The quest for magic: recent advances in C(sp3)–H methylation
-
Kaibo Feng



Abstract
Frequently referred to as the “magic methyl” effect, the introduction of a methyl group into a biologically active molecule has the potential to drastically alter its physical and biological properties and significantly increase potency. This effect is most pronounced when the methyl group is added at the α-position of an aliphatic heterocycle or ortho to a large rotatable group on an aromatic ring. Although seminal developments in C–H activation strategies offered solutions to the latter, until recent years there had been no selective and functional-group-tolerant method for C(sp3)–H methylation at late stages of synthesis. For many years, the lack of a generally applicable methylation strategy necessitated arduous de novo synthesis approaches to access methylated drug candidates, and discouraged further investigation and understandings of the magic methyl effect. This review will provide a summary of the most recent advances that enabled non-directed late-stage C(sp3)–H methylation, including through hydride transfer, chemical or anodic oxidation, and photocatalytic hydrogen atom transfer.
Introduction
Found in more than 67 % of top-selling small-molecule drugs, methyl groups bound to carbon atoms are one of the most prevalent fragments in biologically active molecules [1]. A particularly fascinating feature of the methyl group is that its incorporation has the potential to dramatically improve biological activities, such as increasing potency of a lead compound, even up to over 2000 folds at times [1], [2], [3], [4], [5]. This phenomenon is frequently referred to as the “magic methyl” effect. Several factors that likely contribute to this potency boost include methyl-induced changes in conformation and metabolic stability, and increases in solubility and binding affinity [1, 6], [7], [8], [9], [10]. Accordingly, the most profound magic methyl effect is often observed when a C–Me replaces a C–H at a site α to a heteroatom in a saturated heterocycle, or ortho to a large rotatable substituent on an aromatic ring [1]. Despite these potential alterations in a molecule’s behavior, the addition of a methyl group does not significantly impact a molecule’s polarity, lipophilicity (ΔclogP ∼ 0.5), or molecular weight. These characteristics make methylation a highly desirable approach to the optimization and diversification of drug leads.
Although seminal advances in C–H activation over the past decade innovated diverse and effective strategies to methylate C(sp2)–H bonds and alkylate C(sp3)–H bonds [1, 8], [9], [10], [11], [12], [13], [14], [15], [16], [17], [18], [19], [20], [21], until very recently there had been no general solution to non-directed methylation of C(sp3)–H bonds, especially where such a method is most needed for drug-like molecules with dense functional groups. Consequently, the methylated analogues of interest were often obtained through lengthy and low-yielding de novo synthesis using methyl-containing starting materials (Fig. 1) [3], [4], [5, 22, 23]. Whereas the installation of methyl groups has the potential to result in a potency increase, such an effect is not guaranteed and only around 8 % of methyl introductions led to a boost of potency more than 10 folds, while decrease of potency is also possible [6]. Without a general method to rapidly access these analogues, investigations of these analogues and their magic methyl effects are often neglected. As underscored in a 2013 perspective by Cernak [1], these limitations necessitated the development of generally applicable methylation approaches, especially for the late stages of synthesis, to facilitate the exploration and better understanding of the magic methyl effect. With recent developments and especially since the first report of a general late-stage methylation method in 2020 [24], several powerful strategies began to emerge in this field. This review will describe the recent advances in non-directed C(sp3)–H methylation methods that encompass hydride transfer, oxidative methylation, and photocatalytic hydrogen atom transfer, with an emphasis on their late-stage applications.
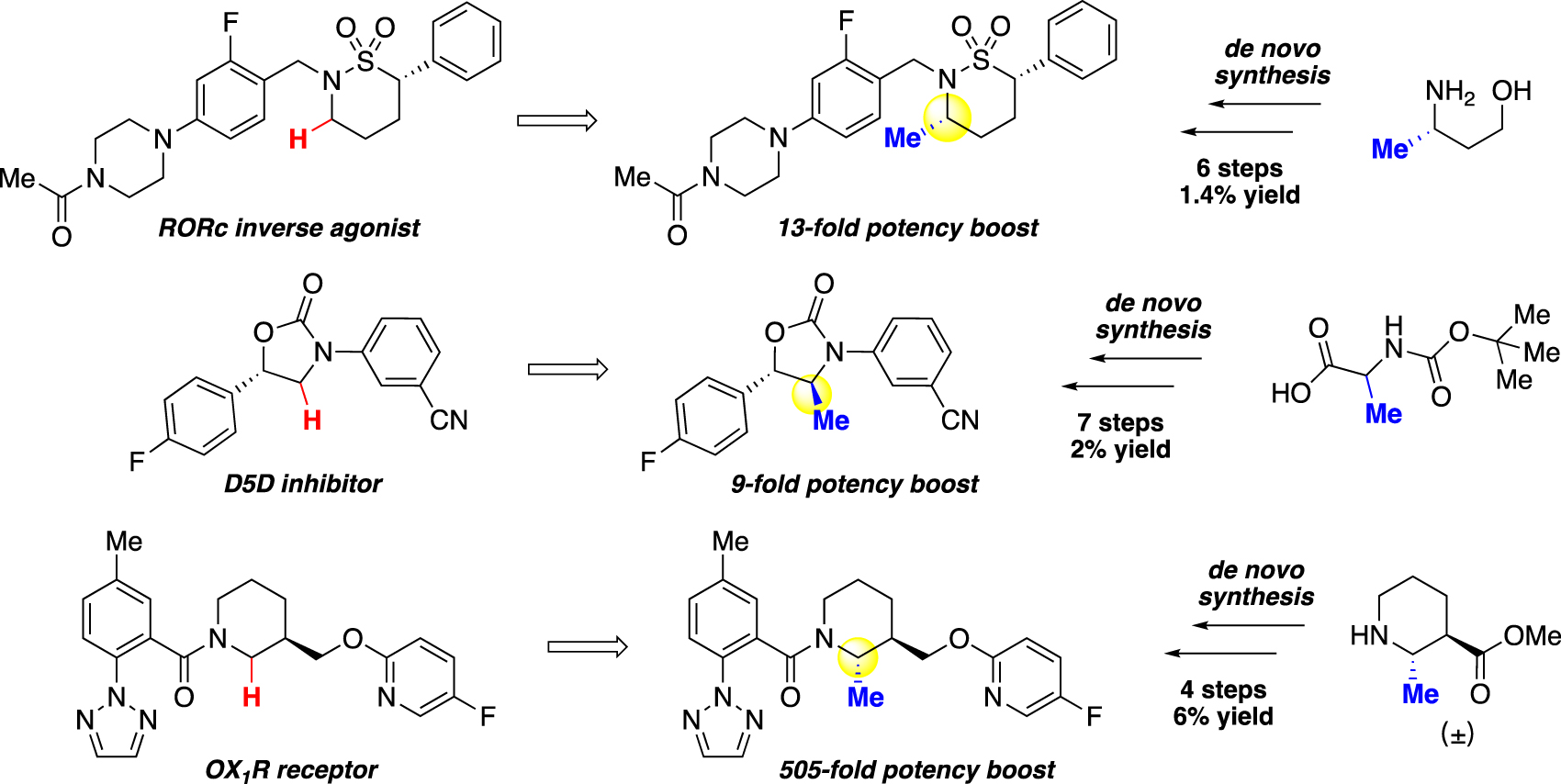
Introduction of methyl groups can lead to substantial increase in potency, but this modification often requires de novo synthesis to access.
Early developments with organolithium reagents
Developed by Beak and coworkers, a traditional approach to introduce alkyl groups on an aliphatic framework relies on deprotonation using a strong base (e.g., sBuLi), followed by reaction with an alkyl electrophile (Fig. 2) [11, 25], [26], [27], [28]. Using dimethyl sulfate, this strategy can be applied to install methyl groups, and produces methylated heterocycles in good yields. By adding a chiral diamine, (–)-sparteine, the methylated product can be obtained in excellent enantioselectivity. However, due to the strong basicity and nucleophilicity of sBuLi, only unfunctionalized azaheterocycles were methylated.
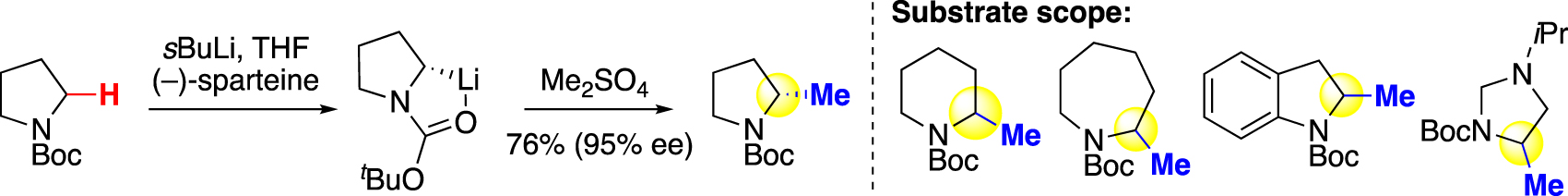
Methylation via deprotonation (Beak).
