The role of brain-derived neurotrophic factor and the neurotrophin receptor p75NTR in age-related brain atrophy and the transition to Alzheimer’s disease
-
Shaun Cade


Abstract
Alzheimer’s disease is a neurodegenerative condition that is potentially mediated by synaptic dysfunction before the onset of cognitive impairments. The disease mostly affects elderly people and there is currently no therapeutic which halts its progression. One therapeutic strategy for Alzheimer’s disease is to regenerate lost synapses by targeting mechanisms involved in synaptic plasticity. This strategy has led to promising drug candidates in clinical trials, but further progress needs to be made. An unresolved problem of Alzheimer’s disease is to identify the molecular mechanisms that render the aged brain susceptible to synaptic dysfunction. Understanding this susceptibility may identify drug targets which could halt, or even reverse, the disease’s progression. Brain derived neurotrophic factor is a neurotrophin expressed in the brain previously implicated in Alzheimer’s disease due to its involvement in synaptic plasticity. Low levels of the protein increase susceptibility to the disease and post-mortem studies consistently show reductions in its expression. A desirable therapeutic approach for Alzheimer’s disease is to stimulate the expression of brain derived neurotrophic factor and potentially regenerate lost synapses. However, synthesis and secretion of the protein are regulated by complex activity-dependent mechanisms within neurons, which makes this approach challenging. Moreover, the protein is synthesised as a precursor which exerts the opposite effect of its mature form through the neurotrophin receptor p75NTR. This review will evaluate current evidence on how age-related alterations in the synthesis, processing and signalling of brain derived neurotrophic factor may increase the risk of Alzheimer’s disease.
Introduction
Dementia is a debilitating condition characterised by a deterioration in cognitive functions that mostly affects elderly people (Alzheimer’s association 2017). The number of people with the condition has increased in recent decades and will likely further increase in future as the population ages (Nichols et al. 2019). A common type of dementia is Alzheimer’s disease (AD), characterised by the presence of two proteinaceous deposits in the brain and wide-spread synapse loss (Abner et al. 2017; Mecca et al. 2020). The protein deposits include amyloid plaques composed of the amyloid beta peptide (Aβ) and neurofibrillary tangles (NFTs) composed of hyperphoshorylated tau (Abner et al. 2017). Current treatments for AD temporarily improve cognitive performance by increasing acetylcholine in the synapses of degenerating neurons, but do not stop its progression (Wattmo et al. 2016). Consequently, research into the underlying causes of AD is necessary to identify drug targets with disease-modifying potential. Evidence suggests that pathological levels of soluble Aβ oligomers cause synapse loss early in AD before the onset of other pathologies (He et al. 2019; Moreno et al. 2009; Wang et al. 2017). However, drugs that reduce Aβ have been unsuccessful in clinical trials (Aisen et al. 2011; Egan et al. 2018; Green et al. 2009; Salloway et al. 2014) – possibly because other age-related mechanisms may work synergistically with Aβ. An understanding of these mechanisms may identify drug targets with disease-modifying potential and halt the progression of AD.
The neurotrophins are a family of growth factors necessary for the survival and dendrite formation of specific neuronal populations during brain development (Baydyuk et al. 2013; Rauskolb et al. 2010). The factors may also protect the aged hippocampus, a region involved in learning and memory, by preventing neuronal death and axonal degeneration (von Bohlen und Halbach et al. 2003). Some of the well-known neurotrophins include nerve growth factor (NGF), brain derived neurotrophic factor (BDNF), neurotrophin-3 (NT-3) and neurotrophin-4 (NT-4) (Schulte-Herbruggen et al. 2008). BDNF plays a role in the mechanisms that underpin learning and memory by stabilising excitatory hippocampal synapses and improving their function following stimulation (Kellner et al. 2014; Petkova-Tuffy et al. 2021). Consequently, age-related alterations in BDNF may increase the risk of AD, which initially manifests as memory impairments (Fang et al. 2019; Weinstein et al. 2014). There are many aspects of BDNF that may be considered when assessing its involvement in brain ageing and the transition to AD. Firstly, BDNF transcription is regulated by complex mechanisms within neurons, so age-related changes in these may affect its expression (Palomer et al. 2016). Secondly, BDNF is synthesised as a precursor (proBDNF), which exerts the opposite effect of its mature form (mBDNF), when bound to the neurotrophin receptor, p75NTR (Yang et al. 2014). Age-related imbalances between these two species, or the expression of p75NTR, may facilitate the production of Aβ and/or mediate its toxic effects (Chen et al. 2017; Shen et al. 2019; Yang et al. 2020). Therefore, alterations in the synthesis, processing and signalling of BDNF should be explored when assessing its role in brain ageing and the transition to AD.
This review will explore the evidence on the role of BDNF and p75NTR in brain ageing and the transition to AD. The basic biology of BDNF will be briefly reviewed first before covering the mechanisms of age-related brain atrophy and the transition to AD. This analysis will be critical to examining how age-related alterations in BDNF may increase the risk of the disease. Finally, treatments currently in clinical trials aimed at modifying the course of AD by targeting neurotrophin signalling will be reviewed.
Transcription, processing and biological effects of BDNF
Transcription of BDNF is augmented by neuronal activity in the brain, suggesting it is involved in adaptive responses (Palomer et al. 2016; Singer et al. 2018; Tuvikene et al. 2021). The BDNF gene contains 9 promoters from which transcription may be initiated, each positioned up-stream from a unique 5′ untranslated region (Maynard et al. 2017; Pruunsild et al. 2007). The use of multiple start sites gives rise to various transcripts (transcripts I-IXa), however, because they are spliced to a common coding exon (exon IX), only one protein is translated (Pruunsild et al. 2007). Under basal conditions, transcription is low in cortical and hippocampal neurons, indicating that constitutive BDNF expression is minimal (Maynard et al. 2017; Singer et al. 2018; Tuvikene et al. 2021). By contrast, transcription is increased following depolarisation in cortical neurons and N-methyl D-aspartate (NMDA) stimulation in hippocampal neurons, a stimulus that mimics glutamate neurotransmission (Palomer et al. 2016). Although BDNF transcription is generally stimulus-dependent, the size and duration of the response differs for each transcript. For example, transcripts II and VI increase more than 2-fold 30 min after low-dose NMDA stimulation in hippocampal neurons before returning to basal levels after 60 min (Palomer et al. 2016). By contrast, transcripts I and IV increase only slightly (∼40%) but remain stably expressed thereafter (Palomer et al. 2016). The recent discovery of an enhancer region within the BDNF gene may further complicate this differential transcription (Tuvikene et al. 2021). In cortical neurons, a +3 kb enhancer downstream of exon III was shown to further augment expression of transcripts I–III, but not IV, VI or IXa following depolarisation (Tuvikene et al. 2021). These same effects were not observed in astrocytes, indicating that the adaptive mechanism is specific to neurons (Tuvikene et al. 2021). The differential expression of BDNF transcripts may be explained by their targeting to different dendritic locations for translation. The dendritic complexity of pyramidal neurons from cornu ammonis 1 and 3 (CA1, CA3) of the hippocampus is differently affected by disruptions to specific transcripts in mice (Maynard et al. 2017). Consequently, differential expression of BDNF transcripts may be due to their distinct roles in the formation of hippocampal dendrites.
Stimulus-driven transcription of BDNF depends on the function of elaborate Ca2+ dependent and independent signalling pathways within neurons (Cohen et al. 2018; Palomer et al. 2016). Coupling excitation to the nucleus in hippocampal neurons requires Ca2+ influx through the activation of NMDA receptor (NMDAR) or L-type voltage-gated Ca2+ channels (LTCCs) (Cohen et al. 2018; Wang et al. 2017). Intracellular Ca2+ activates calmodulin (CaM), which in turn, binds to and activates Ca2+/calmodulin-dependent kinase II (CaMKII). CaMKII is a holoenzyme that consists of multiple subunits, some of which are catalytic and others autoregulatory (Cook et al. 2021). Although multiple isoforms are expressed in the brain, α and β are relatively highly expressed in the hippocampus of adult wild type (WT) mice, suggesting that they are important for learning and memory (Cook et al. 2018). CaMKIIα couples LTCCs to the phosphorylation of nuclear cAMP-responsive element binding protein (pCREB) in hippocampal neurons (Wang et al. 2017), a transcription factor that binds BDNF promoters I, II, IV and VI (Palomer et al. 2016). The enzyme may also function as a molecular switch in hippocampal neurons by sensing changes in stimulation and initiating synaptic alterations in response (Cook et al. 2021). However, CaMKIIγ appears to be critically involved in the stimulus-driven transcription of BDNF, indicating that CaMKII isoforms may have overlapping functions (Cohen et al. 2018). CaMKIIγ translocates Ca2+/CaM across the nucleus in cortical neurons, which in turn, activates nuclear CaMKIV, another CREB kinase, following NMDA stimulation (Cohen et al. 2018). Disrupting this translocation in transgenic mice impairs cognitive performance, suggesting that CaMKIIγ is involved in the mechanisms of learning and memory (Cohen et al. 2018). Stimulus-driven BDNF transcription may depend on more than the function of Ca2+/calmodulin-dependent kinases (Impey et al. 2002). Inhibition of MAPK reduces pCREB, but not pCaMKIV, in cortical and hippocampal neurons, suggesting that MAPK phosphorylates CREB independent from Ca2+ (Cohen et al. 2018; Impey et al. 2002). MAPK is activated by protein kinase A (PKA) and protein kinase C (PKC), which are stimulated by elevations in cAMP (Roberson et al. 1999). The diverse range of pathways that converge on CREB may be due to differential expression of BDNF transcripts. In hippocampal neurons, CREB requires a binding partner, CREB-binding protein (CBP), to initiate transcription at promoters II and VI following NMDA stimulation (Palomer et al. 2016). However, transcription is initiated at promoters I and IV without CBP, suggesting that some transcripts are not as tightly regulated (Palomer et al. 2016). Therefore, the mechanisms which regulate activity-dependent transcription of BDNF may vary, depending on the transcript.
BDNF secretory vesicles are targeted to the regulated secretory pathway in neurons and released in response to synaptic activity (Balkowiec and Katz 2002; Moro et al. 2020). Like other neurotrophins and growth factors, BDNF is translated as a precursor containing an N-terminal pro-domain that is later removed by proteolytic cleavage (Benicky et al. 2019). The pro-domain is required for protein stability (Mowla et al. 2001), regulated secretion (Chen et al. 2005), and possibly also, for protein folding (Rattenholl et al. 2001). Secretion of BDNF depends on the interaction between sorting motifs within the pro- and mature domains of the precursor and intracellular sorting receptors (Chen et al. 2005; Lou et al. 2005). Sortilin, a vesicle-associated sorting receptor (Nykjaer and Willnow 2012), plays a key role in regulated secretion by binding to the pro-domain and targeting proBDNF to secretory vesicles (Chen et al. 2005). Huntington-associated protein 1 may support this process by forming a complex with Sortilin and increasing the efficiency of proBDNF trafficking (Chen et al. 2005; Yang et al. 2011). Regulated secretion also depends on the interaction between a sorting motif contained within the mature domain and carboxypeptidase E (Lou et al. 2005). BDNF is stored in secretory granules that are released in response to depolarisation, suggesting that it is released following synaptic activity (Balkowiec and Katz, 2002; Moro et al. 2020). Similar to gene transcription, this release depends on elevations in intracellular Ca2+.
ProBDNF is converted interchangeably by both intracellular and extracellular proteases following stimulation in hippocampal neurons (Pang et al. 2016). Under basal conditions, small amounts of proBDNF are detectable in hippocampal neurons, whereas mBDNF is absent (Pang et al. 2016). Three hours post stimulation, mBDNF becomes abundant, while proBDNF remains unchanged, indicating that stimulus-driven BDNF transcription is followed by increased conversion (Pang et al. 2016). This stimulus-driven conversion likely takes place both intracellularly and extracellularly (Frazzini et al. 2018; Obiang et al. 2011; Wetsel et al. 2013). Intracellular conversion is mediated by furin (Pang et al. 2016), a ubiquitously expressed pro-protein convertase with a diverse range of substrates (Bennett et al. 2000; Dubois et al. 2001; 1995, Kang et al. 2002; Seidah et al. 1996) and also, by pro-convertase 7 (PC7) (Wetsel et al. 2013). Extracellularly, proBDNF is converted by several proteases such as matrix metalloproteinase 9 (MMP-9) and plasmin (Mizoguchi et al. 2011; Obiang et al. 2011). MMP-9 is a zinc-dependent protease involved in tissue remodelling that increases BDNF maturation following excitation in the hippocampus (Mizoguchi et al. 2011). Reducing MMP-9 activity, through sequestration of zinc, impairs learning and memory in WT mice, indicating that it plays an important role in BDNF-related cognitive functions (Frazzini et al. 2018). Another proBDNF protease, plasmin, is converted from its inactive precursor, plasminogen, by tissue plasminogen activator (tPA). tPA is highly expressed in the granule cell layer of the dentate gyrus (DG) and pyramidal neurons of CA1 and CA3 of the hippocampus in adult mice (Stevenson and Lawrence 2018). However, tPA protein is transported to mossy fiber (MF) axons of dentate granule cells in the hilus and the stratum lucidum lamina, where it likely activates plasmin (Stevenson and Lawrence 2018). tPA is required for the early induction of long-term potentiation (LTP) in CA1 of hippocampal slices, but not for ongoing maintenance (Pang et al. 2016). LTP is a form of excitatory plasticity in the hippocampus that leads to a long-lasting increase in synaptic strength (Minichiello et al. 2002). By contrast, furin and/or PC7 are required for ongoing maintenance, but not for initiation (Pang et al. 2016). These findings suggest that intracellular and extracellular proteases interchangeably regulate activity dependent BDNF maturation at different stages of LTP.
