One-pot oligosaccharide synthesis: latent-active method of glycosylations and radical halogenation activation of allyl glycosides
-
Rita Pal


Abstract
Chemical glycosylations occupy a central importance to synthesize tailor-made oligo- and polysaccharides of functional importance. Generation of the oxocarbenium ion or the glycosyl cation is the method of choice in order to form the glycosidic bond interconnecting a glycosyl moiety with a glycosyl/aglycosyl moiety. A number of elegant methods have been devised that allow the glycosyl cation formation in a fairly stream-lined manner to a large extent. The latent-active method provides a powerful approach in the protecting group controlled glycosylations. In this context, allyl glycosides have been developed to meet the requirement of latent-active reactivities under appropriate glycosylation conditions. Radical halogenation provides a newer route of activation of allyl glycosides to an activated allylic glycoside. Such an allylic halide activation subjects the glycoside reactive under acid catalysis, leading to the conversion to a glycosyl cation and subsequent glycosylation with a number of acceptors. The complete anomeric selectivity favoring the 1,2-trans-anomeric glycosides points to the possibility of a preferred conformation of the glycosyl cation. This article discusses about advancements in the selectivity of glycosylations, followed by delineating the allylic halogenation of allyl glycoside as a glycosylation method and demonstrates synthesis of a repertoire of di- and trisaccharides, including xylosides, with varied protecting groups.
Introduction
Chemical glycosylations provide an optimal route to prepare designed oligo- and polysaccharides, and glycoconjugates of functional importance. The seminal article by Paulsen provided a foundation to this flourishing area in the last few decades [1]. Chemical glycosylations are practiced for more than a century, the first such method which is utilized till date is the Konigs–Knorr glycosylation method [2], [3]. Fischer glycosidation is known even earlier [4], wherein an acid treatment of the hemiacetal provides the electrophilic reactive intermediate, namely, the oxocarbenium ion or the glycosyl cation, the reaction of which with an alcohol nucleophile completes the formation of the glycosidic bond. The glycosyl synthon leading to the formation of the oxocarbenium ion and the nucleophile which is required for reaction with the oxocarbnium ion are referred to as glycosyl donor and glycosyl acceptor, respectively. The notion that the glycosyl cation formation is an intermediate of the reaction has prompted developments of chemical glycosylations in many innovative ways and continues to attract the attention of practitioners even more (Table 1) [5], [6]. As opposed to enzymatic glycosylations, chemical glycosylations pose stringent requirements of protecting the hydroxyl moieties judiciously on both the glycosyl donor and acceptor components [7]. In addition, such protecting groups are being realized increasingly as moieties that influence the course of the glycosylation reactions. Stereoelectronic, steric and conformational features arising from the protecting groups determine the formation and stability of the glycosyl cation, thereby assuming greater importance in the glycosylation reactions. The emphasis is extended further to the role of the solvents in which the glycosylation is conducted, as a result of the solvent coordination abilities on to the evolving glycosyl cation intermediate. Hydrogen bonding abilities of auxiliary substituents in the protecting groups to stabilize the glycosyl cation are also being advanced [8], [9].
The glycosylation reaction involving the formation of oxocarbenium ion intermediate and few commonly used leaving groups on the glycosyl donor component and their activations.
| Leaving group | Examples of activating agents | Leaving group | Examples of activating agents |
|---|---|---|---|
| –F, –Cl, –Br, –I | Halophilic metal | 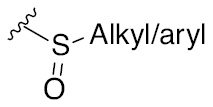 | Br+, I+, Hg+, RS+, Tf2O |
| –OCNHCCl3 | Me3SiOTf BF3·OEt |  | Ph3PAuOTf Ph3PAuNTf2 |
| –SR | Br+, I+ | 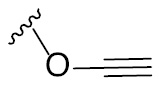 | AuCl3 |
| Glucal | I+ |  | H+, Cu+ |
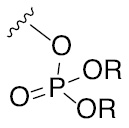 | Me3Si-OTf |  | Br+, I+ |
The ability of the leaving group on the glycosyl donor promoted by a catalyst, namely, the promoter, further adds to the finer details of the oxocarbenium ion stability, through either non-covalent or covalent bond formation. The reactive glycosyl cation formation and reactivity is thus governed by several factors that finally affect the outcome of the glycosylation reaction. The nucleophile entry either through the equatorial or the axial face of the reactive intermediate, leading to the formation of the β- or the α-glycoside product, respectively, relates directly to the stereoselectivity requirement on the target glycoside product. The illustrative reviews by Nielsen and Pedersen [10], Crich et al. [11], Bols and Jensen [12] and Oscarson et al. [13] impart a closer scrutiny of the factors in detail on the glycosyl cation formation and reactivity.
The anomeric halides, thio, trichloroacetimidates, glucal, phosphates/phosphites, esters/carbonates/thiocarbonates and aryloxy groups can be efficiently activated by the corresponding halo-, oxo-, aza- or thiophilic reagents. In addition, anomeric O-derived glycosyl donors, such as, pentenyl, propargyl glycoside can also be activated efficiently using electrophilic halogen reagents or metal ion. Organocatalytic methods also have been developed for stereoselective synthesis of glycosides [5], [6]. One of the pertinent challenges in glycosylations is the regioselective protection-deprotection of the chosen hydroxyl moieties. Further, some of the activations compiled in Table 1 are to be introduced just prior to the glycosylation reaction.
One pot glycosylations attract greater interest, the strategies for the efficient construction of oligosaccharides, wherein two or more glycosylation steps are conducted sequentially without the protecting group manipulation and isolation of the intermediates, yet requiring a pre-activation for the sequential reactivity of the donors. The one-pot glycosylations can be categorised in four major categories: (i) chemoselective; (ii) orthogonal; (iii) pre-activation and (iv) latent-active strategies, based on the factors for the sequential reactivities of the donors. Each of these strategies is described briefly in the following sections.
I. (i) Chemoselective strategy
The influence of the benzyl vs. benzoate protecting groups on the rate of hydrolysis of a glycosyl halide [14] was noticed early by Paulsen, wherein the benzoyl protected glycosyl halide donors underwent slower hydrolysis than that with benzyl protecting groups. A series of elegant work by Fraser-Reid and co-workers advanced the concept of armed-disarmed glycosyl donors, in which protection of hydroxyl moieties with ether protecting groups activated the glycosyl donor ability faster than the glycosyl donor activation in the presence of electron withdrawing group [15]. An early example is the demonstration of this concept as shown in Scheme 1. The O-benzyl protected armed donor 1 and the O-acetyl protected disarmed acceptor 2 reacted, leading to the formation of disaccharide 3, which upon O-deacetylation and installation of benzyl protecting groups to the disaccharide 4 aided a subsequent activation. The reaction of the armed disaccharide 4 with the glycosyl acceptor 5 allowed formation of the trisaccharide 6.
![Scheme 1: Glycosylations leading to the formation of trisaccharide 6 using armed-disarmed glycosyl donors and acceptors [15].](/document/doi/10.1515/pac-2019-0306/asset/graphic/j_pac-2019-0306_scheme_001.jpg)
Glycosylations leading to the formation of trisaccharide 6 using armed-disarmed glycosyl donors and acceptors [15].
Ley and co-workers developed “semi-disarmed” concept [16] wherein torsional effect caused by the presence of a cyclic acetal protection on the glycosyl donor was utilized to develop a one pot glycosylation strategy. Utilizing the concept of semi-disarmed glycosyl donors, the first efforts on tuning the effects of parameters such as the protecting groups, the anomeric leaving group and the nature and stereochemistry of the monosaccharide skeleton were addressed. The sequential glycosylation for the preparation of a trisaccharide 10 from monomers 7–9 on the basis of reactivity of glycoside donor was thus accomplished. The more reactive donor 7 was activated initially, the reaction of which with acceptor 8 afforded the intermediate disaccharide. A repetition of the reaction condition permitted further glycosylation of the disaccharide intermediate with a second acceptor 9, leading to the formation of the trisaccharide 10, in 67% yield (Scheme 2).
![Scheme 2: Synthesis of trisaccharide 10 by a sequential glycosylation [16].](/document/doi/10.1515/pac-2019-0306/asset/graphic/j_pac-2019-0306_scheme_002.jpg)
Synthesis of trisaccharide 10 by a sequential glycosylation [16].
Progress of the concept of ‘armed-disarmed’ glycosyl donor was developed further towards glycosyl donors that are ‘super-armed’. Bols and co-workers [17], [18], and Yamada and coworkers [19] introduced the term super-armed donors, based on installation of sterically bulkier silyl protecting groups on hydroxyl moieties and the evolving altered conformation of the donor permitting the facile formation of glycosyl cation in a twist-boat conformation. A combination of ‘super-armed’ (11) and ‘armed-disarmed’ glycosyl donors and acceptors (12 and 13) was put through to demonstrate the synthesis of a trisaccharide 14 in one-pot, as shown in Scheme 3.
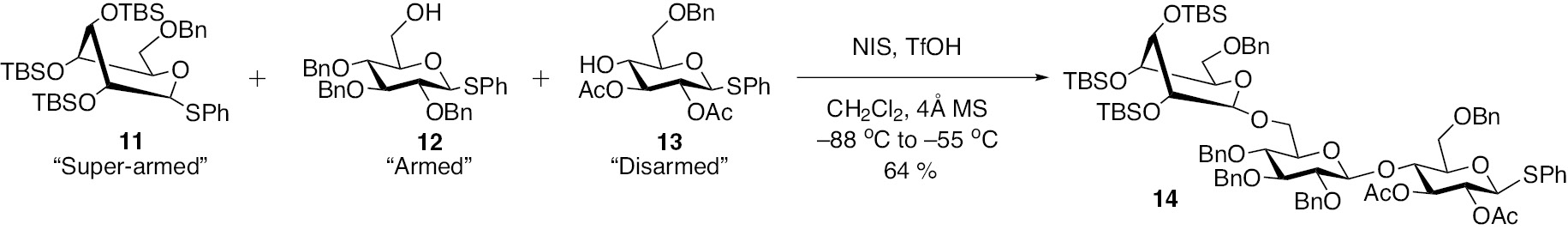
Synthesis of a trisaccharide 14 using synthons that have altered glycosyl donor abilities.
The arming-disarming effect of glycosyl donor is largely relied on the ether and ester protecting groups. Whereas glycosylation with ether protecting groups in the glycosyl donor largely lead to 1,2-cis glycosidic bond, that of with ester protecting groups, 1,2-trans-glycosidic bond forms. In order to overcome this stereoselectivity issue, Zhu and Boons developed C2–C3 cyclic carbonate 16 as a non-participating disarming group, in the synthesis of trisaccharide 19, involving intermediate disaccharide donor 17 (Scheme 4) [20].