Whereas the α-deprotonation method requires a Boc protecting group, in 2019 Seidel and coworkers reported a hydride-transfer-based approach that directly α-functionalizes unprotected cyclic secondary amines [18]. Instead of directly deprotonating the α-C–H, the more acidic amine N–H was deprotonated using nBuLi and the resulting lithium amide was proposed to go through a 6-membered transition state to transfer hydride to a receptor ketone and produce a cyclic imine. This imine was subsequently activated by a Lewis acid (BF3•OEt, TMSOTf) and attacked by an organolithium or Grignard nucleophile to produce the α-functionalized product (Fig. 3a). A wide range of alkyl and aryl groups can be installed on aliphatic N-heterocycles including an intermediate to the commercial drug risperidone. While the majority of this work focused on installing longer alkyl chains and aryl functional groups, one methylation example was demonstrated on azacyclotridecane using MeMgBr and TMSOTf. Seidel and coworkers later expanded this method to enable the functionalization of multiple C–H bonds on the cyclic secondary amine, including one example of methylation on the β-position prior to arylating the α-site of the same piperidine substrate (Fig. 3b) [29]. While not demonstrated on this methylation example, several other β-functionalized imines were reduced by sodium borohydride to render β-monofunctionlized piperidine, showing potential for β-methylation using this strategy.
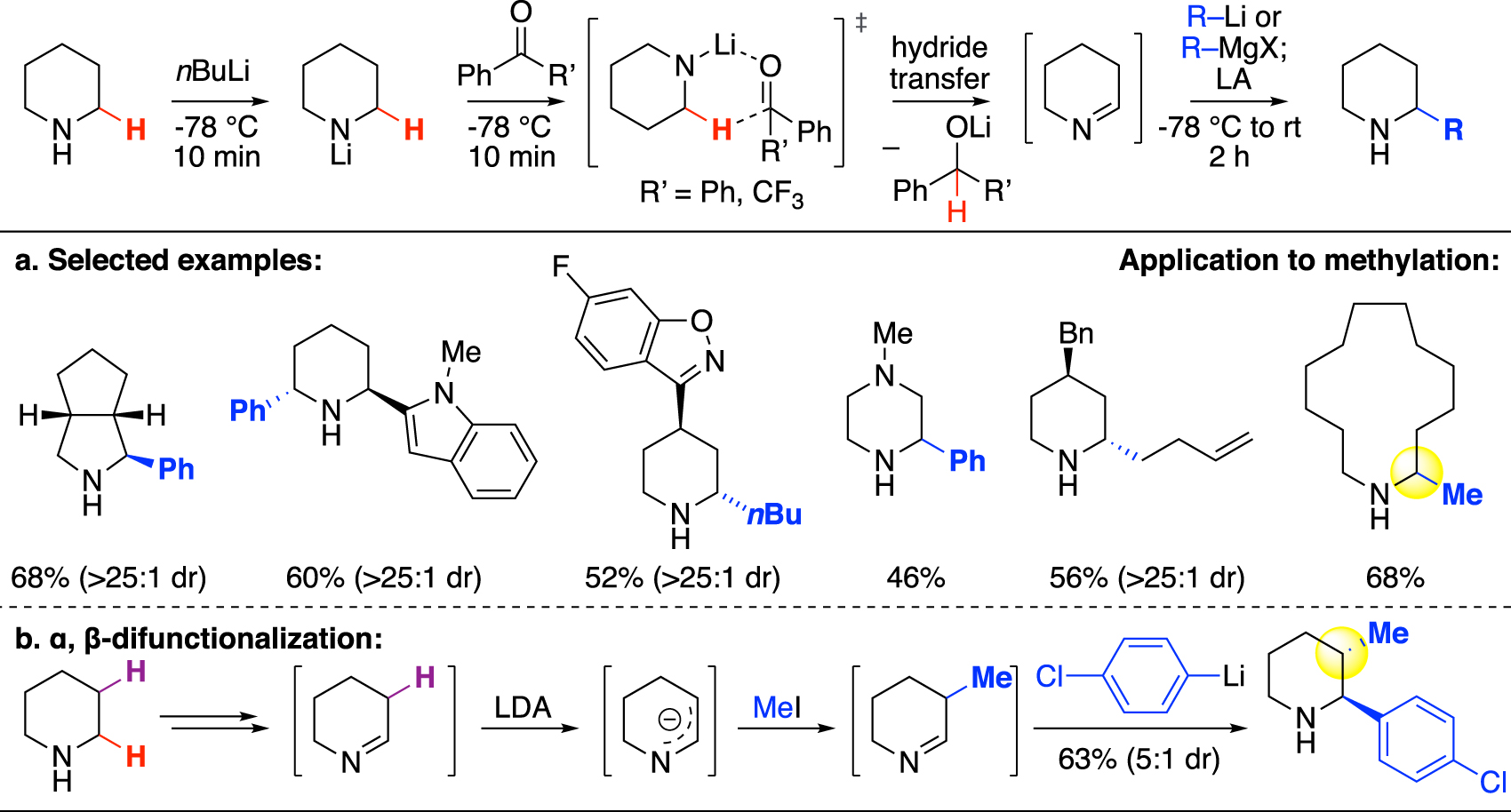
C(sp3)–H functionalization via hydride transfer and application in methylation (Seidel).
In situ imine formation was also exploited for modifying α-branched primary amines. Dixon and coworkers reported in 2019 a quinone-mediated α-functionalization strategy for this type of amine [30]. While this work was mainly focused on installing allyl and aryl groups, a single methylation example was included, in which methyllithium was employed as nucleophile to attack the imine intermediate formed via 1,5-hydrogen shift (Fig. 4).
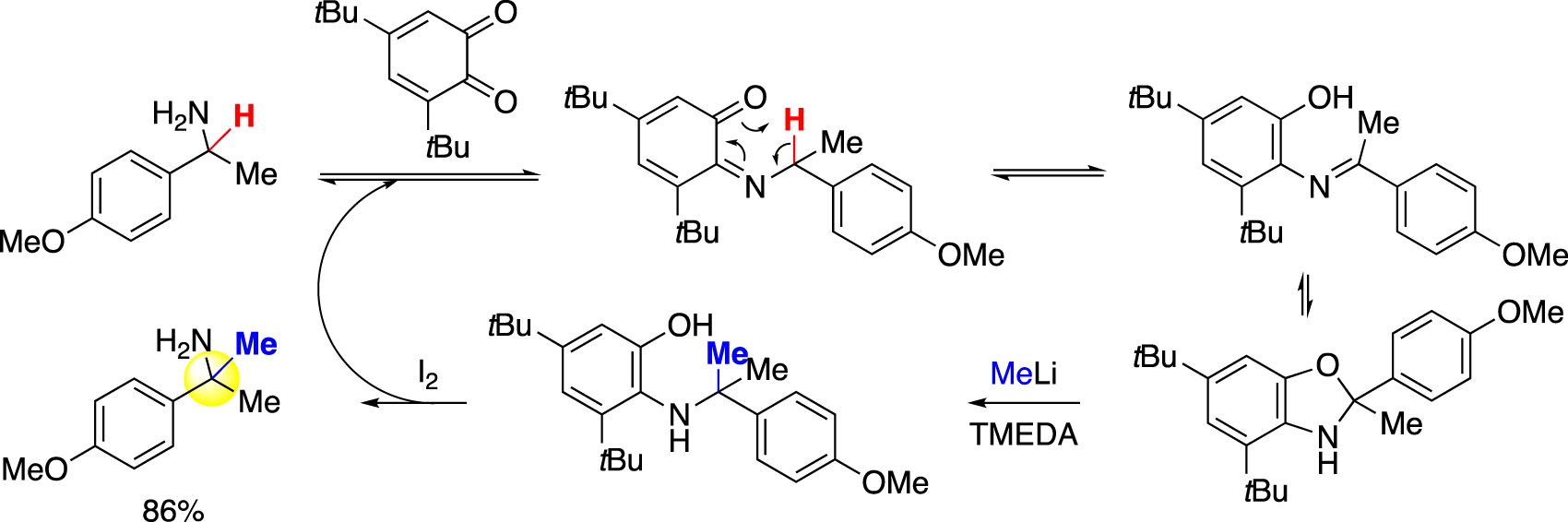
Quinone-mediated α-methylation of α-branched primary amine (Dixon).
Oxidative methylation
The prevalence of acidic or electrophilic groups (e.g., ketone, ester, nitrile, peptides, heteroarenes) in structurally complex bioactive molecules [31] and natural products [32] calls for the development of site- and chemoselective methylation methods with greater functional group tolerance. Additionally, although in general it is highly preferable that late-stage modifications directly lead to the desired product, a unique challenge arises when a methyl group is added. The small size and electron neutrality of the methyl group often cause the product to be inseparable from the unreacted starting material. Isolation difficulties were recorded even for simple azacycles [20], and are likely to be more prevalent in complex molecules where the introduction of a methyl group effects less change on a molecule’s polarity.
In contrast to the traditional metalation strategies, C–H oxidation, including at positions α to heteroatoms, has been demonstrated in far greater substrate scopes, selectivity, and functional group tolerance [23, 33], [34], [35], [36], [37], [38], [39]. An oxidative methylation approach that transforms the resulting oxidized intermediate to the corresponding methylated product can therefore be done with much higher site- and chemoselectivity while circumventing the need to use a strong base [18, 25], [26], [27], [28], [29] or a directing group [17]. Additionally, the formation of a more polar intermediate offers a solution to the product isolation challenge [24]. However, the traditional products of such oxidations are overoxidized amides or esters, given the intermediary hydroxyl group’s strong hyperconjugative activation. Methylation of these intermediates would require undesirable carbonyl reductions detrimental to unsaturated functional groups. Early reports of oxidative functionalization frequently rely on strong oxidants and substrate-controlled selectivity and are thus often limited to functionalizing the privileged tetrahydroisoquinoline or its analogues [11], with only a few examples on methylation (Fig. 5) [40, 41].
