke models. J. Cereb. Blood Flow Metab. [Internet]. 2015;35:993–1004. Available from: http://www.nature.com/doifinder/10.1038/jcbfm.2015.11.10.1038/jcbfm.2015.11Search in Google Scholar PubMed PubMed Central
[6] Edwards AB, Anderton RS, Knuckey NW, et al. Characterisation of neuroprotective efficacy of modified poly-arginine-9 (R9) peptides using a neuronal glutamic acid excitotoxicity model. Mol. Cell. Biochem. 2017;426:75–85.10.1007/s11010-016-2882-zSearch in Google Scholar PubMed
[7] Meloni BP, Milani D, Cross JL, et al. Assessment of the Neuroprotective Effects of Arginine-Rich Protamine Peptides, Poly-Arginine Peptides (R12-Cyclic, R22) and Arginine–Tryptophan-Containing Peptides Following In Vitro Excitotoxicity and/or Permanent Middle Cerebral Artery Occlusion in Rats. NeuroMolecular Med. [Internet]. 2017;1–15. Available from: http://link.springer.com/10.1007/s12017-017-8441-2.10.1007/s12017-017-8441-2Search in Google Scholar PubMed
[8] Milani D, Clark VW, Cross JL, et al. Poly-arginine peptides reduce infarct volume in a permanent middle cerebral artery rat stroke model. BMC Neurosci. [Internet]. 2016;17:19. Available from: http://bmcneurosci.biomedcentral.com/articles/10.1186/s12868-016-0253-z.10.1186/s12868-016-0253-zSearch in Google Scholar PubMed PubMed Central
[9] Milani D, Knuckey NW, Anderton RS, et al. The R18 Polyarginine Peptide Is More Effective Than the TAT-NR2B9c (NA-1) Peptide When Administered 60 Minutes after Permanent Middle Cerebral Artery Occlusion in the Rat. Stroke Res. Treat. [Internet]. 2016;2016:1–9. Available from: http://www.hindawi.com/journals/srt/2016/2372710/.10.1155/2016/2372710Search in Google Scholar PubMed PubMed Central
[10] Milani D, Cross JL, Anderton RS, et al. Neuroprotective efficacy of poly-arginine R18 and NA-1 (TAT-NR2B9c) peptides following transient middle cerebral artery occlusion in the rat. Neurosci. Res. [Internet]. 2017;114:9–15. Available from: http://dx.doi.org/10.1016/j.neures.2016.09.002.10.1016/j.neures.2016.09.002Search in Google Scholar PubMed
[11] Cao F, Jiang Y, Wu Y, et al. Apolipoprotein E-Mimetic COG1410 Reduces Acute Vasogenic Edema following Traumatic Brain Injury. J. Neurotrauma [Internet]. 2016;33:175–182. Available from: http://online.liebertpub.com/doi/10.1089/neu.2015.3887.10.1089/neu.2015.3887Search in Google Scholar PubMed PubMed Central
[12] Corrigan F, Thornton E, Roisman LC, et al. The neuroprotective activity of the amyloid precursor protein against traumatic brain injury is mediated via the heparin binding site in residues 96-110. J. Neurochem. 2014;128:196–204.10.1111/jnc.12391Search in Google Scholar PubMed
[13] Hoane MR, Pierce JL, Holland M a, et al. The novel apolipoprotein E-based peptide COG1410 improves sensorimotor performance and reduces injury magnitude following cortical contusion injury. J. Neurotrauma. 2007;24:1108–1118.10.1089/neu.2006.0254Search in Google Scholar PubMed
[14] Jiang Y, Brody DL. Administration of COG1410 Reduces Axonal Amyloid Precursor Protein Immunoreactivity and Microglial Activation after Controlled Cortical Impact in Mice. J. Neurotrauma. 2012;29:2332–2341.10.1089/neu.2012.2362Search in Google Scholar PubMed PubMed Central
[15] Kaufman NA, Beare JE, Tan AA, et al. COG1410, an apolipoprotein E-based peptide, improves cognitive performance and reduces cortical loss following moderate fluid percussion injury in the rat. Behav. Brain Res. [Internet]. 2010;214:395–401. Available from: http://dx.doi.org/10.1016/j.bbr.2010.06.017.10.1016/j.bbr.2010.06.017Search in Google Scholar PubMed PubMed Central