![Scheme 4: Synthesis of trisaccharide 19, involving a carbonate protected synthon 16 [20].](/document/doi/10.1515/pac-2019-0306/asset/graphic/j_pac-2019-0306_scheme_004.jpg)
Synthesis of trisaccharide 19, involving a carbonate protected synthon 16 [20].
Wong and co-workers developed an approach to the protecting group control on the reactivity of glycosyl donor and acceptor, where an extensive set of experimental results helped to formulate a ‘relative reactivity value (RRV)’ to a number of glycosyl donors and acceptors [21], [22]. The RRV values enabled appropriate choice of donors and acceptors in multiple one-pot oligosaccharides, namely, a programmable one-pot oligosaccharide synthesis. The programming was also made through machine learning methods, wherein a computer programme called Optimer was developed to aid identification of the best possible combination of building blocks based on the RRV. For example, the deactivating ability of protecting groups at C-2 of a given galactosyl core was established to be in the order of –N3>–O(ClAc)>–NPhth>–OBz>–OBn. The effect of the benzoate group in a galactose modifies depending on the carbon site in the order of C-4>C-3>C-2>C-6. Concerning the reactivity of a sugar moiety, the known trend is galactose>mannose>glucose. The RRV values of hundreds of building blocks were used effectively to synthesize several complex oligosaccharides in a one pot fashion by a sequential addition of the thioglycoside building blocks, starting from the most reactive donor at the non-reducing end. Further development of this machine-learning tool is the creation of a new software called Auto-CHO that aids synthesis of complex oligosaccharides.
The programmable one-pot strategy was employed to synthesize complex oligosaccharides, such as, SSEA-4, heparine pentasaccharide, N-glycans with LacNAc repeats. The hexasaccharide SSEA-4 (23) (Scheme 5) was thus synthesized using three building blocks in the decreasing order of reactivity in a one-pot manner. The most reactive S-tolyl sialo-galactoside 20 was reacted with the monosaccharide 21, in the presence of NIS and a catalytic amount of TfOH at −40°C. The resulting trisaccharide was reacted subsequently with another trisaccharide 22 of least RRV, at −20°C, leading to the formation of the target hexasaccharide 23, in 43% yield. Similar methods have been applied for the synthesis of many oligosaccharides. Although computer programming helps in identifying the best possible combination of building blocks, there is an imminent need to have a pool of building blocks with defined reactivities, in both α- and β- anomeric linkages.
![Scheme 5: Synthesis of a hexasaccharide 23, through programmable one-pot oligosaccharide synthesis guided by the RRV [21], [22].](/document/doi/10.1515/pac-2019-0306/asset/graphic/j_pac-2019-0306_scheme_005.jpg)
Utilizing the effect of solvent on the rate of glycosylation, one-pot synthesis of a trisaccharide was demonstrated by Lahman and Oscarson (Scheme 6) [23]. Ethylthio rhamnoside 24 could be activated selectively using NIS–AgOTf in Et2O in the presence of phenylthio mannoside 25 to synthesize the rhamnose-mannose disaccharide. Change of a solvent in the subsequent glycosylation of the disaccharide intermediate with acceptor glucoside 26 in presence of same activator in CH2Cl2 afforded the trisaccharide 27.
![Scheme 6: One-pot synthesis of trisaccharide 27, with differing solvents for the glycosylations [23].](/document/doi/10.1515/pac-2019-0306/asset/graphic/j_pac-2019-0306_scheme_006.jpg)
One-pot synthesis of trisaccharide 27, with differing solvents for the glycosylations [23].
I. (ii) Orthogonal strategy
Figure 1 shows an approach wherein a leaving group ‘X’ in one glycosyl donor I is activated preferentially in the presence of another leaving group ‘Y’ in a competent glycosyl donor II, to afford disaccharide III. This disaccharide, having the leaving group ‘Y’ at the reducing end reacts with another acceptor IV to form trisaccharide V. The fine-tuning of the reactivity of the anomeric leaving groups ‘X’ and ‘Y’ is aided either by changing the steric or the electronic environment and is independent of the nature protecting groups on the glycosyl donor. This strategy developed by Mukaiyama and co-workers [24] relied on anomeric fluoride and carbonate leaving groups in donor 28 (Scheme 7). Glycosylation with the thioglycoside acceptor 29 was promoted by either TfOH or a tetravalent borane reagent to afford a disaccharide intermediate containing the thioglycoside at the reducing end. The reaction of this disaccharide intermediate with the glycosyl acceptor 30, promoted by NIS in TfOH afforded the trisaccharide 31, which is a mucin related F1a antigen, in a one pot manner. Several other oligosaccharides were also synthesized by this protocol.
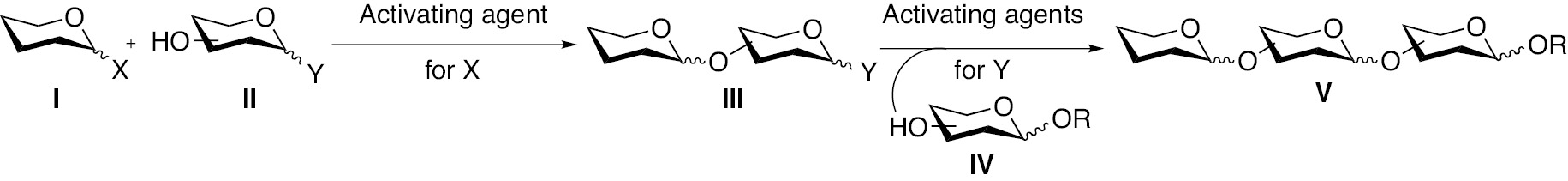
The approach of orthogonality of the leaving groups ‘X’ and ‘Y’ to conduct the one-pot sequential glycosylations.
![Scheme 7: Orthogonal methodology to the synthesis of glucosyl amino acid 31 [24].](/document/doi/10.1515/pac-2019-0306/asset/graphic/j_pac-2019-0306_scheme_007.jpg)
Orthogonal methodology to the synthesis of glucosyl amino acid 31 [24].
Demchenko and De Meo developed a “semi-orthogonal” [25] method to synthesize a tetrasaccharide 35, wherein thio- 32, 34 and pentenyl 33 glycosides were activated sequentially in a one pot manner (Scheme 8). A set of new thioglycosides, such as, S-benzoxazolyl, S-thiazolyl (STaz) and S-benzimidazolyl (SBiz) that could be activated preferentially in the presence of alkyl thioglycosides and these new thioglycosides were implemented to obtain several oligosaccharides. Both STaz and SBiz were used in latent-active glycosylations (vide infra).
![Scheme 8: Synthesis of tetrasaccharide 35 using a semi-orthogonal strategy of glycosylations [25].](/document/doi/10.1515/pac-2019-0306/asset/graphic/j_pac-2019-0306_scheme_008.jpg)
Synthesis of tetrasaccharide 35 using a semi-orthogonal strategy of glycosylations [25].
A combination of chemoselective and orthogonal strategies was used to synthesize an oligosaccharide. As a proof of concept, Ghosh and co-workers [26] reported synthesis of O-antigen of E. coli through mixed one-pot sequential glycosylation. The imidate donor 36 was activated using FeCl3, which was then reacted with the thioglycoside acceptor 37 (Scheme 9). The trisaccharide was activated subsequently with Ph2O/2,4,6-tri-tert-butylpyrimidine (TTBP) – and reacted with thioglycoside acceptor 38. Yet another glycosylation of acceptor 39 with the tetrasaccharide donor intermediate afforded 40, in an impressive overall yield of 72%.
![Scheme 9: Synthesis of pentasacchride 40 through chemoselective and orthogonal strategies [26].](/document/doi/10.1515/pac-2019-0306/asset/graphic/j_pac-2019-0306_scheme_009.jpg)
Synthesis of pentasacchride 40 through chemoselective and orthogonal strategies [26].
I. (iii) Pre-activation strategy
This strategy relies on activation of a glycosyl donor and reaction with an acceptor which possesses the same anomeric leaving group. The resulting glycoside can be activated in situ for further glycosylation in an iterative manner. The method facilitates generating the glycosyl donor and acceptor from common building block. Danishefsky and co-workers demonstrated early on utilizing this approach in an iterative synthesis of oligosaccharides [27]. Figure 2 illustrates the glycal methodology to prepare oligosaccharide in an iterative manner. Activation of glycal VI with an electrophile and the reaction of activated donor VII with an acceptor VIII possessing a glycal functionality occurred to afford disaccharide glycal IX. The iterative glycosylation further afforded the trisaccharide X in an excellent yield.
![Fig. 2: The pre-activation strategy for the synthesis of a oligosaccharide [27].](/document/doi/10.1515/pac-2019-0306/asset/graphic/j_pac-2019-0306_fig_002.jpg)
The pre-activation strategy for the synthesis of a oligosaccharide [27].
Later Gin and co-workers [28] developed chemoselective dehydrative glycosylation and Yamago and co-workers [29] reported selenoglycoside activation with bromine methodology. The glycal method was used for the automated solid phase synthesis of oligosaccharide.
van Boom and co-workers [30], and Yamago and co-workers [31] utilized thioglycoside donors in a pre-activation strategy using 1-benzene sulfinyl piperidine (BSP)/Tf2O as the activating agent. Yamago and co-workers used the BSP–Tf2O mediated activation of thioglycosides for the synthesis of a tetraglucosamines in an iterative manner. In this synthesis, glycosyl donor 41 was activated and reacted with acceptor 42 to afford disaccharide 43. Iteration of glycosylation of the resulting glycoside products with the acceptor 42 afforded tetrasaccharide 44 (Scheme 10).
![Scheme 10: Synthesis of tetrasaccharide 44 by iterative glycosylation using thioglycoside donors and acceptors 41–43 [31].](/document/doi/10.1515/pac-2019-0306/asset/graphic/j_pac-2019-0306_scheme_010.jpg)
Synthesis of tetrasaccharide 44 by iterative glycosylation using thioglycoside donors and acceptors 41–43 [31].
In a similar strategy, Huang and co-workers [32] utilized p-TolSOTf, formed in situ by the reaction of p-TolSCl and AgOTf, as the promoter for the synthesis of oligosaccharide Globo-H 45 (Scheme 11). In this synthesis, a one-pot synthesis consisting of 5 iterative glycosylations, involving donors and acceptors 45–48 was conducted to secure the Globo-H hexasaccharide 49, in overall 47% yield. A stoichiometric amount of the promoter was required to in this iterative glycosylation.