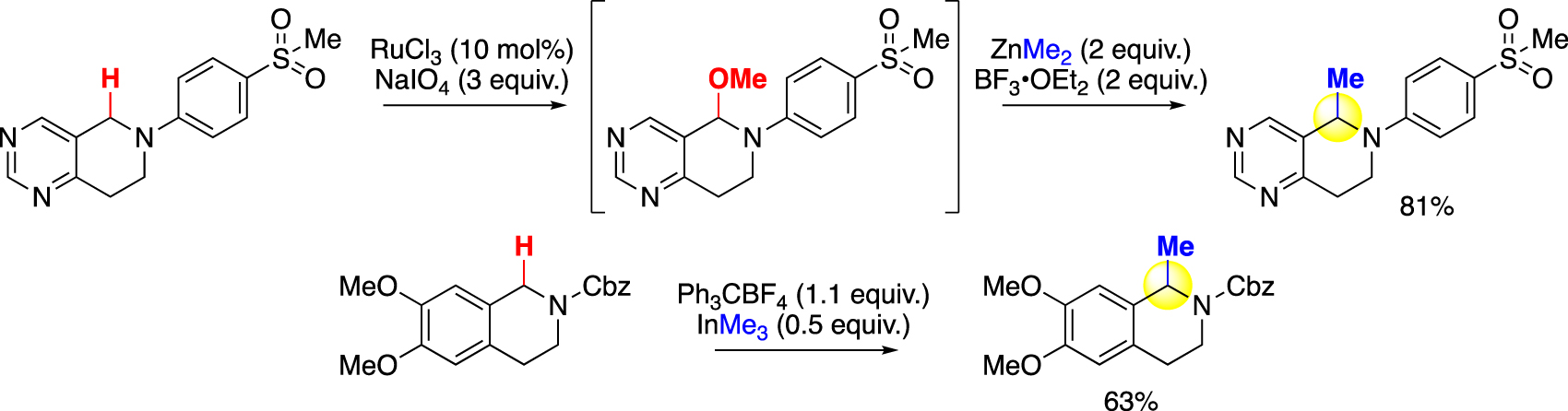
Early examples of oxidative methylation.
For the past two decades, the White group has pioneered in developing catalysts that enable preparative and selective C–H oxidations at the late stages of synthesis [23]. In 2019, White, Zhao, and coworkers reported the innovation of a highly reactive yet chemoselective Mn(CF3PDP)(MeCN)2(SbF6)2 (CF3PDP = 1,1′-bis((5-(2,6-bis(trifluoromethyl)phenyl)pyridin-2-yl)methyl)-2,2′-bipyrrolidine) catalyst capable of oxidizing secondary aliphatic C–H bonds in complex molecules with excellent site-selectivity, while for the first time tolerating electron-poor arenes in the same catalytic system [35]. The oxidation is hypothesized to proceed through hydrogen abstraction by the in situ generated high-valent manganese-oxo species, followed by rapid OH rebound. This putative mechanism is further supported by density functional theory (DFT) calculations recently carried out by Zhang and coworkers (Fig. 6a) [42]. Unlike oxidations led by highly reactive alkoxy radicals [11], the structural feature of Mn(CF3PDP) enables catalyst control of the site of oxidation among unactivated aliphatic C–H bonds while overriding substrate bias [35]. The high selectivity of Mn(CF3PDP), driven by a combinatory effect of electronic, steric, and stereoelectronic properties of the C–H bonds, was crucial to high-yielding oxidations.
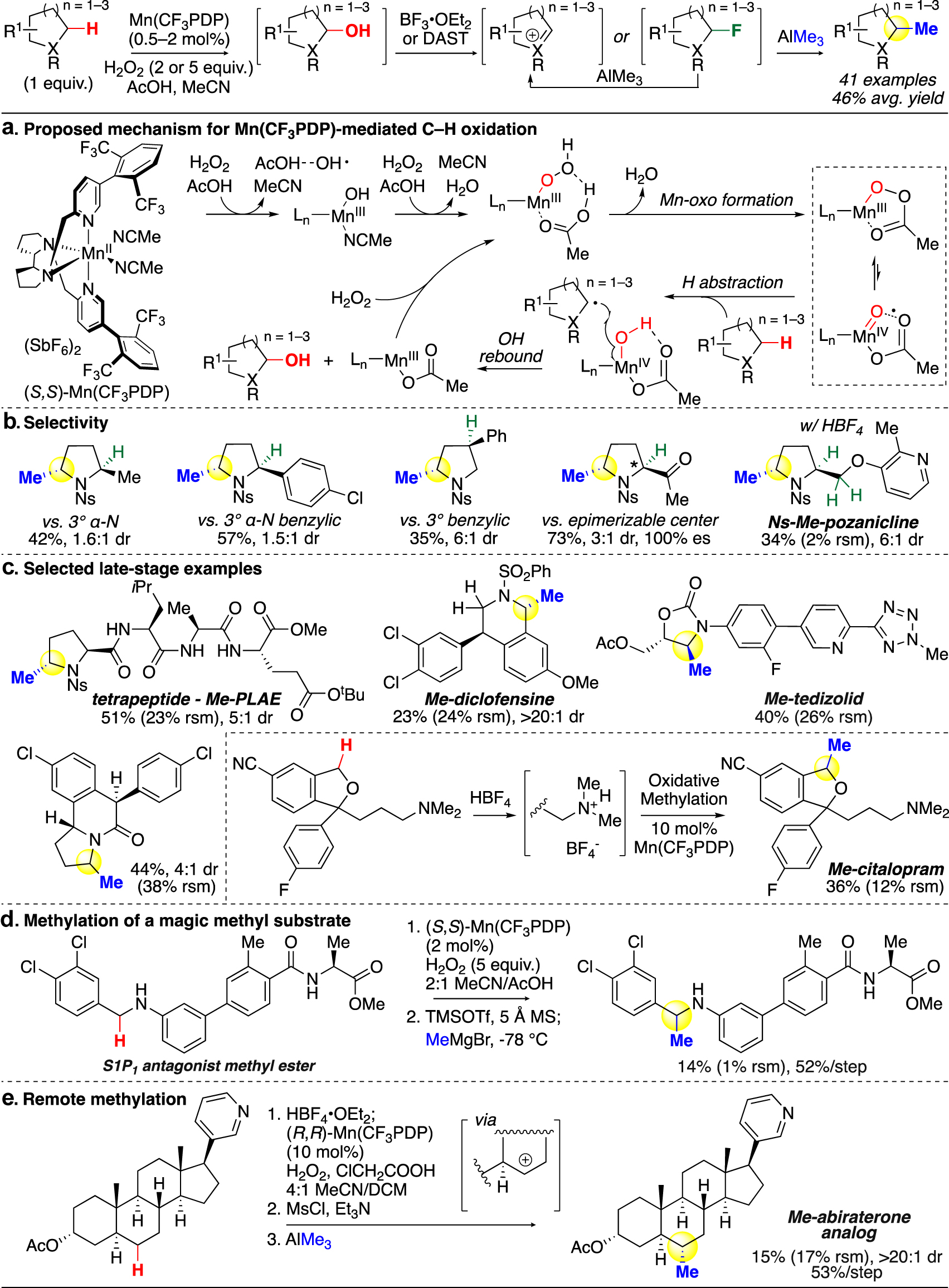
Late-stage oxidative C(sp3)–H methylation mediated by Mn(CF3PDP) (White).
Using the (S,S)-Mn(CF3PDP) catalyst, we established the first general late-stage oxidative methylation strategy in 2020, which enabled facile methyl group incorporation with a broad scope, including 16 pharmaceutically relevant cores [24]. By significantly reducing the catalyst loading, from the previous 10 mol% necessary to oxidize the less reactive aliphatic C–H bonds [35] to 0.5 mol% for the hyperconjugatively activated α-N C–H, we found that undesired overoxidation and off-site oxidation were both suppressed, and unprecedented arene tolerance was achieved in α-hydroxylation.
An additional challenge in developing an oxidative methylation strategy was the sequential activation of the hemiaminal/hemiacetal intermediate and the selection of the nucleophile. Trimethylaluminum, a mild nucleophile, in combination with Lewis acid or fluorinating activators, furnished modestly nucleophilic and non-basic methylating reaction conditions that alkylate reactive iminium or oxonium intermediates while avoiding competing elimination to the undesired enamine or attack on other electrophilic groups on the substrate (e.g. carbonyls, nitriles) [24]. The fluorination strategy is uniquely effective for tolerating electrophilic functional groups during methylation, as it prevents promoting nucleophilic attack by AlMe3 on these groups as observed in activation with Lewis acids. An ethyl group can be likewise incorporated using AlEt3. Because of the excellent selectivity of Mn(CF3PDP) at low catalyst loadings, only the least hindered, most electron-rich secondary sites were methylated while α-tertiary and benzylic sites were left unreacted (Fig. 6b). For substrates containing α-stereocenters, full stereoretention was observed in the methylated products. The significantly reduced catalyst loading also makes this method practical to be carried out on larger scales, as several gram-scale reactions were demosntrated in good yields.
