Analysis of the possibility and molecular mechanism of carbon dioxide consumption in the Diels-Alder processes
-
Karolina Kula


Abstract
The large and significant increase in carbon dioxide concentration in the Earth’s atmosphere is a serious problem for humanity. The amount of CO2 is increasing steadily which causes a harmful greenhouse effect that damages the Earth’s climate. Therefore, one of the current trends in modern chemistry and chemical technology are issues related to its utilization. This work includes the analysis of the possibility of chemical consumption of CO2 in Diels-Alder processes under non-catalytic and catalytic conditions after prior activation of the C=O bond. In addition to the obvious benefits associated with CO2 utilization, such processes open up the possibility of universal synthesis of a wide range of internal carboxylates. These studies have been performed in the framework of Molecular Electron Density Theory as a modern view of the chemical reactivity. It has been found, that explored DA reactions catalyzed by Lewis acids with the boron core, proceeds via unique stepwise mechanism with the zwitterionic intermediate. Bonding Evolution Theory (BET) analysis of the molecular mechanism associated with the DA reaction between cyclopentadiene and carbon dioxide indicates that it takes place thorough a two-stage one-step mechanism, which is initialized by formation of C–C single bond. In turn, the DA reaction between cyclopentadiene and carbon dioxide catalysed by BH3 extends in the environment of DCM, indicates that it takes place through a two-step mechanism. First path of catalysed DA reaction is characterized by 10 different phases, while the second by eight topologically different phases.
Introduction
At the beginning, the natural greenhouse effect was a precursor to life moving from the ocean to the land, and constantly it is important to human beings. It occurs in the lower atmosphere layer, the troposphere. Due to the natural greenhouse effect, the heat is partially accumulated in the atmosphere. That is causing the natural Earth warming which determines the possibility of life existence on the planet. In the absence of natural greenhouse effect, the average temperature on Earth’s surface is estimated around −19 °C, to compare current average temperature is approximately 14 °C [1], [2], [3], [4]. However, human activities are strengthening the natural greenhouse effect causing irreversible climate change [5]. One of the greatest threats, related to the greenhouse effect, is the constantly progressive rise in the temperature of the Earth. Since the beginning of the pre-industrial period, the surface temperature of our planet has already risen by more than 1 °C. Moreover, if the greenhouse effect and global warming will not be reduced, then by 2030 the temperature of the Earth’s surface will greatly rise to even around 2 °C above pre-industrial levels [5], [6].
Human activity is mainly responsible for the growing greenhouse effect [5]. The grow of greenhouse gases (GHG) concentration is caused by burn fossil fuels like coal, oil and gas, which are used on large scale to produce electricity and also for transportation [7]. When the fossil fuels are burnt, the carbon stored inside is released. Sequentially, carbon combines with oxygen, from the air, and create a carbon dioxide. Furthermore, large scale of industrial development has resulted in cutting down many trees and forests. It is well known, that plants intake carbon dioxide and release oxygen, through the process of photosynthesis, which is essential for humans and animals to survive. The wood burning process also produces carbon dioxide [8], [9]. What is more, population growth has become a significant problem. This has resulted in increased demand for food, clothes and homesteads [5]. In the cities, there were created new manufacturing hubs which industry causes release of harmful gases into the atmosphere there by causing greater greenhouse effect. Huge population growth resulted increasing agricultural and industrial waste and landfills and hence concentration rise of methane and nitrous oxide [9], [10].
There are many heat-trapping gases such as methane or water vapor, but carbon dioxide is a key greenhouse gas puts at the greatest risk of irreversible climate changes [8, 11, 12]. Other greenhouse gases which are emitted as a result of human activities are more potent heat-trapping per molecule than CO2, but they are simply far less abundant in the atmosphere and stay therefore much shorter [8], [13]. For example, it takes about a decade for methane emissions to leave the atmosphere and about a century for nitrous oxide [8, 14, 15]. To compare, 40% of the emission of CO2 will remain in the atmosphere for 100 years, 20% will reside for 1000 years, and the last 10% will take 10000 years to leave the atmosphere [15].
Current levels of CO2 in the atmosphere are at the highest level ever recorded. The emission in the last year was 36.81 mld tons [16]. It is about 0.6% more than in 2018 [17] (Fig. 1). Therefore, the attempts to reduce carbon dioxide emissions are still take up. The scientists have been working on various ways to prevent the accumulation of atmospheric CO2 [18] including removal, sequestration, utilization, and also conversion into fuels [19] to reduce our dependence on petrochemicals. One of the main approaches to recycle CO2 is its capture and use (CCU) technique [20], [21]. For this purpose, one of the most popular method to capturing carbon dioxide in industry is an amine scrubbing technology [21]. Carbon dioxide can also useful to production of chemicals [22], [23], [24]. To the most popular research directions related to the use of carbon dioxide can be included production of carboxylic acid e.g. salicylic acid [25] or urea [26], acrylates [27], cyclic carbonates [28] and polypropylene carbonate [29]. On industrial scale CO2 elimination can also be carried out via carbon capture and storage (CCS) technology [30], [31]. The method involves injection of carbon dioxide, captured from large stationary sources, into deep geological formations. So, it is the only viable technology to mitigate carbon emissions while allowing continued large scale use of fossil fuels. Also the CCS determine potentially important methodology in the transition on carbon free energy sources [30], [31].

Global CO2emission from fossil flues and industry.
In the present paper, we carried out theoretical studies about the possibility of the synthesis of six-membered, internal lactones via Diels–Alder (DA) reaction between cyclopentadiene 1 and carbon dioxide 2 (Scheme 1). Generally, DA reaction is one of the most important and widely used method for making six-membered carbo- and hetero- cyclic [32], due to their feasibility to create regio- and/or stereo- selectively cyclic organic molecules [33]. DA reaction is carried out with “full atomic economy” and under mild condition giving high yields [34], [35], [36]. These conditions approach one of the main principles of green chemistry. The advent of computational chemistry at the beginning in the 20th century has gradually attracted theoretical chemists to analyze the reactivity and selectivity of organic molecules. In 2016 Domingo proposed the Molecular Electron Density Theory (MEDT) as anew theoretical outlook for organic reactions after interpretation of a wide spectrum of organic reactivity for 20 years, majority of them dedicated to [3 + 2] cycloaddition and also Diels-Alder reactions [37], [38]. MEDT establishes that changes in the electron density, are responsible for the feasibility of an organic reaction [39]. In MEDT, several quantum-chemical tools, such as the analysis of the Conceptual Density Functional Theory (CDFT) [40] indices, and the topological analysis of the Electron Localization Function (ELF) [41] are used to rigorously characterize the molecular mechanism of the studied reactions.

The reaction between cyclopentadiene 1 and carbon dioxide 2.
Lactones are heterocyclic esters which are widely spread out as biological substances [42]. They exhibit, among others, a cytostatic [43], [44], an antibacterial [45], an antiviral [46] or an antifungal [47] effect, so therefore, they can used in medicine. Moreover, sensory properties make them useful in the production of cosmetics and in the food industry, where they are responsible for the smell and taste of many products [48]. On the other hand, lactones are the repellent activity and they can be used in the production of insect-control agents [49].The natural source of lactones are plants [50] but they are also found in microorganisms [51]and animals [52].Owing to the high costs of extracting lactones from nature, methods for their preparation through chemical synthesis [53], [54] and by using biotechnological methods [55] are still being developed. This interesting group of compounds also attracted our attention.
Results and discussion
In the first part of our study, we analyzed simple case of non-catalyzed reaction between cyclopentadiene 1 and carbon dioxide 2. Formally, this process can be considered as typical DA reaction [56] involving oxygen containing heteroanalog of dienophile (Scheme 1).
It was found, that in the gas phase, the first reaction stage is the formation of molecular, pre-reaction complex (MC) (Fig. 2), which include cyclopentadiene 1 and CO22 substructures. Within MC, new single bonds are not formed (Table 1). Key interatomic distances are equal about 3.3 Å, and are beyond of the range which is typical for new bonds in transition states. At this stage, any global electron density transfer (GEDT [57]) between substructures are not observed (GEDT = 0.00 e). So, the MC can be not classified as electron density transfer complex (EDTC). This transformation is realized without any of the activation barrier, and resulted the reduction of the enthalpy of reaction system by 4.4 kcal·mol−1. Similar, molecular complexes was detected recently in the case of other DA reactions [33, 58, 59].

Views of key structures of non-catalyzed reaction between cyclopentadiene 1 and carbon dioxide 2 in the gas phase in the light of M062X/6-311G(d) calculations.
Key parameters of critical structures of non-catalyzed reaction between cyclopentadiene 1 and carbon dioxide 2 in the light of M062X/6-311G(d) calculations.
| Solvent (ε) | Structure | Interatomic distances [Å] | GEDT [e] | Imaginary frequence [cm−1] | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| C1–C2 | C2–C3 | C3–C4 | C4–O5 | O5–C6 | C6–O7 | C6–C1 | ||||
| Gas phase (1.00) | 1 | 1.342 | 1.470 | 1.342 | ||||||
| 2 | 1.155 | 1.155 | ||||||||
| MC | 1.343 | 1.473 | 1.343 | 3.275 | 1.155 | 1.155 | 3.359 | 0.00 | ||
| TS | 1.435 | 1.382 | 1.404 | 2.086 | 1.238 | 1.200 | 1.753 | 0.24 | −587.42 | |
| DCM (8.93) | 1 | 1.343 | 1.471 | 1.343 | ||||||
| 2 | 1.155 | 1.155 | ||||||||
| MC | 1.344 | 1.473 | 1.344 | 3.284 | 1.155 | 1.155 | 3.375 | 0.00 | ||
| TS | 1.449 | 1.377 | 1.405 | 2.174 | 1.241 | 1.215 | 1.683 | 0.38 | −435.83 | |
| MeNO2 (36.56) | 1 | 1.343 | 1.471 | 1.343 | ||||||
| 2 | 1.155 | 1.155 | ||||||||
| MC | 1.344 | 1.473 | 1.344 | 3.287 | 1.155 | 1.155 | 3.379 | 0.00 | ||
| TS | 1.449 | 1.377 | 1.405 | 2.174 | 1.241 | 1.215 | 1.683 | 0.41 | −409.79 | |
| Water (78.36) | 1 | 1.344 | 1.472 | 1.344 | ||||||
| 2 | 1.155 | 1.155 | ||||||||
| MC | 1.344 | 1.473 | 1.344 | 3.287 | 1.155 | 1.155 | 3.379 | 0.00 | ||
| TS | 1.452 | 1.376 | 1.406 | 2.186 | 1.242 | 1.218 | 1.669 | 0.41 | −405.08 | |
Further transformation of the reaction system lead to the transition state (TS) (Fig. 2). Within TS, interatomic distances C1–C6 and C4–O5 are substantially reduced (up to 1.753 and 2.086 Å respectively) (Table 1). In the first case, the new single bond is formally almost formed [57]. Generally, the reaction course is determined, by the attack of most nucleophilic centre at the cyclopentadiene molecule to most electrophilic centre within CO2 (Scheme 1). This is accompanied with the transfer of the electron density between substructures. This effect (GEDT = 0.24 e) suggest moderately polar nature of the considered structure. The IRC calculations connect this TS with the valley of the MC, and, on the other hand, with the valley of product. This confirm a one-step mechanism of analyzed transformation (Fig. 3, Scheme 2). However, it should be underlined that the considered process should be treatment as kinetically difficult from a experimental point of view, because the activation Gibbs free energy is equal almost 51 kcal·mol−1(!) (Table 4). This exclude the practical sense of these type transformations. In the next step, we analyzed similar process in the presence of solvents characterized by different polarity. It was found, that the geometries of optimized MCs as well as TSs are general close to obtained for the gas phase. This is a consequence of rather moderately polar nature of the considered process. The activation barrier is decreased with increasing solvent polarity. However, not so much that the analyzed process could be considered as allowed from the kinetic point of view.