[16] Laskowitz DT, McKenna SE, Song P, et al. COG1410, a novel apolipoprotein E-based peptide, improves functional recovery in a murine model of traumatic brain injury. J. Neurotrauma. 2007;24:1093–1107.10.1089/neu.2006.0192Search in Google Scholar PubMed
[17] Qin X, You H, Cao F, et al. Apolipoprotein E Mimetic Peptide Increases Cerebral Glucose Uptake by Reducing Blood–Brain Barrier Disruption after Controlled Cortical Impact in Mice: An 18 F-Fluorodeoxyglucose PET/CT Study. J. Neurotrauma [Internet]. 2016;neu.2016.4485. Available from: http://online.liebertpub.com/doi/10.1089/neu.2016.4485.10.1089/neu.2016.4485Search in Google Scholar PubMed PubMed Central
[18] Tukhovskaya EA., Yukin AY, Khokhlova ON, et al. COG1410, a novel apolipoprotein-E mimetic, improves functional and morphological recovery in a rat model of focal brain ischemia. J. Neurosci. Res. 2009;87:677–682.10.1002/jnr.21874Search in Google Scholar PubMed PubMed Central
[19] Chiu LS, Anderton RS, Knuckey NW, et al.The neuroprotective potential of arginine-rich peptides for the acute treatment of traumatic brain injury. Expert Rev. Neurother. [Internet]. 2016;16:361–363. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26840929.10.1586/14737175.2016.1150180Search in Google Scholar PubMed
[20] Chiu LS, Anderton RS, Knuckey NW, et al. Peptide Pharmacological Approaches to Treating Traumatic Brain Injury: a Case for Arginine-Rich Peptides. Mol. Neurobiol. [Internet]. 2016; Available from: http://link.springer.com/10.1007/s12035-016-0287-3.10.1007/s12035-016-0287-3Search in Google Scholar PubMed
[21] Marmarou A, Foda MA, van den Brink W, et al. A new model of diffuse brain injury in rats. Part I: Pathophysiology and biomechanics. J. Neurosurg. 1994;80:291–300.10.3171/jns.1994.80.2.0291Search in Google Scholar PubMed
[22] Barnes CA. Memory deficits associated with senescence: a neurophysiological and behavioral study in the rat. J Comp Physiol Psychol. 1979;93:74–104.10.22215/etd/1977-00330Search in Google Scholar
[23] Bouet V, Boulouard M, Toutain J, et al. The adhesive removal test: a sensitive method to assess sensorimotor deficits in mice. Nat. Protoc. [Internet]. 2009;4:1560–1564. Available from: http://dx.doi.org/10.1038/nprot.2009.125.10.1038/nprot.2009.125Search in Google Scholar PubMed
[24] Alwis DS, Yan EB, Morganti-Kossmann M-C, et al. Sensory Cortex Underpinnings of Traumatic Brain Injury Deficits. PLoS One [Internet]. 2012;7:e52169. Available from: https://doi.org/10.1371/journal.pone.0052169.10.1371/journal.pone.0052169Search in Google Scholar PubMed PubMed Central
[25] Hamm RJ, Pike BR, O’Dell DM, et al. The Rotarod Test : An Evaluation of Its Effectiveness in Assessing Motor Deficits Following Traumatic Brain Injury. J. Neurotrauma [Internet]. 1994;11:187–196. Available from: http://www.ncbi.nlm.nih.gov/pubmed/7932797.10.1089/neu.1994.11.187Search in Google Scholar PubMed
[26] MacDougall G, Anderton RS, Edwards AB, et al. The Neuroprotective Peptide Poly-Arginine-12 (R12) Reduces Cell Surface Levels of NMDA NR2B Receptor Subunit in Cortical Neurons; Investigation into the Involvement of Endocytic Mechanisms. J. Mol. Neurosci. [Internet]. 2016;12:1–12. Available from: http://dx.doi.org/10.1007/s12031-016-0861-1.10.1007/s12031-016-0861-1Search in Google Scholar PubMed