Late-stage methylation was carried out on 18 complex biologically active molecules and drugs (Fig. 6c) [24]. For the first time, electron-rich arenes such as anisole motifs were tolerated with Mn(CF3PDP) at low loadings; previously, attempts to oxidize substrates containing these arenes resulted in only off-site aromatic oxidation [35]. The effectiveness of this strategy is exemplified in the methylation of an antibiotic, tedizolid acetate: a 40 % yield was obtained despite the substrate possessing a mildly basic pyridine, a tetrazole, and an N-methyl.
With nitrogen being incorporated in 84 % of FDA-approved small-molecule drugs [43], a major challenge in late-stage functionalization was the modification of molecules containing basic nitrogens. For transition-metal-mediated transformations, these Lewis-basic nitrogen atoms are prone to bind to the metal center and deactivate the catalyst, and are additionally susceptible to N-oxidation. In 2015, we reported a solution to this challenge by employing a Lewis acid, BF3, or a Brønsted acid, HBF4, to irreversibly complex with or protonate the nitrogen [44]. This protecting strategy eliminates nitrogen’s ability of metal-binding and renders it a strong electron-withdrawing group, thus promoting remote oxidation as catalyzed by Fe(PDP) and Fe(CF3PDP). Using this strategy with Mn(CF3PDP), substrates containing basic nitrogen atoms (e.g., citalopram) can be functionalized without deactivation of the manganese catalyst (Fig. 6b, c) [24].
Some modifications on the standard reaction conditions further expanded the scope of this method. The use of a stronger methylating reagent, MeMgBr, at cryogenic temperatures enabled methylation of a linear secondary aniline via an imine intermediate (Fig. 6d) [24]. The incorporation of a methyl group at the same position on this magic methyl substrate contributed to a 2135-fold potency boost [2]. In addition, since Mn(CF3PDP) has shown extraordinary ability to selectively oxidize unactivated secondary C–H bonds [35], when the resulting alcohol product is transformed into a mesylate, AlMe3 was shown to effectively convert that mesylate into a methyl, affording a remotely methylated carbocycle for the first time (Fig. 6e).
A highly similar approach was subsequently reported by Zhao and Wang in 2021, in which the authors employed N-fluorobenzenesulfonimide (NFSI) as oxidant to promote copper-mediated radical fluorination [45]. The resulting fluorinated products were in situ transformed into hemiaminals by elimination and nucleophilic addition of water. The resulting hemiaminals were then subjected to the same fluorine-activation strategy and transformed back to the fluoride using DAST, followed by in situ conversion to the methylated product by AlMe3 (Fig. 7). Methylation was demonstrated in good yields on a smaller set of N-arylpyrrolidinone and N-aryloxazolidinone substrates with modest complexity. While the substrate scope largely overlaps with the previous work [24], this method presented some orthogonality by showing tolerance of C=C double bonds in the hydroxylation and methylation examples.
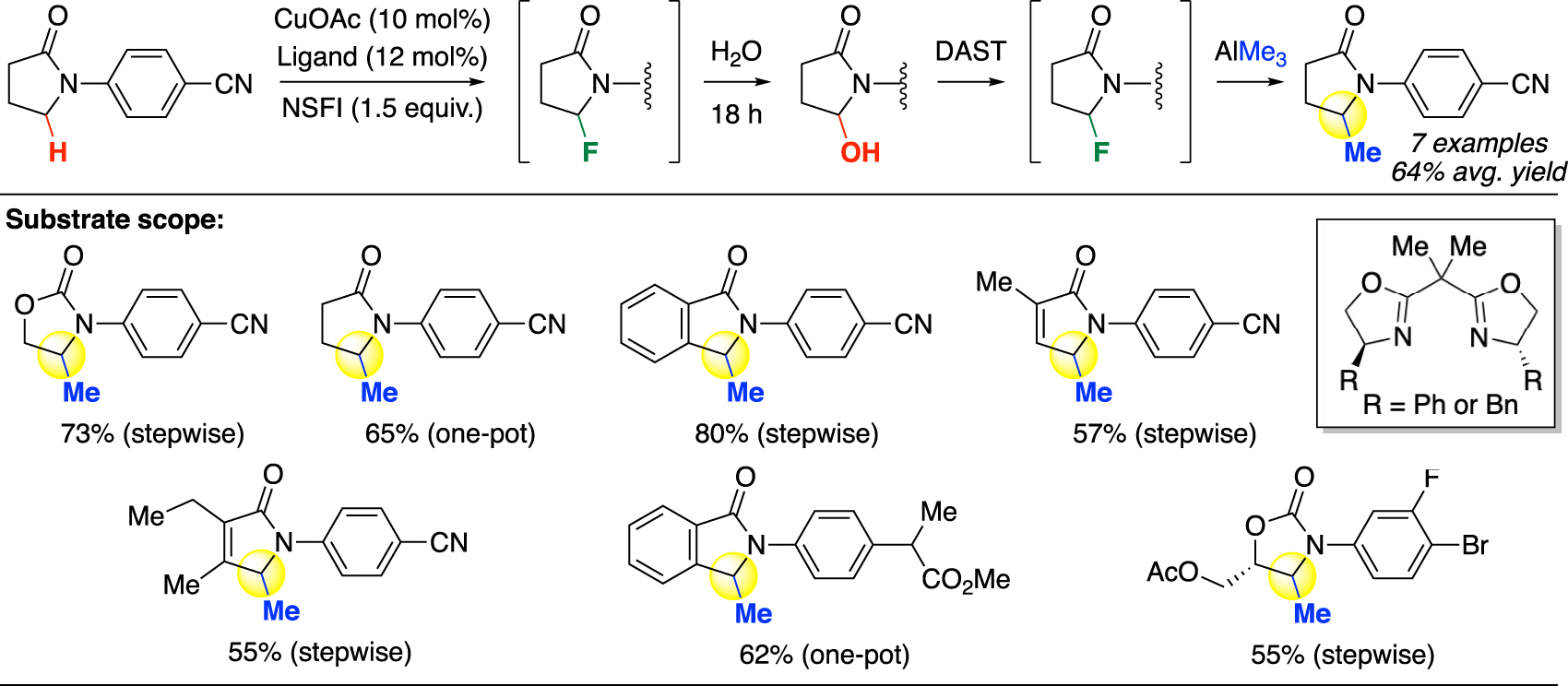
Copper-mediated oxidative C(sp3)–H methylation (Zhao).
Over the past decade, electrochemistry has quickly emerged as a powerful approach to achieve otherwise challenging transformations [46]. Shono oxidation has long been known to oxidize α-C–H in structurally simple carbamates, amides, and sulfonamides [47], [48], [49], but its application had not extended to later stages in synthesis, possibly due to its high overpotential required leading to side reactions in densely functionalized bioactive molecules. As a versatile approach and complementary to chemical oxidative C(sp3)–H methylation, Lin and coworkers recently reported an improved version of the Shono oxidation, applicable at the late stage to furnish hemiaminal ether intermediates with high functional group tolerance [50]. The resulting hemiaminal ether was subsequently methylated using ZnMe2 following a similar Lewis acid activation strategy (Fig. 8). The authors report that by replacing the traditional methanol solvent that was prone to oxidation with trifluoroethanol (TFE), anodic oxidation resistance was significantly improved while product stability remained high. The fluorine-containing alkoxy group introduced also serves as a good leaving group under Lewis-acid activation and facilitated the subsequent methylation step.