Ethalpy profiles of on-catalyzed and LA-catalyzed reaction between cyclopentadiene1 and carbon dioxide 2 in the gas phase and DCM solutions in the light of M062X/6-311G(d) calculations.

The non-catalyzed reaction between cyclopentadiene 1 and carbon dioxide 2 in the gas phase and in solutions.
The ELF topological analysis shed light on the molecular nature of described transformation. The Bonding Evolution Theory (BET) [60], has proven to be a very useful methodological tool to establish the changes along the reaction path. This approach has been used to establish many reaction mechanisms [36, 61–63]. In order to explain the formation of bonding along the DA reaction, a BET study of the DA reaction cyclopentadiene 1 with carbon dioxide 2 was conducted. ELF valence basin populations of the selected structures of the IRC are assembled in Table 2. In Fig. 4, the most important structures involved in the formation of one double and two single bonds was presented. In turn Scheme 3 shows the molecular mechanism of the DA reaction between cyclopentadiene 1 with carbon dioxide 2.
ELF valence basin populations, distances of the forming bonds, M06-2X/6-311G(d) relativea electronic energies of the IRC structures, MC–3, defining the eight phases characterizing the molecular mechanism of the DA reaction between cyclopentadiene 1 and carbon dioxide 2. MC, TS and 3 are also included. Distances are given in angstroms, Å, electron populations in average number of electrons, e, and relative energies in kcal·mol−1.
| Points | 1 | 2 | MC | P1 | P2 | P3 | P4 | P5 | TS | P6 | P7 | 3 | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Phases | I | II | III | IV | V | VI | VII | VIII | ||||||
| d(C2–C3) | 1.470 | 1.462 | 1.434 | 1.431 | 1.420 | 1.408 | 1.382 | 1.358 | 1.355 | 1.331 | ||||
| d(C4–O5) | 3.275 | 2.519 | 2.312 | 2.300 | 2.250 | 2.200 | 2.086 | 1.953 | 1.929 | 1.462 | ||||
| d(C6–C1) | 3.359 | 2.423 | 2.063 | 2.038 | 1.962 | 1.888 | 1.753 | 1.644 | 1.630 | 1.528 | ||||
| ΔE | 0.0 | 9.7 | 18.4 | 28.3 | 36.4 | 41.5 | 43.1 | 39.7 | 29.3 | 19.6 | ||||
| V(C1,C2) | 1.76 | 1.74 | 3.24 | 3.16 | 3.15 | 2.79 | 2.61 | 2.34 | 2.14 | 2.13 | 1.94 | |||
| V′(C1,C2) | 1.63 | 1.60 | ||||||||||||
| V(C2,C3) | 2.23 | 2.21 | 2.23 | 2.39 | 2.43 | 2.47 | 2.58 | 2.90 | 1.70 | 1.65 | 1.75 | |||
| V′(C2,C3) | 1.46 | 1.54 | 1.70 | |||||||||||
| V(C3,C4) | 1.76 | 1.69 | 1.65 | 1.66 | 3.10 | 3.03 | 2.94 | 2.64 | 2.30 | 2.22 | 2.04 | |||
| V′(C3,C4) | 1.63 | 1.68 | 1.64 | 1.47 | ||||||||||
| V(O5,C6) | 3.05 | 2.84 | 2.83 | 2.70 | 2.66 | 2.53 | 2.37 | 2.13 | 1.95 | 1.91 | 1.58 | |||
| V(O5) | 4.68 | 5.03 | 5.06 | 3.74 | 3.57 | 2.93 | 3.13 | 3.93 | 4.96 | 4.98 | 4.68 | |||
| V′(O5) | 1.41 | 1.59 | 2.28 | 2.15 | 1.45 | 0.49 | ||||||||
| V(C6) | 0.17 | 0.21 | 0.38 | |||||||||||
| V(C1) | 0.36 | |||||||||||||
| V(C6,C1) | 1.07 | 1.54 | 1.86 | 1.87 | 2.11 | |||||||||
| V(C4) | 0.02 | |||||||||||||
| V(C4,O5) | 0.58 | 1.48 | ||||||||||||
aRelative to the first point of the IRC, MC.

Attractor positions of the ELF valence basins of the most important structures P2, P4, P5, P6 and P7 participating in the C2–C3 double and C6–C1, C4–O5 single bonds formation along the DA reaction cyclopentadiene 1 with carbon dioxide 2. The electron populations, in average number of electrons, are given in e.

Simplified representation of the molecular mechanism of the DA reaction between cyclopentadiene 1 with carbon dioxide 2 by Lewis-like structures resulting from the topological analysis of the ELF along the reaction path.
BET analysis permitted to distinguish eight different phases (Table 2), associated with rupture and formation of bonds along the DA reaction cyclopentadiene 1 with carbon dioxide 2. Phase I starts at MC, which corresponds with the first point of the IRC. The ELF basin attractor positions of MC features five disynaptic basins, connected with two C1–C2 and C3–C4 double and one C2–C3 single bonds in the most important region derived from molecule 1, one O5–C6 partial double bond and one V(O5) monosynaptic basin relate to molecule 2. Phase II, begins at P1 in which the two V(C1,C2) and V′(C1,C2) disynaptic basins have connected into one new V(C1,C2) disynaptic basin, integrating 3.24 e. This topological change is associated with rupture of C1–C2 double bond (see point P1 at Table 2). In Phase III we follow the creation a new V(C6) monosynaptic basin integrating 0.17 e, as a results of the depollution of V(O5,C6) disynaptic basin. This change is related to formation of a pseudoradical centre at the C6 carbon (Fig. 4). In this phase we also observed the formation of a new V′(O5) monosynaptic basin as a result of division the V(O5) monosynaptic basin present in previous phase. Phase IV, starts at P3 in which the two V(C3,C4) and V′(V′(C3,C4) disynaptic basins have connected into one new V(C3,C4) disynaptic basin, integrating 3.10 e. This topological change is related to rupture of the second C3–C4 double bond in molecule 1. Along the Phase V we remark formation of a new V(C1) monosynaptic basin, integrating 0.36 e, related to formation of a second pseudoradical centre at the C1 carbon (see P4 at Fig. 3). Phase VI, begins at P5 in which the first most relevant change along the IRC path takes place. At this phase, the first C6–C1 bond is formation. The two C6 and C1 pseudoradical centers have merged into new C6–C1 bonding region with an initial population of 1.07 e (see V(C6,C1) in Table 2, Fig. 4 and Scheme 3). Formation of C6–C1 single bond begins at a distance of d(C6–C1) = 1.888 Å (Table 2). In this phase we find a transition state (TS, d(C2–C3) = 1.382 Å, d(C4–O5) = 2.086 Å and d(C6–C1) = 1.753 Å) of the DA reaction cyclopentadiene 1 with carbon dioxide 2, which is bonded with high energy cost of 43.1 kcal·mol−1. The consecutive Phase VII, starts at P6. At this phase, the notable topological change is the split of the single V(C2,C3) disynaptic basin present at the previous phase into two new V(C2,C3) and V′(C2,C3) disynaptic basins, integrating 1.70 e and 1.46 e (Table 2). This topological change is the consequence of an electron density reorganisation within the C2–C3 double bond region. In this phase we also observed formation of a new V(C4) monosynaptic basin, which is connected with formation of a new pseudoradical C4 centre (see P6 at Fig. 4). Finally, the last Phase VIII, begins at P7 and ends at 3. At this phase, while the V(C4) and V′(O5) monosynaptic basins current at P6 are missing, a new V(C4,O5) disynaptic basin is created, integrating 0.58 e. This topological change is associated with formation of a second C4–O5 single bond in molecule 3 (Table 2, Fig. 4 and Scheme 3).
Searching for the conditions for the easier from the kinetic point of view transformation of carbon dioxide 2 in reaction with cyclopentadiene 1, we decided to analyze the processes with the participation of Lewis catalysts with a boron core such as BH3, BCl3, BBr3 (Schemes 4 and 5).These type of catalysts are easy available, and popular in the organic synthesis [64], [65]. For example, they were tested by us regarding to the nitrous acid extrusion from Δ2-nitroisoxazoline systems [66].

The LA-catalyzed reaction between cyclopentadiene 1 and carbon dioxide 2 in the gas phase.

