Characterizations of radicals formed in radical polymerizations and transfer reactions by electron spin resonance spectroscopy
-
Atsushi Kajiwara


Abstract
Electron spin resonance (ESR, aka electron paramagnetic resonance, EPR) investigations have been conducted on radicals formed during radical polymerizations and provide a detailed characterization of the active radical species. Active propagating radicals can be observed during actual radical polymerizations by ESR/EPR. The chain lengths of the observed radicals were estimated by a combination of atom transfer radical polymerization (ATRP) and ESR/EPR. The structures of the chain end radicals were determined by analysis of the ESR/EPR spectra. An increase in the dihedral angles between terminal p-orbital of radical and Cβ–H bonds was observed with increasing chain lengths of methacrylate polymers. Radical transfer reactions were observed during radical polymerization of acrylates. A combination of ATRP and ESR/EPR clarified a 1,5-hydrogen shift mechanism of the radical transfer reactions using model adamantyl acrylate radicals. Penultimate unit effects were also observed. Time-resolved ESR/EPR (TR ESR) spectroscopy clarified the initiation processes of an alternating copolymerization of styrene with maleic anhydride and the copolymerization of styrene with 1,3-butadiene. Several unsolved problems in conventional radical polymerization processes have been clarified using combinations of ATRP with ESR/EPR and TR ESR. Characterization of the radicals in radical polymerizations using various ESR techniques would definitely provide interesting and useful information on conventional radical polymerizations.
Introduction
Electron spin resonance (ESR, aka electron paramagnetic resonance, EPR) is utilizes magnetic resonance spectroscopy for observation of the behavior of unpaired electrons in materials [1], [2], [3], [4], [5], [6]. ESR/EPR spectra of propagating radicals can be observed during actual radical polymerization processes under normal radical polymerization conditions [7], [8], [9], [10], [11], [12], [13], [14]. Clear and well-resolved spectra were observed for n-butyl methacrylate (nBMA), 1,3-butadiene and styrene during their actual radical polymerizations as shown in Fig. 1 [5], [14]. The actual structure of the radicals can be estimated from the values of hyperfine splitting constants (hfc’s) measured from the spectra. The structures of the propagating radicals are clearly assessed from these results, with concrete experimental evidence. For example, the small doublet observed in the spectrum of 1,3-butadiene indicates that a delocalized allyl radical is the structure of the chain end propagating radical.
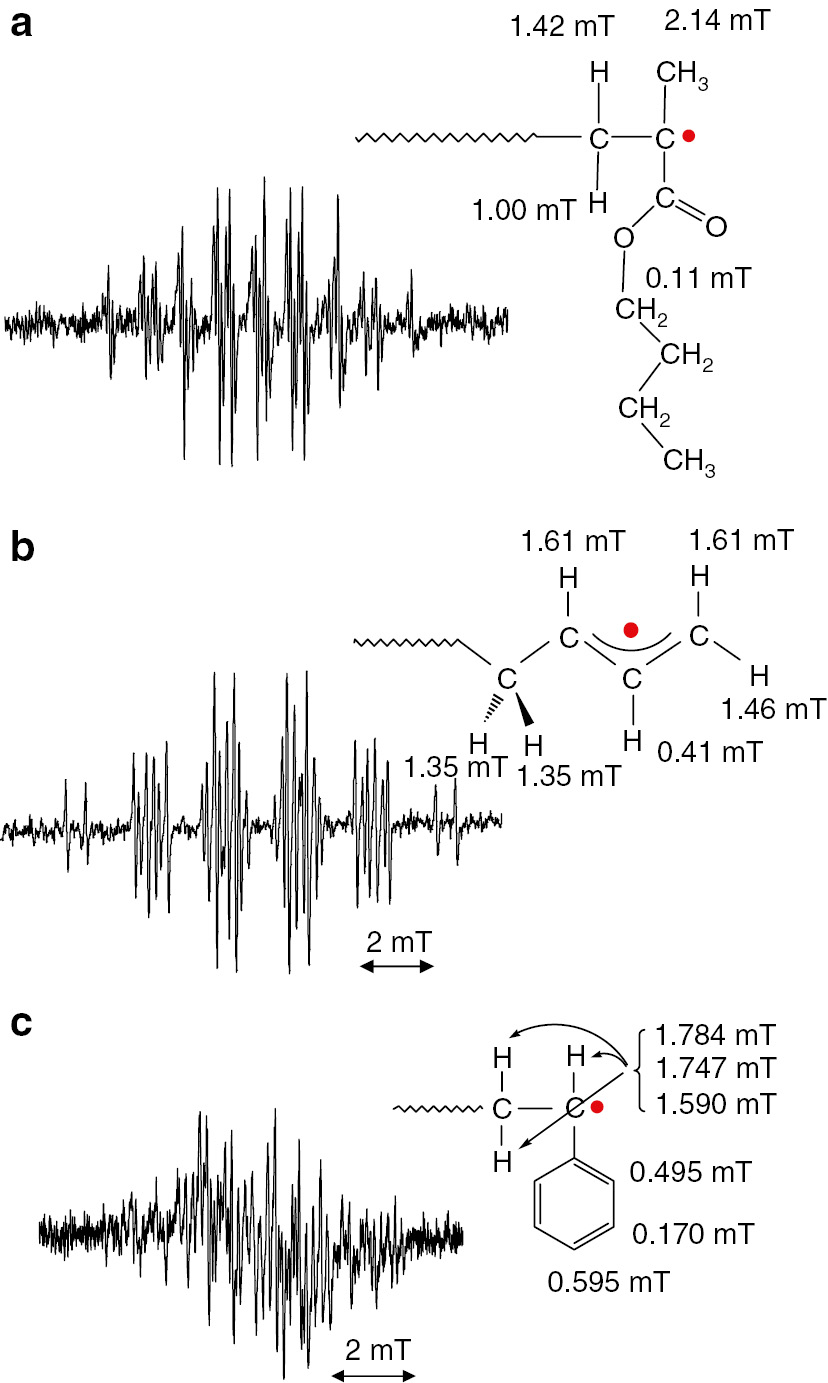
ESR spectra of propagating radicals of (a) n-butyl methacrylate, (b) 1,3-butadiene and (c) styrene with their structures and measured hyperfine splitting constants (hfc’s). Spectra were observed at 120°C (a), 0°C (b) and 25°C (c) in toluene.