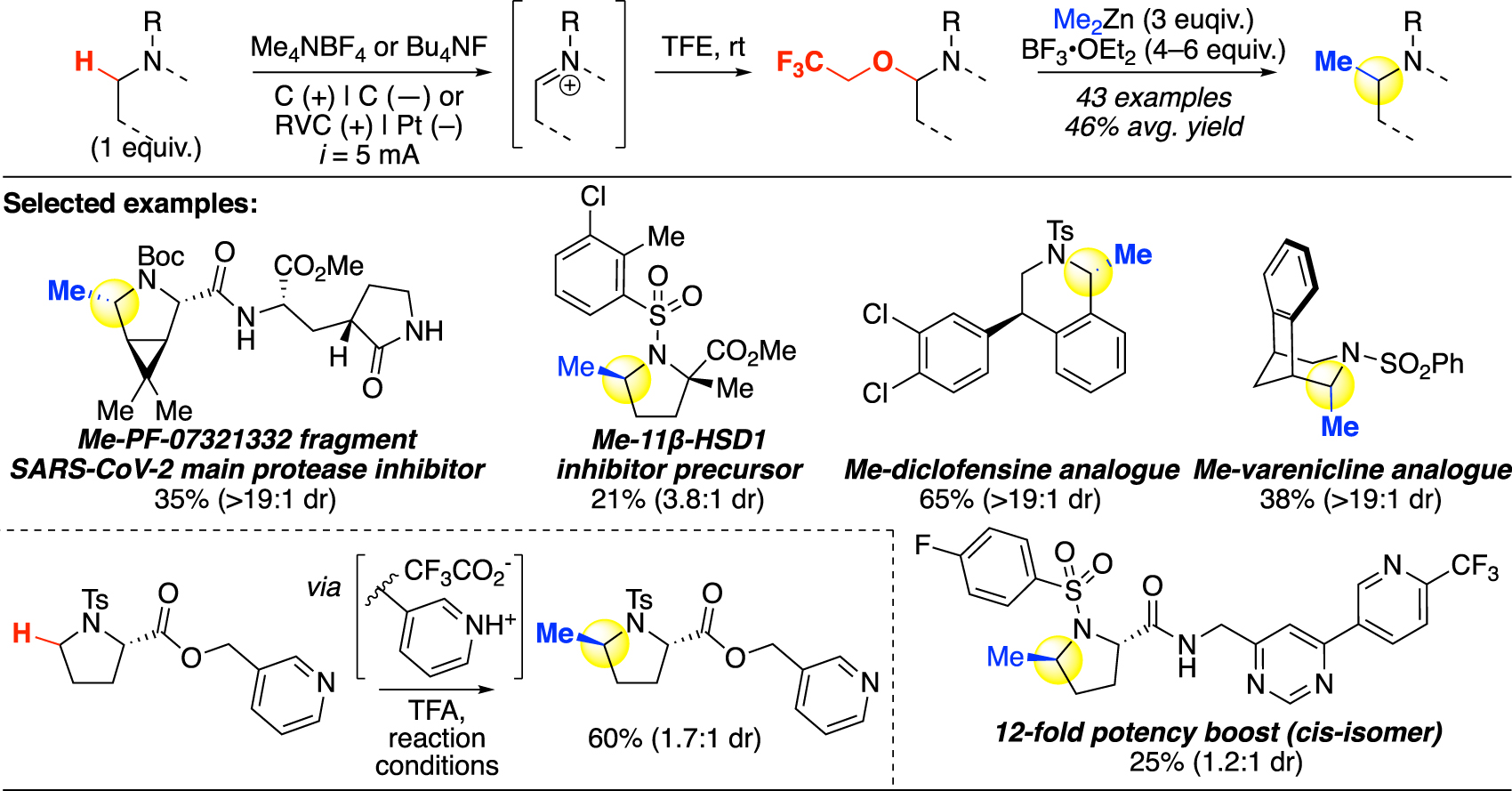
Oxidative C(sp3)–H methylation via anodic oxidation (Lin).
With the newfound conditions, a series of five- to seven-membered heterocycles are oxidized and subsequently methylated in good yields, including a diclofensine analogue with an electron-neutral aryl ring. The selectivity is governed by the electronic and steric environments of the substrate C–H bonds; regioisomers were observed in some heterocycles containing multiple secondary α-N sites. Full stereoretention was seen in the methylation of tosyl-protected methyl L-prolinate containing an epimerizable α-center. Several possible side reactions include benzylic oxidation of the tosyl protecting group and double oxidation for substrates with two secondary α-C–H sites. For six-membered rings, an additional challenge of elimination was observed and was suppressed by running the oxidation at lower temperatures.
Late-stage methylation was demonstrated on several complex molecules, including ones containing basic nitrogens [50]. Comparable to what we and others have demonstrated in transition-metal-mediated oxidations [24, 35, 44, 51], these basic nitrogens can protected via protonation with trifluoroacetic acid, which provides a balanced strength to effectively protonate the nitrogen without corroding the electrode. Notably, a structurally complex magic methyl substrate was methylated in a synthetically useful yield as a mixture of diastereomers. Using corresponding organozinc reagents, Lin and coworkers were also able to incorporate other functional groups including ethyl, allyl, and phenyl for possible expansion of this method’s application.
Methylation through photocatalytic hydrogen atom transfer (HAT)
In the broader arena of C(sp3)–H alkylation, MacMillan and coworkers pioneered in developing a polarity-match-based strategy to selectively α-functionalize aliphatic heterocycles and acyclic amines, ethers, and sulfides via iridium photoredox catalysis and nickel cross-coupling in 2017 [20]. The key to the high selectivity of this method is the use of quinuclidine. Upon its oxidation by *Ir(III), the resulting electrophilic radical can selectively extract the most hydridic C–H bond α to the heteroatom, followed by cross-coupling with nickel to alkylate at the same position (Fig. 9a). With the aid of quinuclidine, this strategy is capable of selectively functionalizing a heterocycle with a broad electrophile scope, though requiring an excess of the substrate (Fig. 9b). One methylation example was demonstrated on the unsubstituted pyrrolidine with the same iridium and nickel catalysts. Whereas quinuclidine governs the reactivity and selectivity of all other alkylations, interestingly, when a methyl electrophile is used, the addition of quinuclidine was found detrimental and the reaction proceeds in its absence. It was postulated that the methyl electrophiles (MeOTs, MeBr) are consumed through an SN2 process by quinuclidine. The authors further suggested that an alternative mechanism is possible for methylation reactions, where the hydrogen abstraction is carried out by a bromide radical (Fig. 9c) [52], [53], [54]. This discrepancy in reactivity exemplifies the unique challenges of installing a methyl group in alkylation.
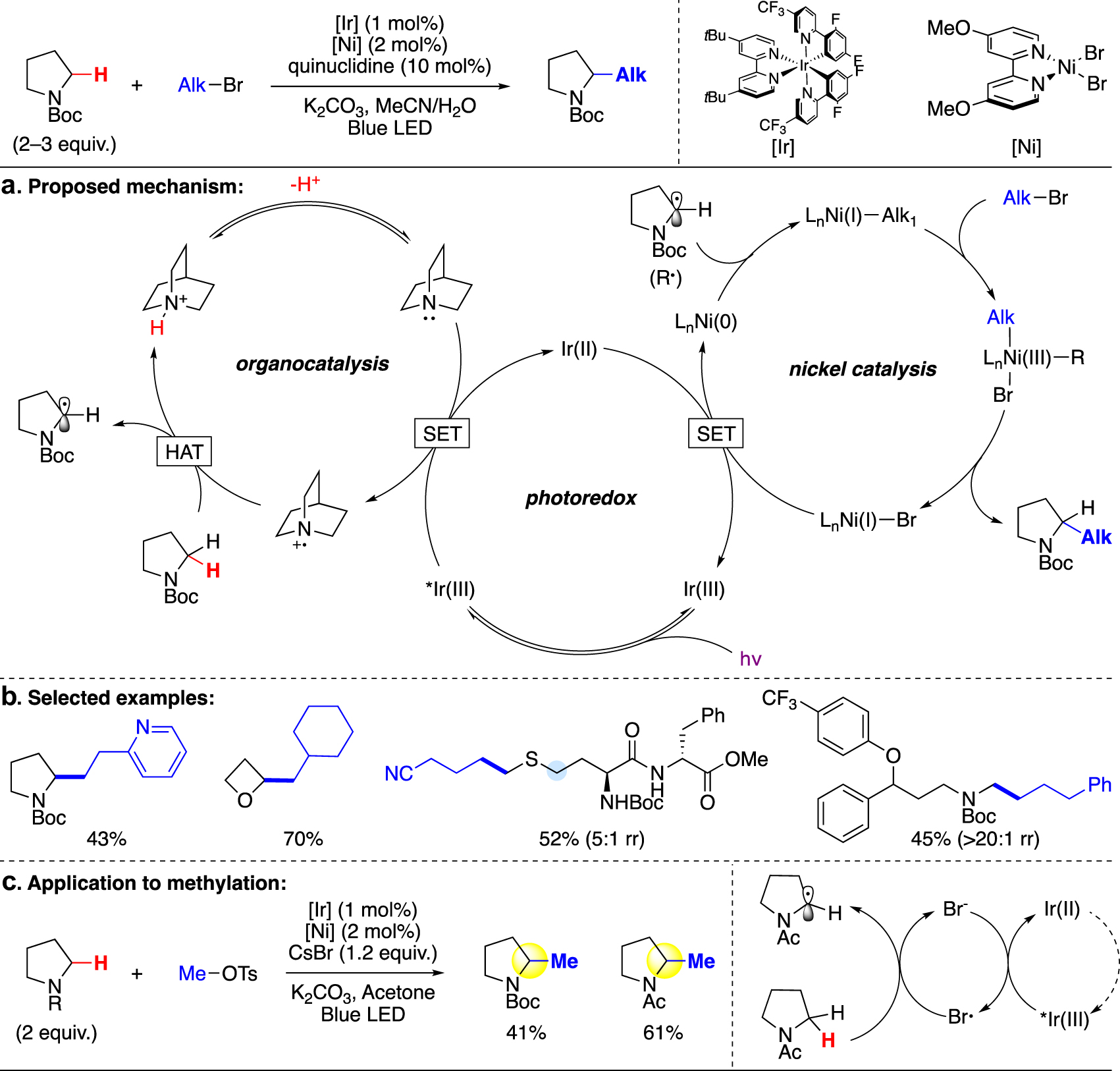
C(sp3)–H alkylation via polarity-matched hydrogen atom transfer (HAT) and nickel-mediated cross-coupling (MacMillan).