The LA-catalyzed reaction between cyclopentadiene 1 and carbon dioxide 2 in the solution.
The BH3-catalysed transformation is started by the formation of 2/BH3 complex. This process is realized without barrier of the activation, and its resulting to reduction of the enthalpy of the reaction system about a few kcal·mol−1. This cycloaddition with the cyclopentadiene 1 molecule proceed firstly via pre-reaction, molecular complex MC (Fig. 5). The nature of optimized MC is similar as in the case of described above non-catalyzed reaction. In particular, MC not exhibits nature of charge-transfer complex (GEDT = 0.00 e). The formation of MC is accompanied with the reduction of the enthalpy of the reaction system about a few kcal·mol−1. The transformation of MC in to product is realized via single transition state (Scheme 4). Dependently of the relatively orientation between [2/BH3] and 1, this can be TSA or TSB transition state (Fig. 5). Within TSA and TSB, key interatomic distances are substantially reduced (Table 3). It should be underlined however, that considered TSs are relatively lower synchronically than TS optimized in non-catalyzed process. Similarly however as in the case of non-catalyzed process, the reaction course is determined, by the attack of most nucleophilic center at the cyclopentadiene 1 molecule to most electrophilic center within CO2 (Scheme 6).

Views of key structures of BH3-catalysed reaction between cyclopentadiene1 and carbon dioxide 2 in the gas phase in the light of M062X/6-311G(d) calculations.
Key parameters of critical structures of LA-catalyzed reaction between cyclopentadiene 1 and carbon dioxide 2 in the gas phase in the light of M062X/6-311G(d) calculations.
| LA catalyst | Structure | Interatomic distances [Å] | GEDT [e] | Imaginary frequence [cm−1] | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C1–C2 | C2–C3 | C3–C4 | C4–O5 | C4–C6 | O5–C6 | C6–O7 | C6–C1 | O7–C1 | ||||
| BH3 | [2/BH3] | 1.150 | 1.159 | |||||||||
| MC | 1.343 | 1.473 | 1.343 | 3.268 | 3.359 | 1.151 | 1.160 | 3.343 | 3.810 | 0.00 | ||
| TSA | 1.447 | 1.381 | 1.398 | 2.254 | 2.697 | 1.214 | 1.238 | 1.670 | 2.420 | 0.42 | −300.21 | |
| TSB | 1.403 | 1.376 | 1.453 | 2.450 | 1.656 | 1.191 | 1.279 | 2.743 | 2.210 | 0.45 | −353.48 | |
| BCl3 | [2/BCl3] | 1.157 | 1.157 | |||||||||
| MC | 1.343 | 1.472 | 1.343 | 3.247 | 3.298 | 1.152 | 1.157 | 3.394 | 3.900 | 0.00 | ||
| TSA | 1.461 | 1.375 | 1.403 | 2.268 | 2.678 | 1.206 | 1.265 | 1.622 | 2.399 | 0.52 | −206.24 | |
| TSB | 1.425 | 1.360 | 1.483 | 2.437 | 1.579 | 1.184 | 1.332 | 2.677 | 2.090 | 0.56 | −283.79 | |
| BBr3 | [2/BBr3] | 1.157 | 1.153 | |||||||||
| MC | 1.343 | 1.472 | 1.343 | 3.266 | 3.368 | 1.157 | 1.153 | 3.347 | 3.821 | 0.00 | ||
| TSA | 1.463 | 1.375 | 1.404 | 2.267 | 2.675 | 1.205 | 1.268 | 1.617 | 2.396 | 0.53 | −199.76 | |
| TSB | 1.429 | 1.358 | 1.487 | 2.436 | 1.571 | 1.183 | 1.340 | 2.662 | 2.063 | 0.54 | −283.86 | |
Key kinetic parameters of non-catalyzed and LA-catalyzed reactions between cyclopentadiene 1 and carbon dioxide 2 in the light of M062x/6-311G(d) calculations.
| LA catalyst | Solvent (ε) | Transition | ΔH [kcal·mol−1] | ΔS [cal·mol−1·K−1] | ΔG [kcal·mol−1] |
|---|---|---|---|---|---|
| – | Gas phase (1.00) | 1 + 2 → MC | −4.4 | −27.1 | 3.7 |
| 1 + 2 → TS | 38.7 | −40.5 | 50.7 | ||
| DCM (8.93) | 1 + 2 → MC | −3.4 | −26.9 | 4.6 | |
| 1 + 2 → TS | 33.3 | −40.6 | 45.4 | ||
| MeNO2 (36.56) | 1 + 2 → MC | −3.3 | −26.7 | 4.7 | |
| 1 + 2 → TS | 32.4 | −40.7 | 44.5 | ||
| Water (78.36) | 1 + 2 → MC | −3.2 | −26.7 | 4.7 | |
| 1 + 2 → TS | 32.2 | −40.7 | 44.3 | ||
| BH3 | Gas phase (1.00) | 1 + [2/BH3] → MC | −4.5 | −32.7 | 5.2 |
| 1 + [2/BH3] → TSA | 22.2 | −54.6 | 38.5 | ||
| 1 + [2/BH3] → TSB | 24.0 | −55.2 | 40.5 | ||
| DCM (8.93) | 1 + [2/BH3] → MC1 | −3.3 | −30.8 | 5.9 | |
| 1 + [2/BH3] → TS1A | 7.1 | −49.6 | 21.9 | ||
| 1 + [2/BH3] → IA | 5.6 | −50.4 | 20.6 | ||
| 1 + [2/BH3] → TS2A | 8.2 | −57.0 | 25.1 | ||
| 1 + [2/BH3] → TS1B | 8.3 | −48.6 | 22.7 | ||
| 1 + [2/BH3] → IB | 6.9 | −49.6 | 21.7 | ||
| 1 + [2/BH3] → TS2B | 12.5 | −53.0 | 28.3 | ||
| BCl3 | Gas phase (1.00) | 1 + [2/BCl3] → MC | −5.3 | −28.3 | 3.1 |
| 1 + [2/BCl3] → TSA | 18.8 | −54.3 | 34.9 | ||
| 1 + [2/BCl3] →TSB | 19.6 | −55.4 | 36.1 | ||
| DCM (8.93) | 1 + [2/BCl3] → MC1 | −4.1 | −28.7 | 4.5 | |
| 1 + [2/BCl3] → TS1A | 9.9 | −43.9 | 23.0 | ||
| 1 + [2/BCl3] → IA | −3.1 | −47.6 | 11.1 | ||
| 1 + [2/BCl3] → TS2A | 1.3 | −55.7 | 17.9 | ||
| 1 + [2/BCl3] → TS1B | 9.1 | −43.8 | 22.2 | ||
| 1 + [2/BCl3] → IB | −2.7 | −47.1 | 11.3 | ||
| 1 + [2/BCl3] → TS2B | 6.5 | −56.3 | 23.3 | ||
| BBr3 | Gas phase (1.00) | 1 + [2/BBr3] → MC | −4.9 | −30.6 | 4.2 |
| 1 + [2/BBr3] → TSA | 17.5 | −53.4 | 33.4 | ||
| 1 + [2/BBr3] →TSB | 17.8 | −54.0 | 33.9 | ||
| DCM (8.93) | 1 + [2/BBr3] → MC1 | −5.4 | −31.6 | 4.0 | |
| 1 + [2/BBr3] → TS1A | 7.4 | −48.47 | 21.8 | ||
| 1 + [2/BBr3] → IA | −6.5 | −46.1 | 7.2 | ||
| 1 + [2/BBr3] → TS2A | −1.3 | −56.1 | 15.4 | ||
| 1 + [2/BBr3] → TS1B | 7.4 | −45.3 | 20.9 | ||
| 1 + [2/BBr3] → IB | −6.3 | −53.7 | 9.7 | ||
| 1 + [2/BBr3] → TS2B | 4.2 | −55.9 | 20.9 |

The local electronic properties of cyclopentadiene 1, carbon dioxide 2 and BX3-catalysed CO2 complexes according to B3LYP/6-31G(d) theory level in the gas phase. The nucleophilic Pk‾ given in blue and the electrophilic Pk+ given in red. The indexes of local nucleophilicity Nk and local electrophilicity ωk giving in brackets.
The less of the synchronicity of new single bond formation is accompanied with the dramatically reduction of the activation Gibbs free energy. This is a logical consequence of the electrophilical activation of CO2 molecule in the complex with LA. The quantitative description of this effect is illustrated clearly by reactivity descriptors collected in the Table 6 and in the Scheme 4.
The kinetic aspects of the non-catalyzed reaction discussed above, showed that the presence of the solvent should to some extent accelerate the cycloaddition process. With this in mind, we decided to check if this principle also applies to the LA catalyzed reaction. Additionally, it was found, that in DCM solution these-type transformation are realized via stepwise mechanism with acyclic intermediate (Scheme 5). The first stage of the reaction is – analogously as in the gas phase – a formation of pre-reaction MC complex. The further transformation of MC proceed however via two transition states. Dependently of the relatively orientation between [2/BH3] and 1 moieties, this can be TS1A + TS2A or TS1B + TS2B transition states (Fig. 6). Maximums connected with the existence of TSs on reaction profiles are separated by valley of intermediate (IA or IB respectively), which exhibit evidently zwitterionic nature (GEDT > 0.6 e). It should be underlined at this point, that some examples of zwitterionic 6-π-electron cycloaddition processes was recently reported [67], [68], [69]. Is most important, that energetically favored reaction path (Scheme 5) is realized via barrier of the activation, which is not exceed twenty-something kcal·mol−1. In the consequence, the described transformation are full allowed under mild conditions. For comparison, very close values of Gibbs free energies of activation was recently measured experimentally regarding to cycloaddition involving electrophilically activated 2-π-components, which are realized easy at room temperature (Table 9).

Views of key structures of BH3-catalysed reaction between cyclopentadiene 1 and carbon dioxide 2 in the DCM solution in the light of M062X/6-311G(d) calculations.
To completely understand the molecular mechanism of the DA reaction between cyclopentadiene 1 and carbon dioxide 2 catalysed by BH3 extends in the environment of DCM, a BET [60] study was performed. ELF valence basin populations of the selected structures of the IRC are assembled in Table 7. In Fig. 7, the most important structures involved in the formation of one C2–C3 double and two C6–C1 and C4–O5 single bonds was presented. In turn Scheme 7 shows the molecular mechanism of the DA reaction between cyclopentadiene 1 and carbon dioxide 2 catalysed by BH3 in the simulated presence of DCM.