On the basis of these observed spectra, the chain end structures of the radicals are clearly identifiable. Moreover, the steady state radical concentrations and the molecular dynamics of the radicals can be investigated from these spectra [3], [12]. On the other hand, other properties like chain lengths of the radicals and penultimate unit effects were difficult to examine solely from these spectra.
In the field of macromolecular chemistry, new techniques were developed, in 1995–1996, for controlling radical polymerization processes allowing preparation of polymers with pre-determined molecular weights with narrow dispersity [15], [16], [17], [18]. A combination of these controlled radical polymerization techniques with ESR/EPR spectroscopy can provide significant information on various chain length dependent phenomena of the propagating radicals. One of the techniques is called atom transfer radical polymerization (ATRP). The generation of model (meth)acrylate propagating radicals, with determined degrees of polymerization and narrow dispersity, from their precursors is shown in Fig. 2. The radical precursors prepared by ATRP have a terminal carbon-bromine bond and the bond can be cleaved homolitically in the presence of organo-tin compound to generate the radicals with pre-determined structures [16], [17].
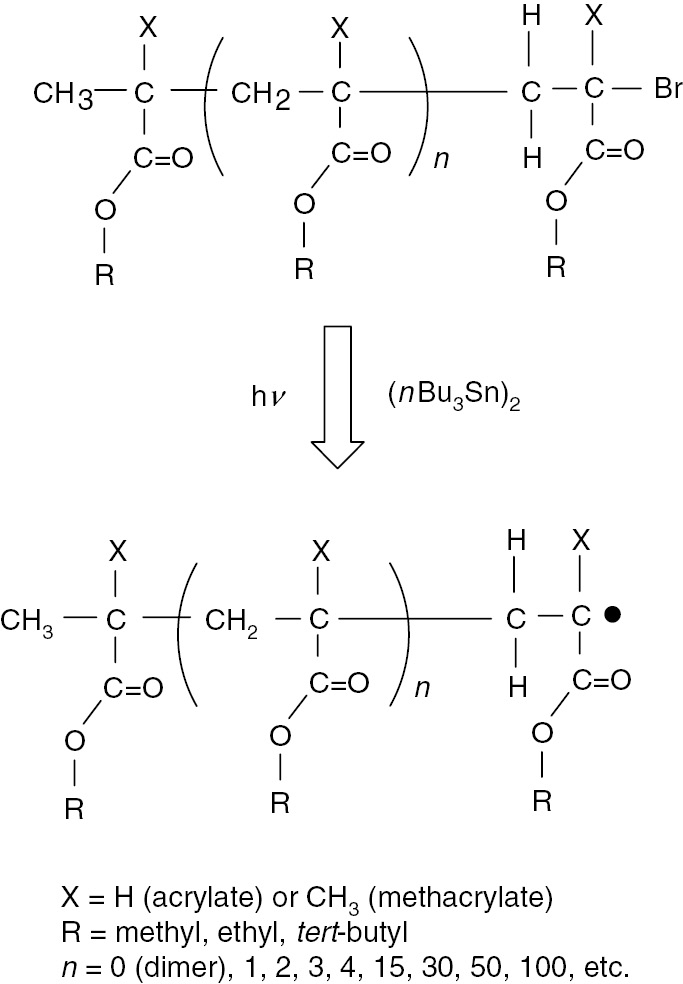
Generation of radicals from radical precursors prepared by atom transfer radical polymerization (ATRP).
The pre-determined degree of polymerization and narrow dispersity are transferred to the radicals at the beginning of the generation procedure. Using this combination technique, ESR/EPR spectra of model propagating radicals with different chain lengths (degrees of polymerization, DP) can be compared and the chain length dependent phenomena in the spectra can be examined [12], [19], [20]. The results provide information on the chain lengths of the observed real propagating radicals during actual radical polymerizations. Methacrylates are the most suitable monomers for evaluation of chain length dependency of the ESR/EPR spectra mainly due to their very sensitive spectroscopic features on molecular dynamics. In the case of acrylates, model propagating radicals showed the occurrence of radical transfer reactions through a 1,5-hydrogen shift mechanism to form mid-chain radicals [13], [21], [22]. In this research work, the results of the case of adamantyl acrylate (AdA) are shown.
