BioChemUDM: a unified data model for compounds and assays
-
Michael A. Kappler
 ,
Christopher T. Lowden
,
Christopher T. Lowden

Abstract
We present a simple, biochemistry data model (BioChemUDM) to represent compounds and assays for the purpose of capturing, reporting, and sharing data, both biological and chemical. We describe an approach to register a compound based solely on a stereo-enhanced sketch, thereby replacing the need for additional user-specified “flags” at the time of compound registration. We describe a convention for string-based labels that enables inter-organizational compound and assay data sharing. By co-adopting the BioChemUDM, we have successfully enabled same-day exchange and utilization of chemical and biological information with various stakeholders.
Background
As described previously [1], we originally approached retrosynthesis based on molecules, reactions, e-notebooks [2] and citations by using a unified data model (UDM) to merge public and private information into an expert-curated chemistry database [3]. The UDM was designed to allow data to be easily shared and integrated between parties by standardizing experimental business processes [4]. With guidance and support from the Pistoia Alliance [5], the original UDM evolved into a robust Extended Markup Language version [6] as a better alternative to the ChemDraw XML-compliant version [7] and the JavaScript Object Notation [8, 9]. As a result, the UDM has accelerated machine-learning of retrosynthetic pathways through merging of information sources [10]. The UDM concept has been applied to other areas of R&D as well [11].
Now, with an increasing number of contract organizations and biotechnology startups, we had begun thinking about the role that the UDM concept can have when applied to compounds and assays. We repeatedly found ourselves registering compounds, defining protocols, capturing assays, visualizing data, and struggling to integrate data from collaborators. This led us to a series of questions that needed to be answered and considered: Why does each group need to define compound registry rules, batch field names, or assay protocol variations; How would that align with experimental results; How does one tackle sharing between research groups? This needed an easy-to-implement, easy-to-understand approach to manage and share data. We need an “Informatics Starter Kit” for small molecule drug discovery groups!
This leads us to present BioChemUDM, a novel approach for integrating compound data and assay results. The original Unified Data Model (UDM) ([12] – this PAC issue) for reaction data integration used the ubiquitous CTfile Formats [13], specifically the RDfile format [14], and we called this prototype the RxnUDM. Similarly, BioChemUDM uses the enhanced SDfile [15] and CSV [16] formats. The techniques described herein enable creation of a small molecule information platform compatible with other organizations without specialized or costly integration services. The philosophy of BioChemUDM is a shift from extensively integrated IT systems towards training bench and data scientists to implement best-of-practices. Lastly, the primary motivation of this study is to improve collaborative data sharing and data sustainability, thus, to foster wide-spread adoption of the BioChemUDM, we encourage the sharing of scripts and templates to enable automation.
Methods
Our data model represents an object, and their relationships are modeled using string labels instead of a markup language such as XML. In the absence of a standard XML parser in commercially available tools, we chose to implement a reader/writer using human readable and easy-to-understand string labels. In the original, RDfile-based RxnUDM, we used the labels MOL (molecule), RXN (reaction), and CITE (citation) to represent objects and constructed a string. For instance, UDM.RXN.MOL.CITE described a citation for a molecule in a reaction. Similarly, in the BioChemUDM, we used the labels MOL (molecule), BAT (batch), SAM (sample), and ASY (assay) to represent objects and constructed a string. For instance, UDM.ASY.MOL.BAT.SAM describes a sample of a batch of a molecule tested in an assay. XML would provide an elegant alternative approach; however, XML reader/writer(s) are commonly absent in commercially available tools. Thus, we find that using string labels to be an effective way to quickly integrate research data with vendor tools and databases. See Appendix A for a list of labels and their relationships to one another.
Compound identification
A fundamental part of a pharmaceutical information platform, in general terms but more specific to this application is the compound registry. The registry provides a unique identifier for a compound, which enables other entities (batch, sample) as a test article in an assay experiment. Establishing a registry involves business rules for drawing sketches, standardizing compounds, normalizing forms, and handling enhanced stereochemistry [17]. As proposed by Hersey and others [18], we subscribe to use a process consistently rather than attempt to curate chemical databases across collaborators. With this in mind, we employed a process-driven registration approach that enables data exchange between organizations where identical compounds are drawn differently.