ProBDNF may play opposing roles in the brain depending on the relative expression of neurotrophin receptors (Fayard et al. 2005). It has been widely demonstrated using primary neuronal cultures, cell lines and animal models that proBDNF is a neurodegenerative ligand in the CNS (Garad et al. 2021; Liu et al. 2018; Sun et al. 2012; Yang et al. 2014). Heterozygous proBDNF knock-in mice show reduced CA1 hippocampal spines and impaired LTP at Schaffer collateral (SC)-CA1 synapses (Yang et al. 2014). Conversely, in hippocampal neurons, proBDNF facilitates long-term depression (LTD) – a form of synaptic plasticity that shrinks dendritic spines – at SC-CA1 hippocampal synapses (Garad et al. 2021). ProBDNF also collapses neurite outgrowth in primary cortical neurons of the CNS and superior cervical ganglion neurons of the PNS (Sun et al. 2012). In several experimental models, proBDNF mediates these toxic effects through the neurotrophin receptor p75NTR (Liu et al. 2018; Sun et al. 2012). However, it may signal independently from p75NTR at some hippocampal synapses. For example, proBDNF does not require p75NTR to mediate LTD at MF-CA3 synapses, suggesting that it may interact with other receptors in certain cell types (Garad et al. 2021). ProBDNF may also promote neurotrophism in some cells through an interaction with the tropomyosin-related kinase receptor (TrkB) (Fayard et al. 2005). ProBDNF increases neurite outgrowth in the rat pheochromocytoma (PC12) cell line by binding to TrkB and activating the downstream extracellular-regulated kinase (ERK) pathway (Fayard et al. 2005). Therefore, proBDNF mediates neurotoxicity when bound to p75NTR, but may promote neurite outgrowth when bound to TrkB. The relative expression of these receptors in hippocampal neurons, therefore, may determine whether proBDNF weakens or strengthens excitatory synapses.
Mature BDNF stabilises excitatory hippocampal synapses and improves their signalling though its interaction with TrkB following stimulation (Petkova-Tuffy et al. 2021). mBDNF/TrkB signalling plays an important role during the late phase of hippocampal LTP through activation of the MAPK, PI-3 K/Akt and PL-Cγ pathways (Minichiello et al. 2002). These pathways lead to changes in the stability and function of excitatory synapses which collectively improves strength. At the pre-synapse, mBDNF/TrkB signalling increases the release probability of synaptic vesicles, suggesting that it promotes neurotransmitter release (Niculescu et al. 2018; Petkova-Tuffy et al. 2021). At the post-synapse, mBDNF/TrkB signalling promotes structural and neurochemical alterations that stabilise the synapse. These changes may be mediated in part by an interaction with neuroligin 1, a post-synaptic cell adhesion protein that binds pre-synaptic neurexin to stabilise excitatory synapses (Petkova-Tuffy et al. 2021). The protein is reduced in the hippocampus of AD brains, suggesting that it is involved in the pathogenesis of the disease (Dufort-Gervais et al. 2020). mBDNF/TrkB signalling also increases the frequency and amplitude of excitatory post-synaptic currents, indicating that it enhances glutamate neurotransmission (Rauti et al. 2020). Taken together, mBDNF strengthens excitatory hippocampal synapse following synaptic activity in the brain.
The transition from mild cognitive impairment to AD
The transition from mild cognitive impairment (MCI) to AD is characterised by a period of accelerated brain atrophy that exceeds normal ageing (Franke and Gaser 2012; Gaser et al. 2013). Brain volume declines with age in humans and is associated with the progressive cognitive decline that is inevitable with age (Fjell et al. 2014; Veldsman et al. 2021). From 65 years onwards, this decline is more pronounced in the hippocampus – a region involved in learning and memory – suggesting that it may be more vulnerable to ageing (Veldsman et al. 2021). While cognitive decline may be inevitable with age, impaired cognition beyond normal ageing can result in MCI (Abner et al. 2017). MCI is an intermediate stage in the transition to AD defined as an impairment in one or more of the four cognitive domains (Overton et al. 2019). However, MCI is not always a prognosis of dementia as some people with the condition never transition to AD (Fang et al. 2019; Franke and Gaser 2012), while others revert to normal (Abner et al. 2017; Overton et al. 2019). People with stable MCI do not show abnormal brain atrophy and are more likely to have moderate AD neuropathologies (Abner et al. 2017; Franke and Gaser 2012; Gaser et al. 2013; Mi et al. 2017). By contrast, people with progressive MCI undergo a period of rapid brain atrophy before the transition to AD and are more likely to have severe AD neuropathologies (Figure 1) (Abner et al. 2017; Fang et al. 2019; Gaser et al. 2013). The development of biomarkers to distinguish progressive from stable MCI is a desirable outcome to identify people most at risk of AD. However, understanding the molecular basis of this early transition could identify drug targets with the potential to halt or reverse the disease in its later stages.
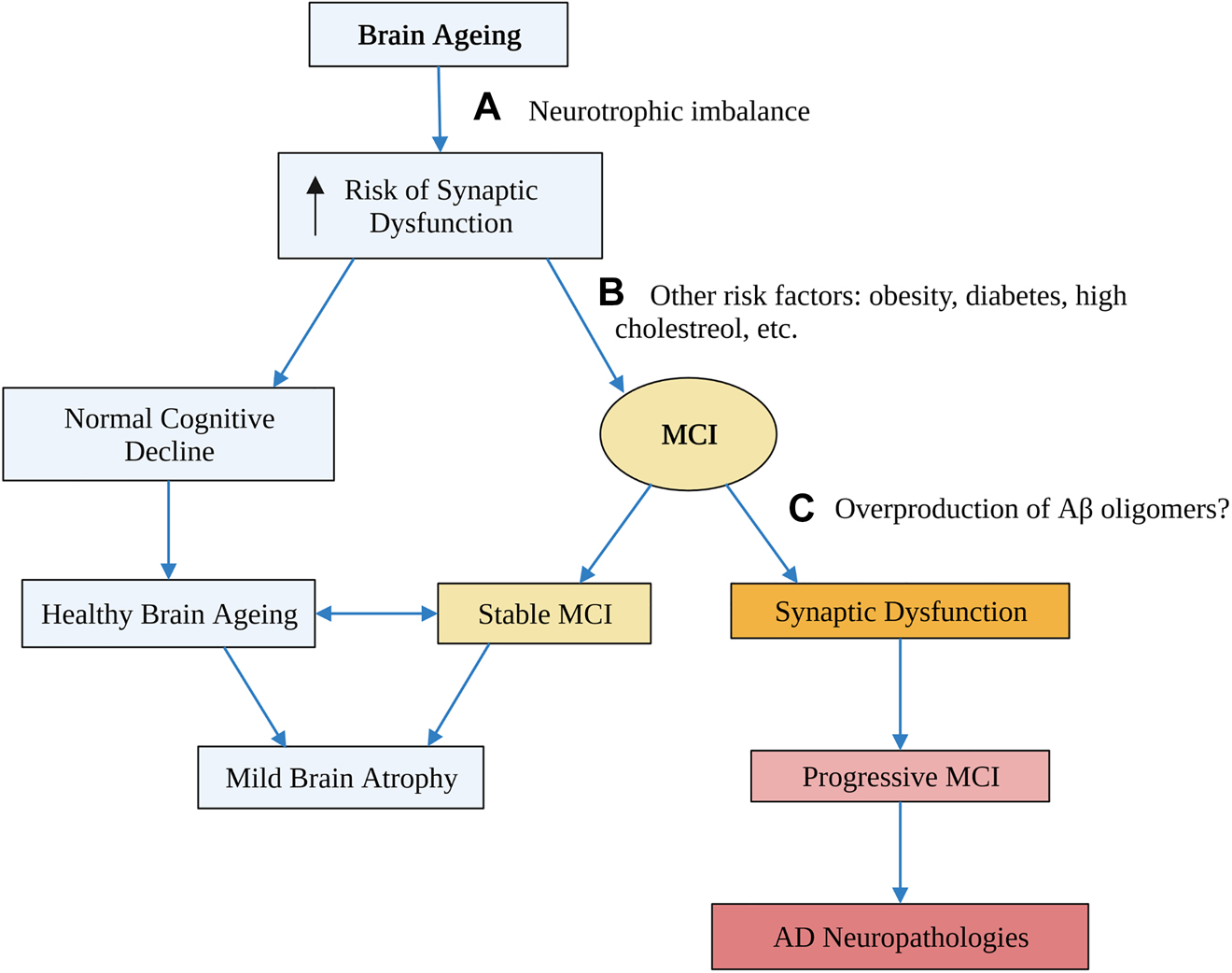
Proposed stages in the transition from brain ageing to AD and potential risk factors for progression from one stage to the next.
An age-related neurotrophic imbalance due to impaired activity-dependent mechanisms may increase the risk of synaptic dysfunction, which is an early event in AD (A). However, many people age healthily and only experience mild brain atrophy, so other risk factors such as obesity and diabetes likely contribute to the development of MCI (B). Finally, an over production of Aβ, or a failure to clear it, may be the final stage that triggers synaptic dysfunction, and begins the path towards relentless neurodegeneration (C). Flow diagram created with BioRender.com. MCI, mild cognitive impairment.
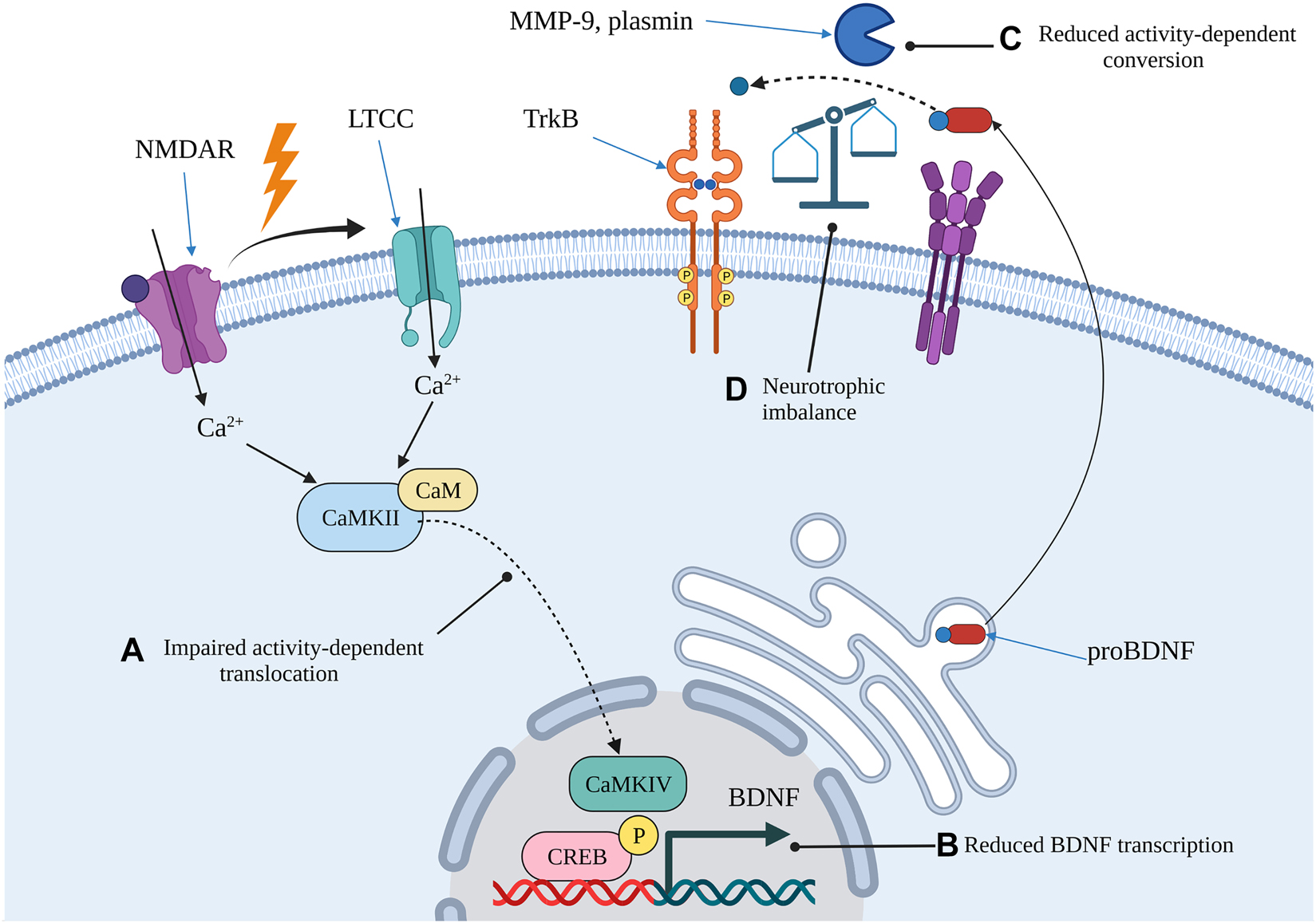
Proposed mechanism of age-related impairments in neurotrophic signalling that may increase the risk of AD in some individuals.
