Does modifying energy metabolism alter cellular signaling and proliferation in KRAS mutant NSCLCs?
-
Sahika Cingir Koker
 und
Irem Dogan Turacli
und
Irem Dogan Turacli


Abstract
Objectives
The capacity to sense and respond to cellular energy stress is an important factor in tumorigenesis. We aimed to investigate the proliferation patterns of KRAS-mutant NSCLC cell lines (A549, Calu-1, H2009) under high (HG) and low glucose (LG) conditions w/wo DCA, which alters metabolic pathways and promotes oxidative phosphorylation.
Methods
Cell viability was detected by MTT assay, protein expressions were determined by Western Blot and RNA expressions were analyzed by RT-qPCR.
Results
NSCLCs exhibited lower proliferation in HG compared to LG conditions. The effect of DCA in reducing the proliferation of the cells, was higher in HG compared to LG condition. We detected lower pRB levels in LG and in DCA treated conditions. LG conditions also showed higher p21 levels w/wo DCA while AMPK activation was more pronounced in 48 h. p-JAK2 was dominant in conditions having LG and p-Stat3 activation varied between all cells. The eIF4e activation was induced in LG w/wo DCA in all the cells. Despite p21-mediated cell cycle arrest, eIF4e and JAK2 signaling promoted proliferation.
Conclusions
Our results clearly showed the potential for increased aggressiveness and the delicate balance of cellular metabolic adjustments in response to energy stress in KRAS-mutant NSCLC cells.
Introduction
Lung cancer is one of the leading causes of cancer-related deaths in both men and women in worldwide. Like other cancer types, it is a highly heterogeneous disease. Based on its molecular characteristics and histological properties it is classified as non-small cell lung cancer (NSCLC) and small cell lung cancer (SCLC). NSCLC is further subdivided to the subtypes such as adenocarcinoma, squamous cell carcinoma and large cell carcinoma [1]. Although the developments in treatment and diagnosis, the prognosis of lung cancer still remains poor, only 20 % patients survive for 5 years after the diagnosis [2].
Both the environmental and genetic factors play significant roles in the development of lung cancer. Despite the very well-established role of smoking related KRAS (Kirsten rat sarcoma viral oncogene homologue) mutations in the development of lung cancer [3], non-smoking related cases due to the genetic mutations in EGFR (Epidermal Growth Factor Receptor) and ALK (Anaplastic Lymphoma Kinase) are also recognized [4]. These mutations have a role in proliferation and metastasis which contributes to the tumorigenesis of the disease. KRAS, which was first detected in lung cancer cells, is one of the most common mutated oncogene in all cancer types [5]. KRAS belongs to RAS family GTPase proteins and plays an important role as molecular switch by being bound to GTP in active state or GDP in inactive state. By this way, it regulates signal transduction pathways such as MAPK, PI3K-AKT resulting in cell proliferation, differentiation and apoptosis [6], 7].
Due to its smooth structure and lack of drug-binding pockets, targeting mutated KRAS was long considered impossible. However, in 2013 a switch II pocket in the KRAS G12C isozyme was identified which enables the development of certain inhibitors such as sotorasib and adagrasib [8]. Those are the FDA approved drugs for the treatment of KRAS G12C positive NSCLC. Despite the efficacy of those inhibitors, rapid resistance mechanisms necessitate combinatory treatment strategies.
Besides the genetic drivers, like other cancer types, lung cancer also has metabolic reprogramming such that despite the nutrient availability they highly rely on glycolysis, which is known as ‘Warburg Effect’ [9]. This shift in metabolism provides glucose availability for further tumor growth and survival. Tumor cells prefer glycolysis over OXPHOS (Oxidative phosphorylation) not because that they have defects in their mitochondria, rather with glycolysis they can generate ATP faster than OXPHOS. Another reason is, with the upregulated glycolysis, they can obtain glycolytic intermediates which are needed for the synthesis of proteins, nucleic acids and lipids [10].
There are many enzymes involved in the regulation of glycolysis which cancer cells depend on heavily. One of these enzymes is pyruvate dehydrogenase kinase (PDK) which inhibits the conversion of pyruvate into acetyl-CoA by phosphorylating pyruvate dehydrogenase (PDH). By this way, pyruvate is redirected away from mitochondrial oxidative phosphorylation to glycolysis [11]. The inhibitor of PDK, Dichloroacetate (DCA), is a small molecule used as a therapeutic agent to alter this metabolic shift to restore oxidative phosphorylation hence induce apoptosis in cancer cells [12]. Also, activation of PDH promotes the mitochondrial oxidation of pyruvate, thereby impairing the metabolic advantage typically held by cancer cells. Additionally, by reducing lactate production, DCA helps counteract the acidic conditions of the tumor microenvironment, thereby contributing to the inhibition of tumor growth and spread [13], 14]. DCA counteracts tumor growth, metastasis, and survival: the primary mechanism is the shift in glucose metabolism from glycolysis to oxidation, reversing the Warburg effect, which results in suppressed cell proliferation and the activation of caspase-driven apoptosis [15]. It has been observed an inverse relationship between DCA’s effectiveness cancer cell survival and the mitochondrial respiratory capacity in oral cell carcinomas [16]. Additionally, DCA is effective on mitochondrial function and in slowing cancer progression depends on the cell phenotype and stemness in a xenograft pancreatic cancer model [17]. DCA has also been investigated in clinical trials [18], 19]. Such as in recurrent brain tumor patients, DCA was generally well-tolerated at lower doses, with minimal toxicity, though higher doses caused some adverse events like peripheral neuropathy [20].