![Scheme 11: Iterative glycosylation using a pre-activation strategy to synthesize hexasaccharide 49 [32].](/document/doi/10.1515/pac-2019-0306/asset/graphic/j_pac-2019-0306_scheme_011.jpg)
Iterative glycosylation using a pre-activation strategy to synthesize hexasaccharide 49 [32].
I. (iv) Latent-active glycosylations
Both the preactivation and latent-active methods allow the glycosyl acceptor and the donor to possess identical functionalities at the anomeric carbon. Yet a difference in the terminology emerges between these two strategies. In the case of the latent-active glycosylation strategy, the latent moiety is made active by a transformation, thereby enabling the moiety to become an active glycosyl donor, which in the presence of a promoter undergoes reaction with an acceptor. On the other hand, the preactivation strategy refers to the in situ activation of the donor by a promoter to generate a reactive intermediate, which is then subjected to glycosylation with the acceptor.
An efficient and rapid assembly of oligosaccharides where donors and acceptors can be synthesized from a common building block has the most potential in an iterative one-pot glycosylations. The emphasis is also to minimize multiple protection-deprotection reactions on glycosyl donors and acceptors. In the latent-active glycosylation strategy, a leaving group (LG) is activated selectively over another LG, with the aid of a suitable promoter. The latent LG, in turn, is made active in a subsequent glycosylation on the resulting glycoside. Such an approach has the potential to avoid protection and deprotection reactions, benefitting a complex target oligosaccharide synthesis.
The latent-active concept was advanced by Roy and co-workers [33], by demonstration of thioglycoside donor XI and acceptor XII as glycosyl components (Fig. 3). The reactivity of the donor was tuned by changing the substitution pattern on arylthio-moiety from an electron withdrawing substituent to an electron donating substituent, thereby enabling donor reactivity of the thioglycoside when reacted with an electrophilic promoter to afford a disaccharide XIII.
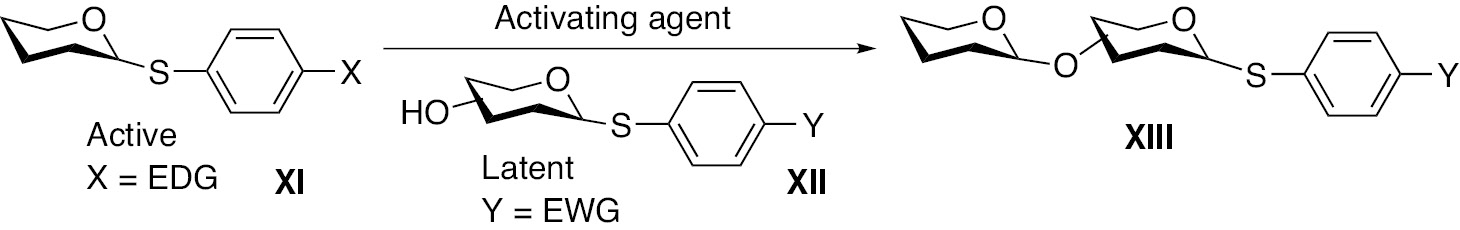
A general scheme of latent-active approach to glycosylation using thioglycosides.
Boons and co-workers [34] reported 1,2-oxathiane ethers that are stable under acidic, basic, and reductive conditions, yet can be converted to an active glycosyl donor upon oxidation to a sulfonium species. This method was employed successfully to synthesize a branched tetrasaccharide 54, originating from Pseudallescheria boydii fungus (Scheme 12). The tetrasaccharide was assembled by a latent-active glycosylation strategy using oxathiane 51 as an acceptor in a glycosylation with a sulfoxide donor 50. The disaccharide 52, in turn, was subjected to oxidation to an active sulfoxide donor 53 for a subsequent glycosylation with appropriate oxathiane glycosyl acceptor.
![Scheme 12: Glycosylation using a bicyclic anomeric sulfonium ion in a latent-active active strategy to synthesize tetrasaccharide 54 [34].](/document/doi/10.1515/pac-2019-0306/asset/graphic/j_pac-2019-0306_scheme_012.jpg)
Glycosylation using a bicyclic anomeric sulfonium ion in a latent-active active strategy to synthesize tetrasaccharide 54 [34].
A new type of latent-active glycosylation reaction by utilizing 2-(benzyloxycarbonyl)benzyl (BCB) glycosides as a latent donor was developed by Kim and co-workers [35]. The BCB glycoside is converted to the corresponding 2-(hydroxycarbonyl)benzyl (HCB) glycoside by hydrogenolysis (Scheme 13). In presence of triflic anhydride and di-tert-butylmethylpyridine, glycosyl donor 55 undergoes glycosylation with acceptor 56, possessing a BCB moiety at the anomeric position, to afford disaccharide 57, with β-mannopyranosyl linkage. The latent BCB-disaccharide was, in turn, converted to an active HCB-disaccharide by hydrogenolysis to acid 58. Repetitive glycosylation of the donor with the same acceptor 56 afforded the BCB-trisaccharide 59.
![Scheme 13: Synthesis of trisaccharide 59 through BCB/HCB donors in a latent-active glycosylation [35].](/document/doi/10.1515/pac-2019-0306/asset/graphic/j_pac-2019-0306_scheme_013.jpg)
Synthesis of trisaccharide 59 through BCB/HCB donors in a latent-active glycosylation [35].
S-Benzimidazolyl (SBiz) glycosides were utilized by Demchenko and co-workers [36] to develop a new latent-active glycosylation, wherein the SBiz donor 60 was selectively activated in the presence of the N-anisoylated SBiz acceptor 61. The initial glycosylation reaction was conducted between the donor and the acceptor, in the presence of MeI to obtain the latent disaccharide 62 (Scheme 14). Deprotection of the N-anisoyl group was achieved on treatment with NaOMe in MeOH to get the active disaccharide 63 which was used to conduct glycosylation under same condition with the acceptor 64 to afford the trisaccharide 65.
![Scheme 14: Synthesis of a trisaccharide 65 initiated from S-benzimidazolyl (SBiz) glycosides 60 and 61, through a latent-active glycosylation method [36].](/document/doi/10.1515/pac-2019-0306/asset/graphic/j_pac-2019-0306_scheme_014.jpg)
Synthesis of a trisaccharide 65 initiated from S-benzimidazolyl (SBiz) glycosides 60 and 61, through a latent-active glycosylation method [36].
A new glycosyl donor ortho-(methyltosylaminoethynyl)benzyl glycoside was developed by Yu and co-workers [37], wherein the glycosides were prepared from the corresponding ortho-iodobenzyl glycosides via a Sonogashira coupling with a ynamide. The glycosides were used in the synthesis of oligosaccharides in a latent-active manner. In the ‘latent-active’ assembly of glycans, the ‘active’ ortho-(methyltosylaminoethynyl)-benzyl glycoside 66 was coupled with the ‘latent’ ortho-iodobenzyl glucoside derivative 67 in the presence of TMSOTf (0.1 eq.) to obtain disaccharide 68 (Scheme 15), which, in turn, was activated via the iterative Sonogashira coupling to derivative 69 and subjected to the glycosylation sequence to afford trisaccharide 70.
![Scheme 15: Synthesis of trisaccharide 70 through activation ortho-(methyltosylaminoethynyl)benzyl glycoside 66, in a latent-active glycosylation methodology [37].](/document/doi/10.1515/pac-2019-0306/asset/graphic/j_pac-2019-0306_scheme_015.jpg)
Synthesis of trisaccharide 70 through activation ortho-(methyltosylaminoethynyl)benzyl glycoside 66, in a latent-active glycosylation methodology [37].
Recently, 2-(2-propylthio)benzyl (PTB) glycosides were exploited as a donor in a new latent-active glycosylation method [38]. An example is shown in Scheme 16. The active glycosyl donor 72, 2-(2-propylsulfinyl)benzyl (PSB) glycoside was synthesised by oxidation of the corresponding PTB glycoside. Treatment of the PSB donor with acceptor 71, in the presence of triflic anhydride provided the desired disaccharide 73 in a good yield. This methodology was extended for the synthesis of natural hepatoprotective glycoside, leonoside F 77, involving intermediates 74 and 75, and acceptor aglycon 76.
![Scheme 16: 2-(2-Propylthiol)benzyl (PTB) glycosides as glycosyl donors to synthesize trisaccharide 77, in latent-active glycosylation methodology [39].](/document/doi/10.1515/pac-2019-0306/asset/graphic/j_pac-2019-0306_scheme_016.jpg)
2-(2-Propylthiol)benzyl (PTB) glycosides as glycosyl donors to synthesize trisaccharide 77, in latent-active glycosylation methodology [39].
The problem associated with the disarmed donor in this method was resolved by introducing a new thioglycoside, namely, S-2-(2-propylsulfinyl)benzyl (SPSB) glycoside as a glycosyl donor that was efficiently activated in a tandem remote mode [39]. The SPSB glycoside was synthesized from the corresponding S-2-(2-propylthio)benzyl (SPTB) glycoside by oxidation.
II. (i) Allyl glycoside in latent-active glycosylations
As a protecting group, allyl moiety is orthogonal to other protecting groups and is deprotected under mild conditions through many methods, such as, the metal-mediated isomerization and acidolytic cleavage. The metal-mediated isomerization of allyl ether to vinyl ether is also used beneficially to conduct a glycosylation reaction with an acceptor alcohol. Further elegant development pertains to establishing such vinyl glycoside intermediate as a glycosyl donor within the preamble of latent-active glycosylation concept. In a latent-active glycosylation, an allyl glycoside is activated as a reactive vinyl glycoside donor, which upon reaction with an allyl glycoside having an acceptor alcohol moiety, leads to a glycoside product. The newly formed glycoside possesses the allyl moiety at the reducing end, which is activated as the isomerized vinyl ether donor for subsequent reaction with a latent allyl glycoside acceptor, leading to the formation of a newer glycoside.
Isomerisation of a latent substituted allyl glycoside to an active vinyl glycoside enabled the glycosylation reaction in the presence of a Lewis acid, as reported early by Boons and co-workers [40], [41]. This protocol was utilized to synthesize saccharide libraries. In the first step of glycosylation, the substituted allyl glycoside 78 is isomerised to the substituted vinyl ethers, using Wilkinson catalyst. The vinyl ether intermediate is used as an active glycosyl donor promoted by TMSOTf to an allyl glycoside acceptor 79, leading to the formation of disaccharide 80, in an excellent yield (89%) (α/β l/20). The allyl moiety in disaccharide is subjected to the metal-mediated activation to an active vinyl glycoside intermediate and the glycosylation is enabled repetitively to secure trisaccharide 81 (Scheme 17).