Impaired activity-dependent mechanisms due to ageing may result in a lack of CaMKII responsiveness following stimulation in hippocampal neurons (A). The impaired translocation could lead to delayed, or attenuated, augmentation of BDNF transcription (B). In addition, age-related reductions in the activity of both intracellular and extracellular proteases could impair the rapid maturation of BDNF that normally occurs following excitatory synaptic activity (C). The net result is an increase in proBDNF relative to mBDNF which skews neurotrophic signalling towards LTD and increases the risk of synaptic dysfunction (D). Image created with BioRender.com. BDNF, brain-derived neurotrophic factor; CaM, calmodulin; CaMKII, Ca2+/calmodulin-dependent kinase II; CaMKVI, Ca2+/calmodulin-dependent kinase IV; CREB, calcium-responsive element binding protein; LTCC, L-type voltage gated Ca2+ channel; MMP-9, matrix metalloproteinase 9; NMDAR, N-methyl d-aspartate receptor; TrkB, tropomyosin-related kinase B.
There is considerable evidence that synapse loss early in AD is a consequence of Aβ-induced synaptic dysfunction (He et al. 2019; Lauren et al. 2009; Moreno et al. 2009; Shankar et al. 2007). Reductions in synaptic proteins, dendritic spine density and synapse loss are well established pathologies in post-mortem AD brains (Mi et al. 2017; Proctor et al. 2010; Scheff et al. 2007; Sze et al. 1997). These pathologies occur early in the disease and correlate with cognitive performance suggesting that synapse loss accurately reflects disease progression (Mecca et al. 2020; Sze et al. 1997). Synapse loss early in AD is likely a consequence of synaptic dysfunction mediated by abnormally high levels of Aβ oligomers (Figure 1) (He et al. 2019; Moreno et al. 2009; Shankar et al. 2007). Aβ oligomers are intermediates between peptide monomers and fibrils that culminate in insoluble plaques. Although Aβ oligomers increase synaptic transmission at physiological levels, pathological levels disrupt synaptic transmission and impair hippocampal LTP (He et al. 2019; Lauren et al. 2009; Moreno et al. 2009; Wang et al. 2017). This synaptic dysfunction precedes synapse loss in AD transgenic mice, suggesting that an overproduction of Aβ, or a failure to clear it, is an early event in AD (He et al. 2019). To identify drug targets for AD, therefore, it is critical to understand the molecular mechanisms that cause Aβ to be overproduced or mediate its toxic effects (Figure 1).
Alterations in BDNF transcription and expression during brain ageing and the transition to AD
Age-related impairments in activity dependent BDNF transcription may underpin excessive cognitive decline in rodents (Hongpaisan et al. 2013). BDNF transcription is reduced in the hippocampus of aged rats and in the cortex of aged mice, suggesting that alterations in its stimulus-driven expression occur with age (Belviranli and Okudan 2018; Caughey et al. 2017). Consistent with this interpretation are findings of reduced pCREB, pTrkB and PKCε expression in the aged rodent hippocampus (Calabrese et al. 2013; Hattiangady et al. 2005; Hongpaisan et al. 2013; Smiljanic et al. 2015). Age-related reductions in BDNF transcription may be due to impairments in the mechanisms that couple excitation to the nucleus of hippocampal neurons. The augmented recruitment of CBP to BDNF promoters II and VI following NMDA stimulation is lost in the hippocampus of aged mice (Palomer et al. 2016). This impairment may result from a lack of CaMKIIα activation following Ca2+ influx, indicating that stimulus driven BDNF transcription is compromised (Palomer et al. 2016). However, impairments in activity-dependent mechanisms may not occur in all aged animals, which could explain why some are resistant to cognitive decline (Hongpaisan et al. 2013). For example, ERK activation and PKCε expression are reduced in cognitively impaired aged rats, but not aged rats with normal cognition (Hongpaisan et al. 2013; Nagahara et al. 2009). These impairments are reversed by increasing PKCε activity, suggesting that they are caused by a functional loss in activity dependent mechanisms (Hongpaisan et al. 2013). Taken together, impairments in the stimulus-driven transcription of BDNF could explain cognitive impairment in aged rodents.
Transcription of BDNF is reduced in MCI before the transition to AD in humans, suggesting an early loss in activity-dependent mechanisms (Ginsberg et al. 2019). BDNF transcription is reduced in CA1 pyramidal neurons and the hippocampus in MCI before reverting to normal in AD (Ginsberg et al. 2019). This early reduction predicts neuritic plaques in CA1 pyramidal neurons, but not diffuse plaques or NFTs, suggesting that down regulation of BDNF may precede some AD pathologies (Ginsberg et al. 2019). TrkB transcription, on the other hand, is reduced from MCI onwards and correlates with cognitive performance, suggesting that it relates with disease severity (Ginsberg et al. 2019). The early down-regulation of BDNF may be due, in part, to alterations in the subcellular localisation of activated CaMKII (pCaMKII) (Reese et al. 2011). pCaMKII increasingly translocates from dendrites to the soma of granule cells in the DG and CA3 pyramidal neurons in the hippocampus from MCI onwards (Reese et al. 2011). This translocation correlates with the mini-mental state examination (MMSE) scores, suggesting that Ca2+ dependent signalling pathway are disrupted during the progression of AD (Reese et al. 2011). Further research in this area may identify other mechanisms involved in the down regulation of BDNF in MCI and its relationship to the progression of AD.
Low BDNF protein levels increase the risk of AD in humans, suggesting that a loss of neurotrophic support occurs early in the disease (Fang et al. 2019; Weinstein et al. 2014). Post-mortem analysis shows reduced BDNF protein levels in brain regions affected by AD, indicating that a loss of neurotrophic support occurs in the disease along with other neuropathologies (Jiao et al. 2016; Lee et al. 2005; Narisawa-Saito et al. 1996). However, these analyses cannot establish whether reduced BDNF protein levels precede the onset of the disease. Prospective studies analyse serum from ageing humans with normal cognition and MCI to determine if reductions in BDNF relate to cognitive impairment and/or the risk of AD (Fang et al. 2019; Weinstein et al. 2014). However, while BDNF may cross the blood–brain barrier, levels in the periphery may not reflect expression in the brain, which limits interpretation of the data. BDNF is expressed by perivascular adipose tissue surrounding arteries into circulation, where it binds receptors on smooth muscle cells (Zierold et al. 2021). This peripheral release of BDNF may mask reductions in the brain, potentially limiting the detection of abnormally low levels. Nevertheless, serum BDNF levels positively correlate with cerebrospinal fluid Aβ42 levels and MMSE scores suggesting that they are related to AD pathologies (Mori et al. 2021). Elderly people (>65 years) with low serum BDNF are more likely to experience worsening cognitive performance than people with higher levels (Fujiwara et al. 2021). In people with MCI, low serum BDNF increases the risk of transitioning to AD, indicating that a loss of neurotrophic support precedes onset of the disease (Fang et al. 2019; Weinstein et al. 2014). These results are consistent with the down regulation of hippocampal BDNF transcription that occurs in MCI (Ginsberg et al. 2019). Taken together, an age-related reduction in BDNF, which may be more severe in some individuals, could increase the risk of AD in humans (Figure 2).
ProBDNF is increased in the aged rodent hippocampus, but it is less clear how mBDNF changes with brain ageing (Buhusi et al. 2017; Perovic et al. 2013; Smiljanic et al. 2015; Wong et al. 2021). It is difficult to establish how total BDNF levels change in the aged rodent hippocampus given the conflicting results of earlier studies (Croll et al. 1998; Hattiangady et al. 2005; Katoh-Semba et al. 1998). Some show that BDNF remains stable (Croll et al. 1998; Silhol et al. 2005), while others show that it increases (Katoh-Semba et al. 1998) and yet others show that it declines (Hattiangady et al. 2005). Nevertheless, with the development of pro and mature BDNF antibodies (Nagappan et al. 2009), most studies now measure each isoform independently (Buhusi et al. 2017; Calabrese et al. 2013; Perovic et al. 2013; Smiljanic et al. 2015; Wong et al. 2021). Increased proBDNF levels have consistently been shown in the aged hippocampus of rodents (Buhusi et al. 2017; Perovic et al. 2013; Smiljanic et al. 2015; Wong et al. 2021), with only one study showing they remain stable (Calabrese et al. 2013). By contrast, proBDNF remains unchanged in the cortex, indicating that these changes are specific to the hippocampus (Table 1). The results for mBDNF are less clear as findings of age-related decreases (Calabrese et al. 2013; Smiljanic et al. 2015), and no changes (Buhusi et al. 2017; Perovic et al. 2013; Wong et al. 2021) have both been reported. Given that maturation of BDNF is dependent on proteolytic processing, it is surprising that age-related changes in the pro/mature BDNF ratio have not been more extensively investigated. Our lab group recently found that proBDNF increases relative to mBDNF with increasing age in the pre-frontal cortex and brain stem, but not in the hippocampus of WT mice (unpublished data). These results suggest that proteolytic processing declines with normal brain ageing, possibly due to age-related decreases in enzymatic activity (Figure 2).
Age-related changes in proBDNF and mature BDNF in the hippocampus and cortex of old rodents relative to adult.
| Hippocampus | Cortex | ||||
|---|---|---|---|---|---|
| Author | Animal | proBDNF | mBDNF | proBDNF | mBDNF |
| Buhusi et al. (2017) | Mouse | ↑ | ↔ | – | – |
| Calabrese et al. (2013) | Rat | ↔ | ↓ | ↔ | ↓ |
| Perovic et al. (2013) | Rat | ↑ | ↔ | ↓ | ↔ |
| Smiljanic et al. (2015) | Rat | ↑ | ↓ | ↔ | ↔ |
| Wong et al. (2021) | Mouse | ↑ | ↔ | - | - |
-
↑, increased; ↓, decreased; ↔, unchanged; -, not measured.
There is limited evidence on how proBDNF relates to AD in humans, but it may be associated with tau pathology (Bharani et al. 2020). The involvement of pro and mature BDNF in the transition to AD is complicated by a technical issue with the ELISA kits used for their detection (Polacchini et al. 2015). The capture antibodies for mBDNF cross-react with proBDNF in some, but not all kits (Lim et al. 2015; Polacchini et al. 2015). However, with recent improvements in the specificity of capture antibodies, both isoforms can usually be reliably measured (Lim et al. 2015; Mizoguchi et al. 2020). Serum proBDNF levels do not correlate with memory scores – as measured by the Rivermead behavioural memory test – in elderly subjects with normal cognition (Mizoguchi et al. 2020). However, the MMSE is often used to detect dementia because performance on this task reflects brain volume (Dinomais et al. 2016). Consequently, it is currently unknown how proBDNF relates to age-related cognitive decline and the transition to AD. Nevertheless, serum proBDNF levels correlate with phospho-tau staining in CA1 of the hippocampus in AD subjects, indicating that it may be related to tau pathology (Bharani et al. 2020). Further studies on how proBDNF changes with age in humans may provide better insights into the molecular basis of the transition to AD.
Age-related increases in hippocampal proBDNF may increase the risk of AD by driving Aβ production and aggregation (Chen et al. 2017). ProBDNF is highly expressed during post-natal brain development and the dominant isoform until early adulthood, when it declines relative to mBDNF (Yang et al. 2014). The high proBDNF levels are accompanied by high expression of p75NTR (Yang et al. 2014), the receptor that mediates its toxic effects (Chen et al. 2017; Sun et al. 2012). Many studies investigate the neurotoxic effects of exogenous proBDNF using neonatal neurons from mice or cell lines (Fleitas et al. 2018; Liu et al. 2018; Sun et al. 2012). However, given that proBDNF is increased in the aged rodent hippocampus (Buhusi et al. 2017; Perovic et al. 2013; Smiljanic et al. 2015), it is critical to understand how endogenous proBDNF contributes to brain ageing and the transition to AD. Endogenous proBDNF facilitates the production of toxic Aβ species in AD transgenic mice indicating that it may have a role in amyloid pathology (Chen et al. 2016). Further research in this area may establish a better understanding of how age-related changes in proBDNF contribute to AD.