[27] Håberg AK, Olsen A, Moen KG, et al. White matter microstructure in chronic moderate-to-severe traumatic brain injury: Impact of acute-phase injury-related variables and associations with outcome measures. J. Neurosci. Res. [Internet]. 2014;1126:n/a-n/a. Available from: http://doi.wiley.com/10.1002/jnr.23534.10.1002/jnr.23534Search in Google Scholar PubMed
[28] Johnson VE, Stewart W, Smith DH. Axonal Pathology in Traumatic Brain Injury. Exp. Neurol. [Internet]. 2013;246:35–43. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22285252.10.1016/j.expneurol.2012.01.013Search in Google Scholar PubMed PubMed Central
[29] Davceva N, Basheska N, Balazic J. Diffuse Axonal Injury—A Distinct Clinicopathological Entity in Closed Head Injuries. Am. J. Forensic Med. Pathol. [Internet]. 2015;0:1. Available from: http://content.wkhealth.com/linkback/openurl?sid=WKPTLP:landingpage&an=00000433-900000000-99459.Search in Google Scholar
[30] Hill CS, Coleman MP, Menon DK. Traumatic Axonal Injury: Mechanisms and Translational Opportunities. Trends Neurosci. [Internet]. 2016;39:311–324. Available from: http://dx.doi.org/10.1016/j.tins.2016.03.002.10.1016/j.tins.2016.03.002Search in Google Scholar PubMed PubMed Central
[31] Büki A, Okonkwo DO, Wang KK, et al. Cytochrome c release and caspase activation in traumatic axonal injury. J. Neurosci. [Internet]. 2000;20:2825–2834. Available from: http://www.ncbi.nlm.nih.gov/pubmed/10751434.10.1523/JNEUROSCI.20-08-02825.2000Search in Google Scholar
[32] Hånell A, Greer JE, McGinn MJ, et al. Traumatic brain injury-induced axonal phenotypes react differently to treatment. Acta Neuropathol. [Internet]. 2015;129:317–332. Available from: http://link.springer.com/10.1007/s00401-014-1376-x.10.1007/s00401-014-1376-xSearch in Google Scholar
[33] Zhao K, Zhao G-M, Wu D, et al. Cell-permeable Peptide Antioxidants Targeted to Inner Mitochondrial Membrane inhibit Mitochondrial Swelling, Oxidative Cell Death, and Reperfusion Injury. J. Biol. Chem. [Internet]. 2004;279:34682–34690. Available from: http://www.jbc.org/cgi/doi/10.1074/jbc.M402999200.10.1074/jbc.M402999200Search in Google Scholar
[34] Nakase I, Niwa M, Takeuchi T, et al. Cellular uptake of arginine-rich peptides: Roles for macropinocytosis and actin rearrangement. Mol. Ther. 2004;10:1011–1022.10.1016/j.ymthe.2004.08.010Search in Google Scholar
[35] Hoane MR, Kaufman N, Vitek MP, et al. COG1410 improves cognitive performance and reduces cortical neuronal loss in the traumatically injured brain. J. Neurotrauma. 2009;26:121–129.10.1089/neu.2008.0565Search in Google Scholar
[36] Plummer S, Van den Heuvel C, Thornton E, et al. The Neuroprotective Properties of the Amyloid Precursor Protein Following Traumatic Brain Injury. Aging Dis. 2016;7:0.10.14336/AD.2015.0907Search in Google Scholar
[37] Wang XS, Gruenstein E. Rapid elevation of neuronal cytoplasmic calcium by apolipoprotein E peptide. J. Cell. Physiol. 1997;173:73–83.10.1002/(SICI)1097-4652(199710)173:1<73::AID-JCP9>3.0.CO;2-GSearch in Google Scholar
[38] Leupold E, Nikolenko H, Beyermann M, et al. Insight into the role of HSPG in the cellular uptake of apolipoprotein E-derived peptide micelles and liposomes. Biochim. Biophys. Acta - Biomembr. [Internet]. 2008;1778:2781–2789. Available from: http://dx.doi.org/10.1016/j.bbamem.2008.09.008.10.1016/j.bbamem.2008.09.008Search in Google Scholar
[39] Misra UK, Adlakha CL, Gawdi G, et al. Apolipoprotein E and mimetic peptide initiate a calcium-dependent signaling response in macrophages. J. Leukoc. Biol. 2001;70:677–683.Search in Google Scholar