In this study, we aimed to investigate the proliferation pattern of KRAS-mutant NSCLC cell lines (A549, Calu-1 and H2009) under high glucose (HG) and low glucose (LG) conditions to understand how glucose availability affects tumor cell growth. Furthermore, since we know the effect of DCA which forces the cells to shift from aerobic glycolysis to OXPHOS, we aim to include DCA in our high and low glucose conditions to decipher its role in altering metabolic pathways and its potential action to suppress tumor cell proliferation.
Materials and methods
Cell line maintenance
A549, Calu1 and H2009 cells were maintained in high glucose DMEM with 10 % FBS at 37 °C, 5 % CO2 and 95 % humidity. When the cultured cells cover 70 % of 75 cm2 flasks, they were passaged to another cell culture flasks by using trypsin.
Cell viability assay
5 × 103 A549 and H2009, 6 × 103 Calu1 cells were seeded in 96-well plates and treated with 30 mM DCA in HG or LG conditions. DCA concentration was chosen based on previous studies [21], [22], [23]. At the end of either 24, 48 and 72 h, MTT (3- (4,5-Dimethyltiazol-2-yl)-2,5-Diphenyltetrazolium Bromide), (Merck, 475989) assay was performed based on manufacturer’s instructions. Color absorbance was measured using microplate reader (SpectraMax iD3) at a wavelength of 570 nm. Results were normalized against the mean measurements from at least six replicates which were not exposed to any treatment. Each experiment was repeated twice.
RNA isolation, cDNA synthesis and RT-qPCR
RNA was isolated from the cell pellets by using GENEALL RNA isolation kit (Catalog number: 305-101) according to the manufacturer’s protocol and RNAs were transcribed into cDNA by using Roche Transcriptor High Fidelity cDNA (Complementary DNA) Synthesis Kit (Catalog number: 5091284001). Followed by cDNA synthesis, RT-qPCR (Real Time Polymerase Chain Reaction) was done by using SYBR green (Biorad) with Biorad CFX Connect Real Time System. Beta Actin was used as a reference gene. Graphs were plotted by using log2 fold changes for each gene which were normalized against control group.
Western blot
At the end of each indicated condition, cell pellets were obtained from the cells which were seeded 2 × 105 per six well plate. After attaching they were treated with 30 mM DCA in HG or LG conditions. Proteins were isolated by using these pellets with freshly prepared RIPA buffer (NaCl, TrisHCl, NP-40), 10 % SDS, protease inhibitor cocktail (CST, 5871S) and phosphatase inhibitor cocktail (CST, 5870S). With BCA protein assay reagent kit (Thermo Scientific, 23227), total protein concentrations were determined. Before loading the gel, proteins were denatured with 4X Loading Dye (Biorad, 161–0747) at 95 °C for 5 min. Proteins were run in 10 % SDS-PAGE (Bio-Rad Acrylamide Kit, 161–0183) and then transferred onto PVDF membrane (Merck, 3010040001) by semi-dry transfer method. Later, membranes were blocked with 5 % milk prepared with Tris-buffered saline with Tween-20 (0.2 %) (TBST) for 1 h at room temperature. Then, membranes were incubated with the following antibodies at 4 °C overnight at a dilution 1:1000 if not indicated otherwise. At the end of 24 h, membranes were first washed 3 times for 10 min with TBST, and then they were incubated with secondary antibodies (1:5000) for 1 h at room temperature. Then, they were again washed 3 times for 10 min with TBST. Indicated proteins were detected by using ECL (Biorad, 170062) and images were captured with Biorad ChemiDoc XRS+. Beta-Actin was used as a loading control. The antibodies used in this study are all from Cell Signaling Technology, Beta Actin (#4967), p-RB (#9308), p21 (#2947), p-JAK2 (#3771), JAK2 (#3230), p-STAT3 (#9145), p-AMPK (#50081), p-eIF4E (#9741), eIF4E (#9742).
Statistics
Graphpad (Prism 8) was used to plot graphs and for statistical analysis. For RT-qPCR, log2 of ΔΔCt values were plotted and analyzed with One-Way ANOVA with multiple comparisons (Dunnett’s multiple comparison tests). MTT results were plotted by using OD values and analyzed by using Two-Way-ANOVA with multiple comparison tests (Tukey’s multiple comparison tests).
Results
DCA selectively targets cancer cells by shifting their metabolism from glycolysis to oxidative phosphorylation. Firstly, we aim to investigate how cancer cells respond when glycolysis is injured with DCA, particularly in relation to changes in glucose concentration in the microenvironment. At 30 mM, DCA has been shown to inhibit cell proliferation and induce metabolic shifts, specifically by promoting oxidative phosphorylation through the inhibition of PDK. This leads to a reduction in glycolysis and a decrease in lactate production, which, under normal conditions, can slow down cellular growth. To investigate the sensitivity of KRAS mutant NSCLC cells to DCA under different glucose conditions, we assessed cell viability using MTT proliferation assay at 24, 48 and 72 h. All OD values were compared according to HG concentration within their respective time intervals in each cell line.