ESR/EPR spectroscopy can be applied not only for investigation of radical homo polymerizations but also radical co-polymerizations [23], [24]. The penultimate unit effects (PUE) have been discussed on the nature of the terminal radicals during kinetic studies of radical co-polymerizations [25], [26], [27], [28]. A combination of ATRP/ESR methods can clarify the nature of the PUE from both electronic and steric effects through examination of the ESR/EPR spectra of model radicals with various terminal monomer structures. In this research work, three kinds of dimeric model radicals with terminal tert-butyl acrylate (tBA) units were examined. These are H-tBA-tBA, H-ethyl methacrylate (EMA)-tBA, and H-styrene (St)-tBA radicals. The hfc’s of the β-methylene protons of the tBA in H-St-tBA radical clearly showed a temperature dependent change due to PUE [20], [22], [29].
Typical ESR/EPR spectroscopy is based on steady-state of spin state of radicals. It is also called steady-state ESR/EPR (SS ESR) spectroscopy. The measurement processes of the SS ESR contains “field modulation” process and the process provides the higher sensitivity and resolution of the spectroscopy. On the other hand, the process utilizes 100 kHz field modulation which limits time resolution. Radical reactions that occur shorter than the 10 μs (=100 kHz) cannot be observed by the SS ESR. However, time-resolved ESR/EPR (TR ESR) spectroscopy provides not only estimations of addition rate constants and activation energy of radical addition reactions in initiation reactions of radical homo-polymerization reactions [20], [30], but also allows investigation of the initial step of copolymerizations [23].
The combination of results from both SS and TR ESR spectroscopy are a strong tool for detailed examination of the mechanism of copolymerization reactions. TR ESR spectroscopy can exclusively observe the first radical addition reaction of a radical, generated from an initiator to a monomer. Laser pulse generated spin polarized radicals relax to a thermally stable state with Boltzmann distribution and the relaxation process can be observed as ESR/EPR signals. 2,4,6-Trimethylbenzoyl diphenylphosphine oxide (TMDPO) has a carbon-phosphorous bond that can be homolytically cleaved by a 355 nm laser pulse [31], [32], [33], [34], [35], [36], [37]. Both C- and P-centered radicals are spin polarized but the P-centered radical addition to monomer is faster than that of C-centered radical. Spin polarization of the P-centered radical is transferred to the chain initiating radical formed by an addition reaction of P-centered radical to a first monomer unit. Thus, polarization and relaxation of the chain initiating radicals can be detected in the ESR/EPR signal by TR ESR spectroscopy. The transferred spin polarization will relax within 2–3 μs which is before second radical addition. That is why the first radical addition reaction is selectively observed. This method has been applied to the polymerizations of (meth)acrylates, styrenes, dienes and other monomers for estimation of addition rate constants and activation energies [34], [35], [36]. In this research work, the TR ESR spectroscopy was applied to the investigation of both alternating and random co-polymerizations.
Experimental
Materials
CuIBr (99%, Sigma-Aldrich Japan, Tokyo, Japan) was used after purification to remove any copper (II) species. (1-Bromoethyl)benzene (PhEtBr) (>95%, TCI, Tokyo, Japan), ethyl 2-bromoisobutyrate (EBriB) (99%, Sigma-Aldrich Japan, Tokyo, Japan), ethyl 2-bromopropionate (EBrP) (99%, Sigma-Aldrich Japan, Tokyo, Japan), methyl 2-bromopropionate (MBrP) (99%, Sigma-Aldrich Japan, Tokyo, Japan) and tert-butyl 2-bromopropionate (tBBrP) (99%, Sigma-Aldrich Japan, Tokyo, Japan) and N,N,N′,N″,N″-pentamethyldiethylenetriamine (PMDETA) (99%, Sigma-Aldrich Japan, Tokyo, Japan) were distilled under reduced pressure before use. tert-Butyl acrylate (tBA, TCI, Tokyo, Japan), dodecyl acrylate (DoA, Wako, Osaka, Japan), methyl methacrylate (MMA, nacalai-tesque, Kyoto, Japan), n-butyl methacrylate (nBMA, TCI, Tokyo, Japan), and styrene (Wako, Osaka, Japan) were distilled over calcium hydride under reduced pressure and stored in the freezer before use. 1,3-Butadiene (TCI, Tokyo, Japan) and maleic anhydride (nacalai-tesque, Kyoto, Japan) were used as received. 1-Adamantyl methacrylate was received from Idemitsu Kosan Co. Ltd. and used as received. 1-Adamantyl acrylate was prepared by a reaction of 1-adamantanol (TCI, Tokyo, Japan) with acryloyl chloride (TCI, Tokyo, Japan) and was purified by recrystallization. Dimethyl 2,2′-azobis(isobutyrate) (MAIB) (>97%, Wako, Osaka, Japan), di tert-butyl peroxide (tBPO) (>98%, Sigma-Aldrich Japan, Tokyo, Japan), and bis(tri-n-butyltin) [(nBu)3Sn)2] (>90%, TCI, Tokyo, Japan) were used as received. Diphenyl (2,4,6-trimethylbenzoyl) phosphine oxide (TMDPO, Wako, Osaka, Japan) was purified by re-crystallization from ethanol. All other reagents were purified before use.
General procedure for preparation of model radical precursor of poly(meth)acrylates
A model radical precursor of a polymeric AdA with a degree of polymerization (DP=50) was prepared by ATRP. CuBr (19 mg, 1.32×10−4 mol), AdA (6.60 g, 2.75×10−2 mol), tBuBrP (0.91 g, 5.02×10−3 mol), PMDETA (23 mg, 1.33×10−4 mol), and degassed acetone (7.0 ml) were mixed under nitrogen in a Schlenk flask. The flask was flushed with nitrogen by standard Schlenk techniques. The resulting mixture was placed in an oil bath at 60°C for 72 h. Formed poly AdA (DP=50) was separated from the mixture and purified by repeated precipitations. Yield was 3.15 g. Mn=14575, Mw/Mn=1.10.