[40] Laskowitz DT, Wang H, Chen T, et al. Neuroprotective pentapeptide CN-105 is associated with reduced sterile inflammation and improved functional outcomes in a traumatic brain injury murine model. Sci. Rep. [Internet]. 2017;7:46461. Available from: http://www.nature.com/articles/srep46461.10.1038/srep46461Search in Google Scholar
[41] Tu TM, Kolls BJ, Soderblom EJ, et al. Apolipoprotein E mimetic peptide, CN-105, improves outcomes in ischemic stroke. Ann. Clin. Transl. Neurol. [Internet]. 2017 [cited 2017 May 25];4:246–265. Available from: http://doi.wiley.com/10.1002/acn3.399.10.1002/acn3.399Search in Google Scholar
[42] Lei B, James ML, Liu J, et al. Neuroprotective pentapeptide CN-105 improves functional and histological outcomes in a murine model of intracerebral hemorrhage. Sci. Rep. [Internet]. 2016;6:34834. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27713572.10.1038/srep34834Search in Google Scholar
© 2017 Li Shan Chiu et al.
This work is licensed under the Creative Commons Attribution-NonCommercial-NoDerivatives 3.0 License.
Articles in the same Issue
- Regular Articles
- ISS-N1 makes the first FDA-approved drug for spinal muscular atrophy
- Regular Articles
- Fragile X syndrome: Lessons learned from the most translated neurodevelopmental disorder in clinical trials
- Regular Articles
- Thymoquinone attenuates brain injury via an antioxidative pathway in a status epilepticus rat model
- Regular Articles
- Personality traits and perception of Müller-Lyer illusion in male Chinese military soldiers and university students
- Regular Articles
- Effects of mTOR on neurological deficits after transient global ischemia
- Regular Articles
- The concomitant association of thyroid disorders and Myasthenia gravis
- Regular Articles
- Dynamic expression of CX36 protein in kainic acid kindling induced epilepsy
- Regular Articles
- Autophagy, endoplasmic reticulum stress and the unfolded protein response in intracerebral hemorrhage
- Regular Articles
- White matter sexual dimorphism of the adult human brain
- Regular Articles
- Toward understanding non-coding RNA roles in intracranial aneurysms and subarachnoid hemorrhage
- Regular Articles
- COLQ-mutant congenital myasthenic syndrome with microcephaly: A unique case with literature review
- Regular Articles
- HILIC-MS rat brain analysis, a new approach for the study of ischemic attack
- Regular Articles
- SSTR4, childhood adversity, self-efficacy and suicide risk in alcoholics
- Regular Articles
- The essentials of a global index for cognitive function
- Regular Articles
- Solitaire stent in the treatment of acute ischemic stroke with large cerebral artery occlusion
- Regular Articles
- Grifolin attenuates white matter lesion in oxygen/glucose deprivation
- Regular Articles
- Caffeoylquinic acid enhances proliferation of oligodendrocyte precursor cells
- Regular Articles
- The arcuate fasciculus network and verbal deficits in psychosis
- Regular Articles
- Childhood adversities are not a predictors of SSTR4met in alcoholics
- Regular Articles
- A Rasch analysis between schizophrenic patients and the general population
- Regular Articles
- Assessment of R18, COG1410, and APP96-110 in excitotoxicity and traumatic brain injury
- Regular Articles
- Postural control and emotion in children with autism spectrum disorders
- Regular Articles
- One-shot synesthesia
- Regular Articles