As illustrated in Figure 1, while significant differences in cell viability were observed at 24 h in all experimental conditions, the reduction in viability was particularly pronounced in both LG and HG conditions following DCA treatment at 48 and 72 h. Notably, a higher rate of proliferation was consistently observed in the LG groups at nearly all time points across the three cell lines tested.
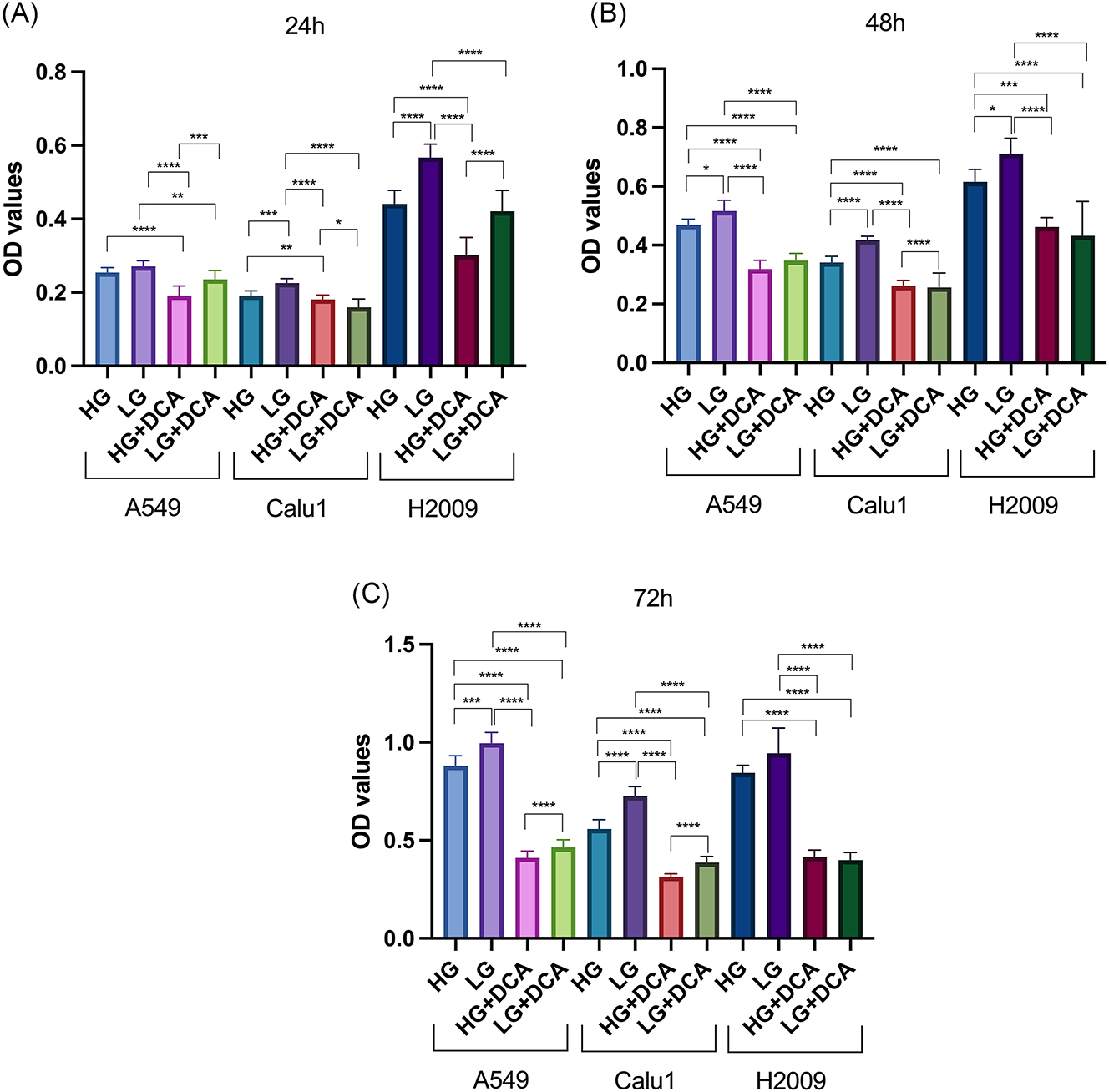
The effects of 30 mM DCA on proliferation rate of KRAS mutant cells under different glucose concentrations at 24 (a), 48 (b) and 72 (c) hours. **** p<0.0001, *** p<0.001, ** p<0.01, * p<0.05.
After observing its effect on proliferation, we investigated the impact of DCA on the expression of genes involved in cell cycle regulation under different glucose concentrations. To this end, the expression levels of CCND1 and TOP2A were analyzed in all cell lines after 24 and 48 h of treatment (Figure 2). All gene expressions were normalized according to HG concentration within their respective time intervals.