![Scheme 17: Synthesis of trisaccharide 81 using allyl glycoside as a latent glycosyl acceptor moiety and the corresponding isomerized vinyl glycoside as an active glycosyl donor [40], [41].](/document/doi/10.1515/pac-2019-0306/asset/graphic/j_pac-2019-0306_scheme_017.jpg)
The use of allyl glycosides for glycosylations in a latent-active fashion was utilized for the synthesis of Shigella flexneri serotype Y O-antigen by Wang and co-workers [42]. Thus, the allyl glycosyl donor 82 was isomerized to its corresponding vinyl glycoside with iridium catalyst, followed by an activation with NIS/TfOH and glycosylation with the allyl glycosyl acceptor 83 led to the disaccharide 84 in 82% (Scheme 18). Repeating the reactions, the disaccharide 87 was obtained from the glycosylation of the donor 85 and the acceptor 86 in 83% yield. Glycosylation of disaccharides 87 as isomerized active glycosyl donor and 84 as latent glycosyl acceptor afforded the desired fully protected tetrasaccharide 88, in 76% yield.
![Scheme 18: Synthesis of tetrasaccharide 88 by the latent-active glycosylation methodology using appropriate ally1 glycosides [42].](/document/doi/10.1515/pac-2019-0306/asset/graphic/j_pac-2019-0306_scheme_018.jpg)
Synthesis of tetrasaccharide 88 by the latent-active glycosylation methodology using appropriate ally1 glycosides [42].
II. (ii) Allylic halide activation of allyl glycosides
A newer allylic glycoside activation was developed recently in our research group [43]. The newly developed synthetic method combines radical-mediated activation of an allyl glycoside to an allyl halide, which upon reaction with a glycosyl and aglycosyl acceptor in the presence of an acid promoter leads to a glycoside product. Development of this new glycosylation methodology is fine-tuned further so as to implement the latent-active concept, in which a reactive allyl glycoside donor undergoes glycosylation with a latent allyl glycoside acceptor to form a glycosylated product. The glycosylated product, in turn, possesses the allyl moiety at its reducing end suitable for further activation. The synthetic scheme initiated with an allyl glycoside as synthon of the present work is shown in Fig. 4.
![Fig. 4: A scheme of latent-active glycosylation involving an allyl glycoside, initiated by a radical-mediated allylic halogenations [43].](/document/doi/10.1515/pac-2019-0306/asset/graphic/j_pac-2019-0306_fig_004.jpg)
A scheme of latent-active glycosylation involving an allyl glycoside, initiated by a radical-mediated allylic halogenations [43].
In this strategy, allyl glycoside XIV is subjected to (i) a free radical-mediated allylic halogenation to afford allylic halide XV; (ii) treatment of the allylic halide intermediate with an acid promoter and (iii) reaction with an acceptor allylic glycoside XVI, thereby leading to the formation of glycoside XVII. Reiteration of the above steps leads to higher glycoside product XVIII. The sequence of converting allyl glycoside XIV to product oligosaccharide allyl glycosides XVII and XVIII is accomplished in one pot.
The anomeric allyl protecting group of the donor was activated by brominating the allylic position using the prototypical reagent system N-bromosuccinimide (NBS)/azobisisobutyronitrile (AIBN) in CCl4. The newly formed allylic halide moiety is highly unstable and upon exposure to moisture leads to the formation of hemiacetal. Among the protecting groups on the glycosyl donor, esters were found to be optimal, whereas benzyl ethers are incompatible due to competitive benzylic halogenation. Upon allylic halogenation of the ester protected allylic glycoside donors, the resulting allylic halide was reacted with acceptor glycosyl/aglycosyl alcohols, in presence of a promoter. Halophiles AgOTf, AgClO4 and AgCO3 were assessed as the promoter of the reaction, so as to derive the oxocarbenium ion and subsequent of the same with alcohols. Initial reactions conducted using MeOH as the acceptor is shown in Scheme 19.
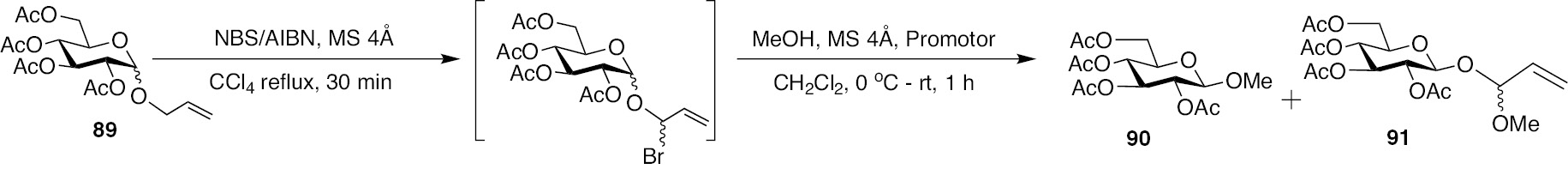
Reaction of allyl tetra-O-acetyl glucopyranoside 89 with MeOH.
It was observed that using AgClO4 as the promotor, the reaction of allyl tetra-O-acetyl glucopyranoside 89 afforded the desired glycosylated product 90, along with a varied product, characterization of which revealed it to be the substitution product 91 as the major product. Further, AgCO3 as promotor also led to only the substitution product. Similar results were observed when the reactions were conducted with allyl tetra-O-acetyl mannopyranoside and allyl tri-O-acetyl fucopyranoside. The higher reactivity of MeOH coupled with ambient temperature might promote the reaction towards substitution of the halide, rather than the formation of the oxocarbenium ion.
An improvement in the glycosylation product 90 was observed when using AgOTf as the promotor. Approximately, 60% yield of 90 was obtained, along with the substitution product 91 in 20% yield. Thus, among the promoters verified, AgOTf was found to be optimal and further initial glycosylations were conducted with this promoter. Relating to the glycosylated product 90, the β-anomer formed exclusively, corresponding C1–C2 trans-configurations, adhering to the nominal expectation of a neighbouring group participation in the glycosidic bond formation.
A change of the protecting group from O-acetyl to O-benzoyl on the donor moiety increased the yield of the glycosylation product. Scheme 20 shows the glycosylation reactions using these two protecting groups in allyl glucopyranosides (89 and 92), promoters AgClO4, AgOTf, acceptors MeOH, n-pentanol and glycosyl acceptor. Whereas n-pentanol afforded pentyl glycoside 93 when AgOTf was used as the promoter, MeOH afforded exclusively substitution product 91. A difference in the reactivities of alcohols thus contributes to the glycosylation vs substitution products. The glycosyl acceptor 94 afforded disaccharides products 95 and 96, in moderate yields.
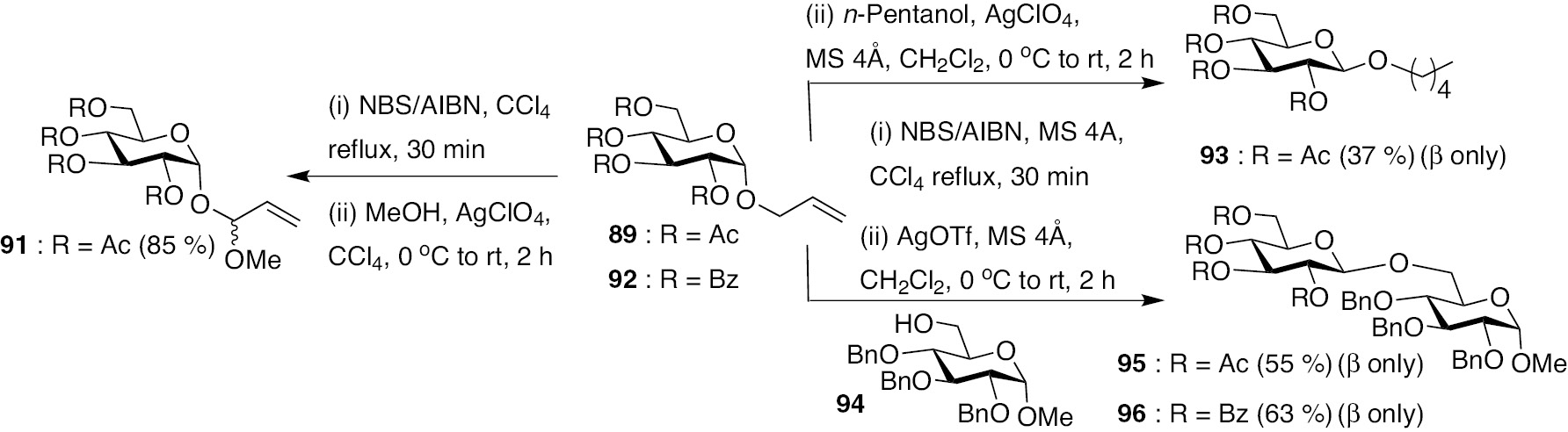
Reactions of allyl glycoside 89 and 92 in the presence of AgClO4 and AgOTf promoters in the glycosylation reaction.
Continuing optimization, a metal-free promoter TfOH was used to mediate the glycosylation. In the event, a significant increase in the yield of the required glycosylation product obtained. The formation of the allylic substitution product was absent in this reaction, implying that the oxocarbenium ion formation is the preferred reaction of the glycosyl donor with TfOH promoter. Rigorous exclusion of moisture is necessary, without which hemiacetal formation competes the glycosylation reaction. It is also pertinent to note that a glycosylation reaction did not proceed in the absence of a promoter reacting with the glycosyl allylic halide intermediate, implying the role of the promoter in the reaction.
The new glycosylation reaction temperature was assessed between −40°C and 0°C, for reactions involving MeOH as the acceptor and glycosyl donor 89, during 2 h of reaction duration. Initial attempts to conduct the reaction at −78°C showed a lack of the reaction and the acceptor alcohol was recovered, along with the hemiacetal arising from the donor. On the other hand, when the reaction was conducted at 0°C, the reaction course preferred a substitution reaction, wherein the halide was substituted with the incoming alcohol, along with the desired glycoside as a minor product, whereas, reaction at ambient temperature led to only a mixture of products. Reactions conducted at −30°C afforded the glycoside as the exclusive product, thereby warranting further reactions to be conducted at this temperature.