In 2021, Stahl and coworkers devised a novel approach using the iridium photocatalyst [55]. A notable innovation was the dual role of di-tert-butyl peroxide (DTBP) or dicumyl peroxide (DCP) as both oxidant and methyl source. Instead of generating the quinuclidine-based oxidant through a photoredox pathway, the iridium catalyst was employed as a photosensitizer to homolytically cleave the peroxide reagent to generate alkoxy radicals as oxidants (Fig. 10a). This homolysis can be carried out under mild conditions, in contrast to the traditional high-energy thermolysis and photolysis pathways. The alkoxy radicals then undergo hydrogen atom transfer (HAT) to abstract the α-C–H, benzylic C–H, or tertiary aliphatic C–H to furnish a radical intermediate. At the same time, part of the alkoxy radicals formed are consumed via a competing β-scission process that generates methyl radical while releasing acetone or acetophenone.
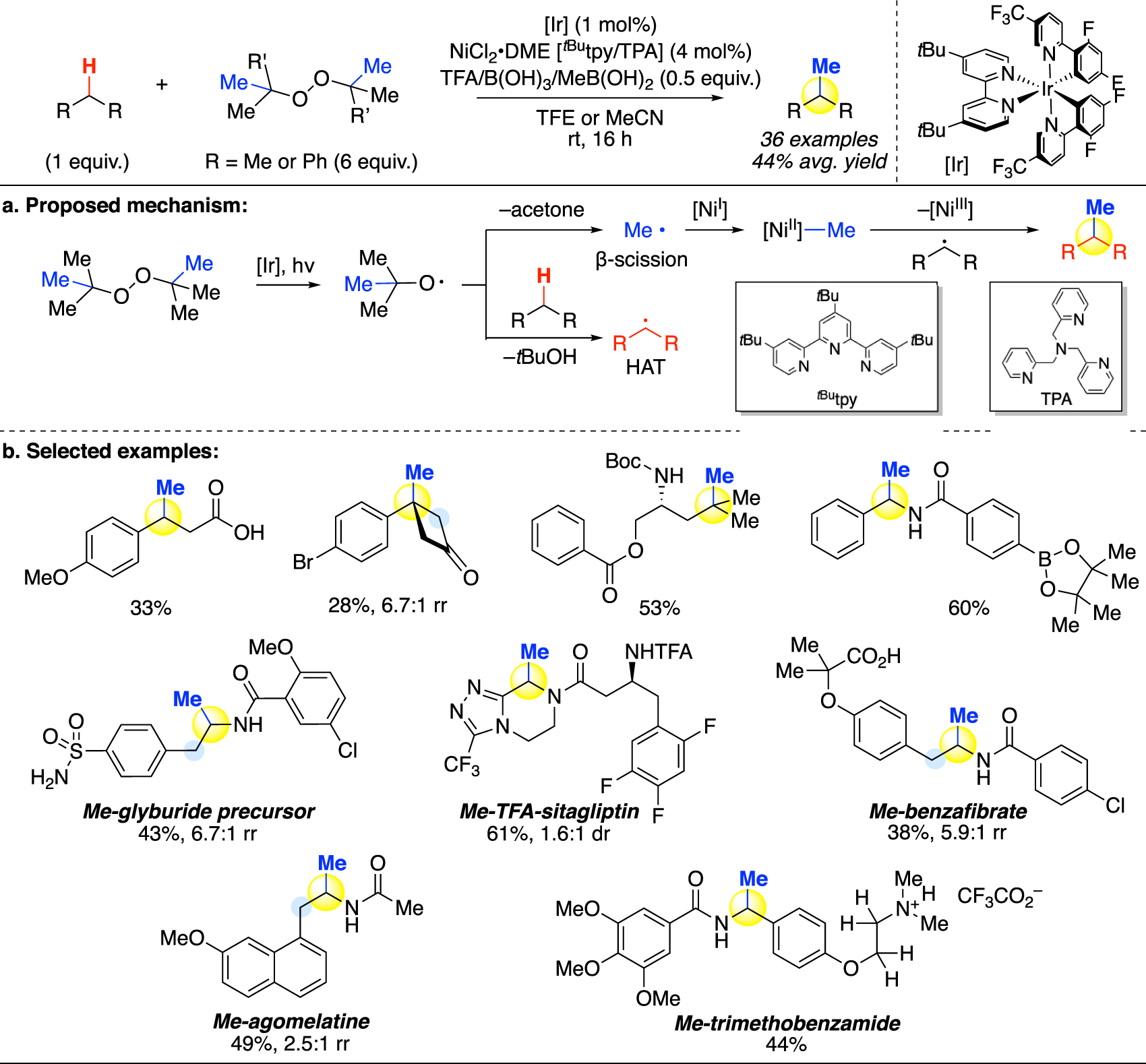
C(sp3)–H methylation via photosensitization-assisted hydrogen atom transfer (HAT) and nickel-mediated cross-coupling (Stahl).
Although oxidations by alkoxy radicals traditionally require a large excess or even solvent quantity of the substrate [11, 56], [57], [58], [59], Stahl reports that by carefully tuning the reaction temperature and the reagents used, a working balance can be achieved between the HAT and β-scission processes, which enables the reaction to be carried out preparatively. Due to the dual function that DTBP or DCP serves, 6 equiv. of the oxidant is used for the reaction.
When the initial attempts to directly cross-couple the substrate radical with the methyl radical produced substantial amounts of byproducts, Stahl and coworkers similarly employed a nickel co-catalyst to mediate the radical-radical coupling (Fig. 10a). Under the Ir/Ni dual catalytic system, preparative methylation was achieved on a diverse series of benzylic, tertiary, and heterocyclic substrates in good yields, including examples of late-stage functionalization (Fig. 10b). Regioisomers were observed in substrates containing both α-N and benzylic sites, with α-N as the favored site for methylation. In order to achieve benzylic oxidation in these substrates, Stahl reports that the previously established acid-protection strategies for remote oxidation [24, 35, 44, 51] could be likewise employed for the HAT. Upon protonation by trifluoroacetic acid, sites proximal to a basic nitrogen atom are deactivated and remote benzylic C–H abstraction is favored to render exclusively benzylic methylation products.
Allylic methylation
While not as prevalent in drug candidates [31], olefins are found in roughly 40 % of natural products as the second most frequently occurring functional group [32]. Unlike methylations at the α-heteroatom and benzylic positions, the analogous transformation for allylic C–H bonds remains largely undeveloped, possibly due to challenges of alkene tolerance, overoxidation, and regioselectivity [23]. In 2019, Tambar and coworkers reported a stereoselective allylic alkylation method [60]. Two allylic methylation examples were demonstrated on simple terminal alkenes through an ene reaction followed by copper-mediated cross-coupling (Fig. 11). Enantioselectivity can be achieved using a chiral ligand; however, only a modest ee was obtained when a methyl group is installed. In order to practically modify natural products at their allylic positions, a late-stage method that incorporates methyl groups onto more complex structures and internal olefins is yet to be developed.
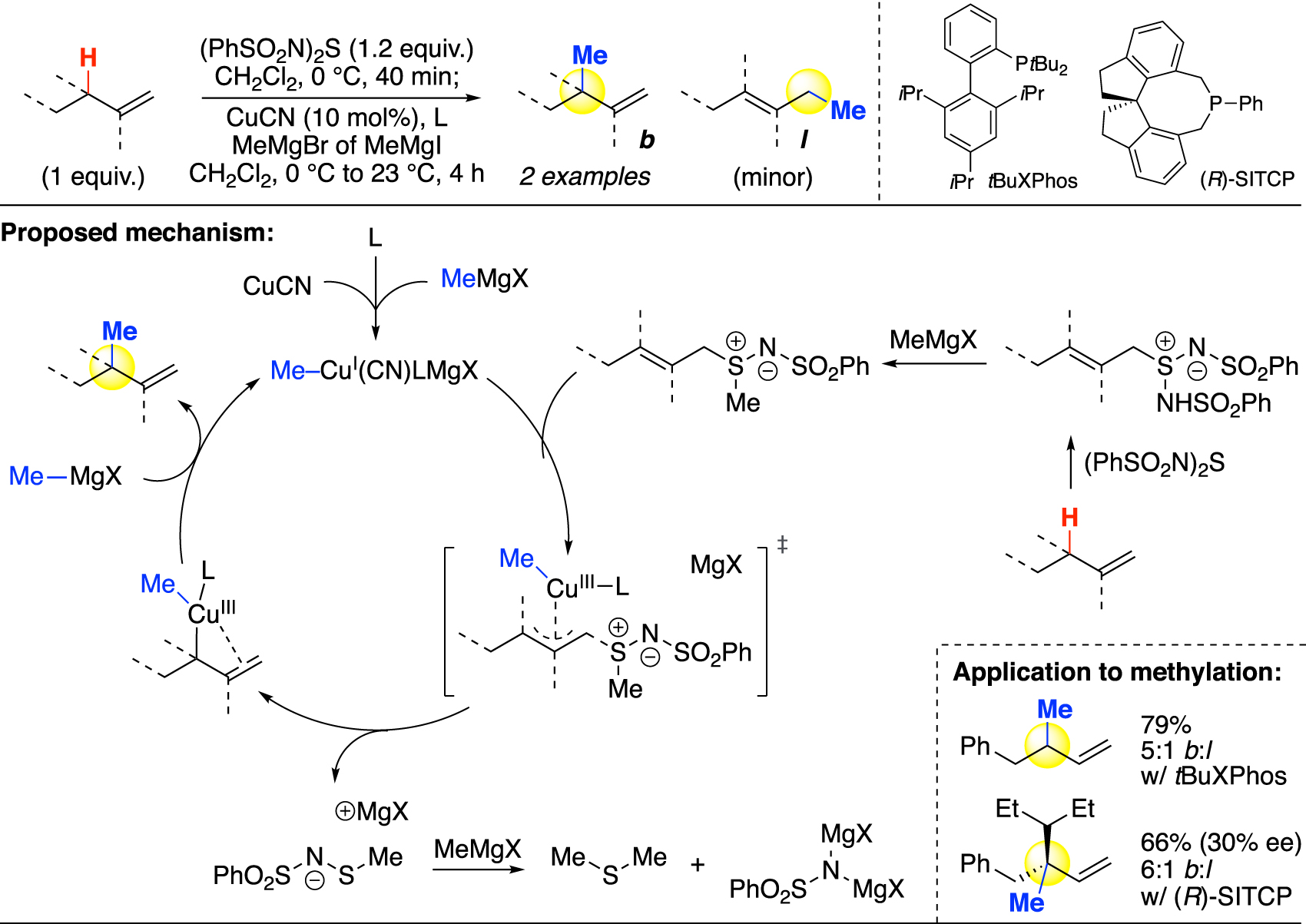
Allylic methylation via ene reaction and copper-catalyzed cross-coupling (Tambar).