The role of p75NTR in age-related brain atrophy and the transition to AD
p75NTR may exacerbate AD by facilitating Aβ production and mediating its toxic effects, but it’s role in age-related brain atrophy is not fully understood (Wong et al. 2021; Xie et al. 2019). p75NTR is widely expressed throughout the developing brain (Hallbook et al. 1990), but then becomes refined to specific brain regions during adulthood (Springer et al. 1990). These regions include cholinergic neurons of the basal forebrain (BFCNs) and neurons and astrocytes of hippocampal CA1, amongst others (Wong et al. 2021). BFCNs develop long projections that extend into a range of cortical areas (Li et al. 2018) such as the entorhinal cortex (McGaughy et al. 2005), amygdala (Jiang et al. 2016) and visual cortex (Pinto et al. 2013). The resulting cholinergic circuity regulates critical brain functions including working memory (McGaughy et al. 2005), visual discrimination (Pinto et al. 2013) and attention. BFCNs are critically important to AD because they degenerate early in the disease and are amongst the worst affected neuronal population (Schmitz et al. 2016). Moreover, their early degeneration predicts the spread of AD neuropathologies, suggesting that the disease originates in BFCNs before emanating to other regions (Schmitz et al. 2016). p75NTR has long been proposed as a mediator of amyloid pathology because it is expressed by BFCNs and facilitates the production of Aβ (Coulson 2006, 2009; Zeng et al. 2011). In addition, p75NTR mediates the down-stream toxic effects of Aβ, such as tau pathology, so alterations in its expression or signalling facilitate the spread of AD neuropathologies (Shen et al. 2019; Yang et al. 2020). However, age-related alterations in the expression of p75NTR and its involvement in the degeneration of BFCNs or other brain regions remain unclear. Although recent research has aimed to bridge this gap in the literature, further work may be required.
Evidence from transgenic mice indicates that p75NTR eliminates developing BFCNs but does not affect their age-related degeneration (Boskovic et al. 2014; Busch et al. 2017; Dokter et al. 2015). p75KO mice are a useful tool for investigating the effects of p75NTR in developing BFCNs and on their survival during brain ageing (Greferath et al. 2000). The original models, p75NTRexonIII, lacked expression of full-length p75NTR, but maintained expression of a truncated isoform, neurotrophin receptor homolog 2 (NRH2) (Kanning et al. 2003). Using these mice, it was shown that p75NTR eliminates BFCNs during development and that these negative effects are long-lasting (Yeo et al. 1997). However, others found, using genetically-matched controls that p75NTR reduces the size of BFCNs but not their number, indicating that it may cause atrophy but not death (Greferath et al. 2000). Despite these contrasting results, the discovery that NRH2 can modulate Trk signalling – potentially countering p75NTR-mediated apoptosis (Murray et al. 2004) – necessitated the development of a new model. The resulting complete knock outs, p75NTRexonIV, lacked both p75NTR receptor protein and its truncated isoform (von Schack et al. 2001). Adult p75NTRexonIV mice show an increased number of BFCNs (Naumann et al. 2002), increased cholinergic fiber density in the amygdala (Busch et al. 2017) and increased hippocampal volume (Dokter et al. 2015; Poser et al. 2015). These same findings were also shown in a conditional p75KO mouse model, suggesting that p75NTR eliminates BFCNs during development (Boskovic et al. 2014). However, the same increases are not observed in aged mice relative to adult, suggesting that the receptor is not involved in age-related BFCN degeneration (Boskovic et al. 2014; Dokter et al. 2015; Poser et al. 2015). A limitation of knock-out mice is that the effects of p75NTR up-regulation during adulthood cannot be investigated. Modulating p75NTR signalling in adulthood or old age, therefore, may provide insights into age-related BFCN degeneration.
p75NTR signalling mediates BFCN degeneration in the aged rodent brain, suggesting that it may contribute to age-related brain atrophy (Xie et al. 2019). Modulation of p75NTR signalling in old age mice (21 months) preserves the size of BFCNs and neurite density of their hippocampal CA1 targets (Xie et al. 2019). These same effects are not observed in moderately aged mice (17 months), indicating that the aged brain is susceptible to p75NTR-mediated neurodegeneration (Xie et al. 2019). The neurodegenerative effects of p75NTR on BFCNs may be mediated by neurotrophic imbalances in the brain regions they innervate. p75NTR triggers apoptosis when bound to all neurotrophins, so alterations in BDNF may only partly explain their degeneration (Friedman 2000). For example, disruptions to the maturation of proNGF, which is retrogradely trafficked to BFCN cell bodies, in the frontal cortex of adult rats causes BFCNs to degenerate (Allard et al. 2018). Further research in this area may lead to a better understanding of the involvement of neurotrophins on the age-related degeneration of BFCNs.
Disease-modifying therapies aimed at restoring the neurotrophic imbalance in AD
Restoring the neurotrophic imbalance is one strategy amongst many aimed at regenerating lost synapses and halting AD progression (Cummings et al. 2020). The number of drugs targeting synaptic plasticity/neuroprotection in clinical trials increased from 2016 to 2020, demonstrating that neurorestoration is an increasingly popular therapeutic approach (Cummings et al. 2020). By contrast, drugs targeting amyloid pathology declined over the same period, indicating that a shift in therapeutic strategies has occurred (Cummings et al. 2020). Amongst neurotrophin drug candidates for AD is an analog of NGF which modulates p75NTR signalling (Simmons et al. 2014; Xie et al. 2019). This drug reduces Aβ-induced synapse loss in AD transgenic mice, suggesting that it has the potential to halt AD progression (Yang et al. 2020). Another approach is to alter p75NTR signalling indirectly by scavenging neurodegenerative ligands, such as Aβ and pro-neurotrophins, with the ectodomain of the receptor (Yao et al. 2015). A chimeric fusion protein of p75ECD and IgG (p75ECD-Fc) reduces amyloid plaque load and improves cognitive performance in AD transgenic mice, suggesting that it alters the course of the disease (Wang et al. 2016; Yao et al. 2015). However, modulation of p75NTR signalling is only one strategy that may potentially restore the neurotrophic imbalance (Figure 3). Another strategy involves promoting neurotrophic signalling by indirectly stimulating the expression of BDNF and regenerating lost synapses (Figure 3). Activation of the hippocampal-specific PKCε with Bryostatin-1 effectively restores BDNF levels, counters Aβ production and regenerates lost synapses in AD transgenic mouse models (Hongpaisan et al. 2011). This drug was shown to improve cognitive performance after completion of dosing in a clinical trial, indicating that it may promote synaptogenesis (Farlow et al. 2019). Further research on the involvement of the neurotrophic imbalance in brain ageing and the risk for AD may identify other therapeutic targets.
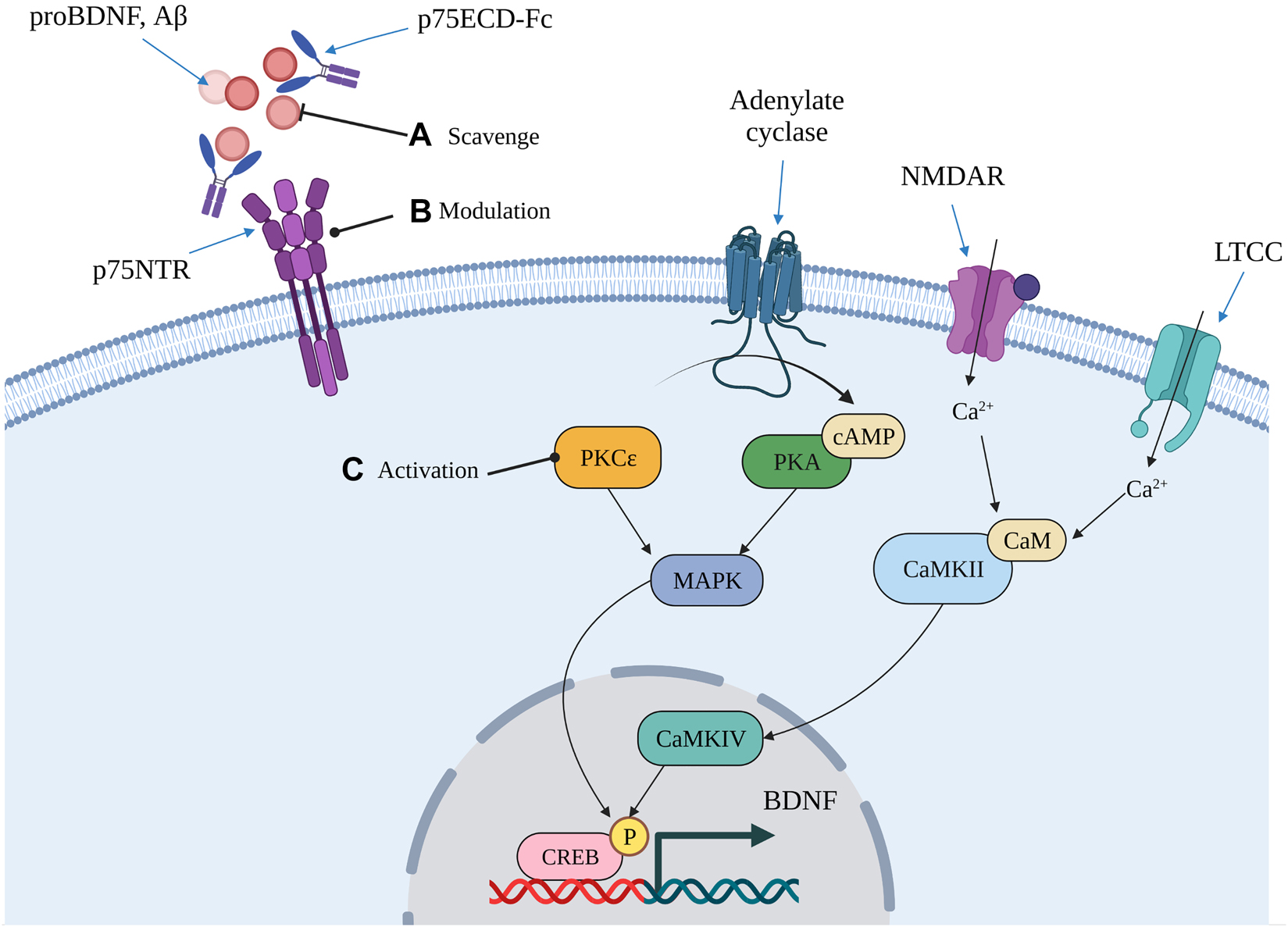
Some therapeutic approaches aimed at restoring the neurotrophic imbalance that may modify the course of AD pathology.
One approach that may alter the course of AD progression involves countering p75NTR-mediated neurodegenerative signalling through either direct or indirect modulation of the receptor (A, B). Indirect modulation involves scavenging neurodegenerative ligands to prevent further amyloid deposition and p75NTR-mediated tau hyperphosphorylation (A). Direct modulation involves using compounds to directly interact with the receptor and alter its signalling (B). Another approach is to stimulate BDNF transcription by increasing the activity of kinases that regulate its activity-dependent transcription (C). This approach may lead to the stabilisation of excitatory synapses and improve their function, thereby slowing or halting AD-related synapse loss. Image created with BioRender.com. BDNF, brain-derived neurotrophic factor; CaM, Calmodulin; CaMKII, Ca2+/calmodulin-dependent kinase II; CaMKIV, Ca2+/calmodulin-dependent kinase IV; cAMP, cyclic adeno monophosphate; CREB, calcium responsive element binding protein; LTCC, L-type voltage-gated Ca2+ channel; MAPK, mitogen-activated protein kinase; NMDAR, N-methyl-d-aspartate receptor; p75ECD-Fc, fusion protein of p75NTR and human IgG-Fc; P, phosphorylation; PKA, protein kinase A; PKCε, protein kinase C epsilon.
Discussion/conclusion
This review has critically evaluated current evidence on the role of BDNF and p75NTR in brain ageing and the transition to AD. As outlined, research on how these neurotrophins strengthen and collapse dendritic spines respectively has led to the development of drugs currently in clinical trials. However, gaps in the literature still exist and, if filled, may lead to the identification of further drug targets. These gaps primarily relate to proBDNF, though the role of p75NTR in brain ageing is limited, but can now be investigated with the advent of molecules that modulate its function (Xie et al. 2019). A key question regarding proBDNF is how it relates to brain ageing in humans and whether it can predict the transition to AD. This question is technically feasible now that ELISA kits specific to BDNF isoforms have been developed (Bharani et al. 2020; Mizoguchi et al. 2020). Given that proBDNF mediates hippocampal LTD (Garad et al. 2021), age-related increases in its expression would imply an increased risk of synaptic dysfunction. However, precise experiments in mouse models of AD may be required to make this link. In summary, BDNF and p75NTR are promising therapeutic targets with the potential to modify, or reverse, the course of AD pathology.
Acknowledgements
The author would like to acknowledge Isaac Deng for assistance provided during the generation of each figure.
-
Author contributions: All the authors have accepted responsibility for the entire content of this submitted manuscript and approved submission.
-
Research funding: The first author, Shaun Cade, is a recipient of the Australian Government Research Training Program (RTP) Scholarship.