Varying the solvent for glycosylation of glycosyl acceptor 98 with donor 97 was exercised and the reactions were conducted in CH2Cl2, CH3CN and CH3NO2. Among these solvents, CH2Cl2 afforded the glycoside product 99 in better yields than remaining solvents (Scheme 21). Whereas CH3NO2 as solvent led to a complex mixture of the crude reaction mixture, CH3CN mediated the glycosylation to afford the desired glycoside in 65% yield, as 1:1 α/β anomers. The coordination ability of CH3CN is observed by which there is considerable percentage of α-anomer was also obtained along with the β-anomer, implying the role of solvents on the oxocarbenium ion species.
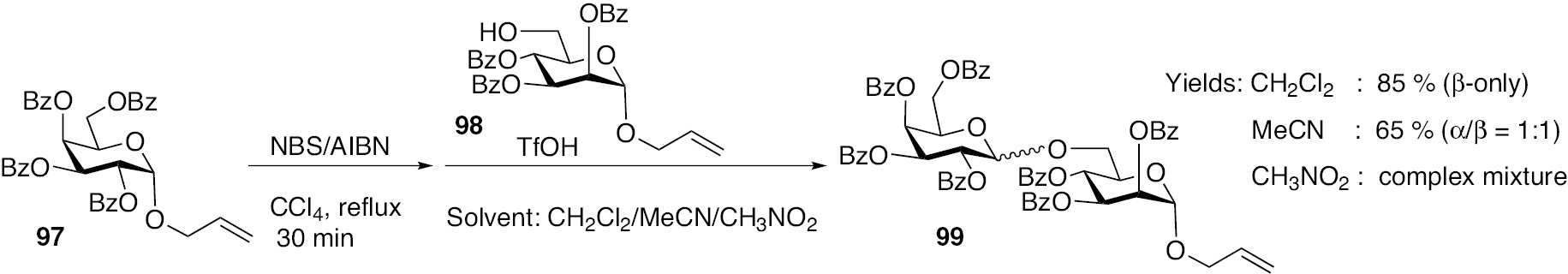
Allylic halide mediated glycosylation, with varying solvents for the glycosylation step.
With the optimised reaction conditions, glycosylation reactions were performed with various glycosyl donors and acceptors, originating from pyranosides of glucose, galactose, mannose and fucose. Further, the glycosylations were conducted so as to secure glycosides with C1–C2–C4 and C1–C6 glycosidic bond connectivities. Among the glycosyl donors, a general observation was that the galactopyranosyl-donor was more reactive, underwent complete activation and glycosylation occurred rather facile. Similarly, primary alcohol as acceptor underwent efficient glycosylation compared to secondary alcohols. Scheme 22 summarizes disaccharide formation from the gluco- and galactopyranosyl donors, reacting with appropriate glycosyl acceptor, using TfOH as the promoter in CH2Cl2 at −40°C. As can be seen, the new glycosylation method is facile, providing the glycoside products in good to excellent yields, covering the glycosidic bond connectivities arising from all secondary and primary alcohols as the acceptor sites.
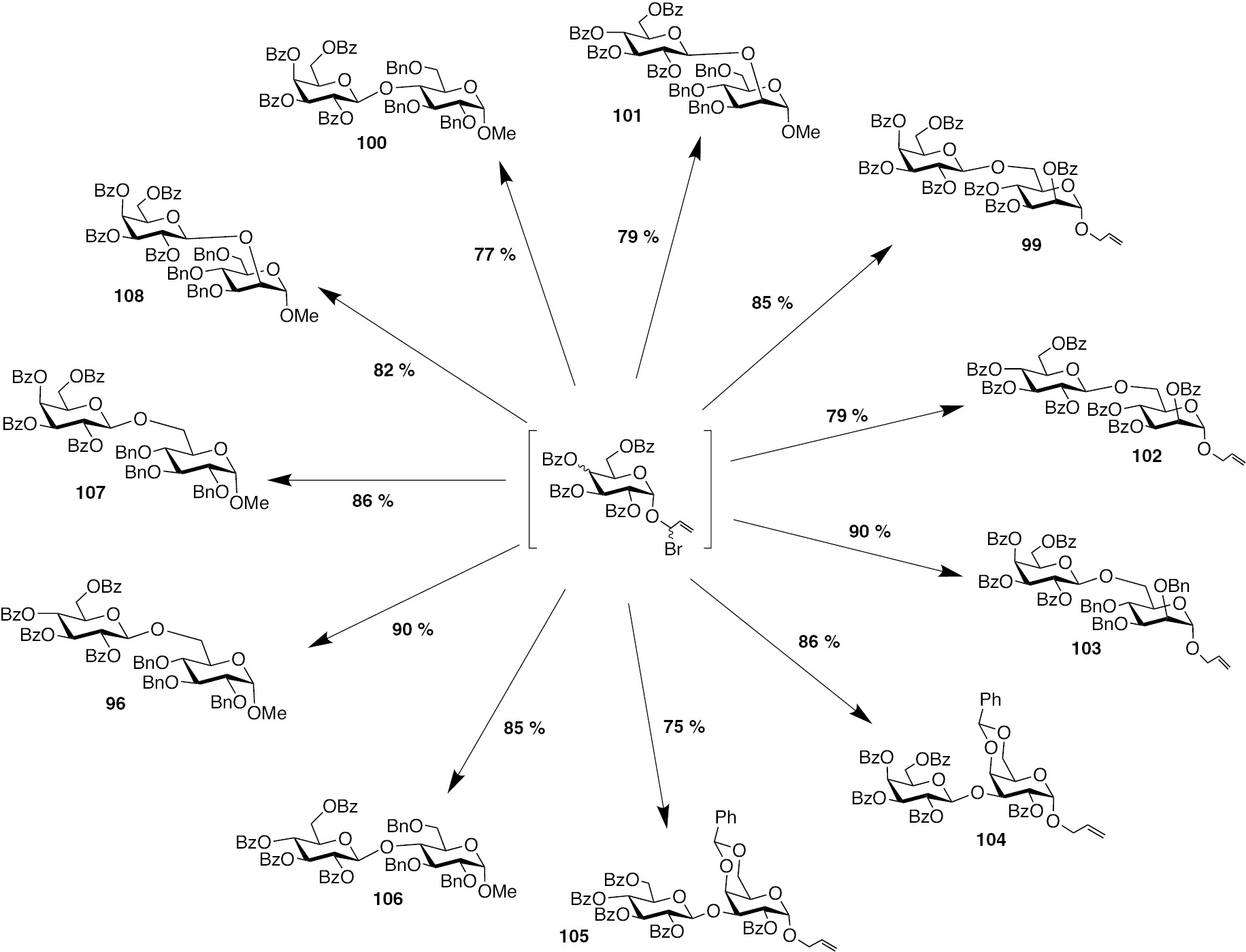
Glycosylation products formed from reactions of glucose and galactose donors with appropriate glycosyl acceptors.
Whereas the ester protecting groups on the glycosyl donors are optimal, glycosyl acceptors might possess varying protecting groups, including benzyl ethers. As can be seen with examples 96, 100, 101, 103 and 106–108 glycosyl acceptors with benzyl ether protecting groups do not encounter limitations under the glycosylation reaction conditions and glycosides form in excellent yields. Similarly, glycosylation with 4,6-benzylidene protected glycosyl acceptors affords the disaccharides in good yields, examples are formation of disaccharides 104 and 105. Secondary hydroxyl functionalities as glycosyl acceptors did not differ greatly in reactivities, as adjudged through the corresponding disaccharide formation with comparable yields. Scaling up of glycosylation reactions to grams could be conducted, without a loss in the efficiency of the reaction, thereby indicating that this new allylic halide activation extends scope for glycosylations in larger quantities of donors and acceptors.
The lability of acetate protecting group under TfOH mediated glycosylation reaction was observed, wherein the acetate moiety underwent deprotection, thereby the newly generated alcohol functionality turn out to be an acceptor site. In the light of this lability, glycosylations involving glycosyl donors and acceptors with acetate protecting groups might not be preferred. Silyl protecting group on the donor component is compatible, as given in Scheme 23 for the preparation of mannose 1–6-linked disaccharide 110, from donor 109.
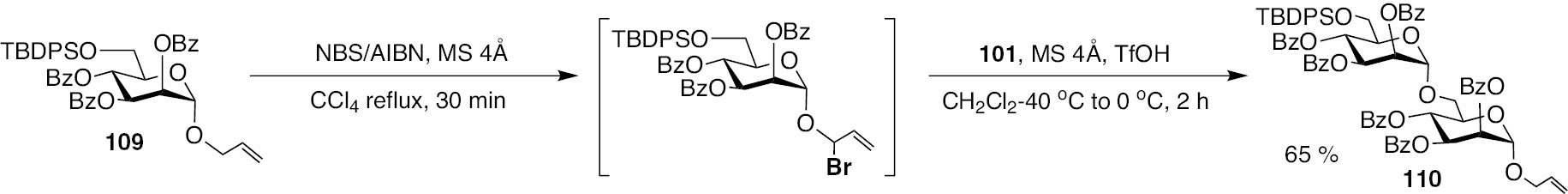
Silyl protecting group in glycosyl donor 109 and the glycosylation leading to the formation of disaccharide 110.
Glycosylations involving the activated mannopyranosyl donor 111 and varied glycosyl acceptors led to disaccharide formation in good yields, depending on the protecting groups on the donor, using reaction conditions of TfOH promoter in CH2Cl2 at −40°C. Synthesis of several disaccharides using a common mannopyranosyl donor with appropriate acceptors is shown in Scheme 24. Thus, with acetate protecting groups, the disaccharide formation is only moderate, as a result of the lability of acetate groups under acidic reaction conditions. Benzyl, benzylidene and benzoate protecting groups on the acceptor moieties are compatible to the reaction and the disaccharides form as the exclusive product. With ester as the protecting groups in the donor component, the reactions are under the manifold of anchimeric or neighbouring group assistance and afford 1,2-trans-configured disaccharides.