The BioChemUDM is based purely on the chemical connection table with enhanced stereochemistry, so a compound identifier is based solely on a sketch . With one assumption about stereocenters (unspecified means mixture), there is no need for complicated business rules or controlled vocabularies to distinguish a compound. Interpretation of sketches using normalization techniques lends itself well to the language of the chemist, who is skilled in drawing molecules. Upon compound registration of a sketch, the following fields are key for data sharing.
The field ‘Compound’ is part of the molecule (MOL) object:
UDM.MOL.Compound – Connection table with enhanced stereochemistry.
The field ‘Num’ is the unique identifier for the compound:
UDM.MOL.Num – Registry number assigned by the system, e.g., 7.
The field ‘Name’ is a globally unique identifier, typically denoted by an organizational prefix:
UDM.MOL.Name – A organizational prefix with registry number, e.g., IDYA-7.
To enable cross-references between multiple organizations, the field ‘Synonyms’ extends the MOL object of the BioChemUDM:
UDM.MOL.Synonyms – An array UDM.MOL.Name values (from other registries).
To allow for verification of the compound, the MOL object represents additional unique identifiers:
UDM.MOL.InChI – A non-proprietary identifier for chemical substances.
UDM.MOL.InChIKey – A compact chemical identifier derived from InChI.
UDM.MOL.IUPAC – A name for organic compounds as recommended by IUPAC.
One aspect of compound identification that deserves attention is handling of tautomers. To wit, a study of commercially available samples shows the same compound may be sold as different products at different costs [21]. Tautomerism is complicated [19] and steps to address tautomers in compound registration have been described previously [20]. Our normalization technique for tautomeric states entering the UDM is based on recent work from Dhaked and others [22]. For each compound, a set of business rules, i.e., SMIRKS patterns [23], is applied to afford a canonical tautomer for registration. These patterns, originally developed for use with the CACTVS tools [24], are adapted for use in RDKit toolkit [25] and Pipeline Pilot™ (PLP) software [26]. Furthermore, recent work by Baker and others demonstrates that use of an energy function to establish these business rules may yield tautomers in better agreement with experimental observations [27]. See Appendix B for details on the canonical tautomer implementation.
Drawing rules
There are three kinds of enhanced stereochemistry sketches: mixture, relative, and absolute.
The sketches shown in Fig. 1 naturally support the kinds of compounds synthesized during compound progression. We may begin with a stereochemical mixture, then progress to a separation of isomers, and end with a determination of chirality. In practice, these three kinds may be implied by the following sketches in Fig. 2.
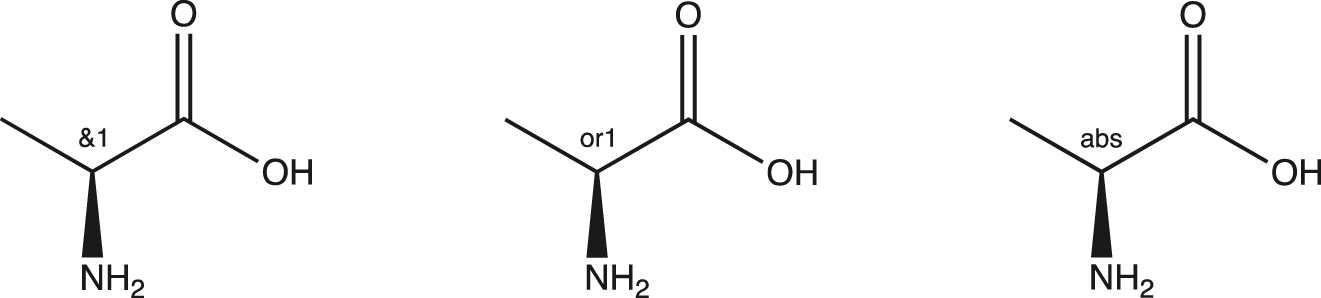
Sketch of mixture (DL), relative (D or L), absolute (L-alanine).
