The exploration of deoxygenation reactions for alcohols and derivatives using earth-abundant reagents
-
Miao Tian


Abstract
In Earth matter evolution, the deoxygenation process plays a central role as plant and animal remains, which are composed by highly oxygenated molecules, were gradually deoxygenated into hydrocarbons to give fossil fuels deep in the Earth crust. The understanding of this process is becoming crucial to the entire world and to the sustainable development of mankind. This review provides a brief summary of the extensive deoxygenation research under mild, potentially sustainable conditions. We also summarize some challenges and opportunities for potential deoxygenation reactions in the future.
Introduction
After the first industrial revolution [1], the invention of steam engines marks the beginning of “fossil era” where the use of fossil energy [2], [3], [4] greatly accelerates economic development [5], [6], [7]. Serving as the industrial foundation for modern human prosperity [8], [9], [10], [11], [12], fossil energy reservoir has enabled many countries and regimes to become superpowers [13], [14], [15], [16], [17]. As fossil fuel generation is naturally achieved by burying the remains of ancient animals and plants, which are composed of highly oxygenated molecules [18], [19], [20], [21], [22], [23], [24] and essentially transformed into hydrocarbons in the Earth crust [25], [26], [27], [28], [29], [30], [31], [32], the transformation from animal and plant remains to fossil energy in Earth crust is technically a chemical process where oxygenated organic molecules are deoxygenated and recombined into hydrocarbons [33]. Therefore, the deoxygenation process plays a critical role in the preservation of Earth energy resources [34]. To further understand the detailed mechanism of this process, and to reproduce this process in relatively short period of time, is of great significance to the entire human society [35], [36], [37], [38], [39], [40], [41].
Generally speaking, the deoxygenation reaction involves the decrease in chemical valence for atoms in the organic molecule [42, 43]. Therefore, reductive reaction conditions are usually required [44, 45]. Although this type of reaction generally falls in the larger research area of C-O bond activation or reductive coupling/rearrangement, which have been reviewed thoroughly by many excellent literatures [46], [47], [48], [49], [50], many of these reactions require relatively harsh reaction conditions such as stoichiometric amount of metallic or phosphine/boron reagents [51], [52], [53], [54], [55], [56]. To the best of our knowledge, reviews on specific deoxygenation processes, which target inactivated substrates and earth-abundant conditions, is rarely found. In this review, we highlighted the recent progress in deoxygenation reactions of inactivated alcohols and their derivatives, which involves the use of earth-abundant reagents. Reactions involving certain organometallic reagents or reagents that are not stable in the Earth crust will not be covered. In general, we have characterized the desired deoxygenation reactions into: (1) Direct deoxygenation strategies for alcohols [57], [58], [59], [60], [61], [62], [63], [64], [65]; (2) The study of lignin model compounds derived from the degradation and transformation of lignin [66], [67], [68], [69], [70], [71], [72].
Deoxygenation of alcohols with potentially earth-abundant reagents
As a relatively stable class of compounds which can be widely found in nature [73], alcohols, known to be inert to many reductive conditions thanks to their generally saturated bond structure and hard-to-leave –OH group [74], [75], [76], can now be readily deoxygenated by many mild reduction conditions using potentially Earth-abundant reagents, generating the corresponding deoxygenated alkanes. Those impressive research have also broadened our knowledge regarding deoxygenation processes, offering us new tools for the facile transformations of oxygenated compounds.
Since phenols are oxygen-containing derivatives as important as alcohols, their wide practicability and structural diversity make them the most extensive substrates in transition metal catalysis. In 2012, Han’s group developed a highly stable PyBrop-NiCl2(dppp) catalytic system to build a one-pot construction of C–P bonds from electron-deficient phenols via the in situ aryl C–O activation. The method shows a wide range of feasibility, not only for various phenol derivatives, but also for different types of phosphorus nucleophiles, opening a new avenue for the C–P bond forming reaction and more efficient protocols for C–P coupling of phenols (Scheme 1) [77].
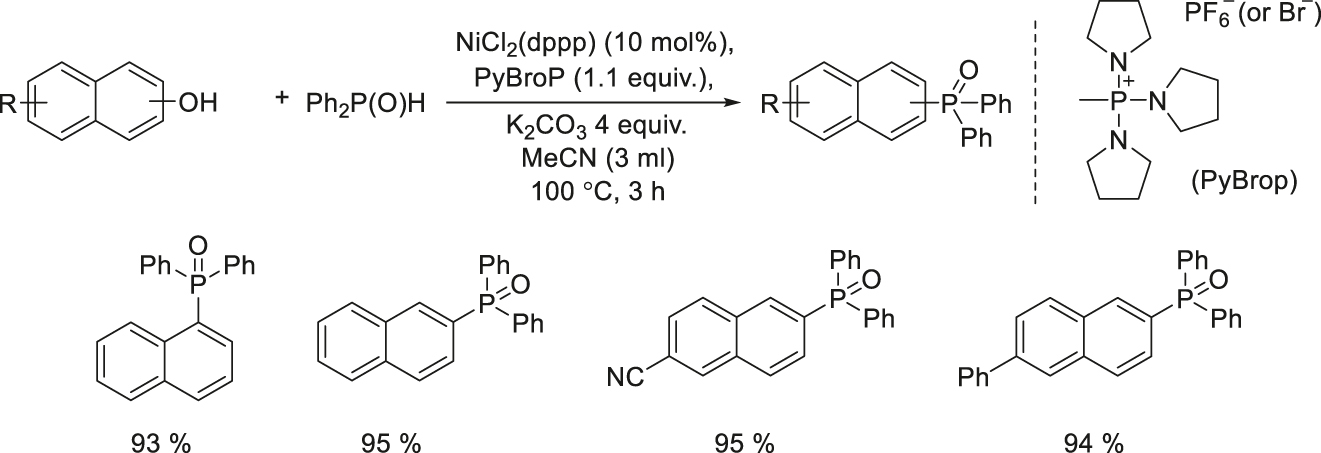
Ni-catalyzed one-pot deoxygenation of phenols via the C–O activations.
In 2014, Ollivier and coworkers reported that a novel photocatalyzed deoxygenation method based on the activation of Ir(ppy)3 under reduced conditions by visible light. The method realizes the photocatalytic redox deoxygenation reaction with the thiocarbamate precursor under activation of visible light to convert secondary and tertiary aliphatic and benzyl alcohols into corresponding alkanes. This approach features high efficiency and good functional group tolerance, it will open new perspectives to transformations in the fields of organic synthesis and polymer sciences (Scheme 2) [78].