General procedures for generation of model radicals
Uniform oligo model radicals
Model radicals were generated by reduction of the corresponding alkyl bromides [37]. For example, generation of the radical from H-EA-methyl methacrylate (MMA)-Br was performed according to the following procedure. H-EA-MMA-Br (20 mg, 9.61×10−5 mol) and [(nBu)3Sn]2 (20 mg, 1.15×10−4 mol) were dissolved in toluene (200 mg) in a quartz tube (o.d. 5.0 mm). The tube was flushed with argon. After setting into an ESR cavity, the mixture was irradiated with UV light at a temperature of 30°C. Irradiation was carried out with a 500 W ultra high-pressure mercury lamp (Ushio USH-500D) at a distance of ca. 15 cm from the tube in the cavity. An IR-cut filter (Toshiba IRA-25S) was used.
Polymeric model radicals
Poly(AdA)-Br (DP=10) (50 mg, ca. 2.5×10−5 mol), [(nBu)3Sn]2 (4.5 mg, 2.60×10−5 mol), and tBPO (3.7 mg, 2.58×10−5 mol) were dissolved in toluene (150 mg) in a quartz tube (o.d. 5.0 mm). The solution was flushed with dry argon for several minutes and was placed in an ESR cavity. Radicals were generated under UV-irradiation at −30°C.
General procedure for in situ ESR detection of (meth)acrylates propagating radicals
Dodecyl acrylate (DoA) (30 mg, 1.25×10−4 mol) and tBPO (2.0 mg, 1.36×10−5 mol) were dissolved in toluene (170 mg) in a quartz tube (o.d. 5 mm). The solution was irradiated with UV-light in an ESR cavity to initiate polymerizations at −30°C. The formed propagating radicals were observed by ESR/EPR.
Measurements
SS ESR
The ESR/EPR spectra of the radicals were recorded on a JEOL JES RE-2X spectrometer operating in the X-band, utilizing a 100 kHz field modulation, and a microwave power of 1 mW. A TE011 mode cavity was used. Temperature was controlled by JEOL DVT2 variable-temperature accessory. ESR/EPR measurements were performed at 90, 60, 30, 0, −15 and −30°C in toluene and at +120 and +150°C in mesitylene. Spectroscopic simulations were carried out by JEOL IPRIT Data Analysis System. Toluene and mesitylene can be used as a solvent for ESR/EPR measurements of acrylate polymerizations. Polymerization results of acrylates were almost the same in toluene and in benzene. No trace of ESR/EPR spectra of benzyl radicals generated from hydrogen abstraction from toluene (or mesitylene) by initiating or propagating radicals were observed.
TR ESR
TMDPO (Sigma-Aldrich Japan, Tokyo, Japan) was purified by recrystallization from ethanol before use. Benzyl benzodithioate (BTBA, Sigma-Aldrich Japan, Tokyo, Japan) was used without further purification. (Meth)acrylate monomers were purified by distillation just before use. A toluene or benzene solution of TMDPO (0.1 M) containing various concentrations of monomers was taken in an ESR/EPR sample cell. JEOL JES-LC11 flat flow cell and micro tube pump (Tokyo Rikakikai Co. Ltd., EYELA) were used for flow method. Laser pulses were irradiated by using a Q-switched Nd:YAG laser (Spectra Physics Quantaray DCR-2) operated at the third harmonic (10 mJ/flash at 355 nm with a 6-ns fwhm). For the measurements of the TR ESR, a JEOL JES RE-2X spectrometer, equipped with a WBPA2 wide band pre-amplifier, was operated without magnetic field modulation, and the data were stored in a Tektronix TDS520A digital oscilloscope. Magnetic fields at resonance signals were determined by an Echo Electronics ES-FC5 NMR field meter. Measurement temperature was controlled by a JEOL DVT2 variable-temperature accessory. Data analysis was conducted by CIDEP software provided by JEOL Ltd.
SEC
Molecular weights and molecular weight distributions were estimated by size exclusion chromatography (SEC) using a TOSOH CCP&8020 series GPC system with TSK-gel columns. Linear combination of two G2000HHR and two GMHXL columns was employed. Polystyrene standards were used to calibrate the columns.
ESI-MS
ESI-MS was performed using a SCIEX API III+ triple quadrupole mass spectrometer (Perkin-Elmer Sciex) equipped with a nebulizer-assisted electrospray (ionspray) source. Calibration was performed in positive mode using poly(propylene glycol) ions. Mass spectra were analyzed using a Quadra 950 data system (Apple Computer Inc.). MacBioSpec software (Perkin-Elmer Sciex) was used to calculate the mass.
Results and discussion
ATRP/ESR combination technique
Estimation of chain lengths of propagating radicals from ESR/EPR spectra
Model radical precursors of tert-butyl methacrylate (tBMA) were prepared by ATRP. Oligomers were polymerized in one-pot and the resulting mixture contains oligomers from dimeric to around decameric tBMA [12], [38]. Each uniform oligomeric model radical precursor was purified using preparative HPLC and the structures were confirmed by 1H NMR and ESI-MS spectroscopy. Figure 3 shows the ESR/EPR spectra of model propagating radicals of tBMA with DP=2, 4 and 100 along with the spectrum measured in an actual radical polymerization of tBMA. These spectra were recorded at 150°C. At that temperature, molecular rotation around Cα–Cβ bond was very smooth and molecular dynamics due to restricted rotation did not need to be considered [3], [5], [12]. These spectra can also be simulated using an isotropic simulation program. From the resulting hfc’s, dihedral angles between pπ-orbital and Cβ–H bond can be calculated using cosine square law. Rotational motion of the terminal Cβ–Cα bond would have two energy minimums [3], [5].