Conclusion
Recent innovations of diverse and powerful late-stage C(sp3)–H methylation strategies open up new avenues to rapidly access methylated drug candidates and opportunities to explore their magic methyl effects in drug discovery. Complementary approaches encompassing oxidative methylation and photocatalytic HAT now enable the interrogation of a broad set of biologically active molecules and their methylated derivatives. Despite these exciting advances, numerous opportunities remain in the future development of late-stage methylation methods, including their expansion to functionalize allylic and unactivated aliphatic C–H bonds, as well as to incorporate more complex alkyl groups using readily accessible sources. Furthermore, while stereoselective methylation was known for methods employing organolithium [26], [27], [28] and directing groups [17], the development of a non-directed approach that incorporates methyl groups enantioselectively at late stages will be beneficial. It is fully expected that the continued advances in this field will further facilitate the discovery of new pharmaceutical agents with the aid of the magic methyl effect.
Acknowledgments
I thank the IUPAC and Solvay for a 2021 IUPAC-Solvay International Award for Young Chemists, and Pure and Applied Chemistry for an invitation for this review. I thank Prof. M. Christina White for her guidance and support throughout my doctoral study. I am also grateful for all of my coworkers at the University of Illinois, especially Dr. Jennifer Howell, Prof. Joseph Clark, and Dr. Raundi Quevedo, for their immense contributions to our shared works, including those discussed in this review.
References
[1] H. Schönherr, T. Cernak. Angew. Chem. Int. Ed.52, 12256 (2013). doi:https://doi.org/10.1002/anie.201303207.Suche in Google Scholar PubMed
[2] J. Quancard, B. Bollbuck, P. Janser, D. Angst, F. Berst, P. Buehlmayer, M. Streiff, C. Beerli, V. Brinkmann, D. Guerini, P. A. Smith, T. J. Seabrook, M. Traebert, K. Seuwen, R. Hersperger, C. Bruns, F. Bassilana, M. Bigaud. Chem. Biol.19, 1142 (2012), https://doi.org/10.1016/j.chembiol.2012.07.016.Suche in Google Scholar PubMed
[3] B. P. Fauber, O. René, Y. Deng, J. DeVoss, C. Eidenschenk, C. Everett, A. Ganguli, A. Gobbi, J. Hawkins, A. R. Johnson, H. La, J. Lesch, P. Lockey, M. Norman, W. Ouyang, S. Summerhill, H. Wong. J. Med. Chem.58, 5308 (2015), https://doi.org/10.1021/acs.jmedchem.5b00597.Suche in Google Scholar PubMed
[4] J. Fujimoto, R. Okamoto, N. Noguchi, R. Hara, S. Masada, T. Kawamoto, H. Nagase, Y. O. Tamura, M. Imanishi, S. Takagahara, K. Kubo, K. Tohyama, K. Iida, T. Andou, I. Miyahisa, J. Matsui, R. Hayashi, T. Maekawa, N. Matsunaga. J. Med. Chem.60, 8963 (2017), https://doi.org/10.1021/acs.jmedchem.7b01210.Suche in Google Scholar PubMed
[5] P. J. Coleman, J. D. Schreier, C. D. Cox, M. J. Breslin, D. B. Whitman, M. J. Bogusky, G. B. McGaughey, R. A. Bednar, W. Lemaire, S. M. Doran, S. V. Fox, S. L. Garson, A. L. Gotter, C. M. Harrell, D. R. Reiss, T. D. Cabalu, D. Cui, T. Prueksaritanont, J. Stevens, P. Tannenbaum, R. G. Ball, J. Stellabott, S. D. Young, G. D. Hartman, C. J. Winrow, J. J. Renger. ChemMedChem7, 415 (2012), https://doi.org/10.1002/cmdc.201200025.Suche in Google Scholar PubMed
[6] E. J. Barreiro, A. E. Kümmerle, C. A. M. Fraga. Chem. Rev.111, 5215 (2011), https://doi.org/10.1021/cr200060g.Suche in Google Scholar PubMed
[7] C. S. Leung, S. S. F. Leung, J. Tirado-Rives, W. L. Jorgensen. J. Med. Chem.55, 4489 (2012), https://doi.org/10.1021/jm3003697.Suche in Google Scholar PubMed PubMed Central
[8] P. J. Belshaw, J. G. Schoepfer, K.-Q. Liu, K. L. Morrison, S. L. Schreiber. Angew. Chem. Int. Ed. Engl.34, 2129 (1995), https://doi.org/10.1002/anie.199521291.Suche in Google Scholar
[9] T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachal, S. W. Krska. Chem. Soc. Rev.45, 546 (2016); correction 46, 1760 (2017), https://doi.org/10.1039/c5cs00628g.Suche in Google Scholar PubMed
[10] D. Aynetdinova, M. C. Callens, H. B. Hicks, C. Y. X. Poh, B. D. A. Shennan, A. M. Boyd, Z. H. Lim, J. A. Leitch, D. J. Dixon. Chem. Soc. Rev.50, 5517 (2021), https://doi.org/10.1039/d0cs00973c.Suche in Google Scholar PubMed
[11] E. A. Mitchell, A. Peschiulli, N. Lefevre, L. Meerpoel, B. U. W. Maes. Chem. Eur. J.18, 10092 (2012), https://doi.org/10.1002/chem.201201539.Suche in Google Scholar PubMed
[12] S. D. Friis, M. J. Johansson, L. Ackermann. Nat. Chem.12, 511 (2020), https://doi.org/10.1038/s41557-020-0475-7.Suche in Google Scholar PubMed
[13] L. Guillemard, N. Kaplaneris, L. Ackermann, M. J. Johansson. Nat. Rev. Chem.5, 522 (2021), https://doi.org/10.1038/s41570-021-00300-6.Suche in Google Scholar PubMed
[14] C. J. Cordier, R. J. Lundren, G. C. Fu. J. Am. Chem. Soc.135, 10946 (2013), https://doi.org/10.1021/ja4054114.Suche in Google Scholar PubMed PubMed Central
[15] K. R. Campos. Chem. Soc. Rev.36, 1069 (2007), https://doi.org/10.1039/b607547a.Suche in Google Scholar PubMed
[16] J. A. Milligan, J. P. Phelan, S. O. Badir, G. A. Molander. Angew. Chem. Int. Ed.58, 6152 (2019), https://doi.org/10.1002/anie.201809431.Suche in Google Scholar PubMed PubMed Central
[17] P. Jain, P. Verma, G. Xia, J.-Q. Yu. Nat. Chem.9, 140 (2017), https://doi.org/10.1038/nchem.2619.Suche in Google Scholar PubMed PubMed Central
[18] A. Paul, D. Seidel. J. Am. Chem. Soc.141, 8778 (2019), https://doi.org/10.1021/jacs.9b04325.Suche in Google Scholar PubMed PubMed Central
[19] J. H. Paul, S. Kim, D. Daniel, D. Seidel. Angew. Chem. Int. Ed.60, 1625 (2021), https://doi.org/10.1002/anie.202011641.Suche in Google Scholar PubMed PubMed Central
[20] C. Le, Y. Liang, R. W. Evans, X. Li, D. W. C. MacMillan. Nature547, 79 (2017), https://doi.org/10.1038/nature22813.Suche in Google Scholar PubMed PubMed Central
[21] S. Dutta, B. Li, D. R. L. Rickertsen, D. A. Valles, D. Seidel. SynOpen5, 173 (2021).Suche in Google Scholar
[22] C. Blakemore, L. Castro, I. Churcher, D. C. Rees, A. W. Thomas, D. M. Wilson, A. Wood. Nat. Chem.10, 383 (2018), https://doi.org/10.1038/s41557-018-0021-z.Suche in Google Scholar PubMed
[23] M. C. White, J. Zhao. J. Am. Chem. Soc.140, 13988 (2018), https://doi.org/10.1021/jacs.8b05195.Suche in Google Scholar PubMed PubMed Central
[24] K. Feng, R. E. Quevedo, J. T. Kohrt, M. S. Oderinde, U. Reilly, M. C. White. Nature580, 621 (2020), https://doi.org/10.1038/s41586-020-2137-8.Suche in Google Scholar PubMed PubMed Central
[25] P. Beak, W.-K. Lee. Tetrahedron Lett.30, 1197 (1989), https://doi.org/10.1016/s0040-4039(00)72714-6.Suche in Google Scholar