Visible light photocatalytic reduction of O-thiocarbamates.
In the past, most of the methods for reducing C–O bonds to C–C bonds not only require harsh reaction conditions but also add a large amount of reducing agents, which leads to a decrease in the selectivity of the reaction and the accompanying production of large amount of toxic waste. Therefore, catalytic transfer hydrogenolysis is an attractive alternative. In 2018, Fleischer’s group first achieved the homogeneous palladium catalyzed transfer hydrogenolysis of benzylic alcohols, using formic acid as a sustainable reductant agent. The method has broad applicability for primary, secondary, and tertiary benzylic alcohols, at the same time, a more selective and active homogeneous catalyst system has been developed (Scheme 3) [79].

Palladium-catalyzed transfer hydrogenolysis of benzylic alcohols by formic acids.
Since palladium heterogeneous catalysis is simple to recycle in multiple reaction cycles, it has economic and environmental advantages. In this context, Córdova and co-workers developed an asymmetric carbocyclizations reaction. In other cascade reactions, simple chiral amine co-catalysts are used to achieve heterogeneous palladium catalysts. Proposed an effective palladium catalysis transfer hydrogenolysis of primary, secondary, and tertiary benzylic alcohols by formic acid as a hydrogen source. The reaction obtains the corresponding products with high yield and chemical selectivity. It is worth noting that the palladium catalyst can be effectively continuously recycled without loss of catalytic activity. The future research and industrial application of these heterogeneous catalyst systems are of great significance (Scheme 4) [80].

Palladium-catalyzed transfer hydrogenolysis of benzylic alcohols.
The previously reported methods include transition metal-catalyzed hydrogenation and acid-catalyzed reduction or mediated reduction of hydride species. The site selectivity of deoxygenation will vary with conditions and reagents. Although based on oxidative dehydrogenation and subsequent Wolff–Kishner reduction catalyzed by iridium, ruthenium and manganese, the deoxygenation of primary alcohols was selectively achieved. But the scope and site selectivity of the hydroxyl group constitute the main problem that has not yet been completely resolved. In the same year, Xu’s group reported that a highly efficient, and site-selective deoxygenation of alcohols method based on iridium-catalyzed, under the assistance of an N-substituted (amino)aryl DG. The process was in mild conditions, has good functional tolerance, to provide a new route for the deoxygenation of alcohols (Scheme 5) [81].

Iridium-catalyzed highly efficient and site-selective deoxygenation of alcohols.
In 2020, Schafer and co-worker reported the catalytic oxidation/reductive coupling of alcohols with vanadium pyridonate complexes. The involvement of bimetallic species, coupled with ligand hemilability and metal-ligand cooperativity enables deoxygenated transformations of renewable feedstocks, this work provides additional evidence for early mechanical proposals for alcohol reduction coupling (Scheme 6) [82].

Vanadium pyridonate catalyzed reductive coupling of alcohols.
| Entry | R | Time (h) | Product yield (%) | Product yield (%) |
|---|---|---|---|---|
| 1 | Ph | 24 | >99 | 88 |
| 2 | Me | 48 | 65 | 66 |
| 3 | H | 48 | 34 | 31 |
The deoxygenation of lignin under more sustainable conditions
Ever since the realization of fossil fuel depletion crisis, the great social demand of aromatic compounds, which is historically produced from petroleum exclusively [83, 84], is constantly calling for sustainable alternative source. It is important that in the pursuit of the most promising alternative aromatic source, scientist have been extensively targeting lignin, which is mainly composed by polymerized alcohol (tonquinol, synapol, coniferol, etc.) and is where petroleum can be originated from in Earth matter evolution after all [85], [86], [87]. With significant development in alcohol deoxygenation methodology under more and more sustainable conditions, many excellent studies on lignin deoxygenation have been conducted and the utilization of lignin as a promising fossil fuel replacement has been fast advancing in the sustainable avenue.
For lignocellulosic biofuels to be sustainable, lignin depolymerization to easy-to-handle aromatic monomers is of great importance. Lignin depolymerization has become one of the biggest obstacles to making full use of lignocellulosic biofuels as alternatives to fossil fuels. In 2010, Ellman’s group developed a redox neutral approach for tandem catalytic dehydrogenation/C–O bond cleavage to depolymerize lignin. In this method, the secondary alcohol is selectively pre-oxidized, and then the reductive C–O ether bond is broken to cleave the most abundant β-O-4 unit, thereby realizing effective lignin depolymerization (Scheme 7) [88].

Catalytic C–O bond cleavage of 2-aryloxy-1-arylethanols.
Although the conversion of lignin has great potential, the direct dehydroxylation of phenols is still a very challenging chemical conversion process. The most typical method of phenol dehydration requires the derivatization of the Ar–OH group with an electron-withdrawing group (EWG) to weaken the C–O bond with forming the corresponding arenes, but these methods usually produce stoichiometric amounts of non-recyclable waste. Rinaldi and a co-worker reported the first nonderivative method for phenol dehydroxylation to yield aromatics by using Raney Ni with isopropanol and H-BEA-35 as the hydrogen donor solvent under mild conditions. The process provided a new route for the conversion of lignin to arenes (Scheme 8) [89].