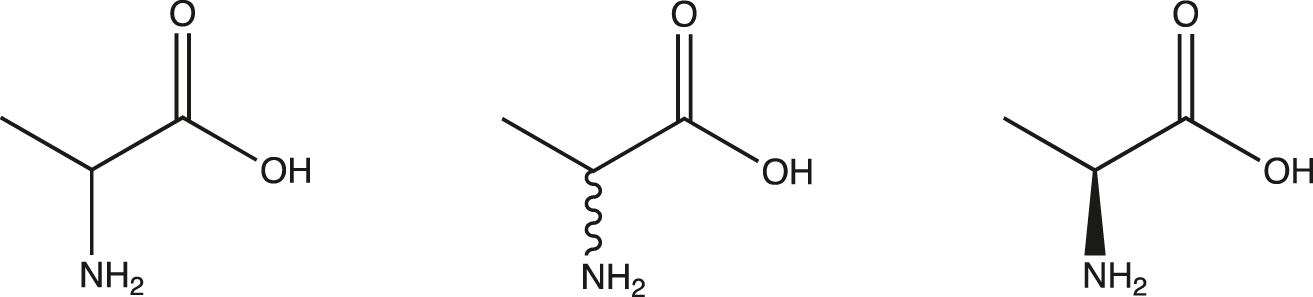
Sketch of straight bond, wiggly bond, wedge/hash bond.
Using an industry standard drawing tool [28], some chemists naturally draw standard sketches (Fig. 1) while others prefer shorthand sketches (Fig. 2). Therefore,
a “straight bond” means a mixed stereocenter
a “wiggly bond” means a stereocenter is enantiopure but not determined
a chiral center without designation means it is absolute.
Compound KeyVal
Using the Morgan Algorithm [29], CIP rules [30], and PLP scripting language [31], we produced a ‘Perceive Structure & Stereo’ component to promote straight bonds, enumerate wiggly bonds, and ensure proper representation of relative stereochemistry, pseudo-stereoisomers, atropisomers, global chirality, and meso compounds. The PP component prevents invalid registrations and is amenable to implementation in KNIME™ script [32]. The component exposes a categorical chirality attribute called StereoKeyVal for informational purposes.
The field ‘StereoKeyVal’ is part of the MOL object:
UDM.MOL.StereoKeyVal – Concatenation of chirality attributes (output of perception).
Format: {chiral} {isomer} {ee} {member} {meso}
{chiral} – T or F, depending on whether the compound is chiral (rotates light).
{isomer} – A, D, E, N
A – Atropisomer
D – Diastereomer
E – Enantiomer
N – Not a stereoisomer
{ee} – 0 or 1, a Boolean depending on whether stereo excess can define a mixture.
{member} – M or P for atropisomers, A or B for pseudo-stereo compounds, otherwise X.
{meso} – T or F, depending on whether the compound is a meso form (no mirror image).
Batch identification
For a given compound, many batches may be synthesized. So, the BioChemUDM for batches depends on the compound and each batch is identified by registration order. For example, the seventh compound registered would be IDYA-7 and the third batch of that compound would be IDYA-7-3.
The field ‘Num’ on the Batch (BAT) object is the unique identifier for the batch (in this registry):
UDM.MOL.BAT.Num – Sequential number assigned by the system, e.g., 3.
The field ‘Name’ is a globally unique batch identifier, by extending the compound identifier.
UDM.MOL.BAT.Name – The compound name and the batch integer, e.g., IDYA-7-3.
The field ‘ExperimentID’ is a reference to an electronic laboratory notebook (ELN) experiment.
UDM.MOL.BAT.ExperimentID – The link to the source ELN experiment.
The field ‘Source’ is the owner/author of the ELN experiment.
UDM.MOL.BAT.Source – The controlled vocabulary (list) of collaborators.
The field ‘Tag’ is the organizational project code to associate a batch with a project.
UDM.MOL.BAT.Project – The controlled vocabulary (list) of projects.
To ensure an immutable batch identifier, we do not include salt form as part of the batch identifier thus avoiding the inevitable problem of an incorrect salt form assignment. Instead, we opt to define salt form as an independent data label (UDM.MOL.BAT.Salt). See Appendix C for the complete list of batch fields.
Sample identification
For a given batch, many samples may be containerized, thus, the BioChemUDM for samples depends on the batch and each sample is identified by registration order. In this way, an immutable sequential number starting at 1 can be used for compounds, batches, and samples and everything else is an attribute thereof.
The field ‘Num’ on the Sample (SAM) object is the unique identifier for the batch (in this registry):
UDM.MOL.BAT.SAM.Num – Sequential number assigned by the system, e.g., 1.