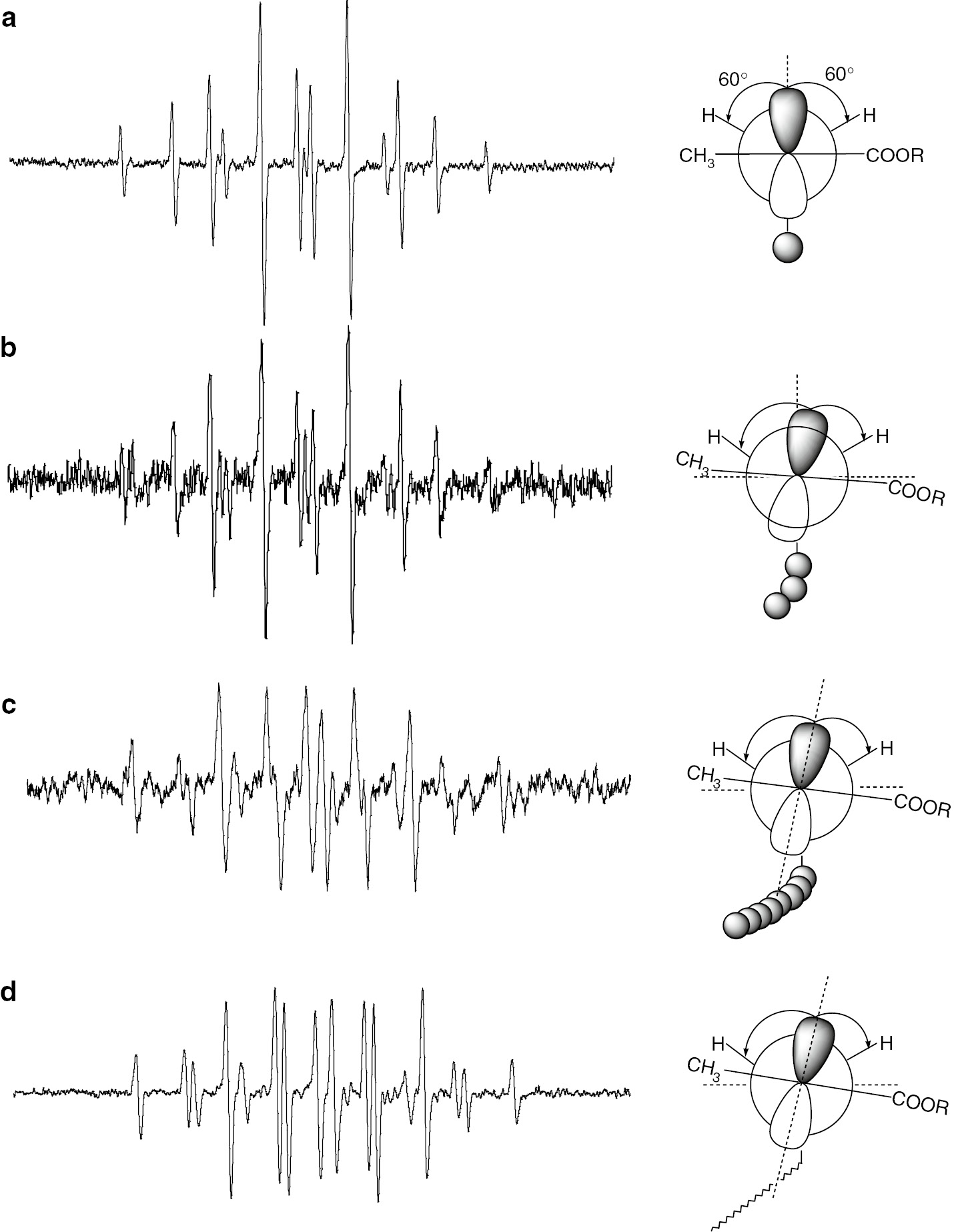
ESR spectra of model propagating radicals of tBMA with different chain lengths (a) DP=2 (b) DP=5, (c) DP=100 and (d) actual propagating radical observed in an actual polymerization system. Newman projections of the radicals with dihedral angles are also shown.
The terminal radical has sp2 type flat structure and the plane just makes a pendulum like motion. Newman projections of each radical are also shown along with the spectra. As we can see, the stabilized structure of each radical is distorted with increasing chain length. Plots of the calculated dihedral angles against the degree of polymerization (DP) are shown in Fig. 4. The dihedral angles show clear chain length dependence.