Dehydroxylation of phenols into arenes via catalytic tandem reactions.
The removal of covalently bonded oxygen in lignin by hydrodeoxygenation not only provides an opportunity to decompose the complex structure of lignin, but also increases the total energy density and the value of the resulting product. The difficulty with this type of reaction is that the strength of the C–O and C–C bonds are similar, which makes selective hydrodeoxygenation challenging relative to aromatic hydrogenation. In 2013, Omar’s and co-workers reported a bimetallic catalytic system based on metallic Pd nanoparticles (NP) on carbon and Zn(II) Lewis acid for the cleavage of β-O-4 lignin molecules. Under relatively mild conditions, the catalyst selectively cleaves the stubborn ether bonds of lignin dimers and polymers, removes alcohol oxygen atoms on the alkyl chain, and maintains the precious aromaticity of the raw materials (Scheme 9) [90].

Cleavage of C–O bonds relevant to lignin by Pd/Zn synergistic catalysis.
The β-O-4′ bonds was the most abundant structural unit in lignin, accounting for about 50–60% of natural lignin. Different pathways and mechanisms to catalyze the C–O bond cleavage of β-O-4′-ether bond have been proposed. In 2014, Samec’s group used formic acid as a reducing agent in the air to develope a selective palladium-on-carbon (Pd/C) catalyzed C–O bond cleavage of the β-O-4′-ether bond of lignin model compounds. the reaction was robust, mild, and efficient. The lignin-derived dimer model was efficiently transformed to the corresponding aryl ketone and phenolic compounds (Scheme 10) [91].

Palladium-catalyzed cleavage of β-O-4′-ether linkages of lignin.
In 2014, Stahl’s group developed a method to depolymerize oxidized lignin in an aqueous solution of formic acid under mild conditions, which achieved the depolymerization of lignin very effectively. Before depolymerization, poplar lignin was subjected to aerobic oxidation, then sodium formate was added to formic acid and reacted at 110 °C for 24 h. Obtained over 60 wt% of low molecular weight aromatic hydrocarbons, and did not consume formic acid during the depolymerization process. Obviously, the reaction pathway is different from hydrogenolysis mediated by metal catalysts. The work has further draws attention to biomass separation methods (Scheme 11) [92].

Depolymerization of oxidized lignin to aromatics by formic acids.
As we all know, the cleavage of high content C–O–C bonds is a key step to promote the depolymerization of lignin. The reduction and depolymerization of lignin using reducing agents such as hydrogen has shown the prospect of converting lignin into high-value aromatic compounds and liquid fuels. Then this process leads to very high infrastructure costs. In 2017, Xu’s group reported that reductive de-polymerization of kraft lignin with formic acid at low temperatures by inexpensive supported Ni-based catalysts. It is possible to use formic acid as an internal hydrogen source without using high-pressure hydrogen, which proves that catalytic reduction de-polymerization in ethanol/formic acid solution may be a promising method for producing liquid oil products (Scheme 12) [93].

Ni-based catalyzed reductive de-polymerization of kraft lignin with formic acids.
Formic acid as an efficient hydrogen donor molecule has been widely used to improve the depolymerization process of lignin. Dai’s group reported a novel method to achieve low-temperature hydrogenolysis of lignin by carbon-supported ruthenium and formic acid as reducing agent, at the same time, the effects of formic acid, Ru/C catalyst and aerobic oxidation conditions on the yield and quality of bio-oil were studied in detail. The method provided a possibility to depolymerize lignin under a low temperature (Scheme 13) [94].

Ruthenium catalyzed hydrogenolysis of lignin by formic acids as reducing agents.
Oxidation is one of the chemical reactions of lignin depolymerization. Catalysts including organometallic and metal oxides have been developed for depolymerization of oxidized lignin. Lu and co-workers developed a composite photocatalyst, CuFe2O4@rGO, to perform a sun-light assisted heterogeneous Fenton process to cleavage the β-O-4 bonds of lignin model compounds for the production of value-added at room temperature and environment pressure in the short reaction time of aromatic chemicals. The process was developed to produce high-value aromatic chemicals and important intermediates in the chemical industry (Scheme 14) [95].

Selective and efficient cleavage of lignin model compounds.
Compared with traditional transition metals, Rhenium has lower electronegativity. The lignin contains a large number of hydroxyl groups, and rhenium has an outstanding ability to remove hydroxyl groups through deoxygenation and dehydration. Because of its special ability to coordinate with diols to form esters, it found that rhenium can promote the deoxygenation and dehydration of polyols and alcohols to form olefins. In 2019, Li’s group used a heterogeneous rhenium catalyst to cleavage of lignin C-O bonds with hydrogen transfer reactions. It was shown that the potential applications with Re-based catalysts for the conversion of biomass (Scheme 15) [96].

Cleavage of lignin C–O bonds over heterogeneous rhenium catalysts.
In previous studies, although various functionalization and depolymerization methods of lignin have been reported, however, achieving high yield and selectivity under environmental conditions remains a challenge. In 2020, Stephenson’s group developed an organocatalytic method for photochemical C–O bond cleavage of lignin model compounds. The strategy of using organic photocatalysts to decompose oxidized lignin under mild conditions is of great value for the conversion of renewable biomass raw materials into useful chemicals for lignin research (Scheme 16) [97].

Photochemical lignin fragmentation by an organocatalytic approach.
In order to more effectively utilize the lignin produce valuable aromatic derivatives, Han’s group find that 4-ethyltoluene is a very valuable chemical, currently produced from fossil raw feedstocks. In 2020, they developed a one-pot catalytic process to produce 4-ethyltoluene from lignin or lignocellulose. This process was using an RhCl3-LiI-LiBF4 catalytic system through demethoxylation, depolymerization, and transmethylation to produce high value-added chemicals, it provides a new strategy for utilizing renewable resources to produce valuable aromatic compounds (Scheme 17) [98].