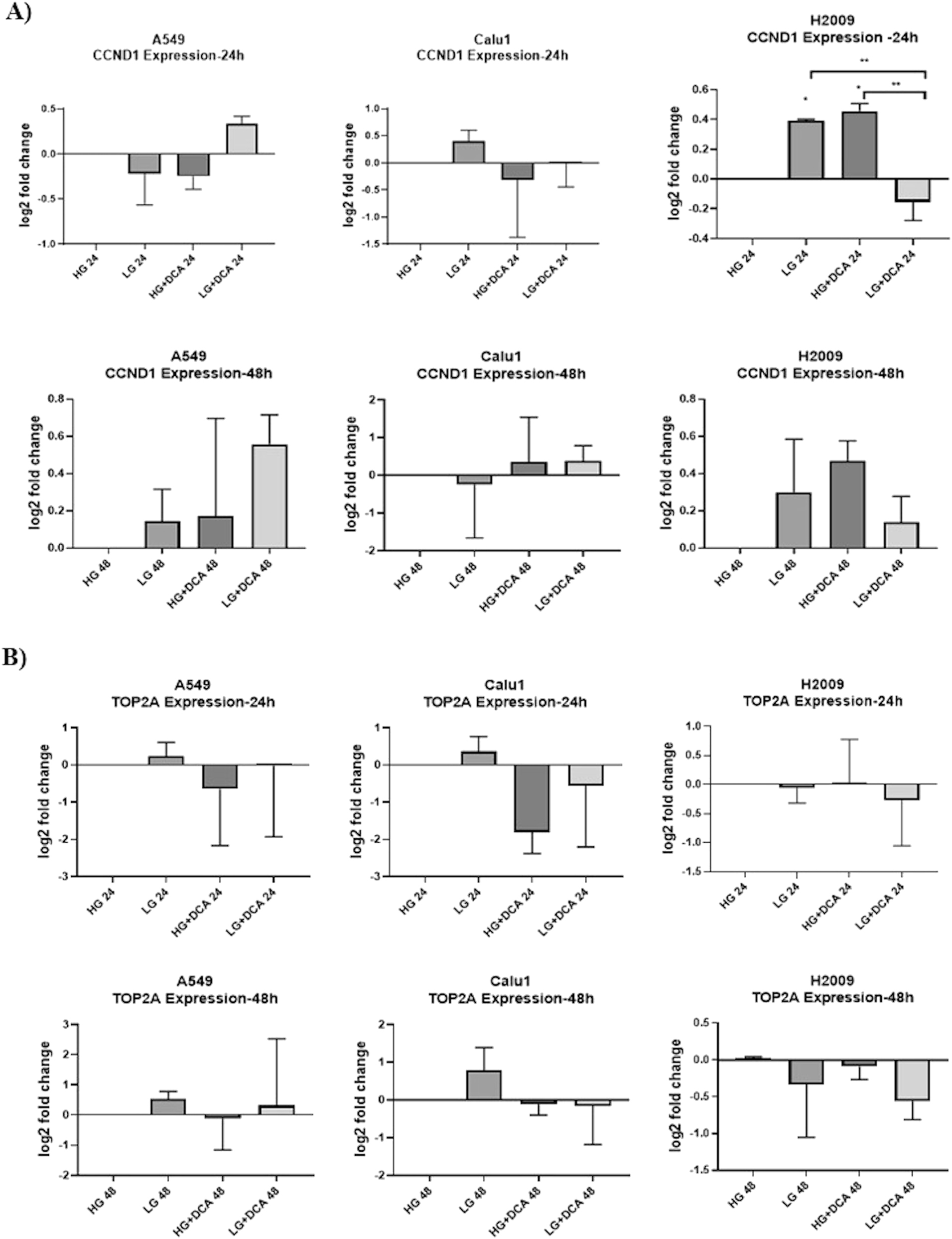
The effects of 30 mM DCA on CCND1 (A) and TOP2A (B) expression under different glucose concentrations at 24 and 48 h in KRAS mutant cells. ** p<0.01, * p<0.05.

Cellular signaling changes under 30 mM DCA and high/low glucose conditions at 24 and 48 h in A549 (A), Calu1 (B) and H2009 (C) cells.
As shown in Figure 2, upon examining gene expression profiles across all cell lines at 24 and 48 h, with attention to consistency, it was observed that only the H2009 cells exhibited a significant trend of increased CCND1 expression in both LG and HG+DCA conditions. These discrepant results may be attributed to sudden changes in cellular energy levels, RNA stability, and modifications. To obtain more definitive conclusions, we focused on the analysis of protein expression over time in all three cell lines.
In western blot analysis of A549 and Calu1 cells, we observed elevated levels of p21 under LG conditions compared to HG both in 24 and 48 h. This was concomitant with a reduction in p-RB protein levels. The JAK/STAT pathway plays an important role in regulating energy metabolism in cancer cells and is thought to be more activated in HG conditions. However, the activation can be context-dependent and influenced by various other factors such as the cancer cell line type and the presence of inflammatory cytokines or growth factors. Although these conditions are typically associated with promotion of cell cycle arrest, we detected increased activation of JAK2 in A549 and Calu1 cells in LG conditions. However, this upregulation of JAK2 activation did not correlate with changes in p-STAT3 levels, which were downregulated in the LG condition compared to the HG condition in A549 and Calu1 cells (Figure 3). The lack of correlation between JAK activation and STAT3 activation in cancer cells could be due to several potential mechanisms, such as crosstalk with other signaling pathways which reflect the complexity of cancer biology.