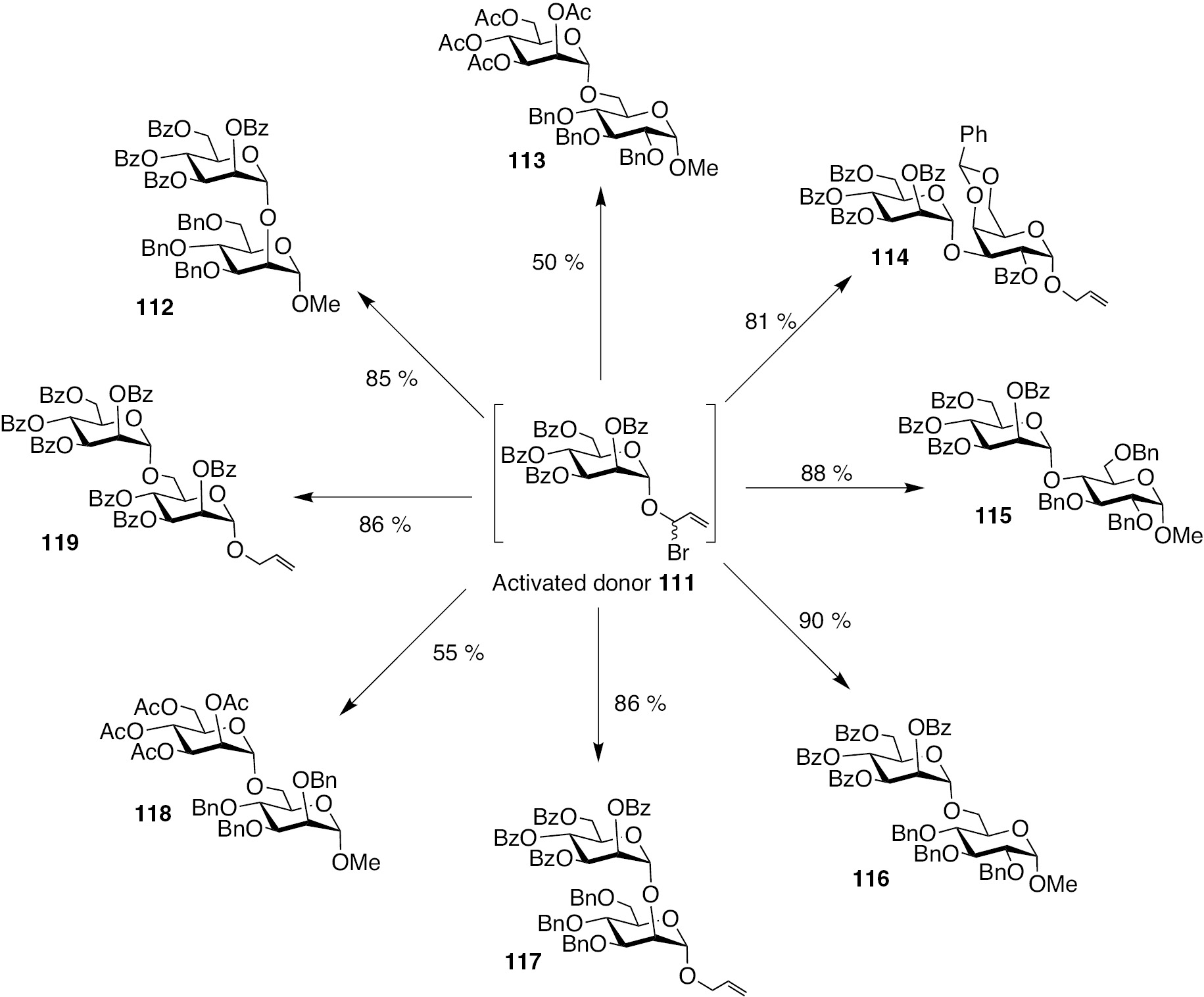
Glycosylation of allylic halide activated mannopyranosyl donor with varying glycosyl acceptors and the formation of corresponding disaccharides.
Following synthesis of disaccharides, the new glycosylation method was subjected to double glycosylation possibilities. In these instances, glycosyl acceptors having two hydroxyl functionalities were planned. The reaction sequence leading to a trisaccharide 121 is shown in Scheme 25. The mannopyranosyl donor 111 was activated to an allylic halide with the aid of NBS/AIBN reagent system. Reaction of allylic halide intermediate with diol acceptor 120, in the presence of catalytic TfOH, leads to the formation of the double glycosylated product, namely, the (1,3), (1,6)-linked trisaccharide 121, in a moderate yield. The disaccharide 122 also formed as a minor product in the reaction.
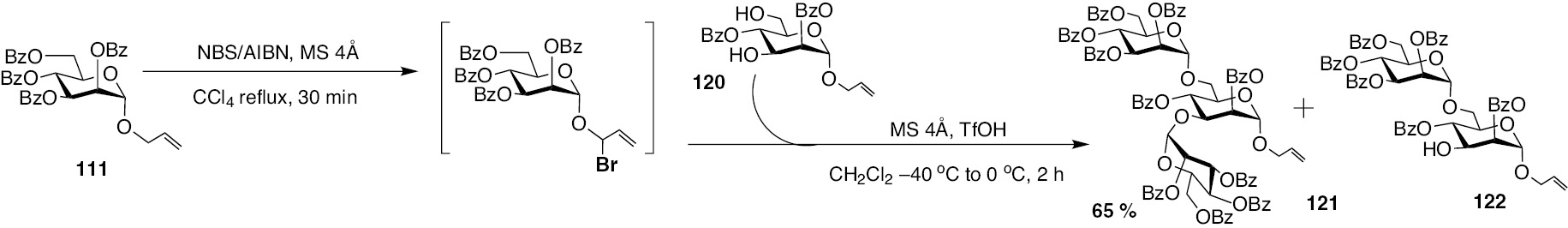
Preparation of trisaccharide by double glycosylation of glycosyl donor 111, with acceptor 122.
A step-wise glycosylation of the synthesis of a trisaccharide was also feasible, wherein mannosyl donor 111 reacts with acceptor 98 to afford disaccharide 119, having the allyl moiety at the reducing end (Scheme 26). Iteration of the glycosylation reaction of halide activated 119 with the acceptor 98 subsequently affords the (1–6), (1–6)-linked trisaccharide 123, in a moderate yield after two-stage glycosylation starting from donor 111. Stereoselectivity of the reaction remains 1,2-trans-selective. Similarly, the two-stage glycosylation could also be accomplished on galactosyl donor 97, which upon allylic halide activation and reaction with allyl mannopyranoside 98 in the first step to afford disaccharide 99, followed by another iteration on 99 leads to formation of trisaccharide 124, in a good yield.
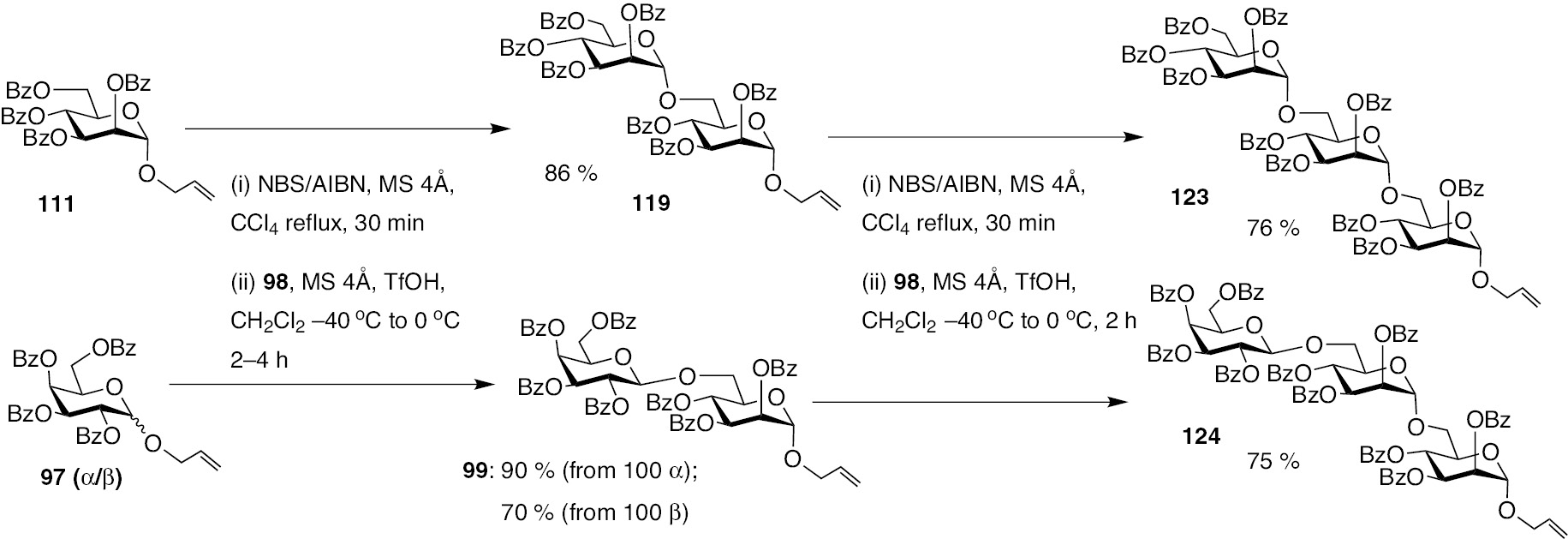
Preparation of trisaccharides 123 and 124 by the latent-active glycosylation method.
The above reactions illustrate that allylic halide activation mediated glycosylation method is suitable to sequential reactions, expanding the principle of latent-active glycosylation methodology.
The β-allyl galactopyranoside anomer (97β) was reacted as a glycosyl donor, similar to that of the corresponding α-anomer. Whereas the halogenation occurred in the first step within 30 min., subsequent glycosylation with acceptor alcohol for 4 h afforded the disaccharide 99 in a good yield, with 1,2-trans-configuration at the newly formed glycosidic bond.
The newly developed allylic halide mediated glycosylation method was utilized towards synthesis of β-(1→3)-linked xylan. Xylans being one of the major constituents of the plant hemicelluloses [44] and are also known to play a major role in initiating the biosynthesis of GAG [45], there have been efforts to synthesize xylo-oligosaccharides. The reactivities of three secondary equatorial hydroxyl groups in xylose is dependent on the C-1 configuration [46]. For example, regioselectivity of protection among the 3 secondary hydroxyl groups are dependent on whether the anomer is α- of β-. Further, in homo- and heteroxylans, the xylose moieties can be linked to one another in either β-(1→3) or β-(1→4) linkage [47]. Figure 5 represents a few early works reported on the synthesis of homoxylans.