The production of 4-ethyltoluene from lignin by the directional valorization.
Copyright obtained from Wiley-VCH [98]
In addition, inspired by the achievements of lignin derivatives to synthesis amines, Han’s group also reported a direct strategy to synthesize 4-cyclohexylmorpholines from morpholines and aryl ethers (including several lignin model compounds) over Pd/C by H2 as the hydrogen resource. This method offers a new reference to developing robust catalytic for efficient lignin transformation (Scheme 18) [99].

Palladium-catalyzed synthesis of 4-cyclohexylmorpholines from the reductive coupling of lignin model compounds.
Photocatalytic alcohol deoxygenation with photosensitizing semiconductor
Ever since the emergence of photo-driven reactions, photochemistry has been serving as a powerful arsenal to overcome the activation energy barrier of reactions [100, 101] that can only be performed under less sustainable conditions traditionally. However, alcohols, as a generally saturated compound consists of mainly sp3 hybridized atoms, require photons with very short wavelength to afford the cleavage of their inert C–O bonds theoretically. Meanwhile, more options are being proposed as interdisciplinary collaborations are constantly being promoted nowadays. With new technology being introduced [102], [103], [104], photo-driven alcohol deoxygenation reactions can now be achieved under milder and ‘greener’ conditions.
In 2019, Liu and co-workers reported the application of photosensitizing semiconductor in the direct conversion of methanol to ethanol catalyzed by the c-plane of GaN NWs. This process is not required solvent, ligand, additive, heating, atmosphere, or pressurization, the mechanistic studies suggested the generation of a methyl carbene intermediate on the polar c-plane of the GaN NWs surface in this novel catalytic process. The direct conversion of the readily available methanol to the more user-friendly, less toxic, and broadly applicable ethanol would greatly contribute to solving the problems that the advancement of sustainable fuels (Scheme 19) [105].

Direct catalytic methanol-to-ethanol photoconversion via methyl carbene.
With the research on the mechanism of methyl carbene, in 2020, Liu and co-workers reported that methylations of simple alkanes and arenes using GaN NWs as a robust catalyst and methanol as the solvent to get the reaction high efficiency. Mechanistic studies suggested that the methanol is dehydrated into methyl carbene (:CH2) intermediate via a bimolecular mechanism. Making the originally complicated C–H methylation to simpler and cleaner, and provides new ideas for the research of C–H functionalization reaction (Scheme 20) [106].