Although they are often considered to be separate in their functions, there is significant crosstalk and regulation between the AMPK and JAK/STAT pathways, especially in the context of cellular stress and metabolic changes. Therefore, at this stage, we investigated the activation of AMPK in these cells which is a master regulator of cellular energy status. In A549 cells, AMPK activation decreased at 24 h under LG conditions but increased upon addition of DCA. At 48 h, AMPK activation was elevated in LG and further increased in the HG+DCA condition. In Calu1 cells, as expected, AMPK activity was higher at both 24 and 48 h in the LG and LG+DCA conditions compared to their peers.
eIF4E (eukaryotic translation initiation factor 4E) is a critical factor involved in protein synthesis, particularly in the initiation phase of translation which is also involved in cellular stress responses. It can also be attributed as an oncogenic marker for cancer cell lines. Also, there is a crosstalk mechanism between AMPK and eIF4E. AMPK activation can upregulate eIF4E expression and activation [24]. In the A549 cell line, p-eIF4E was observed to increase at 24 h under LG+DCA conditions, while the most significant increase in activity was seen at 48 h under LG conditions. In the Calu1 cell line, an increase in p-eIF4E was only observed at 24 h under LG conditions, and no significant differences were observed across conditions at 48 h (Figure 3).
On the other hand, compared to A549 and Calu1 cells, we observed more obvious effects in H2009 cells. Due to the absence of RB in H2009 cells, and because of cellular energy stress as seen in the two other cell lines, p21 levels were elevated under LG conditions. p21 can function as a brake on proliferation. However, the effect of p21 can be context dependent and can promote cancer cell survival. Thus, we aimed to see the effects of energy stress related changes on this specific cell line. JAK2 activation was consistently higher under HG conditions. However, at 24 h, a decrease in JAK2 activity was observed under HG + DCA condition compared to HG alone. This suggests that the induced energy stress may also affect oncogenic pathways. The STAT3 activation was correlated with JAK2 activation, showing a decrease under LG condition. In line with these signs of energy stress, the most evident indicator was the increase in AMPK activity at all time points under LG, indicating that the cells were experiencing significant energy stress. Notably, eIF4E activation increased in parallel with AMPK activity in the LG group (Figure 3). The observed parallel activation of AMPK and eIF4E in response to energy stress in the LG condition suggests a specific regulatory mechanism which can indicate selective translation of stress-responsive proteins that are crucial for cell survival and adaptation under metabolic stress. This process demonstrates how energy sensing and translation regulation interact dynamically under cellular stress.
Discussion
In NCSLC, most prevalent mutations occur in KRAS and its incidence changes according to the lung cancer’s ethnicity, smoking history and pathological type [25]. These mutations generally occur at chromosome 12p12.1 and most of them are found at codon 12. Because of this mutation, glycine is exchanged with other amino acids (except proline) which results in disruption of GAP binding and promotion of GTP hydrolysis. By this way, KRAS-GTP accumulates and promotes oncogenic signaling [25].
Despite the existence of debates, different types of mutation have different prognosis among the patients as well, such that G12D mutation has worse prognosis compared to G12C mutation [26]. Moreover, different types of mutations require mutation-specific treatment strategies in KRAS-mutated NSCLC to get better therapeutic outcomes. For instance, cell lines having p.G12C and p.G12CV affect the downstream signaling pathways in a different way such that RAS-related protein (RAL) A/B signaling increase with a decrease in phosphorylated Protein Kinase B (AKT) levels [27]. This is completely different compared to the WT KRAS cell line and cell lines bearing other types of KRAS mutations. In contrast to this, PI3K-AKT pathway is highly activated in the cell lines having KRAS G12D mutation [28], 29]. In the cell lines we studied, due to both the genomic differences between them and the distinct mutations in the KRAS gene, we observed different response mechanisms across our treatments. Some exhibited more sensitive responses, while others showed stronger responses. This variation may be attributed to alterations in certain critical genes, such as p53, RB and energy sensing proteins. In other words, not all cells possess the same level of response for the environment or activation/inactivation of the normal tumor suppressor proteins such as p53. Also, the capacity to sense and respond to energy stress levels varies between cells. Indeed, our study provides a compelling example of how responses can differ even in isogenic cells within the same cancer subtype carrying the KRAS mutation.
While it is generally true that cancer cells thrive in HG conditions, some cancer cells can exhibit increased proliferation in LG conditions due to the activation of survival pathways. For example, the AMPK pathway can promote cell survival and metabolic adaptation in LG conditions just seen in the cells we studied [30], [31], [32]. Particularly in our study, when we treated the cells with DCA and shifted their cellular energy metabolism towards oxidative phosphorylation, the increased levels of AMPK, along with a concurrent rise in survival proteins, serve as a clear example of this phenomenon. Additionally, we observed a negative correlation between p-STAT3 and p-JAK2 under the treatment conditions in the A459 and Calu-1 cell lines. This may be due to the fact that STAT3 can be activated by kinases other than JAK2, such as SRC or RTKs, or through crosstalk with other cellular pathways [33]. However, we did not observe similar results in H2009 cells which again proves different cell types have different cellular mechanisms under stress conditions and may express other signaling proteins, affecting the interplay between these pathways. Also, H2009 cells are RB null, which suggests that the loss of functional RB may lead to increased JAK2/STAT3 signaling, contributing to tumor growth.
p21 can stop cell division by blocking the cell cycle. As a defense against uncontrolled growth, p21 is generated in healthy cells in response to stress or DNA damage. This mechanism is frequently turned off by cancer cells, enabling them to proliferate more quickly. Under stressful conditions, such as low glucose, certain cancer cells may, nevertheless, upregulate p21, possibly as a survival strategy or in reaction to metabolic stress [34]. As seen in our study p21 is induced in response to glucose deprivation and DCA treatment and contributed to increased cellular proliferation in KRAS mutant cells.