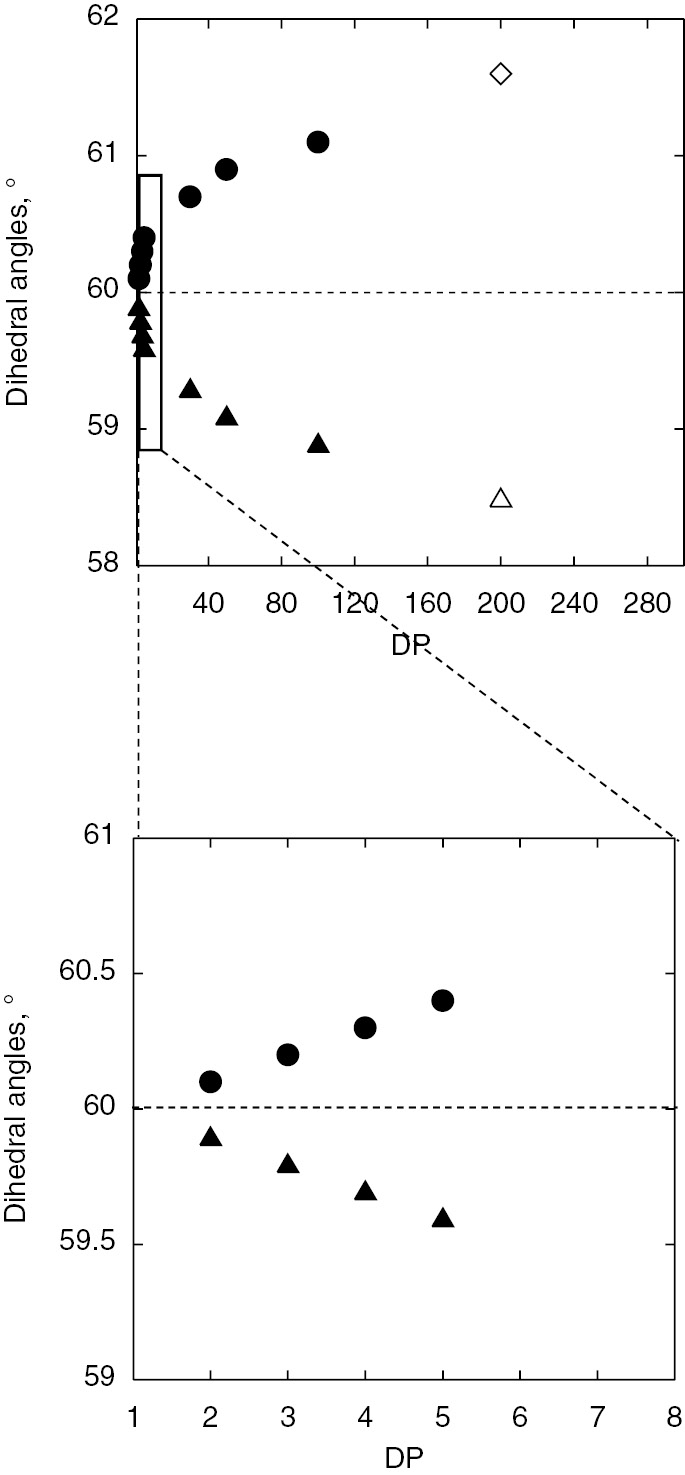
Chain length dependence of dihedral angles between p-orbital of radical and Cβ–H bonds (above) of tBMA model (filled circle and triangle) and actual propagating radicals (open circle and triangle). The oligomer region was expanded in the bottom plot.
Although the reason for the increasing distortion from the symmetrical structure with increasing chain length is not clear yet, this correlation could be used for estimation of the chain lengths of the radicals observed during actual radical polymerizations. The increasing distortion means the increasing the difference between hfc’s of two β-methylene protons and the increase of the splitting constants reach steady values. In the extrapolation of the plots, the hfc values of actual propagating radicals are located around DP=200. The Mn of the resulting polymer after ESR/EPR measurements were estimated by SEC and they were around 30 000. These results are consistent. Judging from the results, observed propagating radicals have a chain length around DP=200 [12], [22], [38].
Before development of ATRP, there was no method to estimate the chain lengths of the propagating radicals observed by ESR/EPR. Now the chain lengths can be estimated with experimental evidences. Methacrylates show very clear chain length dependent spectroscopic change in ESR/EPR spectra and the correlations were confirmed for not only tBMA but also for nBMA, adamantyl methacrylate (AdMA), and MMA (Figs. 1a and 5).