Attractor positions of the ELF valence basins of the most important structures TS1A, P3A, P4A, P5A, P6A and P7A participating in the C2–C3 double and C6–C1 and C4–O5 single bonds formation along the DA reaction cyclopentadiene 1 with carbon dioxide 2 catalyzed by BH3 in the simulated presence of DCM. The electron populations, in average number of electrons, are given in e.

Simplified representation of the molecular mechanism of the DA reaction cyclopentadiene 1 with carbon dioxide 2 catalyzed by BH3, in the simulated presence of DCM, by Lewis-like structures resulting from the topological analysis of the ELF along the reaction path.
BET analysis allows to highlight 10 topological different phases (see Table 7), related to rupture and formation of bonds along the DA reaction between cyclopentadiene 1 and carbon dioxide 2 catalysed by BH3. Phase I begins at MC1, where the interacting reagents are far apart from each other, the ELF picture of MC1 is very similar to those of individual 1 and 2/BH3. Phase II starts at P1A in which the two V(O5,C6) and V′(O5,C6) disynaptic basins, presented in previous phase, have connected into one new V(O5,C6) disynaptic basin, integrating 3.03 e. This topological change is connected with rupture of the O5–C6 double bond in 2/BH3. At this point we also note that two V(O5) and V′(O5) monosynaptic basins, related to nonbonding electron pair on the O5, have merge into one new V(O5) monosynaptic basin, integrating 4.86 e (see point P1A at Table 7). In Phase III, which starts at P2A, the two V(C1,C2) and V′(C1,C2) disynaptic basins present at P1A have merged into a new V(C1,C2) disynaptic basin, whose valence basin population at P2A is 3.26 e. The described topological change is related to rupture of C1–C2 double bond in the molecule of cyclopentadiene 1. The next Phase IV, starts at first transition state (TS1A) of the DA reaction cyclopentadiene 1 with carbon dioxide 2 catalysed by BH3,with energy cost of 10.3 kcal·mol−1 (TS1A (d(C2–C3) = 1.439 Å, d(C4–O5) = 3.017 Å and d(C6–C1) = 2.014 Å). In this phase, a new V(C6) monosynaptic basin is created on the C6 carbon atom with a population of 0.30 e. The electron density form creation of V(C6) monosynaptic basin comes from the O5–C6 bonding region which experiences depopulation from 3.08 e at P2A to 2.85 e at TS1A. This topological change is linked to formation of a pseudoradical centre at C6 carbon atom (Fig. 7). Subsequent Phase V, starts at P3A, which is linked with formation of a new V(C1) monosynaptic basin integrating 0.36 e. In this phase, we observed the formation a second pseudoradical centre at C1 carbon atom, as a result of depopulation of V(C1,C2) disynaptic basin. Formation the first C6–C1 single bond occurs in Phase VI, through the merged two V(C6) and V(C1) monosynaptic basins, with an initial population 1.04 e (see P4A in Table 7, Fig. 7 and Scheme 7). Formation of C6–C1 single bond begins at a distance of d(C6–C1) = 1.760 Å (Table 7). Phase VII, starts at IA, is associated with a minor change related to split the V(O5) monosynaptic basin into two new V(O5) and V′(O5) monosynaptic basins, integrating 2.72 e and 2.52 e. In this phase, we find second transition state (TS2A) of the DA reaction between cyclopentadiene 1 and carbon dioxide 2 catalysed by BH3,with energy cost of 11.4 kcal·mol−1 (TS2A (d(C2–C3) = 1.368 Å, d(C4–O5) = 2.221 Å and d(C6–C1) = 1.592 Å). Phase VIII, begins at P5A and is related with division of one V(C2,C3) disynaptic basins into two new V(C2,C3) and V′(C2,C3) disynaptic basins, integrating 1.59 e and 1.52 e, respectively. This topological change is related to formation of a double C2–C3 bond (see P5A in Fig. 7). The formation of a new V(C4) monosynaptic basin takes place in Phase IX, which is a connect with formation a pseudoradical centre at C4 carbon atom (Fig. 7 and Scheme 7) as a consequence of the depopulation of V(C3,C4) disynaptic basin. Finally, the last Phase X, is located between points P7A and final product 3. At this phase, while the V(C4) and V′(O5) monosynaptic basins current at P6A are lacking, a new V(C4,O5) disynaptic basin is formed, integrating 0.78 e. This topological change is related to formation a second C4–O5 single bond with distance of d(C4–O5) = 1.939 Å (Fig. 7, Table 7 and Scheme 7).
A BET study of the second path of the DA reaction cyclopentadiene 1 and carbon dioxide 2 catalysed by BH3, was also carried out, in order to perceive the bonding changes in this reaction and compare it with the antecedent reaction path. ELF valence basin populations of the selected structures of the IRC are assembled in Table 8. In Fig. 8, the most important structures involved in the formation of one C2–C3 double and two C4–C6 and C1–O7 single bonds was presented. In turn Scheme 8 shows the proposed second path of molecular mechanism of the DA reaction between cyclopentadiene 1 and carbon dioxide 2 catalyzed by BH3.

Attractor positions of the ELF valence basins of the most important structures P2B, TS1B,P3B, P4B and P5B participating in the C2–C3 double and C4–C6 and C1–O7 single bonds formation along DA reaction cyclopentadiene 1with carbon dioxide 2 catalyzed by BH3 in the simulated presence of DCM. The electron populations, in average number of electrons, are given in e.