The field ‘Name’ is an extension of the globally unique compound identifier:
UDM.MOL.BAT.SAM.Name – The batch name and sample integer, e.g., IDYA-7-3-1.
The field ‘ID’ is the barcode associated with the sample container.
UDM.MOL.BAT.SAM.ID – The barcode associated with the sample container.
The field ‘Type’ is a controlled vocabulary (list) of container types (e.g., dram1, dram4).
UDM.MOL.BAT.SAM.Type – The type of sample container
The field ‘Location’ is a controlled vocabulary (list) of inventory endpoints.
UDM.MOL.BAT.SAM.Location – The current location of the container.
The field ‘Form’ is a controlled vocabulary (list) describing the formulation of the sample (e.g., neat, solution).
UDM.MOL.BAT.SAM.Form – The formulation of the sample.
The field ‘Amount’ is the quantity of a neat sample (without weight of the container).
UDM.MOL.BAT.SAM.Amount (mg) – The sample quantity measured in milligrams.
The field ‘Volume’ is the quantity of a solution sample.
UDM.MOL.BAT.SAM.Volume (mL) – The sample quantity measured in milliliters, used when UDM.MOL.BAT.SAM.Form=solution.
The field ‘Conc’ is the concentration of a solution sample.
UDM.MOL.BAT.SAM.Conc (uM) – The sample concentration measured in micromoles/liter, used when UDM.MOL.BAT.SAM.Form=solution.
Assay identification
Another fundamental part of the information platform is the assay definition. Ontology concepts serve as intentionally broad categories for organizing assays. We propose the following protocol categories for organizing pharmaceutical data:
Activation
Binding
Induction
Inhibition
Oxidation
Permeability
Pharmacokinetics
PhysChemProperty
Stability
Toxicity
Protocol categories are beneficial because similar assays can be treated in the same way. Within each category are fields to distinguish one assay from another. For example, two kinds of activation are distinguished by the conditional field called ‘Target’ (set to AhR or PXR). To simplify the problem, we represent unpivoted data within the categories.
Each protocol category includes a common set of fields:
UDM.ASY.Name – The controlled vocabulary (list) of protocol names (shown above).
UDM.ASY.Protocol – The author and version number of the protocol, e.g., IDYA/v1.
UDM.ASY.Date – The run date of the experiment. For multi-day experiments, the start day.
UDM.DOC.Query – A query string reference to documents associated with the experiment.
UDM.ELN.Query – A query string reference to the source ELN experiment.
UDM.COM.Contact – A controlled vocabulary (list) of internal points of contacts.
UDM.PRJ.Code – A controlled vocabulary of project identifiers.
UDM.ASY.Identifier – A controlled vocabulary (list) of assay identifiers.
UDM.ASY.TST.Article – A test article, e.g., a compound, batch, or sample identifier.
UDM.ASY.DAT.Control – A reference to a control test article.
UDM.ASY.DAT.Value – The readout for the control.
UDM.ASY.DAT.Comment – A description of the control value or unit of measure.
UDM.ASY.CND.Format – A controlled vocabulary (list) of assay formats.
UDM.ASY.CND.Target – A controlled vocabulary (list) of assay targets.
Each protocol contains result types and specific conditions. For example, the remainder of the ‘Activation’ protocol is:
UDM.ASY.CND.Conc (uM) – The concentration of the additive/stimulant (specified in the UDM.ASY.CND.Format field)
UDM.ASY.RES.Emax (%) – The maximum fold activation.
UDM.ASY.RES.FoldActive – The fold-change over baseline.
UDM.ASY.RES.EC50 (uM) – The concentration associated with half-maximal effectiveness.
The ‘Activation’ protocol defines two assays: AhR and PXR. See Appendix D for the complete list of protocol fields, controlled vocabularies, and templates.
Discussion
Reducing compound identity to its enhanced stereochemical connection table has benefits and consequences. By basing the identity on a sketch only, we eliminate user-specified terms to describe stereochemistry and register compounds based purely on chemical graph theory [33]. This means that we can register compounds from disparate data sources using the extended connection table (V3000) format [15] or a canonical identifier derived from it. Consequently, this requires adoption of the above drawing rules and commitment to representing stereo-enhanced sketches in the V3000 format. Additionally, ChemAxon CXSMILES [34] was amenable to a canonical identifier, whereas InChI [35] was not due to issues regarding tautomers [36] and lack of support of enhanced stereochemistry logic [37].