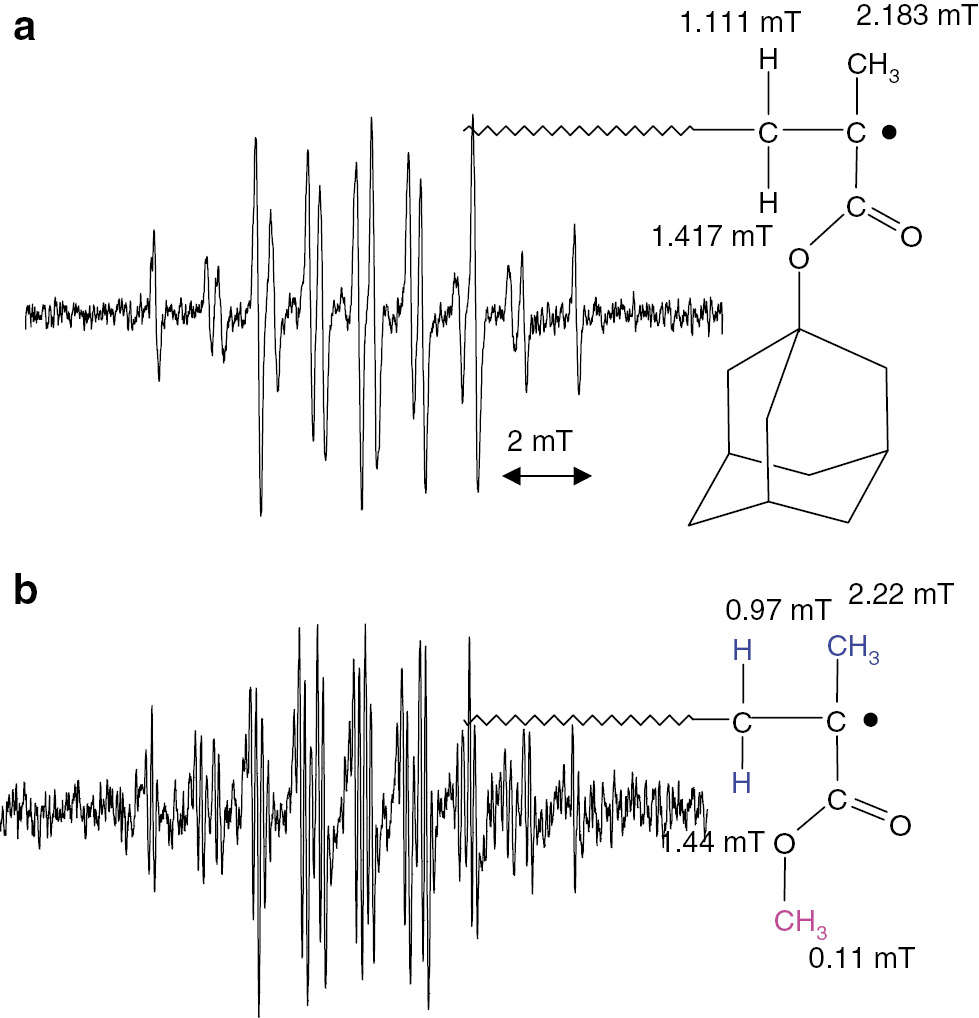
ESR spectra of propagating radicals of (a) adamantyl methacrylate and (b) methyl methacrylate, both observed in toluene at 120°C.
In these cases, 16-line signals due to inequivalent β-protons were clearly observed. The same combination technique of ATRP/ESR has been applied to other monomers including acrylates, dienes and styrenes. They also show chain length dependent changes in their ESR/EPR spectra, however these differences are much smaller than the case of methacrylates. Based on these results, ESR/EPR spectra of propagating radicals observed during actual radical polymerizations can be used for kinetics treatments as the spectra of real propagating radicals with sufficiently long chain lengths.
Clarification of radical transfer mechanism in acrylate polymerizations
Interpretation of ESR/EPR spectra observed in acrylate radical polymerizations have been very difficult [39], [40], [41], [42], [43], [44], [45], [46], [47], [48]. The ESR spectra observed during radical polymerization of acrylates are very different from those from methacrylates, even under almost identical conditions. In the acrylate radical polymerizations, ESR/EPR signal of propagating radicals was observed at lower temperature (below −10°C) and that of mid-chain radicals was observed at higher temperature (>60°C) [20], [49]. Figure 6 shows a 1,5-hyrdrogen shift reaction for terminal propagating radicals of acrylates [38]. The radical transfer reaction occurring through six-membered ring structure had been discussed for long time for ethylene [50], acrylate [51], and vinyl ester [52], [53], [54], [55], [56] polymerizations but there had been no clear experimental evidence for the mechanism. The combination of ATRP/ESR provided evidence of the mechanism of this reaction for various acrylates [13], [19], [20], [38], [49].
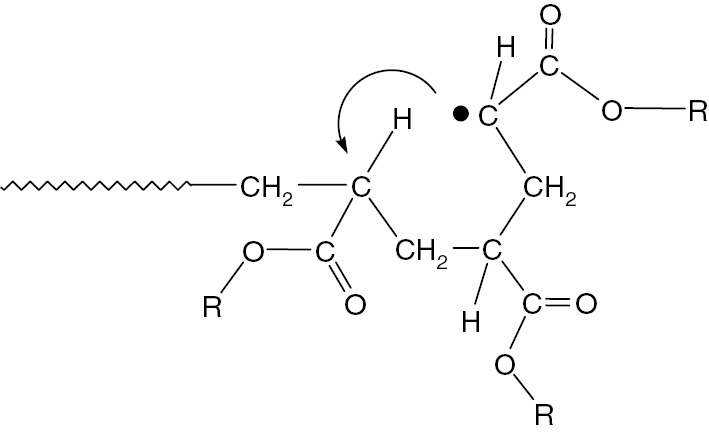
1,5-Hydrogen shift reaction of chain end propagating acrylate radicals.
The method employed is very similar to the case of methacrylates discussed above. A mixture of oligomers of adamantyl acrylate was prepared by ATRP and each oligomer from dimer to pentamer was purified using preparative HPLC as shown in Fig. 7. Model propagating radicals were generated from each purified oligomer and were measured by ESR/EPR at various temperatures.