Simplified representation of the molecular mechanism of the DA reaction cyclopentadiene 1 with carbon dioxide 2 catalyzed by BH3 in the simulated presence of DCM, by Lewis-like structures resulting from the topological analysis of the ELF along the reaction path.
BET analysis mentioned reaction path, allows to describe this path by eight topological different phases (Table 8), associated with rupture and formation of bonds along the DA reaction between cyclopentadiene 1 and carbon dioxide 2, catalysed by BH3. Phase I begins at MC1, which is the first point along the IRC path. In this phase, the ELF picture of MC1 is looks like to those of individuals 1 and 2/BH3. The next Phase II, which starts at P1B, is associated with rupture of the first C1–C2 double bond in 1 molecule. The two V(C1,C2) and V′(C1,C2) disynaptic basins, have merged into one new V(C1,C2) disynaptic basin, integrating 3.29 e. In P2B, Phase III begins. In this phase a new V(C6) monosynaptic basin is created on the C6 carbon atom with a population of 0.24 e. The electron density form creation of V(C6) monosynaptic basin comes from the C6–O7 bonding region which experiences depopulation from 2.76 e at P1B to 2.35 e at P2B. This topological change is connected with formation of a pseudoradical centre at C6 carbon atom (Fig. 8). Phase IV, begins at first transition state (TS1B) of the DA reaction cyclopentadiene 1 and carbon dioxide 2, catalysed by BH3 with energy cost of 11.5 kcal·mol−1 (TS1B (d(C2–C3) = 1.436 Å, d(O7–C1) = 3.033 Å and d(C4–C6) = 1.979 Å). In this point, we observed the formation a new V(C1) monosynaptic basin, integrating 0.34 e. This topological change is related to formation of a second pseudoradical centre at C1 carbon atom as a result of depopulation of V(C1,C2) disynaptic basin (see TS1B in Fig. 8). Formation the first C4–C6 single bond occurs in Phase V, through the merged two V(C4) and V(C6) monosynaptic basins, with an initial population 0.93 e (see P3B in Table 8, Fig. 8 and Scheme 8). Formation of C4–C6 single bond begins at a distance of d(C4–C6) = 1.925 Å (Table 8). Phase VI, begins at IB in which the two V(C3,C4) and V′(C3,C4) disynaptic basins have connected into one new V(C3,C4) disynaptic basin, integrating 2.84 e. This topological change is associated with rupture of C3–C4 double bond. In this phase, we find second transition state (TS2B) of the reaction cyclopentadiene 1 and carbon dioxide 2 with energy cost of 15.8 kcal·mol−1 (TS2B (d(C2–C3) = 1.363 Å, d(O7–C1) = 2.141 Å and d(C4–C6) = 1.579 Å). The ELF picture of next point P4B, in which Phase VII begins, presents formation of a C2–C3 double bond. The V(C2,C3) monosynaptic basin, present in previous point, have split into two new V(C2,C3) and V′(C2,C3) disynaptic basins, integrating 1.66 e and 1.55 e (Fig. 8). Finally, the last Phase VIII, is located between points P5B and 3. In this phase, we observed formation of a new V(O7,C1) disynaptic basin, integrating 0.96 e, as a results of the depopulation of V(O7) monosynaptic basin. This topological change is related to formation a second O7–C1 single bond with distance of d(O7–C1) = 1.943 Å (Fig. 8, Table 8 and Scheme 8).
Finally, on similar way we examined, analogous processes in the simulated presence of other borium-core LA catalysts (Tables 3–5). It was found, that in all cases, similar acceleration of the cycloaddition process are observed. Next, all of these reactions are realized via stepwise mechanism with the participation of zwitterionic intermediate. Our extended studies showed additionally, that according to similar manner, react with CO2 also functionalized analogs of cyclopentadiene. In the consequence, the presented methodology can be treated as general and universal method for the preparation of internal lactones with the consumption of carbon dioxide.
Key parameters of critical structures of LA-catalyzed reaction between cyclopentadiene1 and carbon dioxide 2 in DCM solution in the light of M062X/6-311G(d) calculations.
| LA catalyst | Structure | Interatomic distances [Å] | GEDT [e] | Imaginary frequence [cm−1] | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C1–C2 | C2–C3 | C3–C4 | C4–O5 | C4–C6 | O5–C6 | C6–O7 | C6–O1 | O7–C1 | ||||
| BH3 | 1 | 1.343 | 1.471 | 1.343 | ||||||||
| [2/BH3] | 1.159 | 1.150 | ||||||||||
| MC1 | 1.344 | 1.473 | 1.344 | 3.271 | 3.363 | 1.159 | 1.150 | 3.371 | 3.851 | 0.00 | ||
| TS1A | 1.384 | 1.439 | 1.354 | 3.017 | 3.254 | 1.169 | 1.223 | 2.014 | 2.586 | 0.38 | −237.72 | |
| IA | 1.444 | 1.395 | 1.378 | 2.963 | 2.963 | 1.200 | 1.265 | 1.648 | 2.393 | 0.64 | ||
| TS2A | 1.473 | 1.368 | 1.412 | 2.221 | 2.650 | 1.223 | 1.259 | 1.592 | 2.408 | 0.58 | −240.73 | |
| TS1B | 1.355 | 1.436 | 1.388 | 2.619 | 1.979 | 1.174 | 1.223 | 3.348 | 3.033 | 0.41 | −271.04 | |
| IB | 1.378 | 1.394 | 1.448 | 2.429 | 1.638 | 1.200 | 1.267 | 3.259 | 3.027 | 0.68 | ||
| TS2B | 1.419 | 1.363 | 1.483 | 2.438 | 1.579 | 1.201 | 1.297 | 2.682 | 2.141 | 0.62 | −284.67 | |
| BCl3 | [2/BCl3] | 1.157 | 1.152 | |||||||||
| MC1 | 1.344 | 1.473 | 1.344 | 3.273 | 3.321 | 1.157 | 1.153 | 3.423 | 3.943 | 0.00 | ||
| TS1A | 1.361 | 1.460 | 1.346 | 3.118 | 3.460 | 1.144 | 1.205 | 2.420 | 2.829 | 0.18 | −239.98 | |
| IA | 1.467 | 1.386 | 1.384 | 3.215 | 3.226 | 1.193 | 1.306 | 1.581 | 2.367 | 0.78 | ||
| TS2A | 1.488 | 1.360 | 1.424 | 2.117 | 2.564 | 1.218 | 1.283 | 1.562 | 2.411 | 0.64 | −238.18 | |
| TS1B | 1.348 | 1.455 | 1.366 | 2.863 | 2.329 | 1.148 | 1.210 | 3.443 | 2.995 | 0.24 | −241.89 | |
| IB | 1.385 | 1.383 | 1.472 | 2.419 | 1.572 | 1.192 | 1.312 | 3.324 | 3.109 | 0.81 | ||
| TS2B | 1.446 | 1.350 | 1.505 | 2.447 | 1.540 | 1.185 | 1.368 | 2.584 | 1.943 | 0.67 | −296.22 | |
| BBr3 | [2/BBr3] | 1.157 | 1.153 | |||||||||
| MC1 | 1.344 | 1.473 | 1.344 | 3.274 | 3.381 | 1.157 | 1.153 | 3.274 | 3.381 | 0.00 | ||
| TS1A | 1.360 | 1.461 | 1.346 | 3.142 | 3.484 | 1.142 | 1.206 | 2.456 | 2.845 | 0.17 | −241.26 | |
| IA | 1.474 | 1.383 | 1.384 | 3.944 | 3.547 | 1.193 | 1.318 | 1.549 | 2.343 | 0.81 | ||
| TS2A | 1.489 | 1.359 | 1.426 | 2.106 | 2.555 | 1.217 | 1.285 | 1.560 | 2.412 | 0.64 | −237.86 | |
| TS1B | 1.348 | 1.457 | 1.363 | 2.905 | 2.387 | 1.145 | 1.209 | 3.480 | 3.013 | 0.65 | −248.59 | |
| IB | 1.385 | 1.382 | 1.473 | 2.421 | 1.570 | 1.191 | 1.315 | 3.321 | 3.099 | 0.81 | ||
| TS2B | 1.450 | 1.348 | 1.508 | 2.447 | 1.536 | 1.183 | 1.380 | 2.569 | 1.916 | 0.20 | −295.46 | |
Key electronic properties of carbon dioxide 2 and its complexes with selected LA according to B3LYP/6-31G(d) theory level in the gas phase.
| Component | Electronic potential | Chemical hardness | Global electrophilicity |
|---|---|---|---|
| µ [eV] | ŋ [eV] | ω [eV] | |
| 2 | −4.63 | 10.88 | 0.98 |
| [2/BHr3] | −5.17 | 8.10 | 1.65 |
| [2/BClr3] | −5.23 | 6.96 | 1.97 |
| [2/BBr3] | −4.95 | 5.81 | 2.11 |
ELF valence basin populations, distances of the forming bonds, M06-2X(PCM)/6-311G(d) relativea electronic energies of the IRC structures, MC1–3, defining the 10 phases characterizing the molecular mechanism of the DA reaction between cyclopentadiene 1 and carbon dioxide 2 catalysed by BH3. MC1, TS1A, IA, TS2A and 3 are also included. Distances are given in angstroms, Å, electron populations in average number of electrons, e, and relative energies in kcal·mol−1.
| Points | 1 | 2/BH3 | MC1 | P1A | P2A | TS1A | P3A | P4A | IA | TS2A | P5A | P6A | P7A | 3 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Phases | I | II | III | IV | V | VI | VII | VIII | IX | X | |||||
| d(C2–C3) | 1.471 | 1.468 | 1.467 | 1.439 | 1.427 | 1.411 | 1.395 | 1.368 | 1.357 | 1.353 | 1.349 | 1.332 | |||
| d(C4–O5) | 3.232 | 3.110 | 3.084 | 3.017 | 2.989 | 2.978 | 2.963 | 2.221 | 2.062 | 2.001 | 1.939 | 1.505 | |||
| d(C6–C1) | 2.983 | 2.702 | 2.605 | 2.014 | 1.863 | 1.760 | 1.648 | 1.592 | 1.567 | 1.558 | 1.550 | 1.514 | |||
| ΔE | 0.0 | 1.8 | 7.2 | 10.3 | 9.7 | 9.1 | 8.9 | 11.4 | 8.6 | 2.9 | −0.5 | −1.5 | |||
| V(C1,C2) | 1.74 | 1.76 | 1.78 | 3.26 | 3.05 | 2.54 | 2.32 | 2.26 | 2.14 | 2.08 | 2.08 | 2.06 | 1.93 | ||
| V′(C1,C2) | 1.63 | 1.60 | 1.53 | ||||||||||||
| V(C2,C3) | 2.22 | 2.22 | 2.22 | 2.25 | 2.34 | 2.47 | 2.54 | 2.62 | 2.97 | 1.59 | 1.66 | 1.67 | 1.77 | ||
| V′(C2,C3) | 1.52 | 1.52 | 1.54 | 1.69 | |||||||||||
| V(C3,C4) | 1.74 | 1.68 | 1.69 | 1.70 | 1.64 | 1.56 | 2.90 | 2.85 | 2.54 | 2.43 | 2.26 | 2.21 | 2.05 | ||
| V′(C3,C4) | 1.63 | 1.67 | 1.66 | 1.66 | 1.56 | 1.43 | |||||||||
| V(O5,C6) | 1.69 | 1.66 | 3.03 | 3.08 | 2.85 | 2.73 | 2.69 | 2.50 | 2.34 | 2.25 | 2.21 | 2.18 | 1.85 | ||