-
Conflict of interest statement: The authors declare no conflicts of interest regarding this article.
References
Abner, E.L., Kryscio, R.J., Schmitt, F.A., Fardo, D.W., Moga, D.C., Ighodaro, E.T., Jicha, G.A., Yu, L., Dodge, H.H., Xiong, C., et al.. (2017). Outcomes after diagnosis of mild cognitive impairment in a large autopsy series. Ann. Neurol. 81: 549–559, https://doi.org/10.1002/ana.24903.Search in Google Scholar PubMed PubMed Central
Aisen, P.S., Gauthier, S., Ferris, S.H., Saumier, D., Haine, D., Garceau, D., Duong, A., Suhy, J., Oh, J., Lau, W.C., et al.. (2011). Tramiprosate in mild-to-moderate Alzheimer’s disease - a randomized, double-blind, placebo-controlled, multi-centre study (the Alphase Study). Arch. Med. Sci. 7: 102–111, https://doi.org/10.5114/aoms.2011.20612.Search in Google Scholar PubMed PubMed Central
Allard, S., Jacobs, M.L., Do Carmo, S., and Cuello, A.C. (2018). Compromise of cortical proNGF maturation causes selective retrograde atrophy in cholinergic nucleus basalis neurons. Neurobiol. Aging 67: 10–20, https://doi.org/10.1016/j.neurobiolaging.2018.03.002.Search in Google Scholar PubMed
Alzheimer’s Association (2017). 2017 Alzheimer’s disease facts and figures. Alzheimer’s Dementia 13: 325–373.10.1016/j.jalz.2017.02.001Search in Google Scholar
Balkowiec, A. and Katz, D.M. (2002). Cellular mechanisms regulating activity-dependent release of native brain-derived neurotrophic factor from hippocampal neurons. J. Neurosci. 22: 10399–10407, https://doi.org/10.1523/jneurosci.22-23-10399.2002.Search in Google Scholar
Baydyuk, M., Xie, Y., Tessarollo, L., and Xu, B. (2013). Midbrain-derived neurotrophins support survival of immature striatal projection neurons. J. Neurosci. 33: 3363–3369, https://doi.org/10.1523/jneurosci.3687-12.2013.Search in Google Scholar PubMed PubMed Central
Belviranli, M. and Okudan, N. (2018). Exercise training protects against aging-induced cognitive dysfunction via activation of the hippocampal PGC-1alpha/FNDC5/BDNF pathway. NeuroMolecular Med. 20: 386–400.10.1007/s12017-018-8500-3Search in Google Scholar PubMed
Benicky, J., Sanda, M., Brnakova Kennedy, Z., and Goldman, R. (2019). N-Glycosylation is required for secretion of the precursor to brain-derived neurotrophic factor (proBDNF) carrying sulfated LacdiNAc structures. J. Biol. Chem. 294: 16816–16830, https://doi.org/10.1074/jbc.ra119.009989.Search in Google Scholar
Bennett, B.D., Denis, P., Haniu, M., Teplow, D.B., Kahn, S., Louis, J.C., Citron, M., and Vassar, R. (2000). A furin-like convertase mediates propeptide cleavage of BACE, the Alzheimer’s beta -secretase. J. Biol. Chem. 275: 37712–37717, https://doi.org/10.1074/jbc.m005339200.Search in Google Scholar PubMed
Bharani, K.L., Ledreux, A., Gilmore, A., Carroll, S.L., and Granholm, A.C. (2020). Serum pro-BDNF levels correlate with phospho-tau staining in Alzheimer’s disease. Neurobiol. Aging 87: 49–59, https://doi.org/10.1016/j.neurobiolaging.2019.11.010.Search in Google Scholar PubMed
Boskovic, Z., Alfonsi, F., Rumballe, B.A., Fonseka, S., Windels, F., and Coulson, E.J. (2014). The role of p75NTR in cholinergic basal forebrain structure and function. J. Neurosci. 34: 13033–13038, https://doi.org/10.1523/jneurosci.2364-14.2014.Search in Google Scholar
Buhusi, M., Etheredge, C., Granholm, A.C., and Buhusi, C.V. (2017). Increased hippocampal proBDNF contributes to memory impairments in aged mice. Front. Aging Neurosci. 9: 1–15, https://doi.org/10.3389/fnagi.2017.00284.Search in Google Scholar PubMed PubMed Central
Busch, R., Baldus, M., Vogt, M.A., Berger, S.M., Bartsch, D., Gass, P., and von Bohlen Und Halbach, O. (2017). Effects of p75NTR deficiency on cholinergic innervation of the amygdala and anxiety-like behavior. J. Neurochem. 141: 461–471, https://doi.org/10.1111/jnc.14006.Search in Google Scholar PubMed
Calabrese, F., Guidotti, G., Racagni, G., and Riva, M.A. (2013). Reduced neuroplasticity in aged rats: a role for the neurotrophin brain-derived neurotrophic factor. Neurobiol. Aging 34: 2768–2776, https://doi.org/10.1016/j.neurobiolaging.2013.06.014.Search in Google Scholar PubMed
Caughey, S., Harris, A.P., Seckl, J.R., Holmes, M.C., and Yau, J.L. (2017). Forebrain-specific transgene rescue of 11beta-HSD1 associates with impaired spatial memory and reduced hippocampal brain-derived neurotrophic factor mRNA levels in aged 11beta-HSD1 deficient mice. J. Neuroendocrinol. 29: 1–9, https://doi.org/10.1111/jne.12447.Search in Google Scholar PubMed PubMed Central
Chen, J., Li, C.R., Yang, H., Liu, J., Zhang, T., Jiao, S.S., Wang, Y.J., and Xu, Z.Q. (2016). proBDNF attenuates hippocampal neurogenesis and induces learning and memory deficits in aged mice. Neurotox. Res. 29: 47–53, https://doi.org/10.1007/s12640-015-9568-2.Search in Google Scholar PubMed
Chen, J., Zhang, T., Jiao, S., Zhou, X., Zhong, J., Wang, Y., Liu, J., Deng, J., Wang, S., and Xu, Z. (2017). proBDNF accelerates brain amyloid-beta deposition and learning and memory impairment in APPswePS1dE9 transgenic mice. J. Alzheimers Dis. 59: 941–949, https://doi.org/10.3233/jad-161191.Search in Google Scholar
Chen, Z., Ieraci, A., Teng, H., Dall, H., Meng, C., Herrera, D., Nykjaer, A., Hempstead, B., and Lee, F. (2005). Sortilin controls intracellular sorting of brain-derived neurotrophic factor to the regulated secretory pathway. J. Neurosci. 25: 6156–6166, https://doi.org/10.1523/jneurosci.1017-05.2005.Search in Google Scholar
Cohen, S.M., Suutari, B., He, X., Wang, Y., Sanchez, S., Tirko, N.N., Mandelberg, N.J., Mullins, C., Zhou, G., Wang, S., et al.. (2018). Calmodulin shuttling mediates cytonuclear signaling to trigger experience-dependent transcription and memory. Nat. Commun. 9: 1–12, https://doi.org/10.1038/s41467-018-04705-8.Search in Google Scholar PubMed PubMed Central
Cook, S.G., Bourke, A.M., O’Leary, H., Zaegel, V., Lasda, E., Mize-Berge, J., Quillinan, N., Tucker, C.L., Coultrap, S.J., Herson, P.S., et al.. (2018). Analysis of the CaMKIIalpha and beta splice-variant distribution among brain regions reveals isoform-specific differences in holoenzyme formation. Sci. Rep. 8: 1–15, https://doi.org/10.1038/s41598-018-23779-4.Search in Google Scholar PubMed PubMed Central
Cook, S.G., Buonaroti, O.R., Coultrap, S.J., and Bayer, K.U. (2021). CaMKII holoenzyme mechanisms that govern the LTP versus LTD decision. Sci. Adv. 7: 1–13, https://doi.org/10.1126/sciadv.abe2300.Search in Google Scholar
Coulson, E.J. (2006). Does the p75 neurotrophin receptor mediate Abeta-induced toxicity in Alzheimer’s disease? J. Neurochem. 98: 654–660, https://doi.org/10.1111/j.1471-4159.2006.03905.x.Search in Google Scholar
Coulson, E.J., May, L.M., Sykes, A.M., and Hamlin, A.S. (2009). The role of the p75 neurotrophin receptor in cholinergic dysfunction in Alzheimer’s disease. Neuroscientist 15: 317–323, https://doi.org/10.1177/1073858408331376.Search in Google Scholar
Croll, S.D., Ip, N.Y., Lindsay, R.M., and Wiegand, S.J. (1998). Expression of BDNF and trkB as a function of age and cognitive performance. Brain Res. 812: 200–208, https://doi.org/10.1016/s0006-8993(98)00993-7.Search in Google Scholar
Cummings, J., Lee, G., Ritter, A., Sabbagh, M., and Zhong, K. (2020). Alzheimer’s disease drug development pipeline: 2020. Alzheimers Dement (N Y) 6: 1–29, https://doi.org/10.1002/trc2.12050.Search in Google Scholar
Dinomais, M., Celle, S., Duval, G.T., Roche, F., Henni, S., Bartha, R., Beauchet, O., and Annweiler, C. (2016). Anatomic correlation of the mini-mental state examination: a voxel-based morphometric study in older adults. PLoS One 11: 1–13, https://doi.org/10.1371/journal.pone.0162889.Search in Google Scholar
Dokter, M., Busch, R., Poser, R., Vogt, M.A., von Bohlen Und Halbach, V., Gass, P., Unsicker, K., and von Bohlen Und Halbach, O. (2015). Implications of p75NTR for dentate gyrus morphology and hippocampus-related behavior revisited. Brain Struct. Funct. 220: 1449–1462, https://doi.org/10.1007/s00429-014-0737-5.Search in Google Scholar
Dubois, C.M., Blanchette, F., Laprise, M.H., Leduc, R., Grondin, F., and Seidah, N.G. (2001). Evidence that furin is an authentic transforming growth factor-b1-converting enzyme. Am. J. Pathol. 158: 305–316, https://doi.org/10.1016/s0002-9440(10)63970-3.Search in Google Scholar
Dubois, C.M., Laprise, M.H., Blanchette, F., Gentry, L.E., and Leduc, R. (1995). Processing of transforming growth factor β1 precursor by human furin convertase. J. Biol. Chem. 270: 10618–10624, https://doi.org/10.1074/jbc.270.18.10618.Search in Google Scholar PubMed
Dufort-Gervais, J., Provost, C., Charbonneau, L., Norris, C.M., Calon, F., Mongrain, V., and Brouillette, J. (2020). Neuroligin-1 is altered in the hippocampus of Alzheimer’s disease patients and mouse models, and modulates the toxicity of amyloid-beta oligomers. Sci. Rep. 10: 1–16, https://doi.org/10.2307/j.ctv1h0p2wn.3.Search in Google Scholar
Egan, M.F., Kost, J., Tariot, P.N., Aisen, P.S., Cummings, J.L., Vellas, B., Sur, C., Mukai, Y., Voss, T., Furtek, C., et al.. (2018). Randomized trial of Verubecestat for mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 378: 1691–1703, https://doi.org/10.1056/nejmoa1706441.Search in Google Scholar PubMed PubMed Central
Fang, Y., Du, N., Xing, L., Duo, Y., and Zheng, L. (2019). Evaluation of hippocampal volume and serum brain-derived neurotrophic factor as potential diagnostic markers of conversion from amnestic mild cognitive impairment to Alzheimer disease: a STROBE-compliant article. Medicine (Baltimore) 98: 1–6, https://doi.org/10.1097/MD.0000000000016604.Search in Google Scholar PubMed PubMed Central
Farlow, M.R., Thompson, R.E., Wei, L.J., Tuchman, A.J., Grenier, E., Crockford, D., Wilke, S., Benison, J., and Alkon, D.L. (2019). A randomized, double-blind, placebo-controlled, phase II study assessing safety, tolerability, and efficacy of Bryostatin in the treatment of moderately severe to severe Alzheimer’s Disease. J. Alzheimers Dis. 67: 555–570, https://doi.org/10.3233/jad-180759.Search in Google Scholar
Fayard, B., Loeffler, S., Weis, J., Vogelin, E., and Kruttgen, A. (2005). The secreted brain-derived neurotrophic factor precursor pro-BDNF binds to TrkB and p75NTR but not to TrkA or TrkC. J. Neurosci. Res. 80: 18–28, https://doi.org/10.1002/jnr.20432.Search in Google Scholar PubMed
Fjell, A.M., Westlye, L.T., Grydeland, H., Amlien, I., Espeseth, T., Reinvang, I., Raz, N., Dale, A.M., and Walhovd, K.B., and Alzheimer Disease Neuroimaging Initiative (2014). Accelerating cortical thinning: unique to dementia or universal in aging? Cerebr. Cortex 24: 919–934, https://doi.org/10.1093/cercor/bhs379.Search in Google Scholar PubMed PubMed Central