Photocatalytic methylation of nonactivated sp3 and sp2 C–H bonds by methanol on GaN NWs.
Conclusions and outlook
In conclusion, we have summarized an overview for deoxygenation reactions of alcohols and derivatives. It was shown that alcohol deoxygenations, which were known to require relatively harsh conditions historically, can now be conducted in relatively mild reaction conditions thanks to innovative conditions or powerful catalysts. In addition, the deoxygenation of lignin has achieved great advancement with both various lignin model compounds and real-life lignin obtained from nature successfully deoxygenated to give value-added compounds or readily available biofuels. The deoxygenation reaction for certain small molecules can also be promoted with nano-semiconductor arrays under light.
The inspiring research and their promising result have left us not only enthusiasm for future sustainability of mankind, but also sparkled further scientific curiosity for the chemist community. Firstly, what is the real mechanism behind the Earth deoxygenation process that gave birth to billions of tons of fossil fuel reservoir? Secondly, are we able to readily re-create the deoxygenation reactions (of, for example, biomass, polyester waste, etc.) at industrial scale to readily produce sustainable materials and fuels? To address these remaining challenges, we first need to create a solid-state reaction environment with the design of innovative catalyst that recreate certain conditions in the Earth crust and eliminates the extensive consumption of solvent for traditional reactions. Once the reaction environment has been created, further optimization is necessary to facilitate the scale-up process. At this stage, innovative reaction conditions such as electrochemistry or photochemistry can be introduced to further accelerate the deoxygenation reaction. Those exciting research proposals could be of great interest to the chemist community. Many of them have already underway in our lab.
References
1. M. B. Gorawar, V. G. Balikai, P. P. Revankar, V. H. Khatawate. IOP Conf. Ser. Mater. Sci. Eng.955, 012072 (2020). https://doi.org/10.1088/1757-899x/955/1/012072.Search in Google Scholar
2. S. González-García, B. Mola-Yudego, R. J. Murphy. Int. J. Life Cycle Assess.18, 783 (2013). https://doi.org/10.1007/s11367-012-0536-2.Search in Google Scholar
3. J. Meng, Z. Li, J. Li, L. Shao, M. Han, S. Guo. Front Earth Sci.-Prc.8, 150 (2014). https://doi.org/10.1007/s11707-013-0397-4.Search in Google Scholar
4. B. A. Martin, P. D. Frymier. Appl. Biochem. Biotechnol.183, 503 (2017). https://doi.org/10.1007/s12010-017-2576-3.Search in Google Scholar PubMed
5. J. T. Mathews. Foreign Aff.68, 162 (1989). https://doi.org/10.2307/20043906.Search in Google Scholar
6. K. Hagos, J. Zong, D. Li, C. Liu, X. Lu. Renew. Sustain. Energy Rev.76, 1485 (2017). https://doi.org/10.1016/j.rser.2016.11.184.Search in Google Scholar
7. T. Meng, J. Qin, D. Xu, M. Cao. ACS Appl. Mater. Interfaces11, 9023 (2019). https://doi.org/10.1021/acsami.8b19341.Search in Google Scholar PubMed
8. J. Gong, J. Liang, K. Sumathy. Renew. Sustain. Energy Rev.16, 5848 (2012). https://doi.org/10.1016/j.rser.2012.04.044.Search in Google Scholar
9. E. W. McFarland. Energy Environ. Sci.7, 846 (2014). https://doi.org/10.1039/c3ee43714k.Search in Google Scholar
10. A. J. Hunt, E. H. K. Sin, R. Marriott, J. H. Clark. ChemSusChem3, 306 (2010). https://doi.org/10.1002/cssc.200900169.Search in Google Scholar PubMed
11. B. Dold. J. Geochem. Explor.219, 106638 (2020). https://doi.org/10.1016/j.gexplo.2020.106638.Search in Google Scholar
12. X. Jiang, X. Nie, X. Guo, C. Song, J. G. Chen. Chem. Rev.120, 7984 (2020). https://doi.org/10.1021/acs.chemrev.9b00723.Search in Google Scholar PubMed
13. R. C. Saxena, D. Seal, S. Kumar, H. B. Goyal. Renew. Sustain. Energy Rev.12, 1909 (2008). https://doi.org/10.1016/j.rser.2007.03.005.Search in Google Scholar
14. S. E. Hosseini, M. A. Wahid. Renew. Sustain. Energy Rev.16, 5732 (2012). https://doi.org/10.1016/j.rser.2012.05.025.Search in Google Scholar
15. M. Tobisu, N. Chatani. Acc. Chem. Res.48, 1717 (2015). https://doi.org/10.1021/acs.accounts.5b00051.Search in Google Scholar PubMed
16. E. C. M. Faria, V. S. Duarte, A. M. da Silva, F. S. Fernandes, R. L. G. de Paula, C. G. Alonso, G. R. Oliveira, H. B. Napolitano. Energy Fuel.34, 5958 (2020). https://doi.org/10.1021/acs.energyfuels.0c00322.Search in Google Scholar
17. S. Taneja, A. Parmar. AIP Conf. Proc.2148, 030056 (2019).Search in Google Scholar
18. B. Elliott, A. N. Alexandrova, A. I. Boldyrev. J. Phys. Chem. A107, 1203 (2003). https://doi.org/10.1021/jp026126v.Search in Google Scholar
19. R. E. Johnson, P. D. Cooper, T. I. Quickenden, G. A. Grieves, T. M. Orlando. J. Chem. Phys.123, 184715 (2005). https://doi.org/10.1063/1.2107447.Search in Google Scholar PubMed