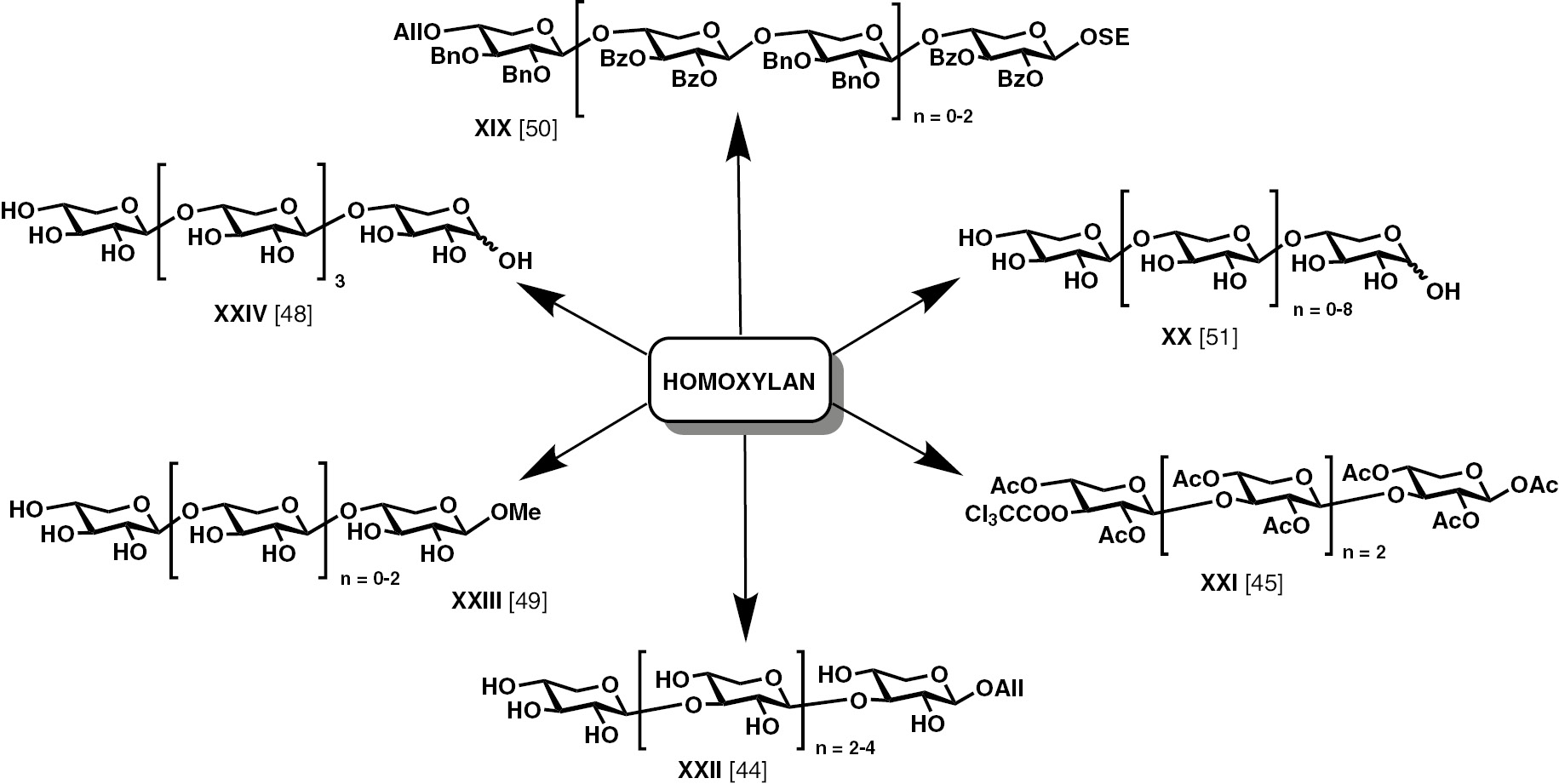
Reports on the synthesis of xylo-oligosaccharides.
Utille and co-workers reported the synthesis of β-(1→3)-linked homoxylan tetrasaccharide, utilising the Koenigs-Knorr method, through glycosylation of the disaccharide acceptor with the halide activated disaccharide glycosyl donor [45]. A similar block condensation of a disaccharide donor and a disaccharide acceptor alcohol was also reported by Kong and Chen [44] to prepare homoxylan up to hexasaccharide by trichloroacetimidate activation method.
As described earlier, the latent-active glycosylation method involves (i) allylic halogenation and (ii) reaction with the acceptor alcohol in the presence of a promoter. Synthesis of the 1,3-linked xylose disaccharide is shown in Scheme 27. Activation of the monosaccharide donor 125, using the prototypical NBS/AIBN/CCl4 reagent system, affords the corresponding allylic halide intermediate. In comparison to other allyl hexopyranosides, an observation was that the allylic halogenation of xyloside 125 required longer duration. Subsequent treatment with TfOH or TMSOTf at 0°C and the acceptor allyl xylopyranoside acceptor 126 afforded the xylobioside 127, as the β-anomer exclusively.
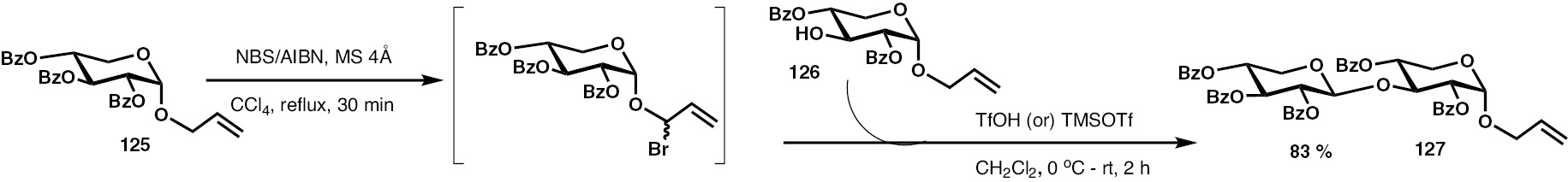
Synthesis of (1–3)-linked xylobioside 127.
Efforts to synthesize (1–3)-linked xylotrioside through glycosylation of acceptor 126 with the activated xylobioside donor 127 by repeating the above reaction sequence, however, did not afford the desired trisaccharide. Rather, intermediate allylic halide afforded only the hydrolysed disaccharide hemiacetal, in spite of repeated attempts conducting the reaction under complete anhydrous conditions. The reactivity of the disaccharide donor 127 to monosaccharide acceptor 126 is inferred to be not efficacious. In order to verify this inefficacy of trisaccharide formation, a glycosylation of the disaccharide donor having the trichloroacetimidate with acceptor 126 was attempted. However, here too, trisaccharide formation was not observed.
Conclusion
Glycosylation chemistry continues to expand addressing a number of issues relating to chemo-, regio- and stereoselectivities. The notion that protecting groups are required to mask the reactivities of otherwise reactive functionalities has been re-engineered and strategies have emerged now to gain the role of the protecting groups in the above selectivity issues. The generation of the oxocarbenium ion with a predictable conformation has the most potential to afford a glycosidic bond with a defined stereoselectivity. Many glycosylation methods involve installation of the leaving group just prior to the glycosylation. Such activated glycosides installed with a leaving group could be unstable also. Strategies that require a chemoselectivity, orthogonality and pre-activation are important in order to develop glycosylation methods beyond the oxocarbenium generation manifold. Utilizing a protecting group to an activated leaving moiety at the anomeric carbon of glycosyl donor allows a latent protecting group to an active glycosyl donor. In this effort, the new development pertains to activating an allyl glycoside to an activated allylic halide donor. Allyl glycosides are acid-base stable and their conversion to a halo-allylic glycoside involves a prototypical radical halogenation reaction. A complete allylic halogenation results, thereby the transformation of the latent moiety to an active glycosyl moiety is ensured fully. The high reactivity of the active halo-allylic glycosyl donor ensures the formation of the oxocarbenium ion and the subsequent reaction with the glycosyl acceptor to afford the desired glycoside. Whereas benzylic moiety is incompatible to the radical halogenation of allylic glycosides, other protecting groups are compatible with the newly developed glycosylation methodology. The presence of the allyl moiety at the reducing end of the growing oligosaccharide enables continuing the glycosylation through activation of the latent allyl moiety, thus adding an advantage of allyl glycosides as donors and acceptors in a oligosaccharide synthesis. Whereas allylic halide formation as a new glycosyl donor moiety is uncovered, issues relating to the formation of 1,2-cis-glycosides, use of benign solvents for radical halogenation and more are to be investigated further.
Article note
A collection of invited papers based on presentations at the 29th International Carbohydrate Symposium (ICS-29), held in the University of Lisbon, Portugal, 14–19 July 2018.
Acknowledgement
We are grateful to DST-SERB and CSIR, New Delhi, for financial support of our research in carbohydrate chemistry. NJ is a J.C. Bose National Fellow. RP is Women Scientist of DST-SERB Scheme. AD is grateful to UGC, New Delhi, for a research fellowship.