Fleitas, C., Pinol-Ripoll, G., Marfull, P., Rocandio, D., Ferrer, I., Rampon, C., Egea, J., and Espinet, C. (2018). ProBDNF is modified by advanced glycation end products in Alzheimer’s disease and causes neuronal apoptosis by inducing p75 neurotrophin receptor processing. Mol. Brain 11: 1–16, https://doi.org/10.1186/s13041-018-0411-6.Search in Google Scholar PubMed PubMed Central
Franke, K. and Gaser, C. (2012). Longitudinal changes in individual brainAGE in healthy aging, mild cognitive impairment, and Alzheimer’s Disease. GeroPsych 25: 235–245, https://doi.org/10.1024/1662-9647/a000074.Search in Google Scholar
Frazzini, V., Granzotto, A., Bomba, M., Massetti, N., Castelli, V., d’Aurora, M., Punzi, M., Iorio, M., Mosca, A., Delli Pizzi, S., et al.. (2018). The pharmacological perturbation of brain zinc impairs BDNF-related signaling and the cognitive performances of young mice. Sci. Rep. 8: 1–12, https://doi.org/10.1038/s41598-018-28083-9.Search in Google Scholar PubMed PubMed Central
Friedman, W.J. (2000). Neurotrophins induce death of hippocampal neurons via the p75 receptor. J. Neurosci. 20: 6340–6346, https://doi.org/10.1523/jneurosci.20-17-06340.2000.Search in Google Scholar
Fujiwara, Y., Ihara, K., Hachisu, M., Suzuki, H., Kawai, H., Sakurai, R., Hirano, H., Chaves, P.H.M., Hashizume, M., and Obuchi, S. (2021). Higher serum brain-derived neurotrophic factor levels are associated with a lower risk of cognitive decline: a 2-Year follow up study in community-dwelling older adults. Front. Behav. Neurosci. 15: 1–9, https://doi.org/10.3389/fnbeh.2021.641608.Search in Google Scholar PubMed PubMed Central
Garad, M., Edelmann, E., and Lessmann, V. (2021). Long-term depression at hippocampal mossy fiber-CA3 synapses involves BDNF but is not mediated by p75NTR signaling. Sci. Rep. 11: 1–14, https://doi.org/10.1038/s41598-021-87769-9.Search in Google Scholar PubMed PubMed Central
Gaser, C., Franke, K., Kloppel, S., Koutsouleris, N., and Sauer, H., and Alzheimer’s Disease Neuroimaging Initiative (2013). BrainAGE in mild cognitive impaired patients: predicting the conversion to Alzheimer’s Disease. PLoS One 8: 1–15, https://doi.org/10.1371/journal.pone.0067346.Search in Google Scholar PubMed PubMed Central
Ginsberg, S.D., Malek-Ahmadi, M.H., Alldred, M.J., Che, S., Elarova, I., Chen, Y., Jeanneteau, F., Kranz, T.M., Chao, M.V., Counts, S.E., et al.. (2019). Selective decline of neurotrophin and neurotrophin receptor genes within CA1 pyramidal neurons and hippocampus proper: correlation with cognitive performance and neuropathology in mild cognitive impairment and Alzheimer’s disease. Hippocampus 29: 422–439, https://doi.org/10.1002/hipo.22802.Search in Google Scholar PubMed PubMed Central
Ginsberg, S.D., Malek-Ahmadi, M.H., Alldred, M.J., Chen, Y., Chen, K., Chao, M.V., Counts, S.E., and Mufson, E.J. (2019). Brain-derived neurotrophic factor (BDNF) and TrkB hippocampal gene expression are putative predictors of neuritic plaque and neurofibrillary tangle pathology. Neurobiol. Dis. 132: 1–12, https://doi.org/10.1016/j.nbd.2019.104540.Search in Google Scholar PubMed PubMed Central
Green, R.C., Schneider, L.S., Amato, D.A., Beelen, A.P., Wilcock, G., Swabb, E.A., and Zavitz, K.H., and Tarenflurbil Phase 3 Study Group (2009). Effect of tarenflurbil on cognitive decline and activities of daily living in patients with mild Alzheimer disease: a randomized controlled trial. JAMA 302: 2557–2564, https://doi.org/10.1001/jama.2009.1866.Search in Google Scholar PubMed PubMed Central
Greferath, U., Bennie, A., Kourakis, A., Bartlett, P.F., Murphy, M., and Barrett, G.L. (2000). Enlarged cholinergic forebrain neurons and improved spatial learning in p75 knockout mice. Eur. J. Neurosci. 12: 885–893, https://doi.org/10.1046/j.1460-9568.2000.00976.x.Search in Google Scholar PubMed
Hallbook, F., Ayer-leleivre, C., Ebendal, T., and Persson, H. (1990). Expression of nerve growth factor receptor mRNA during early development of the chicken embryo: emphasis on cranial ganglia. Development 108: 693–704, https://doi.org/10.1242/dev.108.4.693.Search in Google Scholar PubMed
Hattiangady, B., Rao, M.S., Shetty, G.A., and Shetty, A.K. (2005). Brain-derived neurotrophic factor, phosphorylated cyclic AMP response element binding protein and neuropeptide Y decline as early as middle age in the dentate gyrus and CA1 and CA3 subfields of the hippocampus. Exp. Neurol. 195: 353–371, https://doi.org/10.1016/j.expneurol.2005.05.014.Search in Google Scholar PubMed
He, Y., Wei, M., Wu, Y., Qin, H., Li, W., Ma, X., Cheng, J., Ren, J., Shen, Y., Chen, Z., et al.. (2019). Amyloid beta oligomers suppress excitatory transmitter release via presynaptic depletion of phosphatidylinositol-4,5-bisphosphate. Nat. Commun. 10: 1–18, https://doi.org/10.1038/s41467-019-09114-z.Search in Google Scholar PubMed PubMed Central
Hongpaisan, J., Sun, M.K., and Alkon, D.L. (2011). PKC epsilon activation prevents synaptic loss, Abeta elevation, and cognitive deficits in Alzheimer’s disease transgenic mice. J. Neurosci. 31: 630–643, https://doi.org/10.1523/jneurosci.5209-10.2011.Search in Google Scholar
Hongpaisan, J., Xu, C., Sen, A., Nelson, T.J., and Alkon, D.L. (2013). PKC activation during training restores mushroom spine synapses and memory in the aged rat. Neurobiol. Dis. 55: 44–62, https://doi.org/10.1016/j.nbd.2013.03.012.Search in Google Scholar
Impey, S., Fong, A.L., Wang, Y., Cardinaux, J.R., Fass, D.M., Obrietan, K., Wayman, G.A., Storm, D.R., Soderling, T.R., and Goodman, R.H. (2002). Phosphorylation of CBP mediates transcriptiptional activation by neuronal activity and CaM kinase IV. Neuron 34: 235–244, https://doi.org/10.1016/s0896-6273(02)00654-2.Search in Google Scholar
Jiang, L., Kundu, S., Lederman, J.D., Lopez-Hernandez, G.Y., Ballinger, E.C., Wang, S., Talmage, D.A., and Role, L.W. (2016). Cholinergic signaling controls conditioned fear behaviors and enhances plasticity of cortical-amygdala circuits. Neuron 90: 1057–1070, https://doi.org/10.1016/j.neuron.2016.04.028.Search in Google Scholar
Jiao, S.S., Shen, L.L., Zhu, C., Bu, X.L., Liu, Y.H., Liu, C.H., Yao, X.Q., Zhang, L.L., Zhou, H.D., Walker, D.G., et al.. (2016). Brain-derived neurotrophic factor protects against tau-related neurodegeneration of Alzheimer’s disease. Transl. Psychiatry 6: 1–11, https://doi.org/10.1038/tp.2016.186.Search in Google Scholar
Kang, T., Nagase, H., and Pei, D. (2002). Activation of membrane-type matrix metalloproteinase 3 zymogen by the proprotein convertase furin in the trans-golgi network. Cancer Res. 62: 675–681.Search in Google Scholar
Kanning, K.C., Hudson, M., Amieux, P.S., Wiley, J.C., Bothwell, M., and Schecterson, L.C. (2003). Proteolytic processing of p75NTR and two homologs generates C-terminal fragments with signalling capability. J. Neurosci. 23: 5425–5436, https://doi.org/10.1523/jneurosci.23-13-05425.2003.Search in Google Scholar
Katoh-Semba, R., Semba, R., Takeuchi, I.K., and Kato, K. (1998). Age-related changes in levels of brain-derived neurotrophic factor in selected brain regions of rats, normal mice and senescence accelerated mice: a comparison to those of nerve growth factor and neurotrophin-3. Neurosci. Res. 31: 227–234, https://doi.org/10.1016/s0168-0102(98)00040-6.Search in Google Scholar
Kellner, Y., Godecke, N., Dierkes, T., Thieme, N., Zagrebelsky, M., and Korte, M. (2014). The BDNF effects on dendritic spines of mature hippocampal neurons depend on neuronal activity. Front. Synaptic Neurosci. 6: 1–17, https://doi.org/10.3389/fnsyn.2014.00005.Search in Google Scholar PubMed PubMed Central
Lauren, J., Gimbel, D.A., Nygaard, H.B., Gilbert, J.W., and Strittmatter, S.M. (2009). Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 457: 1128–1132, https://doi.org/10.1038/nature07761.Search in Google Scholar PubMed PubMed Central
Lee, J., Fukumoto, H., Orne, J., Klucken, J., Raju, S., Vanderburg, C.R., Irizarry, M.C., Hyman, B.T., and Ingelsson, M. (2005). Decreased levels of BDNF protein in Alzheimer temporal cortex are independent of BDNF polymorphisms. Exp. Neurol. 194: 91–96, https://doi.org/10.1016/j.expneurol.2005.01.026.Search in Google Scholar PubMed
Li, X., Yu, B., Sun, Q., Zhang, Y., Ren, M., Zhang, X., Li, A., Yuan, J., Madisen, L., Luo, Q., et al.. (2018). Generation of a whole-brain atlas for the cholinergic system and mesoscopic projectome analysis of basal forebrain cholinergic neurons. Proc. Natl. Acad. Sci. U.S.A. 115: 415–420, https://doi.org/10.1073/pnas.1703601115.Search in Google Scholar PubMed PubMed Central
Lim, Y., Zhong, J.H., and Zhou, X.F. (2015). Development of mature BDNF-specific sandwich ELISA. J. Neurochem. 134: 75–85, https://doi.org/10.1111/jnc.13108.Search in Google Scholar
Liu, S., Guo, W., Zhou, H., Tang, L., Feng, S., Zhong, J.H., and Zhou, X.F. (2018). proBDNF inhibits the proliferation and migration of OLN93 oligodendrocytes. Mol. Med. Rep. 18: 3809–3817, https://doi.org/10.3892/mmr.2018.9407.Search in Google Scholar
Lou, H., Kim, S.K., Zaitsev, E., Snell, C.R., Lu, B., and Loh, Y.P. (2005). Sorting and activity-dependent secretion of BDNF require interaction of a specific motif with the sorting receptor carboxypeptidase e. Neuron 45: 245–255, https://doi.org/10.1016/j.neuron.2004.12.037.Search in Google Scholar
Maynard, K.R., Hobbs, J.W., Sukumar, M., Kardian, A.S., Jimenez, D.V., Schloesser, R.J., and Martinowich, K. (2017). BDNF mRNA splice variants differentially impact CA1 and CA3 dendrite complexity and spine morphology in the hippocampus. Brain Struct. Funct. 222: 3295–3307, https://doi.org/10.1007/s00429-017-1405-3.Search in Google Scholar
McGaughy, J., Koene, R.A., Eichenbaum, H., and Hasselmo, M.E. (2005). Cholinergic deafferentation of the entorhinal cortex in rats impairs encoding of novel but not familiar stimuli in a delayed nonmatch-to-sample task. J. Neurosci. 25: 10273–10281, https://doi.org/10.1523/jneurosci.2386-05.2005.Search in Google Scholar
Mecca, A.P., Chen, M.K., O’Dell, R.S., Naganawa, M., Toyonaga, T., Godek, T.A., Harris, J.E., Bartlett, H.H., Zhao, W., Nabulsi, N.B., et al.. (2020). In vivo measurement of widespread synaptic loss in Alzheimer’s disease with SV2A PET. Alzheimers Dement 16: 974–982, https://doi.org/10.1002/alz.12097.Search in Google Scholar
Mi, Z., Abrahamson, E.E., Ryu, A.Y., Fish, K.N., Sweet, R.A., Mufson, E.J., and Ikonomovic, M.D. (2017). Loss of precuneus dendritic spines immunopositive for spinophilin is related to cognitive impairment in early Alzheimer’s disease. Neurobiol. Aging 55: 159–166, https://doi.org/10.1016/j.neurobiolaging.2017.01.022.Search in Google Scholar
Minichiello, L., Calella, A.M., Medina, D.L., Bonhoeffer, T., Klein, R., and Korte, M. (2002). Mechanisms of TrkB-mediated hippocampal long-term potentiation. Neuron 36: 121–137, https://doi.org/10.1016/s0896-6273(02)00942-x.Search in Google Scholar