20. G. Cruz, A. V. S. Silva, J. B. S. Da Silva, R. de Nazaré Caldeiras, M. E. P. de Souza. Biofuel Bioprod. Biorefin.14, 696 (2020). https://doi.org/10.1002/bbb.2089.Search in Google Scholar
21. L. M. Romeo, B. Peña, M. Bailera, P. Lisbona. Int. J. Hydrogen Energy45, 25838 (2020). https://doi.org/10.1016/j.ijhydene.2020.04.095.Search in Google Scholar
22. A. H. Tarighaleslami, A. Ghannadzadeh, M. J. Atkins, M. R. W. Walmsley. J. Clean. Prod.275, 122999 (2020). https://doi.org/10.1016/j.jclepro.2020.122999.Search in Google Scholar
23. Z. Wang, P. Ning, L. Hu, Q. Nie, Y. Liu, Y. Zhou, J. Yang. Renew. Energy160, 211 (2020). https://doi.org/10.1016/j.renene.2020.06.128.Search in Google Scholar
24. T. L. R. Corrêa, J. P. L. Franco Cairo, J. Cota, A. Damasio, L. C. Oliveira, F. M. Squina. Int. J. Biol. Macromol.166, 1188 (2021). https://doi.org/10.1016/j.ijbiomac.2020.11.001.Search in Google Scholar PubMed
25. T. Guo, Q. Xia, Y. Shao, X. Liu, Y. Wang. Appl. Catal. A Gen.547, 30 (2017). https://doi.org/10.1016/j.apcata.2017.07.050.Search in Google Scholar
26. B. P. Pattanaik, R. D. Misra. Renew. Sustain. Energy Rev.73, 545 (2017). https://doi.org/10.1016/j.rser.2017.01.018.Search in Google Scholar
27. I. Shimada, S. Kato, N. Hirazawa, Y. Nakamura, H. Ohta, K. Suzuki, T. Takatsuka. Ind. Eng. Chem. Res.56, 75 (2017). https://doi.org/10.1021/acs.iecr.6b03514.Search in Google Scholar
28. D.-H. Liu, T. J. Marks, Z. Li. ChemSusChem12, 5217 (2019). https://doi.org/10.1002/cssc.201902137.Search in Google Scholar PubMed
29. D. Ma, S. Lu, X. Liu, Y. Guo, Y. Wang. Chin. J. Catal.40, 609 (2019). https://doi.org/10.1016/s1872-2067(19)63317-6.Search in Google Scholar
30. V. L. Yurpalov, V. A. Drozdov, N. V. Antonicheva, A. A. Nepomnyashchiy, E. A. Buluchevskiy, A. V. Lavrenov. Kinet. Catal.60, 231 (2019). https://doi.org/10.1134/s0023158419020149.Search in Google Scholar
31. G. Abdulkareem-Alsultan, N. Asikin-Mijan, G. Mustafa-Alsultan, H. V. Lee, K. Wilson, Y. H. Taufiq-Yap. RSC Adv.10, 4996 (2020). https://doi.org/10.1039/c9ra09516k.Search in Google Scholar PubMed PubMed Central
32. V. Sharma, T. Getahun, M. Verma, A. Villa, N. Gupta. Renew. Sustain. Energy Rev.133, 110280 (2020). https://doi.org/10.1016/j.rser.2020.110280.Search in Google Scholar
33. W. Song, Y. Zhang, Y. Gao, D. Chen, M. Yang. Chemosphere189, 277 (2017). https://doi.org/10.1016/j.chemosphere.2017.09.079.Search in Google Scholar PubMed
34. D. Liu, G. Li, F. Yang, H. Wang, J. Han, X. Zhu, Q. Ge. J. Phys. Chem. C121, 12249 (2017). https://doi.org/10.1021/acs.jpcc.7b03042.Search in Google Scholar
35. J. E. Dennis, E. K. Konstantakos, D. Arm, A. I. Caplan. Biomaterials19, 1323 (1998). https://doi.org/10.1016/s0142-9612(97)00170-1.Search in Google Scholar PubMed
36. B. W. de Jong, S. Shi, V. Siewers, J. Nielsen. Microb. Cell Factories13, 39 (2014). https://doi.org/10.1186/1475-2859-13-39.Search in Google Scholar PubMed PubMed Central
37. S. Shimano, Y. Tokura, Y. Taguchi. Appl. Mater.5, 056103 (2017). https://doi.org/10.1063/1.4983404.Search in Google Scholar
38. A. A. Bhuiyan, A. S. Blicblau, J. Naser. J. Energy Inst.90, 838 (2017). https://doi.org/10.1016/j.joei.2016.08.010.Search in Google Scholar
39. M. Vinod Babu, K. Madhu Murthy, G. Amba Prasad Rao. Renew. Sustain. Energy Rev.78, 1068 (2017).Search in Google Scholar
40. N. R. Scott, H. Chen, H. Cui. J. Agric. Food Chem.66, 6451 (2018). https://doi.org/10.1021/acs.jafc.8b00964.Search in Google Scholar PubMed
41. K. Kümmerer, J. H. Clark, V. G. Zuin. Science367, 369 (2020). https://doi.org/10.1126/science.aba4979.Search in Google Scholar PubMed
42. R. D. McCulla, W. S. Jenks. J. Am. Chem. Soc.126, 16058 (2004). https://doi.org/10.1021/ja045935k.Search in Google Scholar PubMed
43. Y. Shi, J. Zhang, E. Xing, Y. Xie, H. Cao. Ind. Eng. Chem. Res.58, 21341 (2019). https://doi.org/10.1021/acs.iecr.9b04130.Search in Google Scholar
44. G. Kang, A. T. Taguchi, J. Stubbe, C. L. Drennan. Science368, 424 (2020). https://doi.org/10.1126/science.aba6794.Search in Google Scholar PubMed PubMed Central
45. Y. Xiao, Y.-K. Chun, S.-C. Cheng, R. Liu, M.-K. Tse, C.-C. Ko. Org. Biomol. Chem.18, 8686 (2020). https://doi.org/10.1039/d0ob01767a.Search in Google Scholar PubMed
46. M. Tobisu, N. Chatani. Acc. Chem. Res.48, 1717 (2015). https://doi.org/10.1021/acs.accounts.5b00051.Search in Google Scholar PubMed
47. H. Zeng, Z. Qiu, A. Domínguez-Huerta, Z. Hearne, Z. Chen, C.-J. Li. ACS Catal.7, 510 (2017). https://doi.org/10.1021/acscatal.6b02964.Search in Google Scholar
48. T. B. Boit, A. S. Bulger, J. E. Dander, N. K. Garg. ACS Catal.10, 12109 (2020). https://doi.org/10.1021/acscatal.0c03334.Search in Google Scholar PubMed PubMed Central
49. Z. Qiu, C.-J. Li. Chem. Rev.120, 10454 (2020). https://doi.org/10.1021/acs.chemrev.0c00088.Search in Google Scholar PubMed
50. T. Zhou, M. Szostak. Catal. Sci. Technol.10, 5702 (2020). https://doi.org/10.1039/d0cy01159b.Search in Google Scholar PubMed PubMed Central