Constructing string labels to describe data about compounds and assay is simplistic however widespread adoption by multiple companies is noteworthy. Over the past year, the BioChemUDM has been implemented with multiple collaborators using the CDD Vault [38]. With support for mixtures, unknown absolute configuration and atropisomers [39], compound sketches can be systematically registered and assay results can be automatically captured into databases. Given the adoption of the BioChemUDM by several popular contract research organizations (CROs), we anticipate that any new biotech startup or academic group can quickly establish data sharing with compound registration and assay capture. Furthermore, this approach scales well with the addition of new assays and protocol versions.
This work is the result of following the FAIR guiding principles [40]. A persistent identifier for objects (MOL, BAT, SAM) is achieved with a registration system to provide a unique number for an object (e.g., UDM.MOL.Num). Such identifiers become globally unique with an agreed upon organizational prefix. Importantly, metadata clearly and explicitly include the identifier of the data they describe. For interoperability, the use of controlled vocabularies for fields and values follows FAIR principles. Finally, compounds are described with a plurality of accurate and relevant attributes, to enable verification of a compound. As a result, we find collaborators willing to share this approach because it enables machine-actionability leading to practical benefits of saving time and avoiding errors.
We have described an approach to represent compounds and assay data from multiple, disparate sources using an extension of the Unified Data Model (BioChemUDM) concept. This data model, like its predecessor, has been born out of necessity. Compounds from various sources need to be registered and assay data from various sources need to be captured into databases plus collaborators need to update their databases as well. By enlisting collaborators to adopt the BioChemUDM for compounds and assays, coupled with a few powerful scripting components, we can receive data from CROs and share data with other research groups within the same day.
Future directions
It is our hope that the BioChemUDM, like the RxnUDM, will be embraced and incorporated into the UDM. Support for assay data was discussed as part of the Pistoia Alliance UDM project but has not yet materialized due to lack of time and different scope of the project team (Tomczak, J. personal communication, October 14, 2021). We aim to expand the BioChemUDM to include concentration-response assays and surface plasmon resonance experiments, plus integrate raw data from instruments. Additionally, we aim to automate the continuous adaptation of the BioChemUDM with a data lake architecture [41]. Lastly, we aim to configure and launch multiple vendor applications from our data lake based on the BioChemUDM.
Acknowledgments
Special thanks to Sandra Simon (IDEAYA Biosciences) and Jacob Spiegel (Workflow Informatics) for supporting the effort to launch the platform based on the BioChemUDM and assistance with writing this manuscript.
References
[1] F. Agnetti, M. Bensch, H. Biller, M. Blapp, B. Cheikh, G. Blanke, J. Degen, B. Dienon, T. Doerner, G. Doernen, F. Farshchian, W. Gotzeina, P. Hilty, R. Horstmoeller, T. Jeker, B. Jones, M. Kappler, A. Momin, A. Regoli, D. Ribaud, B. Starck, D. Stoffler, K. Weymann, P. Udupa. Intuitive and integrated browsing of reactions, structures, and citations: The Roche experience. In 245th National Meeting of the American Chemical Society, New Orleans, LA, April 7–11, (2013), https://www.nextmovesoftware.com/news/KapplerPoster2013.pdf (accessed Sep 9, 2021).Search in Google Scholar