Like in many cancer types, acquired resistance is one of the most important obstacles in the treatment of NSCLS [35]. For example, targeting growth factor receptors together with immune checkpoint inhibitors improved the results for NSCLC patients with treatments like osimertinib having better efficacy in EGFR-mutant cases [36], 37]. But, due to the acquired or intrinsic resistance to these therapies present an obstacle in their effectiveness and underline the importance of understanding the resistance mechanisms and improvement of new treatment options [38], 39].
Therefore, for many tumor types, conventional chemotherapeutics are still used as the first-line treatment in clinical practice. It is becoming increasingly clear that changes in metabolism such as higher mitochondrial function and expression of mitochondrial genes which accompanied with higher activity of OXPHOS are some of the main reasons for chemotherapy resistance in many cancer types [40]. Based on this, a better understanding of the altered cellular metabolism in drug resistance is needed to further improve cancer therapy; as shown in our study, aggressiveness can be changed even with glucose condition.
In conclusion our data suggests that these cancer cells are adapting to low glucose conditions by activating stress response pathways such as p21 and promoting survival and protein production such as p-JAK2 and p-eIF4E. This could indicate that these KRAS mutant cancer cells are particularly aggressive and have a metabolic advantage in low-nutrient environments. Our study demonstrates that when metabolic stress is induced on cancer cells, they activate multiple pathways to survive, becoming even more aggressive in the process. Therefore, in the future therapeutic approaches, we must consider the potential for increased aggressiveness and the delicate balance of cellular metabolic adjustments, as this could lead to unexpected outcomes.
Acknowledgments
We would like to sincerely thank Mrs. Naime Tomek for making it possible for us to set up our laboratory. Without her generous support, we could not have carried out this study.
-
Research ethics: Not applicable.
-
Informed consent: Not applicable.
-
Author contributions: Conceptualization (IDT), Methodology, Material preparation, Data collection, Analysis (IDT, SCK), Writing-Original Draft (IDT, SCK), Writing-Review & Editing (IDT, SCK). All authors have accepted responsibility for the entire content of this manuscript and approved its submission.
-
Use of Large Language Models, AI and Machine Learning Tools: None declared.
-
Conflict of interest: The author states no conflict of interest.
-
Research funding: None declared.
-
Data availability: Not applicable.
References
1. Forde, PM, Ettinger, DS. Targeted therapy for non-small-cell lung cancer: past, present and future. Expert Rev Anticancer Ther 2013;13:745–58. https://doi.org/10.1586/era.13.47.Suche in Google Scholar PubMed PubMed Central
2. Vicidomini, G. Current challenges and future advances in lung cancer: genetics, instrumental diagnosis and treatment. Cancers (Basel) 2023;15. https://doi.org/10.3390/cancers15143710.Suche in Google Scholar PubMed PubMed Central
3. Walser, T, Cui, X, Yanagawa, J, Lee, JM, Heinrich, E, Lee, G, et al.. Smoking and lung cancer: the role of inflammation. Proc Am Thorac Soc 2008;5:811–15. https://doi.org/10.1513/pats.200809-100th.Suche in Google Scholar
4. Paik, PK, Johnson, ML, D’Angelo, SP, Sima, CS, Ang, D, Dogan, S, et al.. Driver mutations determine survival in smokers and never-smokers with stage IIIB/IV lung adenocarcinomas. Cancer 2012;118:5840–7. https://doi.org/10.1002/cncr.27637.Suche in Google Scholar PubMed PubMed Central
5. Chang, EH, Gonda, MA, Ellis, RW, Scolnick, EM, Lowy, DR. Human genome contains four genes homologous to transforming genes of Harvey and Kirsten murine sarcoma viruses. Proc Natl Acad Sci U S A 1982;79:4848–52. https://doi.org/10.1073/pnas.79.16.4848.Suche in Google Scholar PubMed PubMed Central
6. Hall, BE, Bar-Sagi, D, Nassar, N. The structural basis for the transition from Ras-GTP to Ras-GDP. Proc Natl Acad Sci U S A 2002;99:12138–42. https://doi.org/10.1073/pnas.192453199.Suche in Google Scholar PubMed PubMed Central
7. Zhou, Y, Hancock, JF. Ras nanoclusters: versatile lipid-based signaling platforms. Biochim Biophys Acta 2015;1853:841–9. https://doi.org/10.1016/j.bbamcr.2014.09.008.Suche in Google Scholar PubMed