51. M. Tobisu, A. Yasutome, K. Yamakawa, T. Shimasaki, N. Chatani. Tetrahedron68, 5157 (2012). https://doi.org/10.1016/j.tet.2012.04.005.Search in Google Scholar
52. B. Yu, H. Sun, Z. Xie, G. Zhang, L.-W. Xu, W. Zhang, Z. Gao. Org. Lett.17, 3298 (2015). https://doi.org/10.1021/acs.orglett.5b01466.Search in Google Scholar PubMed
53. D. B. Larsen, A. R. Petersen, J. R. Dethlefsen, A. Teshome, P. Fristrup. Chem. Eur J.22, 16621 (2016). https://doi.org/10.1002/chem.201603028.Search in Google Scholar PubMed
54. J. Xiao, J. Yang, T. Chen, L.-B. Han. Adv. Synth. Catal.358, 816 (2016). https://doi.org/10.1002/adsc.201500822.Search in Google Scholar
55. M. Tobisu, K. Yasui, Y. Aihara, N. Chatani. Angew. Chem. Int. Ed.56, 1877 (2017). https://doi.org/10.1002/anie.201610409.Search in Google Scholar PubMed
56. X. Zhang, F. Jordan, M. Szostak. Org. Chem. Front.5, 2515 (2018). https://doi.org/10.1039/c8qo00585k.Search in Google Scholar
57. S. M. Alia, K. Duong, T. Liu, K. Jensen, Y. Yan. ChemSusChem7, 1739 (2014). https://doi.org/10.1002/cssc.201400129.Search in Google Scholar PubMed
58. H. Dang, N. Cox, G. Lalic. Angew. Chem. Int. Ed.53, 752 (2014). https://doi.org/10.1002/anie.201307697.Search in Google Scholar PubMed
59. J. R. Bernardo, A. C. Fernandes. Green Chem. 18, 2675 (2016). https://doi.org/10.1039/c5gc02777b.Search in Google Scholar
60. Y. Wang, Z. Shao, K. Zhang, Q. Liu. Angew. Chem. Int. Ed.57, 15143 (2018). https://doi.org/10.1002/anie.201809333.Search in Google Scholar PubMed
61. T. Kawamura, H. Moriya, M. Shibuya, Y. Yamamoto. J. Org. Chem.84, 12508 (2019). https://doi.org/10.1021/acs.joc.9b02017.Search in Google Scholar PubMed
62. J. Wu, R. M. Bär, L. Guo, A. Noble, V. K. Aggarwal. Angew. Chem. Int. Ed.58, 18830 (2019). https://doi.org/10.1002/anie.201910051.Search in Google Scholar PubMed
63. H.-W. Chen, F.-D. Lu, Y. Cheng, Y. Jia, L.-Q. Lu, W.-J. Xiao. Chin. J. Chem.38, 1671 (2020). https://doi.org/10.1002/cjoc.202000309.Search in Google Scholar
64. M. S. Gamal, N. Asikin-Mijan, W. N. A. W. Khalit, M. Arumugam, S. M. Izham, Y. H. Taufiq-Yap. Fuel Process. Technol.208, 106519 (2020). https://doi.org/10.1016/j.fuproc.2020.106519.Search in Google Scholar
65. C. Bandari, K. M. Nicholas. Synthesis53, 267 (2021).10.1055/s-0040-1707269Search in Google Scholar
66. S. M. Leckie, G. J. Harkness, M. L. Clarke. ChemComm50, 11511 (2014). https://doi.org/10.1039/c4cc04939j.Search in Google Scholar PubMed
67. K. Alharbi, W. Alharbi, E. F. Kozhevnikova, I. V. Kozhevnikov. ACS Catal.6, 2067 (2016). https://doi.org/10.1021/acscatal.6b00096.Search in Google Scholar
68. L. Chen, R. S. Smith, B. D. Kay, Z. Dohnalek. ACS Catal.7, 2002 (2017). https://doi.org/10.1021/acscatal.6b03225.Search in Google Scholar
69. C. Ju, M. Li, Y. Fang, T. Tan. Green Chem.20, 4492 (2018). https://doi.org/10.1039/c8gc01960f.Search in Google Scholar
70. S. Mukundan, J. Beltramini, K. G. Kumar, D. S. Ravindran. Appl. Catal. A-Gen.606, 117811 (2020). https://doi.org/10.1016/j.apcata.2020.117811.Search in Google Scholar
71. K. Wu, W. Wang, H. Guo, Y. Yang, Y. Huang, W. Li, C. Li. ACS Energy Lett.5, 1330 (2020). https://doi.org/10.1021/acsenergylett.0c00411.Search in Google Scholar
72. A. Zheng, Z. Huang, G. Wei, K. Zhao, L. Jiang, Z. Zhao, Y. Tian, H. Li. iScience23, 100814 (2020). https://doi.org/10.1016/j.isci.2019.100814.Search in Google Scholar PubMed PubMed Central
73. N. U. D. Reshi, J. K. Bera. Coord. Chem. Rev.422, 213334 (2020). https://doi.org/10.1016/j.ccr.2020.213334.Search in Google Scholar
74. A. K. S, N. Chandrasekaran. ACS Sustain. Chem. Eng.7, 15197 (2019). https://doi.org/10.1021/acssuschemeng.9b02007.Search in Google Scholar
75. W.-Q. Wang, Z.-Q. Wang, W. Sang, R. Zhang, H. Cheng, C. Chen, D.-Y. Peng. Polyhedron195, 114979 (2021). https://doi.org/10.1016/j.poly.2020.114979.Search in Google Scholar
76. W.-L. Yang, T. Ni, W.-P. Deng. Org. Lett.23, 588 (2021). https://doi.org/10.1021/acs.orglett.0c04132.Search in Google Scholar PubMed
77. Y.-L. Zhao, G.-J. Wu, F.-S. Han. ChemComm48, 5868 (2012). https://doi.org/10.1039/c2cc31718d.Search in Google Scholar PubMed
78. L. Chenneberg, A. Baralle, M. Daniel, L. Fensterbank, J.-P. Goddard, C. Ollivier. Adv. Synth. Catal.356, 2756 (2014). https://doi.org/10.1002/adsc.201400729.Search in Google Scholar
79. B. Ciszek, I. Fleischer. Chem. Eur J.24, 12259 (2018). https://doi.org/10.1002/chem.201801466.Search in Google Scholar PubMed
80. S. Afewerki, C. Palo-Nieto, A. Córdova. Synthesis52, 2330 (2020). https://doi.org/10.1055/s-0040-1707398.Search in Google Scholar
81. S. Yang, W. Tang, Z. Yang, J. Xu. ACS Catal.8, 9320 (2018). https://doi.org/10.1021/acscatal.8b02495.Search in Google Scholar
82. S. E. Griffin, L. L. Schafer. Inorg. Chem.59, 5256 (2020). https://doi.org/10.1021/acs.inorgchem.0c00071.Search in Google Scholar PubMed
83. B. N. Barman, V. L. Cebolla, L. Membrado. Crit. Rev. Anal. Chem.30, 75 (2000). https://doi.org/10.1080/10408340091164199.Search in Google Scholar