[2] R. Sayle, D. Lowe, N. O’Boyle, M. Kappler, A. Pelliccioli, N. Tomkinson, D. Stoffler. Extraction, analysis, atom mapping, classification and naming of reactions from pharmaceutical ELNs. 6th Joint Sheffield Conference on Cheminformatics, July 22–24, (2013), https://cisrg.shef.ac.uk/shef2013/showabstract.php?id=56 (accessed Oct 10, 2021).Search in Google Scholar
[3] Elsevier Press Release. Elsevier and Roche Collaborate to Integrate Proprietary Chemistry Data in Reaxys®, Elsevier GmbH, Frankfurt, Germany (2012), https://www.elsevier.com/about/press-releases/archive/science-and-technology/elsevier-and-roche-collaborate-to-integrate-proprietary-chemistry-data-in-reaxys. (accessed Apr 19, 2020).Search in Google Scholar
[4] T. Hoctor, M. Kappler. Making Dollars and Sense from Large Data, Global Drug Discovery Informatics Summit, Princeton, NJ (2013).Search in Google Scholar
[5] Elsevier Press Release. Elsevier Donates Unified Data Model to The Pistoia Alliance, Facilitating Data Sharing and Accelerating Research in the Life Sciences, Elsevier GmbH, Frankfurt, Germany (2017), https://www.elsevier.com/about/press-releases/archive/clinical-solutions/elsevier-donates-unified-data-model-to-the-pistoia-alliance,-facilitating-data-sharing-and-accelerating-research-in-the-life-sciences (accessed Oct 10, 2021).Search in Google Scholar
[6] XML 1.0 Specification. World Wide Web Consortium (2008), https://www.w3.org/TR/REC-xml/https://en.wikipedia.org/wiki/XML (accessed May 23, 2022). “The Extensible Markup Language is a markup language and file format for storing, transmitting, and reconstructing arbitrary data. It defines a set of rules for encoding documents in a format that is both human-readable and machine-readable.” (accessed May 23, 2022).Search in Google Scholar
[7] CDXML Format. CDXML is the XML Analogue of the Binary CDX File Type used by CambridgeSoft Corporation’s ChemDraw Chemical Structure Application (2020), https://en.wikipedia.org/wiki/CDXML (accessed May 23, 2022).Search in Google Scholar
[8] JSON Format. JSON is an open standard file format and data interchange format that uses human-readable text to store and transmit data objects consisting of attribute–value pairs and arrays (or other serializable values) (2001), https://en.wikipedia.org/wiki/JSON (accessed May 23, 2022).Search in Google Scholar
[9] Pistoia Alliance News. The Pistoia Alliance announces major milestone in unified data model project to promote life sciences collaboration, The Pistoia Alliance is a Global Not-for-Profit Members’ Organization Collaborating to Lower Barriers to Innovation in Life Science and Healthcare R&D, Elsevier, Boston, MA (2018), https://www.pistoiaalliance.org/news/udm-release-v5/ (accessed Oct 10, 2021).Search in Google Scholar
[10] T. Struble, J. Alvarez, S. Brown, M. Chytil, J. Cisar, R. DesJarlais, O. Engkvist, S. Frank, D. Greve, D. Griffin, X. Hou, J. Johannes, C. Kreatsoulas, B. Lahue, M. Mathea, G. Mogk, C. Nicolaou, A. Palmer, D. Price, R. Robinson, S. Salentin, L. Xing, T. Jaakkola, W. Green, R. Barzilay, C. Coley, K. Jensen. J. Med. Chem. 63, 8667 (2020), https://doi.org/10.1021/acs.jmedchem.9b02120.Search in Google Scholar PubMed PubMed Central
[11] E. Cascade, C. Sears. Leveraging a Unified Data Model to Drive Collaboration and Clinical Trial Efficiency, Applied Clinical Trials (2018), https://www.appliedclinicaltrialsonline.com/view/leveraging-unified-data-model-drive-collaboration-and-clinical-trial-efficiency (accessed Oct 10, 2021).Search in Google Scholar
[12] J. Tomczak, E. Herzog, M. Fisher, J. Swienty-Busch, F. van den Broek, G. Whittick, M. Kappler, B. Jones, G. Blanke. UDM (Unified Data Model) for Chemical Reactions – Past, Present and Future, this issue.Search in Google Scholar