| V′(O5,C6) | 1.32 | 1.34 | |||||||||||||
| V(O5) | 3.18 | 3.21 | 4.86 | 4.83 | 4.95 | 5.08 | 5.21 | 2.72 | 3.77 | 4.41 | 4.52 | 4.54 | 4.48 | ||
| V′(O5) | 1.60 | 1.65 | 2.52 | 1.43 | 0.76 | 0.70 | |||||||||
| V(C6) | 0.30 | 0.52 | |||||||||||||
| V(C1) | 0.36 | ||||||||||||||
| V(C6,C1) | 1.04 | 1.63 | 1.88 | 1.96 | 1.99 | 2.01 | 2.15 | ||||||||
| V(C4) | 0.05 | ||||||||||||||
| V(C4,O5) | 0.78 | 1.45 | |||||||||||||
aRelative to the first point of the IRC, MC1.
ELF valence basin populations, distances of the forming bonds, M06-2X(PCM)/6-311G(d) relativea electronic energies of the IRC structures, MC1–3, defining the eight phases characterizing the molecular mechanism of the DA reaction between cyclopentadiene 1 and carbon dioxide 2 catalysed by BH3, MC1, TS1B, IB, TS2B and 3 are also included. Distances are given in angstroms, Å, electron populations in average number of electrons, e, and relative energies in kcal·mol−1.
| Points | 1 | 2/BH3 | MC1 | P1B | P2B | TS1B | P3B | IB | TS2B | P4B | P5B | 3 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Phases | I | II | III | IV | V | VI | VII | VIII | |||||
| d(C2–C3) | 1.470 | 1.467 | 1.446 | 1.436 | 1.431 | 1.394 | 1.363 | 1.351 | 1.349 | 1.332 | |||
| d(O7–C1) | 3.254 | 3.105 | 3.042 | 3.035 | 3.032 | 3.027 | 2.141 | 1.964 | 1.943 | 1.494 | |||
| d(C4–C6) | 2.948 | 2.578 | 2.087 | 1.979 | 1.925 | 1.638 | 1.579 | 1.552 | 1.549 | 1.519 | |||
| ΔE | 0.0 | 3.5 | 7.9 | 11.5 | 10.9 | 10.2 | 15.8 | 8.7 | 4.6 | 3.2 | |||
| V(C1,C2) | 1.74 | 1.68 | 1.68 | 1.61 | 1.59 | 1.59 | 2.84 | 2.57 | 2.32 | 2.30 | 2.07 | ||
| V′(C1,C2) | 1.63 | 1.67 | 1.67 | 1.60 | 1.58 | 1.53 | |||||||
| V(C2,C3) | 2.22 | 2.21 | 2.24 | 2.33 | 2.37 | 2.39 | 2.61 | 3.01 | 1.66 | 1.68 | 1.75 | ||
| V′(C2,C3) | 1.53 | 1.54 | 1.71 | ||||||||||
| V(C3,C4) | 1.74 | 1.76 | 3.29 | 3.08 | 2.72 | 2.62 | 2.27 | 2.11 | 2.04 | 2.02 | 1.92 | ||
| V′(C3,C4) | 1.63 | 1.58 | |||||||||||
| V(C6,O7) | 3.01 | 2.89 | 2.76 | 2.35 | 2.30 | 2.25 | 2.03 | 1.87 | 1.74 | 1.73 | 1.50 | ||
| V(O7) | 3.78 | 3.80 | 3.91 | 3.75 | 3.76 | 3.75 | 3.72 | 3.72 | 3.26 | 2.29 | 2.17 | ||
| V(C6) | 0.24 | 0.42 | |||||||||||
| V(C4) | 0.34 | ||||||||||||
| V(C4,C6) | 0.93 | 1.66 | 1.94 | 2.03 | 2.04 | 2.14 | |||||||
| V(O7,C1) | 0.96 | 1.47 | |||||||||||
aRelative to the first point of the IRC, MC1.
Gibbs free energies of the activation for selected cycloaddition processes involving electrophilically activated 2-π-components.
| Components | Conditions | ΔG≠ [kcal·mol−1] | Ref. |
|---|---|---|---|
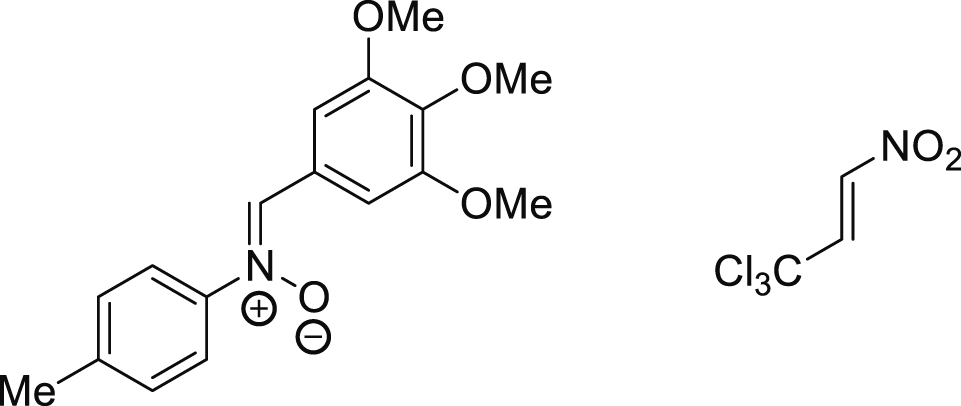 | 25 °C toluene | 22.6 | [70] |
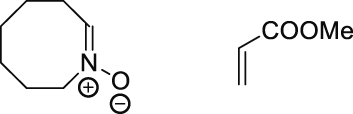 | 25 °C chloroform | 21.3 | [71] |
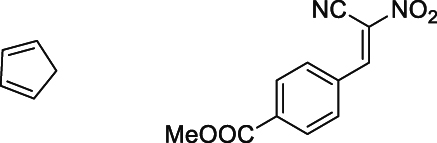 | 25 °C chloroform | 21.0 | [72] |
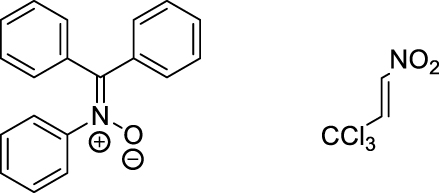 | 25 °C toluene | 20.3 | [73], [74] |
It is worth mentioning that the conversion of CO2 into various heterocycle constitute a very important area of research, e.g. carbon dioxide can be used in a reaction with epoxides into various cyclic carbonates using by boronic acids together with onium salts, which provide highly efficient [75].
Conclusions
MEDT computational study shed valuable light on the possibility of carbon dioxide consumption in the Diels-Alder (DA) involving very popular and good available cyclopentadiene. The simple, non-catalytic reaction in the gas phase is realized via high barrier of activation, which exclude the experimental sense of realization of this type transformation. On the other hand, similar processes in the presence of good known Lewis acids (LA) with the boron core, proceed in the solutions via about 60% lower activation barriers. In the light of our previous experimental study on the kinetic aspects of different type cycloaddition reactions, this level of values of activation parameters are sufficient for the realization of the process at room temperature.
When different Lewis acids are included, the barriers and the activation Gibbs free energies are lower than in the gas phase, but unreachable yet. The LA-catalyzed reactions in the solution are however allowed from kinetic point of view. Is very interesting, that the introduction of LA catalysts to the reaction environment in the solution change dramatically the molecular mechanism of the reagents transformations. In particular, under these-type conditions, the stepwise zwitterionic mechanism successfully replaces the one-step mechanism observed generally regarding to most examples of DA reactions. The detailed study about the reorganization of the electron density in the cycloaddition course was examined in the framework of BET analysis. Analysis of the molecular mechanism associated with the DA reaction between cyclopentadiene and carbon dioxide indicates that it takes place thorough a two-stage one-step mechanism [76], which is initialized by formation of C6–C1 single bond through the connection of two pseudoradical centers. The second C4–O5 single bond was formed in Phase V by joining the V(C4) and V′(O5) monosynaptic basins. BET analysis of the DA reaction cyclopentadiene and carbon dioxide catalyzed by BH3 extends in the environment of DCM, indicates that it takes place through a two-step mechanism. First path of the catalyzed DA reaction is characterized by 10 different phases, while the second by eight topologically different phases. In the first path, we observed the formation a C6–C1 single bond, through the merged two V(C6) and V(C1) monosynaptic basins. While the formation of the second C4–O5 single bond takes place in the last phase, in effect of merging two V(C4) and V(O5) monosynaptic basins. In the second path, we observed the formation of the C4–C6 single bond through the merging of two pseudoradical centers at C4 and C6 carbon and the formation of the second O7–C1 single bond occurs in Phase VII. Based on the topological analysis of the ELF along the reaction paths we can notice that the C–C single bonds formation takes place by sharing the while the new C–O single bonds are formed by the donation.
It should be finally underlined, that the proposed strategy of the CO2 consumption is proceed with full atomic economy, which is one of most important postulate of modern Green Chemistry. Additionally, regarding to similar reactions, we tested also some substituted analogs of the cyclopentadiene. Our results, suggest similar mechanism and kinetic conditions for all considered transformations. In the consequence, the proposed synthetic strategy can be applied for to a wider range of different functionalized internal lactones.
Computational procedure
All quantum chemical calculations were performed using ‘Prometheus’ cluster (CYFRONET regional computational centre). The M06-2x functional [77] included in the GAUSSIAN 09 package [78] and the 6-311G(d) basis set including polarization functions for all relevant atoms was used. Similar level of the theory was recently applied for the resolving of some similar structural/energetical problems (such as synthesis of heterocycle via [3 + 2] and Diels-Alder cycloadditions reactions [79], [80], [81], [82], [83]). All localised stationary points have been characterized using vibrational analysis [84]. It was found that starting molecules as well as products had positive Hessian matrices. On the other hand, all transition states (TS) showed only one negative eigenvalue in their Hessian matrices. For all optimized transition states, intrinsic reaction coordinate (IRC) calculations have been performed. The presence of the solvent in the reaction environment has been included using IEFPCM algorithm [85], with full optimisation of all key structures.
The global reactivity indices (electronic potential μ, chemical hardness η and global electrophilicity ω) were estimated by using theoretical reactivity indices based on the conceptual density functional theory (CDFT) according to the equations recommended by Parr [86] and Domingo [87], [88], [89]. In the calculation are used the correlation-exchange functional B3LYP on the basis 6-31G(d) level set in the gas phase [87], [88]. In particular, the electronic chemical potential μand chemical hardnessη of the reactants studied here were evaluated in terms of the one-electron energies of the frontier molecular orbitals using the following equations [40, 87–89]:
and
where εH and εL may be approached in terms of the one-electron energies of the frontier MOs respectively HOMO and LUMO.