84. M. Bernardin, A. L. Masle, F. Bessueille-Barbier, C.-P. Lienemann, S. Heinisch. J. Chromatogr. A.1611, 460605 (2020). https://doi.org/10.1016/j.chroma.2019.460605.Search in Google Scholar PubMed
85. Y. Hasegawa, K. Shikinaka, Y. Katayama, S. Kajita, E. Masai, M. Nakamura, Y. Otsuka, S. Ohara, K. Shigehara. Seni Gakkai Shi65, 359 (2009). https://doi.org/10.2115/fiber.65.359.Search in Google Scholar
86. J. S. Mahajan, R. M. O’Dea, J. B. Norris, L. T. J. Korley, T. H. Epps. ACS Sustain. Chem. Eng.8, 15072 (2020). https://doi.org/10.1021/acssuschemeng.0c04817.Search in Google Scholar
87. V. Sharma, T. Getahun, M. Verma, A. Villa, N. Gupta. Renew. Sustain. Energy Rev.133, 110280 (2020). https://doi.org/10.1016/j.rser.2020.110280.Search in Google Scholar
88. J. M. Nichols, L. M. Bishop, R. G. Bergman, J. A. Ellman. J. Am. Chem. Soc.132, 12554 (2010). https://doi.org/10.1021/ja106101f.Search in Google Scholar PubMed PubMed Central
89. X. Wang, R. Rinaldi. Angew. Chem. Int. Ed.52, 11499 (2013). https://doi.org/10.1002/anie.201304776.Search in Google Scholar PubMed
90. T. H. Parsell, B. C. Owen, I. Klein, T. M. Jarrell, C. L. Marcum, L. J. Haupert, L. M. Amundson, H. I. Kenttämaa, F. Ribeiro, J. T. Miller, M. M. Abu-Omar. Chem. Sci.4, 806 (2013). https://doi.org/10.1039/c2sc21657d.Search in Google Scholar
91. M. V. Galkin, S. Sawadjoon, V. Rohde, M. Dawange, J. S. M. Samec. ChemCatChem6, 179 (2014). https://doi.org/10.1002/cctc.201300540.Search in Google Scholar
92. A. Rahimi, A. Ulbrich, J. J. Coon, S. S. Stahl. Nature515, 249 (2014). https://doi.org/10.1038/nature13867.Search in Google Scholar PubMed
93. S. Huang, N. Mahmood, Y. Zhang, M. Tymchyshyn, Z. Yuan, C. Xu. Fuel209, 579 (2017). https://doi.org/10.1016/j.fuel.2017.08.031.Search in Google Scholar
94. W. Yang, X. Li, X. Du, Y. Deng, H. Dai. Catal. Commun.126, 30 (2019). https://doi.org/10.1016/j.catcom.2019.04.025.Search in Google Scholar
95. Y.-Y. Lin, S.-Y. Lu. J. Taiwan Inst. Chem. E97, 264 (2019). https://doi.org/10.1016/j.jtice.2019.02.007.Search in Google Scholar
96. B. Zhang, Z. Qi, X. Li, J. Ji, L. Zhang, H. Wang, X. Liu, C. Li. Green Chem.21, 5556 (2019). https://doi.org/10.1039/c9gc01710k.Search in Google Scholar
97. C. Yang, M. D. Kärkäs, G. Magallanes, K. Chan, C. R. J. Stephenson. Org. Lett.22, 8082 (2020). https://doi.org/10.1021/acs.orglett.0c03029.Search in Google Scholar PubMed
98. X. Shen, Q. Meng, Q. Mei, J. Xiang, H. Liu, B. Han. Green Chem.22, 2191 (2020). https://doi.org/10.1039/d0gc00587h.Search in Google Scholar
99. B. Zheng, J. Song, H. Wu, S. Han, J. Zhai, K. Zhang, W. Wu, C. Xu, M. He, B. Han. Green Chem.23, 268 (2021). https://doi.org/10.1039/d0gc03188g.Search in Google Scholar
100. S. Samanta, R. Srivastava. Mater. Adv.1, 1506 (2020). https://doi.org/10.1039/d0ma00293c.Search in Google Scholar
101. H. Li, Y. Liu, Y. Liu, L. Wang, R. Tang, P. Deng, Z. Xu, B. Haynes, C. Sun, J. Huang. Appl. Catal. B281, 119476 (2021). https://doi.org/10.1016/j.apcatb.2020.119476.Search in Google Scholar
102. N. Armaroli, V. Balzani. Angew. Chem. Int. Ed.46, 52 (2007). https://doi.org/10.1002/anie.200602373.Search in Google Scholar PubMed
103. M. Liu, Y. Wang, X. Kong, L. Tan, L. Li, S. Cheng, G. Botton, H. Guo, Z. Mi, C.-J. Li. iScience17, 208 (2019). https://doi.org/10.1016/j.isci.2019.06.032.Search in Google Scholar PubMed PubMed Central
104. M. Liu, L. Tan, R. T. Rashid, Y. Cen, S. Cheng, G. Botton, Z. Mi, C.-J. Li. Chem. Sci.11, 7864 (2020). https://doi.org/10.1039/d0sc02718a.Search in Google Scholar PubMed PubMed Central
105. M. Liu, Y. Wang, X. Kong, R. T. Rashid, S. Chu, C.-C. Li, Z. Hearne, H. Guo, Z. Mi, C.-J. Li. Chem.5, 858 (2019). https://doi.org/10.1016/j.chempr.2019.01.005.Search in Google Scholar
106. M. Liu, Z. Qiu, L. Tan, R. T. Rashid, S. Chu, Y. Cen, Z. Luo, R. Z. Khaliullin, Z. Mi, C.-J. Li. ACS Catal.10, 6248 (2020). https://doi.org/10.1021/acscatal.0c00881.Search in Google Scholar
© 2021 IUPAC & De Gruyter. This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License. For more information, please visit: http://creativecommons.org/licenses/by-nc-nd/4.0/
Articles in the same Issue
- Frontmatter
- Invited papers
- Rational design of nanocatalysts for ambient ammonia electrosynthesis
- The exploration of deoxygenation reactions for alcohols and derivatives using earth-abundant reagents
- IUPAC Technical Report
- Reference materials for phase equilibrium studies. 1. Liquid–liquid equilibria (IUPAC Technical Report)
Articles in the same Issue
- Frontmatter
- Invited papers
- Rational design of nanocatalysts for ambient ammonia electrosynthesis
- The exploration of deoxygenation reactions for alcohols and derivatives using earth-abundant reagents
- IUPAC Technical Report
- Reference materials for phase equilibrium studies. 1. Liquid–liquid equilibria (IUPAC Technical Report)