[13] A. Dalby, J. Nourse, W. Hounshell, A. Gushurst, D. Grier, B. Leland, J. Laufer. J. Chem. Inf. Comput. Sci. 32, 244 (1992), https://doi.org/10.1021/ci00007a012.Search in Google Scholar
[14] CTfile Formats, Elsevier (2005), http://c4.cabrillo.edu/404/ctfile.pdf (accessed Oct 10, 2021).Search in Google Scholar
[15] CTfile Formats. Dassault Systemes (2016), http://help.accelrysonline.com/ulm/onelab/1.0/content/ulm_pdfs/direct/reference/ctfileformats2016.pdf (accessed Oct 10, 2021).Search in Google Scholar
[16] Y. Shafranovich. RFC 4180: Common Format and MIME Type for CSV Files, IETF (2005).Search in Google Scholar
[17] E. Martin, A. Monge, J.-A. Duret, F. Gualandi, M. Peitsch. J. Cheminform. 4, 11 (2012).10.1186/1758-2946-4-11Search in Google Scholar PubMed PubMed Central
[18] A. Hersey, J. Chambers, L. Bellis, A. Bento, A. Gaulton, J. Overington. Drug Discov. Today Technol. 14, 17 (2015).10.1016/j.ddtec.2015.01.005Search in Google Scholar PubMed PubMed Central
[19] R. Sayle. J. Comput. Aided Mol. Des. 24, 485 (2010).10.1007/s10822-010-9329-5Search in Google Scholar PubMed
[20] A. Gobbi, M.-L. Lee. J. Chem. Inf. Model. 52, 285 (2012), https://doi.org/10.1021/ci200330x.Search in Google Scholar PubMed
[21] L. Guasch, W. Yapamudiyansel, M. Peach, J. Kelley, J. JBachiJr, M. Nicklaus. J. Chem. Inf. Model. 56, 2149 (2016).10.1021/acs.jcim.6b00338Search in Google Scholar PubMed PubMed Central
[22] D. Dhaked, M. Nicklaus. Tautomeric conflicts in forty small-molecule databases (2021), ChemRxiv Cambridge Open Engage. This content is a working paper (preprint) and has not been peer-reviewed.10.26434/chemrxiv.14779254Search in Google Scholar
[23] SMIRKS – A Reaction Transform Language. Daylight Theory Manual (1997), https://www.daylight.com/dayhtml/doc/theory/theory.smirks.html (accessed May 24, 2022).Search in Google Scholar
[24] W. Ihlenfeldt, Y. Takahashi, H. Abe, S. Sasaki. J. Chem. Inf. Comput. Sci. 34, 109 (1994).10.1021/ci00017a013Search in Google Scholar
[25] RDKit. Open-Source Cheminformatics, https://www.rdkit.org.Search in Google Scholar
[26] BIOVIA Pipeline Pilot. Release 21.2.0.2574, Dassault Systèmes, San Diego (2021).Search in Google Scholar
[27] C. Baker, N. Kidley, K. Papachristos, M. Hotson, R. Carson, D. Gravestock, M. Pouliot, J. Harrison, A. Dowling. J. Chem. Inf. Model. 60, 3781 (2020).10.1021/acs.jcim.0c00232Search in Google Scholar PubMed
[28] PerkinElmer Announcement. ChemDraw/ChemOffice+ Cloud v20.0 (2020), https://informatics.perkinelmer.com/Support/SupportNews/details/?SupportNews=290 (accessed Oct 10, 2021).Search in Google Scholar
[29] H. Morgan. J. Chem. Doc. 5, 107 (1965), https://doi.org/10.1021/c160017a018.Search in Google Scholar
[30] R. Cahn, C. Ingold, V. Prelog. Angew. Chem. Int. Ed. 5, 385 (1966), https://doi.org/10.1002/anie.196603851.Search in Google Scholar
[31] BIOVIA Pilotscript. Release 2016, Dassault Systèmes, San Diego (2016), http://help.accelrysonline.com/ulm/onelab/1.0/content/ulm_pdfs/pipeline%20pilot/other/pilotscript.pdf (accessed Oct 10, 2021).Search in Google Scholar
[32] Scripting Integrations. KNIME Community (2019), https://www.knime.com/community/scripting (accessed Oct 10, 2021).Search in Google Scholar
[33] D. Bonchev. Chemical Graph Theory: Introduction and Fundamentals, Routledge (1991).Search in Google Scholar
[34] ChemAxon Documentation. ChemAxon Extended SMILES and SMARTS – CXSMILES and CXSMARTS, https://docs.chemaxon.com/display/docs/chemaxon-extended-smiles-and-smarts-cxsmiles-and-cxsmarts.md (accessed Oct 10, 2021).Search in Google Scholar
[35] S. Heller, A. McNaught, I. Pletnev, S. Stein, D. Tchekhovskoi. Journal of Cheminformatics 7, 23 (2015), https://doi.org/10.1186/s13321-015-0068-4.Search in Google Scholar PubMed PubMed Central