Mizoguchi, H., Nakade, J., Tachibana, M., Ibi, D., Someya, E., Koike, H., Kamei, H., Nabeshima, T., Itohara, S., Takuma, K., et al.. (2011). Matrix metalloproteinase-9 contributes to kindled seizure development in pentylenetetrazole-treated mice by converting pro-BDNF to mature BDNF in the hippocampus. J. Neurosci. 31: 12963–12971, https://doi.org/10.1523/jneurosci.3118-11.2011.Search in Google Scholar
Mizoguchi, Y., Yao, H., Imamura, Y., Hashimoto, M., and Monji, A. (2020). Lower brain-derived neurotrophic factor levels are associated with age-related memory impairment in community-dwelling older adults: the Sefuri study. Sci. Rep. 10: 1–9, https://doi.org/10.1038/s41598-020-73576-1.Search in Google Scholar PubMed PubMed Central
Moreno, H., Yu, E., Pigino, G., Hernandez, A.I., Kim, N., Moreira, J.E., Sugimori, M., and Llinas, R.R. (2009). Synaptic transmission block by presynaptic injection of oligomeric amyloid beta. Proc. Natl. Acad. Sci. U.S.A. 106: 5901–5906, https://doi.org/10.1073/pnas.0900944106.Search in Google Scholar
Mori, Y., Tsuji, M., Oguchi, T., Kasuga, K., Kimura, A., Futamura, A., Sugimoto, A., Kasai, H., Kuroda, T., Yano, S., et al.. (2021). Serum BDNF as a potential biomarker of Alzheimer’s Disease: verification through assessment of serum, cerebrospinal fluid, and medial temporal lobe atrophy. Front. Neurol. 12: 1–8, https://doi.org/10.3389/fneur.2021.653267.Search in Google Scholar
Moro, A., van Woerden, G.M., Toonen, R.F., and Verhage, M. (2020). CaMKII controls neuromodulation via neuropeptide gene expression and axonal targeting of neuropeptide vesicles. PLoS Biol. 18: 1–30, https://doi.org/10.1371/journal.pbio.3000826.Search in Google Scholar
Mowla, S.J., Farhadi, H.F., Pareek, S., Atwal, J.K., Morris, S.J., Seidah, N.G., and Murphy, R.A. (2001). Biosynthesis and post-translational processing of the precursor to brain-derived neurotrophic factor. J. Biol. Chem. 276: 12660–12666, https://doi.org/10.1074/jbc.m008104200.Search in Google Scholar
Murray, S.S., Perez, P., Lee, R., Hempstead, B.L., and Chao, M.V. (2004). A novel p75 neurotrophin receptor-related protein, NRH2, regulates nerve growth factor binding to the TrkA receptor. J. Neurosci. 24: 2742–2749, https://doi.org/10.1523/jneurosci.3960-03.2004.Search in Google Scholar
Nagahara, A.H., Merrill, D.A., Coppola, G., Tsukada, S., Schroeder, B.E., Shaked, G.M., Wang, L., Blesch, A., Kim, A., Conner, J.M., et al.. (2009). Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer’s disease. Nat. Med. 15: 331–337, https://doi.org/10.1038/nm.1912.Search in Google Scholar
Nagappan, G., Zaitsev, E., Senatorov, J., V.V., Yang, J., Hempstead, B.L., and Lu, B. (2009). Control of extracellular cleavage of proBDNF by high frequency neuronal activity. Proc. Natl. Acad. Sci. U.S.A. 106: 1267–1272, https://doi.org/10.1073/pnas.0807322106.Search in Google Scholar
Narisawa-Saito, M., Wakabayashi, K., Tsuji, S., Takahashi, H., and Nawa, H. (1996). Regional specificty of alterations in NGF, BDNF and NT-3 levels in Alzheimer’s disease. Neuroreport 7: 2925–2928, https://doi.org/10.1097/00001756-199611250-00024.Search in Google Scholar
Naumann, T., Casademunt, E., Hollerbach, E., Hofmann, J., Dechant, G., Frotscher, M., and Barde, Y.A. (2002). Complete deletion of the neurotrophin receptor p75NTR leads to long-lasting increases in the number of basal forebrain cholinergic neurons. J. Neurosci. 22: 2409–2418, https://doi.org/10.1523/jneurosci.22-07-02409.2002.Search in Google Scholar
Nichols, E., Szoeke, C.E.I., Vollset, S.E., Abbasi, N., Abd-Allah, F., Abdela, J., Aichour, M.T.E., Akinyemi, R.O., Alahdab, F., Asgedom, S.W., et al.. (2019). Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 18: 88–106, https://doi.org/10.1016/S1474-4422(18)30403-4.Search in Google Scholar
Niculescu, D., Michaelsen-Preusse, K., Guner, U., van Dorland, R., Wierenga, C.J., and Lohmann, C. (2018). A BDNF-mediated push-pull plasticity mechanism for synaptic clustering. Cell Rep. 24: 2063–2074, https://doi.org/10.1016/j.celrep.2018.07.073.Search in Google Scholar PubMed
Nykjaer, A. and Willnow, T.E. (2012). Sortilin: a receptor to regulate neuronal viability and function. Trends Neurosci. 35: 261–270, https://doi.org/10.1016/j.tins.2012.01.003.Search in Google Scholar PubMed
Obiang, P., Maubert, E., Bardou, I., Nicole, O., Launay, S., Bezin, L., Vivien, D., and Agin, V. (2011). Enriched housing reverses age-associated impairment of cognitive functions and tPA-dependent maturation of BDNF. Neurobiol. Learn. Mem. 96: 121–129, https://doi.org/10.1016/j.nlm.2011.03.004.Search in Google Scholar PubMed
Overton, M., Pihlsgard, M., and Elmstahl, S. (2019). Diagnostic stability of mild cognitive impairment, and predictors of reversion to normal cognitive functioning. Dement. Geriatr. Cogn. Disord. 48: 317–329, https://doi.org/10.1159/000506255.Search in Google Scholar PubMed
Palomer, E., Carretero, J., Benvegnu, S., Dotti, C.G., and Martin, M.G. (2016). Neuronal activity controls BDNF expression via Polycomb de-repression and CREB/CBP/JMJD3 activation in mature neurons. Nat. Commun. 7: 1–12, https://doi.org/10.1038/ncomms11081.Search in Google Scholar PubMed PubMed Central
Palomer, E., Martin-Segura, A., Baliyan, S., Ahmed, T., Balschun, D., Venero, C., Martin, M.G., and Dotti, C.G. (2016). Aging triggers a repressive chromatin state at BDNF promoters in hippocampal neurons. Cell Rep. 16: 2889–2900, https://doi.org/10.1016/j.celrep.2016.08.028.Search in Google Scholar PubMed
Pang, P.T., Nagappan, G., Guo, W., and Lu, B. (2016). Extracellular and intracellular cleavages of proBDNF required at two distinct stages of late-phase LTP. NPJ Sci. Learn. 1: 1–10, https://doi.org/10.1038/npjscilearn.2016.3.Search in Google Scholar PubMed PubMed Central
Perovic, M., Tesic, V., Mladenovic Djordjevic, A., Smiljanic, K., Loncarevic-Vasiljkovic, N., Ruzdijic, S., and Kanazir, S. (2013). BDNF transcripts, proBDNF and proNGF, in the cortex and hippocampus throughout the life span of the rat. Age 35: 2057–2070, https://doi.org/10.1007/s11357-012-9495-6.Search in Google Scholar PubMed PubMed Central
Petkova-Tuffy, A., Godecke, N., Viotti, J., Korte, M., and Dresbach, T. (2021). Neuroligin-1 mediates presynaptic maturation through brain-derived neurotrophic factor signaling. BMC Biol. 19: 1–20, https://doi.org/10.1186/s12915-021-01145-7.Search in Google Scholar PubMed PubMed Central
Pinto, L., Goard, M.J., Estandian, D., Xu, M., Kwan, A.C., Lee, S.H., Harrison, T.C., Feng, G., and Dan, Y. (2013). Fast modulation of visual perception by basal forebrain cholinergic neurons. Nat. Neurosci. 16: 1857–1863, https://doi.org/10.1038/nn.3552.Search in Google Scholar PubMed PubMed Central
Polacchini, A., Metelli, G., Francavilla, R., Baj, G., Florean, M., Mascaretti, L.G., and Tongiorgi, E. (2015). A method for reproducible measurements of serum BDNF: comparison of the performance of six commercial assays. Sci. Rep. 5: 1–10, https://doi.org/10.1038/srep17989.Search in Google Scholar PubMed PubMed Central
Poser, R., Dokter, M., von Bohlen Und Halbach, V., Berger, S.M., Busch, R., Baldus, M., Unsicker, K., and von Bohlen Und Halbach, O. (2015). Impact of a deletion of the full-length and short isoform of p75NTR on cholinergic innervation and the population of postmitotic doublecortin positive cells in the dentate gyrus. Front. Neuroanat. 9: 1–11, https://doi.org/10.3389/fnana.2015.00063.Search in Google Scholar PubMed PubMed Central
Proctor, D.T., Coulson, E.J., and Dodd, P.R. (2010). Reduction in post-synaptic scaffolding PSD-95 and SAP-102 protein levels in the Alzheimer inferior temporal cortex is correlated with disease pathology. J. Alzheimers Dis. 21: 795–811, https://doi.org/10.3233/jad-2010-100090.Search in Google Scholar
Pruunsild, P., Kazantseva, A., Aid, T., Palm, K., and Timmusk, T. (2007). Dissecting the human BDNF locus: bidirectional transcription, complex splicing, and multiple promoters. Genomics 90: 397–406, https://doi.org/10.1016/j.ygeno.2007.05.004.Search in Google Scholar PubMed PubMed Central
Rattenholl, A., Ruoppolo, M., Flagiello, A., Monti, M., Vinci, F., Marino, G., Lilie, H., Schwarz, E., and Rudolph, R. (2001). Pro-sequence assisted folding and disulfide bond formation of human nerve growth factor. J. Mol. Biol. 305: 523–533, https://doi.org/10.1006/jmbi.2000.4295.Search in Google Scholar PubMed
Rauskolb, S., Zagrebelsky, M., Dreznjak, A., Deogracias, R., Matsumoto, T., Wiese, S., Erne, B., Sendtner, M., Schaeren-Wiemers, N., Korte, M., et al.. (2010). Global deprivation of brain-derived neurotrophic factor in the CNS reveals an area-specific requirement for dendritic growth. J. Neurosci. 30: 1739–1749, https://doi.org/10.1523/jneurosci.5100-09.2010.Search in Google Scholar PubMed PubMed Central
Rauti, R., Cellot, G., D’Andrea, P., Colliva, A., Scaini, D., Tongiorgi, E., and Ballerini, L. (2020). BDNF impact on synaptic dynamics: extra or intracellular long-term release differently regulates cultured hippocampal synapses. Mol. Brain 13: 1–16, https://doi.org/10.1186/s13041-020-00582-9.Search in Google Scholar PubMed PubMed Central
Reese, L.C., Laezza, F., Woltjer, R., and Taglialatela, G. (2011). Dysregulated phosphorylation of Ca(2+)/calmodulin-dependent protein kinase II-alpha in the hippocampus of subjects with mild cognitive impairment and Alzheimer’s disease. J. Neurochem. 119: 791–804, https://doi.org/10.1111/j.1471-4159.2011.07447.x.Search in Google Scholar PubMed PubMed Central
Roberson, E.D., English, J.D., Adams, J.A., Selcher, J.C., Kondratick, C., and Sweatt, J.D. (1999). The mitogen activated protein kinase cascade couples PKA and PKC to cAMP-response element binding protein phosphorylation in area CA1 in hippocampus. J. Neurosci. 19: 4337–4348, https://doi.org/10.1523/jneurosci.19-11-04337.1999.Search in Google Scholar
Salloway, S., Sperling, R., Fox, N.C., Blennow, K., Klunk, W., Raskind, M., Sabbagh, M., Honig, L.S., Porsteinsson, A.P., Ferris, S., et al.. (2014). Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 370: 322–333, https://doi.org/10.1056/nejmoa1304839.Search in Google Scholar PubMed PubMed Central
Scheff, S.W., Price, D.A., Schmitt, F.A., DeKosky, S.T., and Mufson, E.J. (2007). Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment. Neurology 68: 1501–1508, https://doi.org/10.1212/01.wnl.0000260698.46517.8f.Search in Google Scholar PubMed
Schmitz, T.W., and Spreng, N.R., and Alzheimer’s Disease Neuroimaging Initiative (2016). Basal forebrain degeneration precedes and predicts the cortical spread of Alzheimer’s pathology. Nat. Commun. 7: 1–14, https://doi.org/10.1038/ncomms13249.Search in Google Scholar PubMed PubMed Central
Schulte-Herbruggen, O., Eckart, S., Deicke, U., Kuhl, A., Otten, U., Danker-Hopfe, H., Abramowski, D., Staufenbiel, M., and Hellweg, R. (2008). Age-dependent time course of cerebral brain-derived neurotrophic factor, nerve growth factor, and neurotrophin-3 in APP23 transgenic mice. J. Neurosci. Res. 86: 2774–2783, https://doi.org/10.1002/jnr.21704.Search in Google Scholar PubMed