8. Ostrem, JM, Peters, U, Sos, ML, Wells, JA, Shokat, KM. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013;503:548–51. https://doi.org/10.1038/nature12796.Suche in Google Scholar PubMed PubMed Central
9. Sha, L, Lv, Z, Liu, Y, Zhang, Y, Sui, X, Wang, T, et al.. Shikonin inhibits the Warburg effect, cell proliferation, invasion and migration by downregulating PFKFB2 expression in lung cancer. Mol Med Rep 2021;24. https://doi.org/10.3892/mmr.2021.12199.Suche in Google Scholar PubMed PubMed Central
10. Lunt, SY, Vander Heiden, MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol 2011;27:441–64. https://doi.org/10.1146/annurev-cellbio-092910-154237.Suche in Google Scholar PubMed
11. Lu, J, Tan, M, Cai, Q. The Warburg effect in tumor progression: mitochondrial oxidative metabolism as an anti-metastasis mechanism. Cancer Lett 2015;356:156–64. https://doi.org/10.1016/j.canlet.2014.04.001.Suche in Google Scholar PubMed PubMed Central
12. James, MO, Jahn, SC, Zhong, G, Smeltz, MG, Hu, Z, Stacpoole, PW. Therapeutic applications of dichloroacetate and the role of glutathione transferase zeta-1. Pharmacol Ther 2017;170:166–80. https://doi.org/10.1016/j.pharmthera.2016.10.018.Suche in Google Scholar PubMed PubMed Central
13. Stacpoole, PW, Nagaraja, NV, Hutson, AD. Efficacy of dichloroacetate as a lactate-lowering drug. J Clin Pharmacol 2003;43:683–91. https://doi.org/10.1177/0091270003254637.Suche in Google Scholar
14. Tataranni, T, Piccoli, C. Dichloroacetate (DCA) and cancer: an overview towards clinical applications. Oxid Med Cell Longev 2019;2019:8201079. https://doi.org/10.1155/2019/8201079.Suche in Google Scholar PubMed PubMed Central
15. Kankotia, S, Stacpoole, PW. Dichloroacetate and cancer: new home for an orphan drug? Biochim Biophys Acta 2014;1846:617–29. https://doi.org/10.1016/j.bbcan.2014.08.005.Suche in Google Scholar PubMed
16. Ruggieri, V, Agriesti, F, Scrima, R, Laurenzana, I, Perrone, D, Tataranni, T, et al.. Dichloroacetate, a selective mitochondria-targeting drug for oral squamous cell carcinoma: a metabolic perspective of treatment. Oncotarget 2015;6:1217–30. https://doi.org/10.18632/oncotarget.2721.Suche in Google Scholar PubMed PubMed Central
17. Tataranni, T, Agriesti, F, Pacelli, C, Ruggieri, V, Laurenzana, I, Mazzoccoli, C, et al.. Dichloroacetate affects mitochondrial function and stemness-associated properties in pancreatic cancer cell lines. Cells 2019;8. https://doi.org/10.3390/cells8050478.Suche in Google Scholar PubMed PubMed Central
18. Khan, A, Marier, D, Marsden, E, Andrews, D, Eliaz, I. A novel form of dichloroacetate therapy for patients with advanced cancer: a report of 3 cases. Altern Ther Health Med 2014;20:21–8.Suche in Google Scholar
19. Chu, QS, Sangha, R, Spratlin, J, Vos, LJ, Mackey, JR, McEwan, AJ, et al.. A phase I open-labeled, single-arm, dose-escalation, study of dichloroacetate (DCA) in patients with advanced solid tumors. Invest N Drugs 2015;33:603–10. https://doi.org/10.1007/s10637-015-0221-y.Suche in Google Scholar PubMed
20. Dunbar, EM, Coats, BS, Shroads, AL, Langaee, T, Lew, A, Forder, JR, et al.. Phase 1 trial of dichloroacetate (DCA) in adults with recurrent malignant brain tumors. Invest N Drugs 2014;32:452–64. https://doi.org/10.1007/s10637-013-0047-4.Suche in Google Scholar PubMed PubMed Central
21. Skeberdyte, A, Sarapiniene, I, Aleksander-Krasko, J, Stankevicius, V, Suziedelis, K, Jarmalaite, S. Dichloroacetate and salinomycin exert a synergistic cytotoxic effect in colorectal cancer cell lines. Sci Rep 2018;8:17744. https://doi.org/10.1038/s41598-018-35815-4.Suche in Google Scholar PubMed PubMed Central
22. Al-Azawi, A, Sulaiman, S, Arafat, K, Yasin, J, Nemmar, A, Attoub, S. Impact of sodium dichloroacetate alone and in combination therapies on lung tumor growth and metastasis. Int J Mol Sci 2021;22. https://doi.org/10.3390/ijms222212553.Suche in Google Scholar PubMed PubMed Central
23. Madhok, BM, Yeluri, S, Perry, SL, Hughes, TA, Jayne, DG. Dichloroacetate induces apoptosis and cell-cycle arrest in colorectal cancer cells. Br J Cancer 2010;102:1746–52. https://doi.org/10.1038/sj.bjc.6605701.Suche in Google Scholar PubMed PubMed Central