[36] D. Dhaked, W. Ihlenfeldt, H. Patel, V. Delannee, M. Nicklaus. J. Chem. Inf. Model. 60, 1253 (2020).10.1021/acs.jcim.9b01080Search in Google Scholar PubMed PubMed Central
[37] W. DeGruyter. Chem. Int. 42, 1, 30 (2020).Search in Google Scholar
[38] USPTO Reg. No. 3,884,839, CDD Vault, registered December 7, 2010. https://tsdr.uspto.gov/documentviewer?caseId=sn77791158&docId=ORC20101207004753.Search in Google Scholar
[39] L. Fisher. CDD Support: Advanced Stereochemistry Registration: Atropisomers, Mixtures, Unknowns and Non-Tetrahedral Chirality, https://support.collaborativedrug.com/hc/en-us/articles/360020872171-Advanced-Stereochemistry-Registration-Atropisomers-Mixtures-Unknowns-and-Non-Tetrahedral-Chirality (accessed Oct 10, 2021).Search in Google Scholar
[40] M. Wilkinson, M. Dumontier, I. Aalbersberg, G. Appleton, M. Axton, A. Baak, N. Blomberg, J. W. Boiten, L. da Silva Santos, P. Bourne, J. Bouwman, A. Brookes, T. Clark, M. Crosas, I. Dillo, O. Dumon, S. Edmunds, C. Evelo, R. Finkers, A. Gonzalez-Beltran, A. Gray, P. Groth, C. Goble, J. Grethe, J. Heringa, P. Hoen, R. Hooft, T. Kuhn, R. Kok, J. Kok, S. Lusher, M. Martone, A. Mons, A. Packer, B. Persson, P. Rocca-Serra, M. Roos, R. van Schaik, S.-A. Sansone, E. Schultes, T. Sengstag, T. Slater, G. Strawn, M. Swertz, M. Thompson, J. van der Lei, E. van Mulligen, I. Velterop, A. Waagmeester, P. Wittenburg, K. Wolstencroft, J. Zhao, B. Mons. Sci. Data 3, 160018 (2016), https://doi.org/10.1038/sdata.2016.18.Search in Google Scholar PubMed PubMed Central
[41] AWS Lake Formation. What is a Data Lake? https://aws.amazon.com/big-data/datalakes-and-analytics/what-is-a-data-lake/ (accessed Oct 10, 2021).Search in Google Scholar
Supplementary Material
The online version of this article offers supplementary material (https://doi.org/10.1515/pac-2021-1004).
© 2022 IUPAC & De Gruyter. This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License. For more information, please visit: http://creativecommons.org/licenses/by-nc-nd/4.0/
Articles in the same Issue
- Frontmatter
- Editorial
- Cheminformatics: data and standards a Pure and Applied Chemistry special issue
- Invited papers
- Ontologies4Chem: the landscape of ontologies in chemistry
- IUPAC specification for the FAIR management of spectroscopic data in chemistry (IUPAC FAIRSpec) – guiding principles
- Launching a materials informatics initiative for industrial applications in materials science, chemistry, and engineering
- Analysing a billion reactions with the RInChI
- Reaction SPL – extension of a public document markup standard to chemical reactions
- RSC CICAG Open Chemical Science meeting: integrating chemical data from two symposia and a series of workshops
- UDM (Unified Data Model) for chemical reactions – past, present and future
- An overview of the JCAMP-DX format
- Data format standards in analytical chemistry
- BioChemUDM: a unified data model for compounds and assays
Articles in the same Issue
- Frontmatter
- Editorial
- Cheminformatics: data and standards a Pure and Applied Chemistry special issue
- Invited papers
- Ontologies4Chem: the landscape of ontologies in chemistry
- IUPAC specification for the FAIR management of spectroscopic data in chemistry (IUPAC FAIRSpec) – guiding principles
- Launching a materials informatics initiative for industrial applications in materials science, chemistry, and engineering
- Analysing a billion reactions with the RInChI
- Reaction SPL – extension of a public document markup standard to chemical reactions
- RSC CICAG Open Chemical Science meeting: integrating chemical data from two symposia and a series of workshops
- UDM (Unified Data Model) for chemical reactions – past, present and future
- An overview of the JCAMP-DX format
- Data format standards in analytical chemistry
- BioChemUDM: a unified data model for compounds and assays