Seidah, N.G., Benjannet, S., Pareek, S., Savaria, D., Hamelin, J., Goulet, B., Lalibert, J., Lazure, C., Chretien, M., and Murphy, R.A. (1996). Cellular processing of the nerve growth factor precursor by the mammalian pro-protein convertases. Biochem. J. 314: 951–960, https://doi.org/10.1042/bj3140951.Search in Google Scholar PubMed PubMed Central
Shankar, G.M., Bloodgood, B.L., Townsend, M., Walsh, D.M., Selkoe, D.J., and Sabatini, B.L. (2007). Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J. Neurosci. 27: 2866–2875, https://doi.org/10.1523/jneurosci.4970-06.2007.Search in Google Scholar
Shen, L.L., Li, W.W., Xu, Y.L., Gao, S.H., Xu, M.Y., Bu, X.L., Liu, Y.H., Wang, J., Zhu, J., Zeng, F., et al.. (2019). Neurotrophin receptor p75 mediates amyloid beta-induced tau pathology. Neurobiol. Dis. 132: 1–12, https://doi.org/10.1016/j.nbd.2019.104567.Search in Google Scholar PubMed
Silhol, M., Bonnichon, V., Rage, F., and Tapia-Arancibia, L. (2005). Age-related changes in brain-derived neurotrophic factor and tyrosine kinase receptor isoforms in the hippocampus and hypothalamus in male rats. Neuroscience 132: 613–624, https://doi.org/10.1016/j.neuroscience.2005.01.008.Search in Google Scholar PubMed
Simmons, D.A., Knowles, J.K., Belichenko, N.P., Banerjee, G., Finkle, C., Massa, S.M., and Longo, F.M. (2014). A small molecule p75NTR ligand, LM11A-31, reverses cholinergic neurite dystrophy in Alzheimer’s disease mouse models with mid- to late-stage disease progression. PLoS One 9: 1–12, https://doi.org/10.1371/journal.pone.0102136.Search in Google Scholar PubMed PubMed Central
Singer, W., Manthey, M., Panford-Walsh, R., Matt, L., Geisler, H.S., Passeri, E., Baj, G., Tongiorgi, E., Leal, G., Duarte, C.B., et al.. (2018). BDNF-live-exon-visualization (BLEV) allows differential detection of BDNF transcripts in vitro and in vivo. Front. Mol. Neurosci. 11: 1–19, https://doi.org/10.3389/fnmol.2018.00325.Search in Google Scholar PubMed PubMed Central
Smiljanic, K., Pesic, V., Mladenovic Djordjevic, A., Pavkovic, Z., Brkic, M., Ruzdijic, S., and Kanazir, S. (2015). Long-term dietary restriction differentially affects the expression of BDNF and its receptors in the cortex and hippocampus of middle-aged and aged male rats. Biogerontology 16: 71–83, https://doi.org/10.1007/s10522-014-9537-9.Search in Google Scholar PubMed
Springer, J.E., Robbins, E., Meyer, S., Baldino, F., and Lewis, M.E. (1990). Localization of nerve growth factor receptor mRNA in the rat basal forebrain with in situ hybridization histochemistry. Cell. Mol. Neurobiol. 10: 33–39, https://doi.org/10.1007/bf00733633.Search in Google Scholar PubMed
Stevenson, T.K. and Lawrence, D.A. (2018). Characterization of tissue plasminogen activator expression and trafficking in the adult murine brain. eNeuro 5: 1–18, https://doi.org/10.1523/ENEURO.0119-18.2018.Search in Google Scholar PubMed PubMed Central
Sun, Y., Lim, Y., Li, F., Liu, S., Lu, J.J., Haberberger, R., Zhong, J.H., and Zhou, X.F. (2012). ProBDNF collapses neurite outgrowth of primary neurons by activating RhoA. PLoS One 7: 1–12, https://doi.org/10.1371/journal.pone.0035883.Search in Google Scholar PubMed PubMed Central
Sze, C.I., Troncoso, J.C., Kawas, K., Mouton, P., Price, D.L., and Martin, L.J. (1997). Loss of the presynaptic vesicle protein synaptophysin in hippocampus correlates with cognitive decline in AD. J. Neuropathol. Exp. Neurol. 56: 933–944, https://doi.org/10.1097/00005072-199708000-00011.Search in Google Scholar PubMed
Tuvikene, J., Esvald, E.E., Rahni, A., Uustalu, K., Zhuravskaya, A., Avarlaid, A., Makeyev, E.V., and Timmusk, T. (2021). Intronic enhancer region governs transcript-specific Bdnf expression in rodent neurons. Elife 10: 1–28, https://doi.org/10.7554/elife.65161.Search in Google Scholar
Veldsman, M., Nobis, L., Alfaro-Almagro, F., Manohar, S., and Husain, M. (2021). The human hippocampus and its subfield volumes across age, sex and APOE e4 status. Brain Commun. 3: 1–12, https://doi.org/10.1093/braincomms/fcaa219.Search in Google Scholar PubMed PubMed Central
von Bohlen und Halbach, O., Minichiello, L., and Unsicker, K. (2003). Haploinsufficiency in trkB and/or trkC neurotrophin receptors causes structural alterations in the aged hippocampus and amygdala. Eur. J. Neurosci. 18: 2319–2325, https://doi.org/10.1046/j.1460-9568.2003.02953.x.Search in Google Scholar PubMed
von Schack, D., Casademunt, E., Schweigreiter, R., Meyer, M., Bibel, M., and Dechant, G. (2001). Complete ablation of the neurotrophin receptor p75NTR causes defects both in the nervous and the vascular system. Nat. Neurosci. 4: 977–978, https://doi.org/10.1038/nn730.Search in Google Scholar PubMed
Wang, Q.H., Wang, Y.R., Zhang, T., Jiao, S.S., Liu, Y.H., Zeng, F., Li, J., Yao, X.Q., Zhou, H.D., Zhou, X.F., et al.. (2016). Intramuscular delivery of p75NTR ectodomain by an AAV vector attenuates cognitive deficits and Alzheimer’s disease-like pathologies in APP/PS1 transgenic mice. J. Neurochem. 138: 163–173, https://doi.org/10.1111/jnc.13616.Search in Google Scholar PubMed
Wang, X., Marks, C.R., Perfitt, T.L., Nakagawa, T., Lee, A., Jacobson, D.A., and Colbran, R.J. (2017). A novel mechanism for Ca(2+)/calmodulin-dependent protein kinase II targeting to L-type Ca(2+) channels that initiates long-range signaling to the nucleus. J. Biol. Chem. 292: 17324–17336, https://doi.org/10.1074/jbc.m117.788331.Search in Google Scholar
Wattmo, C., Minthon, L., and Wallin, A.K. (2016). Mild versus moderate stages of Alzheimer’s disease: three-year outcomes in a routine clinical setting of cholinesterase inhibitor therapy. Alzheimer’s Res. Ther. 8: 1–15, https://doi.org/10.1186/s13195-016-0174-1.Search in Google Scholar PubMed PubMed Central
Weinstein, G., Beiser, A.S., Choi, S.H., Preis, S.R., Chen, T.C., Vorgas, D., Au, R., Pikula, A., Wolf, P.A., DeStefano, A.L., et al.. (2014). Serum brain-derived neurotrophic factor and the risk for dementia: the Framingham Heart Study. JAMA Neurol. 71: 55–61, https://doi.org/10.1001/jamaneurol.2013.4781.Search in Google Scholar PubMed PubMed Central
Wetsel, W.C., Rodriguiz, R.M., Guillemot, J., Rousselet, E., Essalmani, R., Kim, I.H., Bryant, J.C., Marcinkiewicz, J., Desjardins, R., Day, R., et al.. (2013). Disruption of the expression of the proprotein convertase PC7 reduces BDNF production and affects learning and memory in mice. Proc. Natl. Acad. Sci. U.S.A. 110: 17362–17367, https://doi.org/10.1073/pnas.1314698110.Search in Google Scholar PubMed PubMed Central
Wong, L.W., Chong, Y.S., Lin, W., Kisiswa, L., Sim, E., Ibanez, C.F., and Sajikumar, S. (2021). Age-related changes in hippocampal-dependent synaptic plasticity and memory mediated by p75 neurotrophin receptor. Aging Cell 20: 1–21, https://doi.org/10.1111/acel.13305.Search in Google Scholar PubMed PubMed Central
Xie, Y., Meeker, R.B., Massa, S.M., and Longo, F.M. (2019). Modulation of the p75 neurotrophin receptor suppresses age-related basal forebrain cholinergic neuron degeneration. Sci. Rep. 9: 1–13, https://doi.org/10.1038/s41598-019-41654-8.Search in Google Scholar PubMed PubMed Central
Yang, J., Harte-Hargrove, L.C., Siao, C.J., Marinic, T., Clarke, R., Ma, Q., Jing, D., Lafrancois, J.J., Bath, K.G., Mark, W., et al.. (2014). proBDNF negatively regulates neuronal remodeling, synaptic transmission, and synaptic plasticity in hippocampus. Cell Rep. 7: 796–806, https://doi.org/10.1016/j.celrep.2014.03.040.Search in Google Scholar PubMed PubMed Central
Yang, M., Lim, Y., Li, X., Zhong, J.H., and Zhou, X.F. (2011). Precursor of brain-derived neurotrophic factor (proBDNF) forms a complex with Huntingtin-associated protein-1 (HAP1) and sortilin that modulates proBDNF trafficking, degradation, and processing. J. Biol. Chem. 286: 16272–16284, https://doi.org/10.1074/jbc.m110.195347.Search in Google Scholar
Yang, T., Tran, K.C., Zeng, A.Y., Massa, S.M., and Longo, F.M. (2020). Small molecule modulation of the p75 neurotrophin receptor inhibits multiple amyloid beta-induced tau pathologies. Sci. Rep. 10: 1–17, https://doi.org/10.1038/s41598-020-77210-y.Search in Google Scholar PubMed PubMed Central
Yao, X.Q., Jiao, S.S., Saadipour, K., Zeng, F., Wang, Q.H., Zhu, C., Shen, L.L., Zeng, G.H., Liang, C.R., Wang, J., et al.. (2015). p75NTR ectodomain is a physiological neuroprotective molecule against amyloid-beta toxicity in the brain of Alzheimer’s disease. Mol. Psychiatry 20: 1301–1310, https://doi.org/10.1038/mp.2015.49.Search in Google Scholar PubMed PubMed Central
Yeo, T.T., Chua-Couzens, J., Butcher, L.L., Bredesen, D.E., Cooper, J.D., Valletta, J.S., Mobley, W.C., and Longo, F.M. (1997). Absence of p75NTR causes increased basal forebrain cholinergic neuron size, choline acetyltransferase activity, and target innervation. J. Neurosci. 17: 7594–7605, https://doi.org/10.1523/jneurosci.17-20-07594.1997.Search in Google Scholar
Zeng, F., Lu, J.J., Zhou, X.F., and Wang, Y.J. (2011). Roles of p75NTR in the pathogenesis of Alzheimer’s disease: a novel therapeutic target. Biochem. Pharmacol. 82: 1500–1509, https://doi.org/10.1016/j.bcp.2011.06.040.Search in Google Scholar PubMed
Zierold, S., Buschmann, K., Gachkar, S., Bochenek, M.L., Velmeden, D., Hobohm, L., Vahl, C.F., and Schafer, K. (2021). Brain-derived neurotrophic factor expression and signaling in different perivascular adipose tissue depots of patients with coronary artery disease. J. Am. Heart Assoc. 10: 1–19, https://doi.org/10.1161/jaha.120.018322.Search in Google Scholar
© 2022 Walter de Gruyter GmbH, Berlin/Boston
Articles in the same Issue
- Frontmatter
- Second near-infrared (NIR-II) imaging: a novel diagnostic technique for brain diseases
- Contralateral C7 nerve transfer in the treatment of upper-extremity paralysis: a review of anatomical basis, surgical approaches, and neurobiological mechanisms
- The role of brain-derived neurotrophic factor and the neurotrophin receptor p75NTR in age-related brain atrophy and the transition to Alzheimer’s disease
- Reproducibility of developmental neuroplasticity in in vitro brain tissue models
- Exercise to spot the differences: a framework for the effect of exercise on hippocampal pattern separation in humans
Articles in the same Issue
- Frontmatter
- Second near-infrared (NIR-II) imaging: a novel diagnostic technique for brain diseases
- Contralateral C7 nerve transfer in the treatment of upper-extremity paralysis: a review of anatomical basis, surgical approaches, and neurobiological mechanisms
- The role of brain-derived neurotrophic factor and the neurotrophin receptor p75NTR in age-related brain atrophy and the transition to Alzheimer’s disease
- Reproducibility of developmental neuroplasticity in in vitro brain tissue models
- Exercise to spot the differences: a framework for the effect of exercise on hippocampal pattern separation in humans