References
[1] H. Paulsen. Angew. Chem. Int. Ed. Engl. 21, 155 (1982).10.1002/anie.198201553Search in Google Scholar
[2] W. Koenigs, E. Knorr. Ber. Dtsch. Chem. Ges. 34, 957 (1901).10.1002/cber.190103401162Search in Google Scholar
[3] K. Igarashi. Adv. Carb. Chem. Biochem. 34, 243 (1977).10.1016/S0065-2318(08)60326-1Search in Google Scholar
[4] E. Fischer. Ber. Dtsch. Chem. Ges. 28, 1145 (1895).10.1002/cber.189502801248Search in Google Scholar
[5] R. Das, B. Mukhopadhyay. ChemistryOpen5, 401 (2016).10.1002/open.201600043Search in Google Scholar PubMed PubMed Central
[6] S. C. Ranade, A. V. Demchenko. J. Carbohydr. Chem. 32, 1 (2013).10.1080/07328303.2012.749264Search in Google Scholar
[7] “Protecting Groups: Strategies and Applications in Carbohydrate Chemistry”, S. Vidal (Ed.), Wiley-VCH, 2018. ISBN: 978-3-527-69702–1.Search in Google Scholar
[8] J. P. Yasomanee, A. V. Demchenko. J. Am. Chem. Soc. 134, 20097 (2012).10.1021/ja307355nSearch in Google Scholar PubMed
[9] K. Le Mai Hoang, X.-W. Liu. Nature Commun. 5, 5051 (2014).10.1038/ncomms6051Search in Google Scholar PubMed
[10] M. M. Nielsen, C. M. Pedersen. Chem. Rev. 118, 8285 (2018).10.1021/acs.chemrev.8b00144Search in Google Scholar PubMed
[11] P. O. Adero, H. Amarasekara, P. Wen, L. Bohé, D. Crich. Chem. Rev. 118, 8242 (2018).10.1021/acs.chemrev.8b00083Search in Google Scholar PubMed PubMed Central
[12] H. H. Jensen, M. Bols. Acc. Chem. Res. 39, 259 (2006).10.1021/ar050189pSearch in Google Scholar
[13] H. M. Christensen, S. Oscarson, H. H. Jensen. Carbohydr. Res. 408, 51 (2015).10.1016/j.carres.2015.02.007Search in Google Scholar
[14] H. Paulsen, A. Richter, V. Sinnwell, W. Stenzel. Carbohydr. Res.38, 312 (1974).10.1016/S0008-6215(00)82362-9Search in Google Scholar
[15] B. Fraser-Reid, U. E. Udodong, Z. F. Wu, H. Ottosson, J. R. Merritt, C. S. Rao, C. Roberts, R. Madsen. Synlett. 927 (1992).10.1055/s-1992-21543Search in Google Scholar
[16] N. L. Douglas, S. V. Ley, U. Lücking and S. L. Warriner. J. Chem. Soc., Perkin Trans. 1, 51 (1998).10.1039/a705275hSearch in Google Scholar
[17] H. H. Jensen, C. M. Pedersen, M. Bols. Chem. Eur. J.13, 7577 (2007).10.1002/chem.200700947Search in Google Scholar
[18] C. M. Pedersen, L. U. Nordstrom, M. Bols. J. Am. Chem. Soc.129, 9222 (2007).10.1021/ja071955lSearch in Google Scholar PubMed
[19] Y. Okada, O. Nagata, M. Taira, H. Yamada. Org. Lett.9, 2755 (2007).10.1021/ol070720bSearch in Google Scholar PubMed
[20] T. Zhu, G.-J. Boons. Org. Lett.3, 4201 (2001).10.1021/ol016869jSearch in Google Scholar PubMed
[21] Z. Zhang, I. R. Ollman, X.-S. Ye, R. Wischnat, T. Baasov, C.-H. Wong. J. Am. Chem. Soc.121, 734 (1999).10.1021/ja982232sSearch in Google Scholar
[22] C.-W. Cheng, Y. Zhou, W.-H. Pan, S. Dey, C.-Y. Wu, W. L. Hsu, C.-H. Wong. Nat. Commun.9, 5202 (2018).10.1038/s41467-018-07618-8Search in Google Scholar PubMed PubMed Central
[23] M. Lahmann, M.; S. Oscarson. Org. Lett.2, 3881 (2000).10.1021/ol006621eSearch in Google Scholar
[24] T. Mukaiyama K. Ikegai, H. Jona, T. Hashihayata, K. Takeuchi. Chem. Lett.30, 840 (2001).10.1246/cl.2001.840Search in Google Scholar
[25] A. V. Demchenko, C. De Meo. Tetrahedron Lett.43, 8819 (2002).10.1016/S0040-4039(02)02235-9Search in Google Scholar
[26] M. M. Mukherjee, R. Ghosh. J. Org. Chem.82, 5751 (2017).10.1021/acs.joc.7b00561Search in Google Scholar
[27] S. J. Danishefsky, K. F. McClure, J. T. Randolph, R. R. B. Ruggeri. Science260, 1307 (1993).10.1126/science.8493573Search in Google Scholar
[28] H. M. Nguyen, J. L. Poole, D. Y. Gin. Angew. Chem. Int. Ed.40, 414 (2001).10.1002/1521-3773(20010119)40:2<414::AID-ANIE414>3.0.CO;2-6Search in Google Scholar
[29] S. Yamago, T. Yamada, O. Hara, H. Ito, Y. Mino, J. Yoshida. Org. Lett.3, 3867 (2001).10.1021/ol016713jSearch in Google Scholar
[30] J. D. C. Codee, B. Stubba, M. Schiattarella, H. S. Overkleeft, C. A. A. van Boeckel, J. H. van Boom, G. A. van der Marel. J. Am. Chem. Soc.127, 3767 (2005).10.1021/ja045613gSearch in Google Scholar
[31] S. Yamago, T. Yamada, T. Maruyama, J-i. Yoshida. Angew. Chem. Int. Ed.43, 2145 (2004).10.1002/anie.200353552Search in Google Scholar
[32] Z. Wang, L. Zhou, K. El-Boubbou, X-S. Ye, X. Huang. J. Org. Chem. 72, 6409 (2007).10.1021/jo070585gSearch in Google Scholar
[33] R. Roy, F. O. Andersson, M. Letellier. Tetrahedron Lett.33, 6053 (1992).10.1016/S0040-4039(00)60004-7Search in Google Scholar
[34] T. T. Fang, K. F. Mo, G. -J. Boons. J. Am. Chem. Soc.134, 7545 (2012).10.1021/ja3018187Search in Google Scholar
[35] K. S. Kim, J. H. Kim, Y. J. Lee, Y. J. Lee, J. J. Park. J. Am. Chem. Soc.123, 8477 (2001).10.1021/ja015842sSearch in Google Scholar
[36] S. J. Hasty, M. A. Kleine, A. V. Demchenko. Angew. Chem. Int. Ed. 50, 4197 (2011); Angew. Chem. 123, 4283 (2011).10.1002/anie.201007212Search in Google Scholar
[37] X. Chen, D. Shen, Q. Wang, Y. Yang, B. Yu. Chem. Commun. 51, 13957 (2015).10.1039/C5CC05651ASearch in Google Scholar
[38] P. Shu, X. Xiao, Y. Zhao, Y. Xu, W. Yao, J. Tao, H. Wang, G. Yao, Z. Lu, J. Zeng, Q. Wan. Angew. Chem., Int. Ed. 54, 14432 (2015).10.1002/anie.201507861Search in Google Scholar
[39] X. Xiao, Y. Zhao, P. Shu, X. Zhao, Y. Liu, J. Sun, Q. Zhang, J. Zeng, Q. Wan. J. Am. Chem. Soc. 138, 13402 (2016).10.1021/jacs.6b08305Search in Google Scholar
[40] G. J. Boons, S. Isles. Tetrahedron Lett.35, 3593 (1995).10.1016/S0040-4039(00)73249-7Search in Google Scholar
[41] G.-J. Boons, B. Heskamp, F. Hout. Angew. Chem. Int. Ed. Engl. 35, 2845 (1996).10.1002/anie.199628451Search in Google Scholar
[42] P. Wang, P. Haldar, Y. Wang, H. Hu. J. Org. Chem. 72, 5870 (2007).10.1021/jo070512xSearch in Google Scholar PubMed
[43] R. Pal, A. Das, N. Jayaraman. Chem. Commun. 54, 588 (2018).10.1039/C7CC07332ASearch in Google Scholar
[44] F. Kong, L. Chen. Carbohydr. Res. 337, 2335 (2007).10.1016/S0008-6215(02)00285-9Search in Google Scholar
[45] J-F. Utille, G. Excoffier, D. Dupeyre. Carbohydr. Res.135, C1 (1984).10.1016/0008-6215(84)85016-8Search in Google Scholar
[46] K. Takeo, Y. Ohguchi, R. Hasegawa, S. Kitamura. Carbohydr. Res.278, 301 (1995).10.1016/0008-6215(95)00259-6Search in Google Scholar
[47] P. Kovac, J. Hirsch, V. Kovacik, P. Kocis. Carbohydr. Res.85, 419 (1980).10.1016/S0008-6215(00)84562-0Search in Google Scholar
[48] J. Hirsch, P. Kovac, E. Petrakova. Carbohydr. Res. 106, 203 (1982).10.1016/S0008-6215(00)81074-5Search in Google Scholar
[49] P. Kovac, J. Hirsch. Carbohydr. Res. 90, C5 (1981).10.1016/S0008-6215(00)85935-2Search in Google Scholar
[50] K. Takeo, Y. Murata, S. Kitamura. Carbohydr. Res. 224, 311 (1992).10.1016/0008-6215(92)84118-CSearch in Google Scholar
[51] K. Takeo, Y. Ohguchi, R. Hasegawa, S. Kitamura. Carbohydr. Res. 278, 301 (1995).10.1016/0008-6215(95)00259-6Search in Google Scholar
© 2019 IUPAC & De Gruyter. This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License. For more information, please visit: http://creativecommons.org/licenses/by-nc-nd/4.0/
Articles in the same Issue
- Frontmatter
- In this issue
- Preface
- ICS-29: The 29th International Carbohydrate Symposium
- Conference papers
- 1-(2-Aminoethyl)-3-methyl-1H-imidazol-3-ium tetrafluoroborate: synthesis and application in carbohydrate analysis
- One-pot oligosaccharide synthesis: latent-active method of glycosylations and radical halogenation activation of allyl glycosides
- Nanoparticles for the multivalent presentation of a TnThr mimetic and as tool for solid state NMR coating investigation
- Preface
- Papers from the 1st International Conference on Chemistry for Beauty and Health (Beauty-Torun’2018)
- Conference papers
- In vivo electrical impedance measurement in human skin assessment
- Formulation and characterization of some oil in water cosmetic emulsions based on collagen hydrolysate and vegetable oils mixtures
- Preparation of polylactide scaffolds for cancellous bone regeneration – preliminary investigation and optimization of the process
- Effect of molecular weight of polyvinylpyrrolidone on the skin irritation potential and properties of body wash cosmetics in the coacervate form
- Ortho-substituted azobenzene: shedding light on new benefits
- Erratum
- Erratum to: Efforts to improve Japanese women’s status in STEM fields
Articles in the same Issue
- Frontmatter
- In this issue
- Preface
- ICS-29: The 29th International Carbohydrate Symposium
- Conference papers
- 1-(2-Aminoethyl)-3-methyl-1H-imidazol-3-ium tetrafluoroborate: synthesis and application in carbohydrate analysis
- One-pot oligosaccharide synthesis: latent-active method of glycosylations and radical halogenation activation of allyl glycosides
- Nanoparticles for the multivalent presentation of a TnThr mimetic and as tool for solid state NMR coating investigation
- Preface
- Papers from the 1st International Conference on Chemistry for Beauty and Health (Beauty-Torun’2018)
- Conference papers
- In vivo electrical impedance measurement in human skin assessment
- Formulation and characterization of some oil in water cosmetic emulsions based on collagen hydrolysate and vegetable oils mixtures
- Preparation of polylactide scaffolds for cancellous bone regeneration – preliminary investigation and optimization of the process
- Effect of molecular weight of polyvinylpyrrolidone on the skin irritation potential and properties of body wash cosmetics in the coacervate form
- Ortho-substituted azobenzene: shedding light on new benefits
- Erratum
- Erratum to: Efforts to improve Japanese women’s status in STEM fields