24. Zhu, X, Dahlmans, V, Thali, R, Preisinger, C, Viollet, B, Voncken, JW, et al.. AMP-activated protein kinase up-regulates mitogen-activated protein (MAP) kinase-interacting serine/threonine kinase 1a-dependent phosphorylation of eukaryotic translation initiation factor 4E. J Biol Chem 2016;291:17020–7. https://doi.org/10.1074/jbc.c116.740498.Suche in Google Scholar PubMed PubMed Central
25. Zhao, X, Zheng, Y, Wang, Y, Zhang, M, Dong, Z, Liu, Y, et al.. The potential treatment options and combination strategies of KRAS-mutated lung cancer. OncoTargets Ther 2024;17:1041–57. https://doi.org/10.2147/ott.s484209.Suche in Google Scholar
26. Amanam, I, Mambetsariev, I, Gupta, R, Achuthan, S, Wang, Y, Pharaon, R, et al.. Role of immunotherapy and co-mutations on KRAS-mutant non-small cell lung cancer survival. J Thorac Dis 2020;12:5086–95. https://doi.org/10.21037/jtd.2020.04.18.Suche in Google Scholar PubMed PubMed Central
27. Nadal, E, Beer, DG, Ramnath, N. KRAS-G12C mutation is associated with poor outcome in surgically resected lung adenocarcinoma. J Thorac Oncol 2015;10:e9–10. https://doi.org/10.1097/jto.0000000000000438.Suche in Google Scholar PubMed PubMed Central
28. Xu, S, Long, BN, Boris, GH, Chen, A, Ni, S, Kennedy, MA. Structural insight into the rearrangement of the switch I region in GTP-bound G12A K-Ras. Acta Crystallogr D Struct Biol 2017;73:970–84. https://doi.org/10.1107/s2059798317015418.Suche in Google Scholar
29. Cruz-Migoni, A, Canning, P, Quevedo, CE, Bataille, CJR, Bery, N, Miller, A, et al.. Structure-based development of new RAS-effector inhibitors from a combination of active and inactive RAS-binding compounds. Proc Natl Acad Sci U S A 2019;116:2545–50. https://doi.org/10.1073/pnas.1811360116.Suche in Google Scholar PubMed PubMed Central
30. Liang, J, Mills, GB. AMPK: a contextual oncogene or tumor suppressor? Cancer Res 2013;73:2929–35. https://doi.org/10.1158/0008-5472.can-12-3876.Suche in Google Scholar PubMed PubMed Central
31. Laderoute, KR, Amin, K, Calaoagan, JM, Knapp, M, Le, T, Orduna, J, et al.. 5’-AMP-activated protein kinase (AMPK) is induced by low-oxygen and glucose deprivation conditions found in solid-tumor microenvironments. Mol Cell Biol 2006;26:5336–47. https://doi.org/10.1128/mcb.00166-06.Suche in Google Scholar
32. Jeon, SM, Chandel, NS, Hay, N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature 2012;485:661–5. https://doi.org/10.1038/nature11066.Suche in Google Scholar PubMed PubMed Central
33. Xue, C, Yao, Q, Gu, X, Shi, Q, Yuan, X, Chu, Q, et al.. Evolving cognition of the JAK-STAT signaling pathway: autoimmune disorders and cancer. Signal Transduct Target Ther 2023;8:204. https://doi.org/10.1038/s41392-023-01468-7.Suche in Google Scholar PubMed PubMed Central
34. Gorospe, M, Wang, X, Holbrook, NJ. Functional role of p21 during the cellular response to stress. Gene Expr 1999;7:377–85.Suche in Google Scholar
35. Cao, Z, Zhu, J, Chen, X, Chen, Z, Wang, W, Zhou, Y, et al.. Resistance mechanisms of non-small cell lung cancer and improvement of treatment effects through nanotechnology: a narrative review. J Thorac Dis 2024;16:8039–52. https://doi.org/10.21037/jtd-24-1078.Suche in Google Scholar PubMed PubMed Central
36. Soria, JC, Ohe, Y, Vansteenkiste, J, Reungwetwattana, T, Chewaskulyong, B, Lee, KH, et al.. Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer. N Engl J Med 2018;378:113–25. https://doi.org/10.1056/nejmoa1713137.Suche in Google Scholar PubMed
37. Topalian, SL, Hodi, FS, Brahmer, JR, Gettinger, SN, Smith, DC, McDermott, DF, et al.. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012;366:2443–54. https://doi.org/10.1056/nejmoa1200690.Suche in Google Scholar
38. Gainor, JF, Shaw, AT. Emerging paradigms in the development of resistance to tyrosine kinase inhibitors in lung cancer. J Clin Oncol 2013;31:3987–96. https://doi.org/10.1200/jco.2012.45.2029.Suche in Google Scholar
39. Nagasaki, J, Ishino, T, Togashi, Y. Mechanisms of resistance to immune checkpoint inhibitors. Cancer Sci 2022;113:3303–12. https://doi.org/10.1111/cas.15497.Suche in Google Scholar PubMed PubMed Central
40. Zaal, EA, Berkers, CR. The influence of metabolism on drug response in cancer. Front Oncol 2018;8:500. https://doi.org/10.3389/fonc.2018.00500.Suche in Google Scholar PubMed PubMed Central
© 2025 the author(s), published by De Gruyter, Berlin/Boston
This work is licensed under the Creative Commons Attribution 4.0 International License.