- Chlorogenic acid prevents alcohol-induced brain damage in neonatal rat
- Regular Articles
- Task performance changes the amplitude and timing of the BOLD signal
- Regular Articles
- Effects of calcium on drinking fluorosis-induced hippocampal synaptic plasticity impairment in the offspring of rats
- Regular Articles
- Depression is associated with CRP SNPs in patients with family history
- Regular Articles
- Keyhole surgery of pineal area tumors - personal experience in 22 patients
- Regular Articles
- Models for preterm cortical development using non invasive clinical EEG
Articles in the same Issue
- Regular Articles
- ISS-N1 makes the first FDA-approved drug for spinal muscular atrophy
- Regular Articles
- Fragile X syndrome: Lessons learned from the most translated neurodevelopmental disorder in clinical trials
- Regular Articles
- Thymoquinone attenuates brain injury via an antioxidative pathway in a status epilepticus rat model
- Regular Articles
- Personality traits and perception of Müller-Lyer illusion in male Chinese military soldiers and university students
- Regular Articles
- Effects of mTOR on neurological deficits after transient global ischemia
- Regular Articles
- The concomitant association of thyroid disorders and Myasthenia gravis
- Regular Articles
- Dynamic expression of CX36 protein in kainic acid kindling induced epilepsy
- Regular Articles
- Autophagy, endoplasmic reticulum stress and the unfolded protein response in intracerebral hemorrhage
- Regular Articles
- White matter sexual dimorphism of the adult human brain
- Regular Articles
- Toward understanding non-coding RNA roles in intracranial aneurysms and subarachnoid hemorrhage
- Regular Articles
- COLQ-mutant congenital myasthenic syndrome with microcephaly: A unique case with literature review
- Regular Articles
- HILIC-MS rat brain analysis, a new approach for the study of ischemic attack
- Regular Articles
- SSTR4, childhood adversity, self-efficacy and suicide risk in alcoholics
- Regular Articles
- The essentials of a global index for cognitive function
- Regular Articles
- Solitaire stent in the treatment of acute ischemic stroke with large cerebral artery occlusion
- Regular Articles
- Grifolin attenuates white matter lesion in oxygen/glucose deprivation
- Regular Articles
- Caffeoylquinic acid enhances proliferation of oligodendrocyte precursor cells
- Regular Articles
- The arcuate fasciculus network and verbal deficits in psychosis
- Regular Articles
- Childhood adversities are not a predictors of SSTR4met in alcoholics
- Regular Articles
- A Rasch analysis between schizophrenic patients and the general population
- Regular Articles
- Assessment of R18, COG1410, and APP96-110 in excitotoxicity and traumatic brain injury
- Regular Articles
- Postural control and emotion in children with autism spectrum disorders
- Regular Articles
- One-shot synesthesia
- Regular Articles
- Chlorogenic acid prevents alcohol-induced brain damage in neonatal rat
- Regular Articles
- Task performance changes the amplitude and timing of the BOLD signal
- Regular Articles
- Effects of calcium on drinking fluorosis-induced hippocampal synaptic plasticity impairment in the offspring of rats
- Regular Articles
- Depression is associated with CRP SNPs in patients with family history
- Regular Articles
- Keyhole surgery of pineal area tumors - personal experience in 22 patients
- Regular Articles
- Models for preterm cortical development using non invasive clinical EEG