The global electrophilicity ω is given by the following expression [87], [88], [89]:
Electron Localization Function (ELF) [41] research were carried out with the TopMod package [90] considering the standard cubical grid of step size of 0.1 Bohr. The changes in bonds along the respective reaction paths were analyzed in accordance with the Bonding Evolution Theory [60], by carrying out the topological analysis of the ELF for DA reaction between cyclopentadiene 1 and carbon dioxide 2 in gas phase for 154 nuclear configurations along the IRC path and for two path of the DA reaction between cyclopentadiene 1 and carbon dioxide 2 catalyzed by BH3 in environment of DCM for 267 and 240 nuclear configurations, respectively. A comparable attitude has been successfully used to study the molecular mechanism of different types of reactions [36, 61–63, 91].
Acknowledgments
The generous allocation of computing time by the regional computer centre “Cyfronet” in Cracow is gratefully acknowledged.
Article note: A collection of peer-reviewed articles dedicated to Chemical Research Applied to World Needs (CHEMRAWN).
References
[1] G. A. Schmidt, R. A. Ruedy, R. L. Miller, A. A. Lacis. J. Geophys. Res.115, D20106 (2010).10.1029/2010JD014287Suche in Google Scholar
[2] G. Marx, F. Miskolci. Adv. Space Res.1, 5 (1981).10.1016/0273-1177(81)90238-6Suche in Google Scholar
[3] E. Bard. Compt. Rendus Geosci.336, 603 (2004).10.1016/j.crte.2004.02.005Suche in Google Scholar
[4] M. C. Serreze. Conserv. Biol.24, 10 (2010).10.1111/j.1523-1739.2009.01408.xSuche in Google Scholar PubMed
[5] M. C. MacCracken, Strateg. Plan. Energy Environ.28, 8 (2009).10.1080/10485230909509197Suche in Google Scholar
[6] T. R. Anderson, E. Hawkins, P. D. Jones. Endeavour40, 178 (2016).10.1016/j.endeavour.2016.07.002Suche in Google Scholar PubMed
[7] W. Fulkerson, R. R. Judkins, M. K. Sanghvi. Sci. Am.263, 128 (1990).10.1038/scientificamerican0990-128Suche in Google Scholar
[8] J. K. Casper. Greenhouse Gases: Worldwide Impacts (Global Warming), Infobase Publishing, New York (2010).Suche in Google Scholar
[9] M. C. MacCracken, J. Air. Waste. Manag. Assoc.58, 735 (2008).10.3155/1047-3289.58.6.735Suche in Google Scholar PubMed
[10] D. J. A. Johansson. Climatic Change110, 123 (2012).10.1007/s10584-011-0072-2Suche in Google Scholar
[11] T. S. Ledley, E. T. Sundquist, S. E. Schwartz, D. K. Hall, J. D. Fellows, T. L. Killeen. Eos Trans. AGU80, 453 (1999).10.1029/99EO00325Suche in Google Scholar
[12] D. A. Stainforth, T. Aina, C. Christensen, M. Collins, N. Faull, D. J. Frame, J. A. Kettleborough, S. Knight, A. Martin, J. M. Murphy, C. Piani, D. Sexton, L. A. Smith, R. A. Spicer, A. J. Thorpe, M. R. Allen. Nature433, 403 (2005).10.1038/nature03301Suche in Google Scholar PubMed
[13] A. V. E. Ollila. Energy Environ.23, 781 (2012).10.1260/0958-305X.23.5.781Suche in Google Scholar
[14] S. A. Montzka, E. J. Dlugokencky. J. H. Butler. Nature476, 43 (2011).10.1038/nature10322Suche in Google Scholar PubMed
[15] R. K. Pachauri, L. A. Meyer (Eds.), Core Writing Team, IPCC: Climate Change 2014: Synthesis Report. Contribution of Working Groups I, II and III to the Fifth Assessment Report of theIntergovernmental Panel on Climate Change, WMO, UNEP, Geneva, Switzerland (2014).Suche in Google Scholar
[16] P. Friedlingstein, M. W. Jones, M. O’Sullivan, R. M. Andrew, J. Hauck, G. P. Peters, W. Peters, J. Pongrat, S. Sitch, C. Le Quéré, D. C. E. Bakker, J. G. Canadell, P. Ciais, R. B. Jackson, P. Anthoni, L. Barbero, A. Bastos, V. Bastrikov, M. Becker, L. Bopp, E. Buitenhuis, N. Chandra, F. Chevallier, L. P. Chini, K. I. Currie, R. A. Feely, M. Gehlen, D. Gilfillan, T. Gkritzalis, D. S. Goll, N. Gruber, S. Gutekunst, I. Harris, V. Haverd, R. A. Houghton, G. Hurtt, T. Ilyina, A. K. Jain, E. Joetzjer, J. O. Kaplan, E. Kato, K. K. Goldewijk, J. I. Korsbakken, P. Landschützer, S. K. Lauvset, N. Lefèvre, A. Lenton, S. Lienert, D. Lombardozzi, G. Marland, P. C. McGuire, J. R. Melton, N. Metz, D. R. Munro, J. E. M. S. Nabel, S-Ich Nakaoka, C. Neill, A. M. Omar, T. Ono, A. Aeregon, D. Pierrot, B. Poulter, G. Rehder, L. Resplandy, E. Robertson, Ch. Rödenbeck, R. Séférian, J. Schwinger, N. Smith, P. P. Tans, H. Tian, B. Tilbrook, F. N. Tubiello, G. R. van der Werf, A. J. Wiltshire, S. Zaehle. Earth Syst. Sci. Data11, 1783 (2019).10.5194/essd-11-1783-2019Suche in Google Scholar
[17] C. Le Quéré, R. M. Andrew, P. Friedlingstein, S. Sitch, J. Hauck, J. Pongratz, P. A. Pickers, J. I. Korsbakken, G. P. Peters, J. G. Canadel, A. Arneth, V. K. Arora, L. Barbero, A. Bastos, L. Bopp, F. Chevallier, L. P. Chini, P. Ciais, S. C. Doney, T. Gkritzalis, D. S. Goll, I. Harris, V. Haverd, M. Hoffman, M. Hoppema, R. A. Houghton, G. Hurtt, T. Ilyina, A. K. Jain, T. Johannessen, Ch. D. Jones, E. Kato, R. F. Keeling, K. Klein Goldewijk, P. Landschützer, N. Lefèvre, S. Lienert, Z. Liu, D. Lombardozzi, N. Metzl, D. R. Munro, J. E. M. S. Nabel, S-i Nakaoka, C. Neill, A. Olsen, T. Ono, P. Patra, An. Peregon, W. Peters, P. Peylin, B. Pfeil, D. Pierrot, B. Poulter, G. Rehder, L. Resplandy, E. Robertson, M. Rocher, Ch. Rödenbeck, U. Schuster, J. Schwinger, R. Séférian, I. Skjelvan, T. Steinhoff, A. Sutton, P. P. Tans, H. Tian, B. Tilbrook, F. N. Tubiello, I. T. van der Laan-Luijkx, G. R. van der Werf, N. Viovy, A. P. Walker, A. J. Wiltshire, R. Wright, S. Zaehle, Bo Zheng. Earth Syst. Sci. Data10, 2141 (2018).10.5194/essd-10-2141-2018Suche in Google Scholar
[18] C. M. Sánchez-Sánchez, V. Montiel, D. A. Tryk, A. Aldaz, A. Fujishima. Pure Appl. Chem.73, 1917 (2001).10.1351/pac200173121917Suche in Google Scholar
[19] H. Xu, D. Rebollar, H. He, L. Chong, Y. Liu, C. Liu, Ch-J. Sun, T. Li, J. V. Muntean, R. E. Winans, Di-Jia Liu, T. Xu. Nat. Energy5, 623 (2020).10.1038/s41560-020-0666-xSuche in Google Scholar
[20] S. H. Kim, K. H. Kim, S. H. Hong. Angew. Chem. Int. Ed.53, 771 (2014).10.1002/anie.201308341Suche in Google Scholar PubMed
[21] M. Z. Jacobson. Energy Environ. Sci.12, 3567 (2019).10.1039/C9EE02709BSuche in Google Scholar
[22] P. Michorczyk, K. Zeńczak-Tomera, B. Michorczyk, A. Węgrzyniak, M. Basta, Y. Millot, L. Valentin, S. Dzwigaj. J. CO2 Util.36, 54 (2020).10.1016/j.jcou.2019.09.018Suche in Google Scholar
[23] P. Michorczyk, J. Ogonowski. React. Kinet. Catal. Lett.87, 177 (2005).10.1007/s11144-006-0024-4Suche in Google Scholar
[24] L. N. He, J. Q. Wang, J. L. Wang. Pure Appl. Chem.81, 2069 (2009).10.1351/PAC-CON-08-10-22Suche in Google Scholar
[25] T. Iijima, T. Yamaguchi. Appl. Catal. Gen.345, 12 (2008).10.1016/j.apcata.2008.03.037Suche in Google Scholar
[26] F. Shi, Y. Deng, T. SiMa, J. Peng, Y. Gu, B. Qiao. Angew. Chem.115, 3379 (2003).10.1002/ange.200351098Suche in Google Scholar
[27] A. Vavasori, L. Calgaro, L. Pietrobon, L. Ronchin. Pure Appl. Chem.90, 315 (2018).10.1515/pac-2017-0706Suche in Google Scholar
[28] M. Cokoja, M. E. Wilhelm, M. H. Anthofer, W. A. Herrmann, F. E. Kühn. ChemSusChem8, 2436 (2015).10.1002/cssc.201500161Suche in Google Scholar PubMed
[29] H. Yasuda, L. N. He, T. Sakakura. J. Catal.209, 547 (2002).10.1006/jcat.2002.3662Suche in Google Scholar
[30] M. Liu, D. Grana. Adv. Water Resour.142, 103634 (2020).10.1016/j.advwatres.2020.103634Suche in Google Scholar
[31] M. E. Boot-Handford, et al.. Energy Environ. Sci.7, 130 (2014).10.1039/C3EE42350FSuche in Google Scholar
[32] R. Jasiński. J. Mol. Graph. Model.94, 107461 (2020).10.1016/j.jmgm.2019.107461Suche in Google Scholar PubMed
[33] A. Kącka-Zych, R. Jasiński. J. Mol. Graph.101, 107714 (2020).10.1016/j.jmgm.2020.107714Suche in Google Scholar PubMed
[34] M. Osano, D. P. Jhaveri, P. Wipf. Org. Lett.22, 2215 (2020).10.1021/acs.orglett.0c00417Suche in Google Scholar PubMed
[35] R. Jasiński. React. Kinet. Mech. Catal.119, 49 (2016).10.1007/s11144-016-1038-1Suche in Google Scholar
[36] P. Woliński, A. Kącka-Zych, O. M. Demchuk, A. Łapczuk-Krygier, B. Mirosław, R. Jasiński. J. Clean. Prod.275, 122086 (2020).10.1016/j.jclepro.2020.122086Suche in Google Scholar
[37] L. R. Domingo. Molecules21, 1319 (2016).10.3390/molecules21101319Suche in Google Scholar PubMed PubMed Central
[38] L. R. Domingo, M. Ríos-Gutiérrez, P. Pérez. Org. Biomol. Chem.18, 292 (2020).10.1039/C9OB02467KSuche in Google Scholar PubMed
[39] L. R. Domingo, N. Acharjee. in Frontiers in Computational Chemistry, Z. Ul-Haq, J. D. Madura (Eds.), pp. 174–227, Bentham Science Publishers, Singapore (2020).10.2174/9789811457791120050007Suche in Google Scholar