[26] S. T. Kerrick, P. Beak. J. Am. Chem. Soc.113, 9708 (1991), https://doi.org/10.1021/ja00025a066.Suche in Google Scholar
[27] P. Beak, S. T. Kerrick, S. Wu, J. Chu. J. Am. Chem. Soc.116, 3231 (1994), https://doi.org/10.1021/ja00087a008.Suche in Google Scholar
[28] P. Beak, A. Basu, D. J. Gallagher, Y. S. Park, S. Thayamanavan. Acc. Chem. Res.29, 552 (1996), https://doi.org/10.1021/ar950142b.Suche in Google Scholar
[29] W. Chen, A. Paul, K. A. Abboud, D. Seidel. Nat. Chem.12, 545 (2020), https://doi.org/10.1038/s41557-020-0438-z.Suche in Google Scholar PubMed PubMed Central
[30] D. Vasu, A. L. Fuentes de Arriba, J. A. Leitch, A. de Gombert, D. J. Dixon. Chem. Sci.10, 3401 (2019), https://doi.org/10.1039/c8sc05164j.Suche in Google Scholar PubMed PubMed Central
[31] P. Ertl, E. Altmann, J. M. McKenna. J. Med. Chem.63, 8408 (2020), https://doi.org/10.1021/acs.jmedchem.0c00754.Suche in Google Scholar PubMed
[32] P. Ertl, T. Schuhmann. J. Nat. Prod.82, 1258 (2019), https://doi.org/10.1021/acs.jnatprod.8b01022.Suche in Google Scholar PubMed
[33] T. Osberger, D. C. Rogness, J. T. Kohrt, A. F. Stepan, M. C. White. Nature537, 214 (2016), https://doi.org/10.1038/nature18941.Suche in Google Scholar PubMed PubMed Central
[34] M. Milan, G. Carboni, M. Salamone, M. Costas, M. Bietti. ACS Catal.7, 5903 (2017), https://doi.org/10.1021/acscatal.7b02151.Suche in Google Scholar
[35] J. Zhao, T. Nanjo, E. C. de Lucca, M. C. White. Nat. Chem.11, 213 (2019), https://doi.org/10.1038/s41557-018-0175-8.Suche in Google Scholar PubMed PubMed Central
[36] Y. Kawamata, M. Yan, Z. Liu, D.-H. Bao, J. Chen, J. T. Starr, P. S. Baran. J. Am. Chem. Soc.139, 7448 (2017), https://doi.org/10.1021/jacs.7b03539.Suche in Google Scholar PubMed PubMed Central
[37] C. Annese, L. D’Accolti, C. Fusco, G. Licini, C. Zonta. Chem. Eur. J.23, 259 (2017), https://doi.org/10.1002/chem.201604507.Suche in Google Scholar PubMed
[38] S. Murata, M. Miura, M. Nomura. J. Chem. Soc., Perkin Trans. 1, 1259 (1987), https://doi.org/10.1039/p19870001259.Suche in Google Scholar
[39] S. Yoshifuji, K.-I. Takana, T. Kawai, Y. Nitta. Chem. Pharm. Bull.34, 3873 (1986), https://doi.org/10.1248/cpb.34.3873.Suche in Google Scholar
[40] Z. Cheng, Z. Yu, S. Yang, H. C. Shen, W. Zhao, S. Zhong. J. Org. Chem.82, 13678 (2017), https://doi.org/10.1021/acs.joc.7b02382.Suche in Google Scholar PubMed
[41] J. M. Gil-Negrete, J. P. Sestelo, L. A. Sarandeses. J. Org. Chem.84, 9778 (2019), https://doi.org/10.1021/acs.joc.9b00928.Suche in Google Scholar PubMed
[42] A. Feng, Y. Liu, Y. Yang, R. Zhu, D. Zhang. ACS Catal.12, 2290 (2022), https://doi.org/10.1021/acscatal.1c05025.Suche in Google Scholar
[43] D. Vitaku, T. Smith, J. T. Njardarson. J. Med. Chem.57, 10257 (2014), https://doi.org/10.1021/jm501100b.Suche in Google Scholar PubMed
[44] J. M. Howell, K. Feng, J. R. Clark, L. J. Trzepkowski, M. C. White. J. Am. Chem. Soc.137, 14590 (2015), https://doi.org/10.1021/jacs.5b10299.Suche in Google Scholar PubMed PubMed Central
[45] Z. Chang, J. Huang, S. Wang, G. Chen, H. Zhao, R. Wang, D. Zhao. Nat. Commun.12, 4342 (2021), https://doi.org/10.1038/s41467-021-24671-y.Suche in Google Scholar PubMed PubMed Central
[46] L. F. T. Novaes, J. Liu, Y. Shen, L. Lu, J. M. Meinhardt, S. Lin. Chem. Soc. Rev.50, 7941 (2021), https://doi.org/10.1039/d1cs00223f.Suche in Google Scholar PubMed PubMed Central
[47] T. Shono. Tetrahedron40, 811 (1984), https://doi.org/10.1016/s0040-4020(01)91472-3.Suche in Google Scholar
[48] T. Shono, H. Hamaguchi, Y. Matsumura. J. Am. Chem. Soc.97, 4264 (1975), https://doi.org/10.1021/ja00848a020.Suche in Google Scholar
[49] T. Shono, Y. Matsumura, K. Tsubata, K. Uchida, T. Kanazawa, K. Tsuda. J. Org. Chem.49, 3711 (1984), https://doi.org/10.1021/jo00194a008.Suche in Google Scholar
[50] L. F. T. Novaes, J. S. K. Ho, K. Mao, K. Liu, M. Tanwar, M. Neurock, E. Villemure, J. A. Terrett, S. Lin. J. Am. Chem. Soc.144, 1187 (2022), https://doi.org/10.1021/jacs.1c09412.Suche in Google Scholar PubMed
[51] M. Lee, M. S. Sanford. J. Am. Chem. Soc.137, 12796 (2015), https://doi.org/10.1021/jacs.5b09099.Suche in Google Scholar PubMed PubMed Central
[52] P. Zhang, C. Le, D. W. C. MacMillan. J. Am. Chem. Soc.138, 8084 (2016), https://doi.org/10.1021/jacs.6b04818.Suche in Google Scholar PubMed PubMed Central
[53] B. J. Shields, A. G. Doyle. J. Am. Chem. Soc.138, 12719 (2016), https://doi.org/10.1021/jacs.6b08397.Suche in Google Scholar PubMed PubMed Central
[54] D. R. Heitz, J. C. Tellis, G. A. Molander. J. Am. Chem. Soc.138, 12715 (2016), https://doi.org/10.1021/jacs.6b04789.Suche in Google Scholar PubMed PubMed Central
[55] A. Vasilopoulos, S. W. Krska, S. S. Stahl. Science372, 398 (2021), https://doi.org/10.1126/science.abh2623.Suche in Google Scholar PubMed PubMed Central
[56] M. S. Kharasch, G. Sosnovsky. J. Am. Chem. Soc.80, 756 (1958), https://doi.org/10.1021/ja01536a062.Suche in Google Scholar
[57] R. T. Gephart, C. L. McMullinIII, N. G. Sapiezynski, E. S. Jang, M. J. B. Aguila, T. R. Cundari, T. H. Warren. J. Am. Chem. Soc.134, 17350 (2012), https://doi.org/10.1021/ja3053688.Suche in Google Scholar PubMed
[58] B. L. Tran, B. Li, M. Driess, J. F. Hartwig. J. Am. Chem. Soc.136, 2555 (2014), https://doi.org/10.1021/ja411912p.Suche in Google Scholar PubMed PubMed Central
[59] A. Vasilopoulos, S. L. Zultanski, S. S. Stahl. J. Am. Chem. Soc.139, 7705 (2017), https://doi.org/10.1021/jacs.7b03387.Suche in Google Scholar PubMed
[60] L. Qin, M. Sharique, U. K. Tambar. J. Am. Chem. Soc.141, 17305 (2019), https://doi.org/10.1021/jacs.9b08801.Suche in Google Scholar PubMed PubMed Central
© 2022 IUPAC & De Gruyter. This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License. For more information, please visit: http://creativecommons.org/licenses/by-nc-nd/4.0/
Artikel in diesem Heft
- Frontmatter
- Invited papers
- Electrochemiluminescence sensors and forensic investigations: a viable technique for drug detection?
- The quest for magic: recent advances in C(sp3)–H methylation
- IUPAC Recommendations
- Terminology and the naming of conjugates based on polymers or other substrates (IUPAC Recommendations 2021)
- IUPAC Technical Report
- Standard atomic weights of the elements 2021 (IUPAC Technical Report)
Artikel in diesem Heft
- Frontmatter
- Invited papers
- Electrochemiluminescence sensors and forensic investigations: a viable technique for drug detection?
- The quest for magic: recent advances in C(sp3)–H methylation
- IUPAC Recommendations
- Terminology and the naming of conjugates based on polymers or other substrates (IUPAC Recommendations 2021)
- IUPAC Technical Report
- Standard atomic weights of the elements 2021 (IUPAC Technical Report)