[40] L. R. Domingo, M. Ríos-Gutiérrez, P. Pérez. Molecules21, 748 (2016).10.3390/molecules21060748Suche in Google Scholar PubMed PubMed Central
[41] A. D. Becke, K. E. Edgecombe. J. Chem. Phys.92, 5397 (1990).10.1063/1.458517Suche in Google Scholar
[42] H. Hu, F. Teng, J. Liu, W. Hu, S. Luo. Q. Zhu. Angew. Chem. Int. Ed.58, 9225 (2019).10.1002/anie.201904838Suche in Google Scholar PubMed
[43] H. B. Mereyala, M. Joe. Curr. Med. Chem. Anticancer. Agents.1, 293 (2001).10.2174/1568011013354606Suche in Google Scholar PubMed
[44] G. Sabitha, C. N. Reddy, A. Raju, J. S. Yadav. Tetrahedron Asymmetry22, 493 (2011).10.1016/j.tetasy.2011.02.010Suche in Google Scholar
[45] M. Basanagouda, M. Kulkarni, D. Sharma, V. Gupta, P. Sandhyarani, V. Rasal. J. Chem. Sci.121, 485 (2009).10.1007/s12039-009-0058-zSuche in Google Scholar
[46] B. Ozçelik, I. Gürbüz, T. Karaoglu, E. Yeşilada. Microbiol. Res.164, 545 (2009).10.1016/j.micres.2007.05.006Suche in Google Scholar
[47] P. A. Castelo-Branco, M. M. Rubinger, L. C. de Alves, P. M. de Barros, S. G. Pereira, V. J. de Melo, D. Pilo-Veloso, L. Zambolim. Chem. Biodivers.4, 2745 (2007).10.1002/cbdv.200790223Suche in Google Scholar
[48] A. Li, T. Gong, X. Yang, Y. Guo. Int. J. Biol. Macromol.151, 257 (2020).10.1016/j.ijbiomac.2020.02.107Suche in Google Scholar
[49] A. Skrobiszewski, W. Gładkowski, P. Walczak, A. Gliszczyńska, G. Maciejewska, T. Klejdysz, J. Nawrot, C. Wawrzeńczyk. J. Chem. Sci.127, 687 (2015).10.1007/s12039-015-0823-0Suche in Google Scholar
[50] J. Y. Cho, K. U. Baik, J. H. Jung, M. H. Park. Eur. J. Pharmacol.398, 399 (2000).10.1016/S0014-2999(00)00337-XSuche in Google Scholar
[51] C. Fuqua, M. R. Parsek, E. P. Greenberg. Annu. Rev. Genet.35, 439 (2001).10.1146/annurev.genet.35.102401.090913Suche in Google Scholar
[52] D. R. Kelly. Chem. Biol.3, 595 (1996).10.1016/S1074-5521(96)90125-8Suche in Google Scholar
[53] L. Huang, H. Jiang, C. Qi, X. Liu. J. Am. Chem. Soc.132, 17652 (2010).10.1021/ja108073kSuche in Google Scholar PubMed
[54] M. S. Reddy, Y. K. Kumar, N. Thirupathi. Org. Lett.14, 824 (2012).10.1021/ol2033493Suche in Google Scholar PubMed
[55] K. Wińska, M. Grabarczyk, W. Mączka, B. Żarowska, G. Maciejewska, M. Anioł. J. Saudi Chem. Soc.22, 363 (2018).10.1016/j.jscs.2016.04.004Suche in Google Scholar
[56] F. Fringuelli, A. Taticchi. The Diels-Alder Reaction: Selected Practical Methods, John Wiley&Sons, England (2002).10.1002/0470845813Suche in Google Scholar
[57] L. R. Domingo. RSC Adv.4, 32415 (2014).10.1039/C4RA04280HSuche in Google Scholar
[58] R. Jasiński. J. Fluor. Chem.206, 1 (2018).10.1016/j.jfluchem.2017.12.008Suche in Google Scholar
[59] R. Jasiński. J. Mol. Graph. Model.75, 55 (2017).10.1016/j.jmgm.2017.04.008Suche in Google Scholar PubMed
[60] K. Krokidis, S. Noury, B. Silvi. J. Phys. Chem.A101, 7277 (1997).10.1021/jp9711508Suche in Google Scholar
[61] A. Kącka-Zych. Molecules24, 462 (2019).10.3390/molecules24030462Suche in Google Scholar PubMed PubMed Central
[62] A. Kącka-Zych, L. R. Domingo, R. Jasiński. Res. Chem. Intermed.44, 325 (2018).10.1007/s11164-017-3106-1Suche in Google Scholar
[63] A. Kącka-Zych, R. Jasiński. Theor. Chem. Acc.139, 119 (2020).10.1007/s00214-020-02627-7Suche in Google Scholar
[64] P. Vermeeren, T. A. Hamlin, I. Fernández, F. M. Bickelhaupt. Angew. Chem. Int. Ed.132, 6260 (2020).10.1002/ange.201914582Suche in Google Scholar
[65] K. Sakata, H. Fujimoto. ChemistryOpen9, 662 (2020).10.1002/open.202000112Suche in Google Scholar PubMed PubMed Central
[66] A. Łapczuk-Krygier, J. Jaskowska, R. Jasiński. Chem. Heterocycl. Compd.54, 1172 (2018).10.1007/s10593-019-02410-ySuche in Google Scholar
[67] R. Jasiński, E. Dresler. Organics1, 49 (2020).10.3390/org1010005Suche in Google Scholar
[68] S. A. Siadati, E. Vessally, A. Hosseinian, L. Edjlali. Synth. Met.220, 606 (2016).10.1016/j.synthmet.2016.08.003Suche in Google Scholar
[69] S. A. Siadati. Prog. React. Kinet. Mech.41, 331 (2016).10.3184/146867816X14719552202168Suche in Google Scholar
[70] A. Szczepanek, E. Jasińska, A. Kącka, R. Jasiński. Curr. Chem. Lett.4, 33 (2015).10.5267/j.ccl.2014.10.003Suche in Google Scholar
[71] S. S. Al-Jaroudi, H. P. Perzanowski, M. I. M. Wazeer, Sk. A. Ali. Tetrahedron53, 5581 (1997).10.1016/S0040-4020(97)00214-7Suche in Google Scholar
[72] R. Jasiński. Monatsh. Chem.147, 1207 (2016).10.1007/s00706-016-1735-5Suche in Google Scholar PubMed PubMed Central
[73] R. Jasiński, K. Mróz. React. Kinet. Mech. Catal.116, 35 (2015).10.1007/s11144-015-0882-8Suche in Google Scholar
[74] R. Jasiński, K. Mróz, A. Kącka. J. Heterocycl. Chem.53, 1424 (2016).10.1002/jhet.2442Suche in Google Scholar
[75] J. Wang, Y. Zhang. ACS Catal.6, 4871 (2016).10.1021/acscatal.6b01422Suche in Google Scholar
[76] L. R. Domingo, J. A. Saéz, R. J. Zaragozá, M. Arnó. J. Org. Chem.73, 8791 (2008).10.1021/jo801575gSuche in Google Scholar PubMed
[77] Y. Zhao, D. G. Truhlar. Theor. Chem. Acc.120, 215 (2008).10.1007/s00214-007-0310-xSuche in Google Scholar
[78] M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. PeraltaJr., F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, D. J. Fox. Gaussian 09, Gaussian. Inc., Wallingford CT (2013).Suche in Google Scholar
[79] O. Fedyshyn, Y. Bazeľ, M. Fizer, V. Sidey, J. Imrich, M. Vilkova, O. Barabash, Y. Ostapiuk, O. Tymoshuk. J. Mol. Liq.304, 112713 (2020).10.1016/j.molliq.2020.112713Suche in Google Scholar
[80] N. Korol, M. Slivka, M. Fizer, V. Baumer, V. Lendel. Monatsh. Chem.151, 191 (2020).10.1007/s00706-019-02545-wSuche in Google Scholar
[81] L. R. Domingo, M. Ríos-Gutiérrez, P. Pérez. Molecules25, 2535 (2020).10.3390/molecules25112535Suche in Google Scholar
[82] L. R. Domingo, M. Ríos-Gutiérrez. Org. Biomol. Chem.17, 6478 (2019).10.1039/C9OB01031ASuche in Google Scholar
[83] K. Zawadzińska, K. Kula. Organics2, 26 (2021).10.3390/org2010003Suche in Google Scholar
[84] I. Kurzydym. I. Czekaj. Tech. Trans.117 (2020).Suche in Google Scholar
[85] G. Scalmani, M. J. Frisch. J. Chem. Phys.132, 114110 (2010).10.1063/1.3359469Suche in Google Scholar
[86] R. G. Parr, W. Yang. in Horizonsof Quantum Chemistry, K. Fukui, B. Pullman (Eds.), pp. 5–15, Springer, Netherlands (1979).Suche in Google Scholar
[87] L. R. Domingo, M. J. Aurell, P. Pérez, R. Contreras. Tetrahedron58, 4417 (2002).10.1016/S0040-4020(02)00410-6Suche in Google Scholar
[88] P. Pérez, L. R. Domingo, M. Duque-Noreña, E. Chamorro. J. Mol. Struct.895, 86 (2009).10.1016/j.theochem.2008.10.014Suche in Google Scholar
[89] L. R. Domingo, P. Perez. J. A. Saez. RSC Adv.3, 1486 (2013).10.1039/C2RA22886FSuche in Google Scholar
[90] S. Noury, X. Krokidis, F. Fuster, B. Silvi. Comput. Chem.23, 597 (1991).10.1016/S0097-8485(99)00039-XSuche in Google Scholar
[91] A. Kącka-Zych. Organics1, 36 (2020).10.3390/org1010004Suche in Google Scholar
© 2021 IUPAC & De Gruyter. This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License. For more information, please visit: http://creativecommons.org/licenses/by-nc-nd/4.0/
Artikel in diesem Heft
- Frontmatter
- In this issue
- Preface
- Pure and Applied Chemistry Chemical Research Applied to World Needs (CHEMRAWN) issue
- Special topic papers
- Metathetic approach to new NORPHOS-related bisphosphanes: facile synthesis and application in asymmetric hydrogenation
- Analysis of the possibility and molecular mechanism of carbon dioxide consumption in the Diels-Alder processes
- Effect of MnOx modification and template type on the catalytic performance of ceria-zirconia in CO and soot oxidation
- Mechanistic studies on the formation of 5-hydroxymethylfurfural from the sugars fructose and glucose
- The ghost nets phenomena from the chemical perspective
- Well-defined polyindole–Au NPs nanobrush as a platform for electrochemical oxidation of ethanol
Artikel in diesem Heft
- Frontmatter
- In this issue
- Preface
- Pure and Applied Chemistry Chemical Research Applied to World Needs (CHEMRAWN) issue
- Special topic papers
- Metathetic approach to new NORPHOS-related bisphosphanes: facile synthesis and application in asymmetric hydrogenation
- Analysis of the possibility and molecular mechanism of carbon dioxide consumption in the Diels-Alder processes
- Effect of MnOx modification and template type on the catalytic performance of ceria-zirconia in CO and soot oxidation
- Mechanistic studies on the formation of 5-hydroxymethylfurfural from the sugars fructose and glucose
- The ghost nets phenomena from the chemical perspective
- Well-defined polyindole–Au NPs nanobrush as a platform for electrochemical oxidation of ethanol