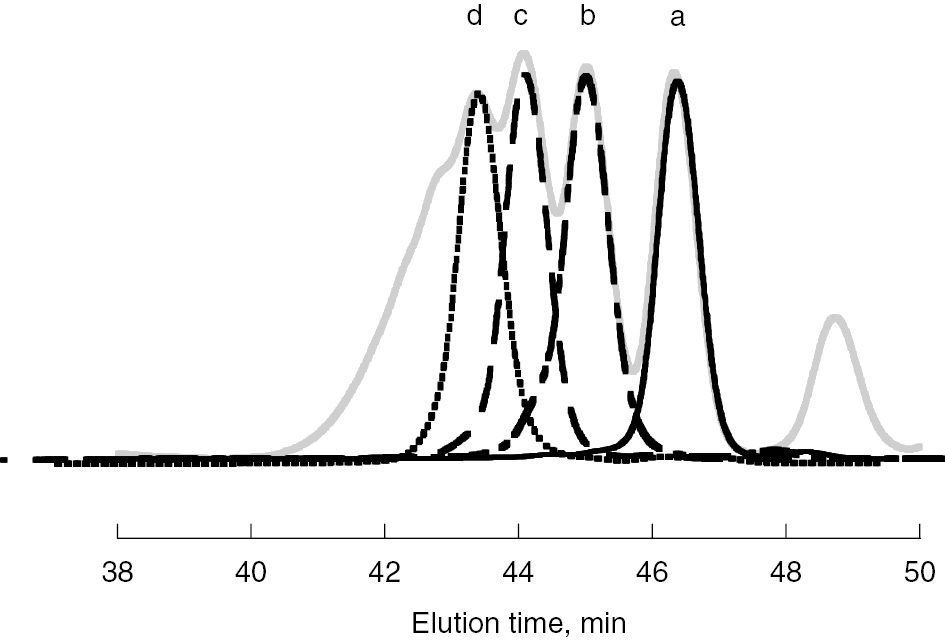
SEC election diagrams of purified oligomeric model radical precursors of AdA. A gray line is original diagram. Diagrams of (a) dimeric (line), (b) trimeric (dot and dashed line), (c) tetrameric (dashed line), and (d) pentameric (dotted line) model radical precursors are shown.
Selected results of ESR/EPR spectra of the radicals are shown in Fig. 8. They are the spectra of dimeric (a), trimeric (b) and tetrameric (c) model propagating radicals measured at 120°C. At 120°C, the radical transfer reactions are completed and the resulting radicals were observed for trimeric and tetrameric model propagating radicals.
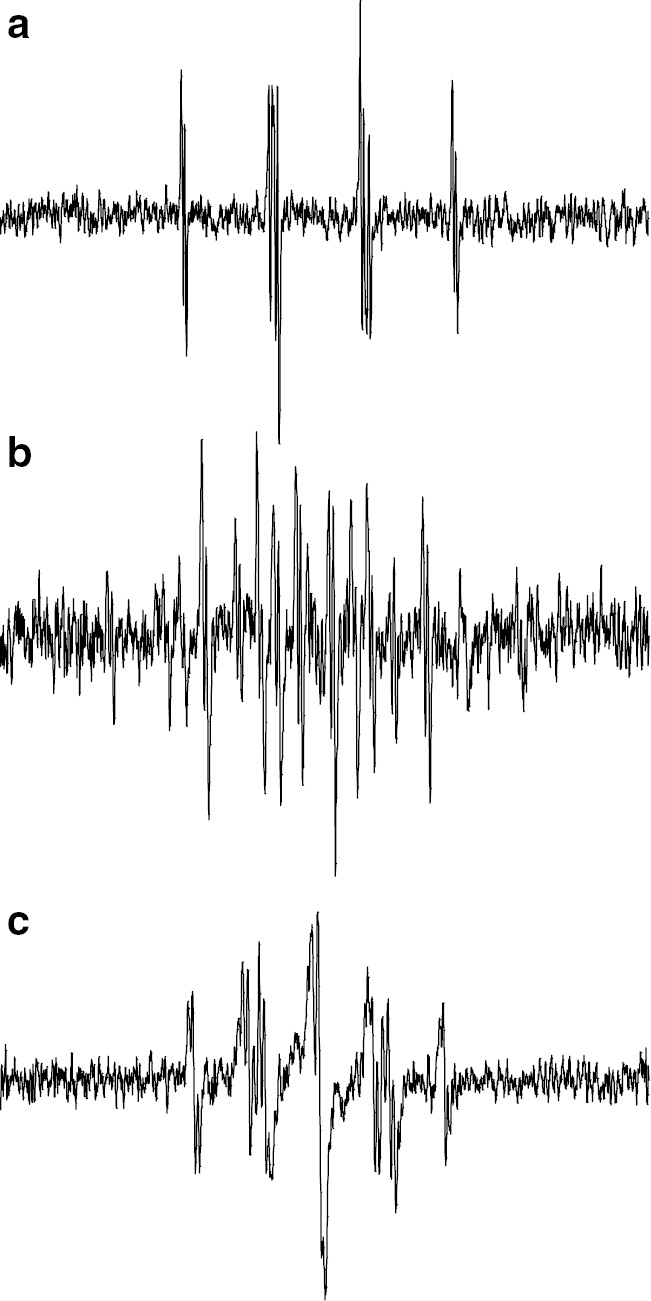
ESR spectra of model propagating radicals of AdA observed in toluene at 120°C. (a) Dimeric, (b) trimeric and (c) tetrameric model propagating radicals.
The dimeric model propagating radical shows almost the same spectra at various temperatures from −30°C to +150°C. The trimeric and tetrameric model propagating radicals showed spectroscopic change with increasing measurement temperatures. At low temperature in the range of −30–0°C, an ESR spectrum just like the dimeric model radical was observed. With increasing the temperature, new and different spectrum appeared as the original signal disappeared. The observed spectra for trimeric (Fig. 8b) and tetrameric (Fig. 8c) model radicals at 120°C can be interpreted the radicals formed through a 1,5-hyrdogen shift reaction via a six-membered ring structure. The proposed structures of the observed radicals in Fig. 8 are summarized in Fig. 9.
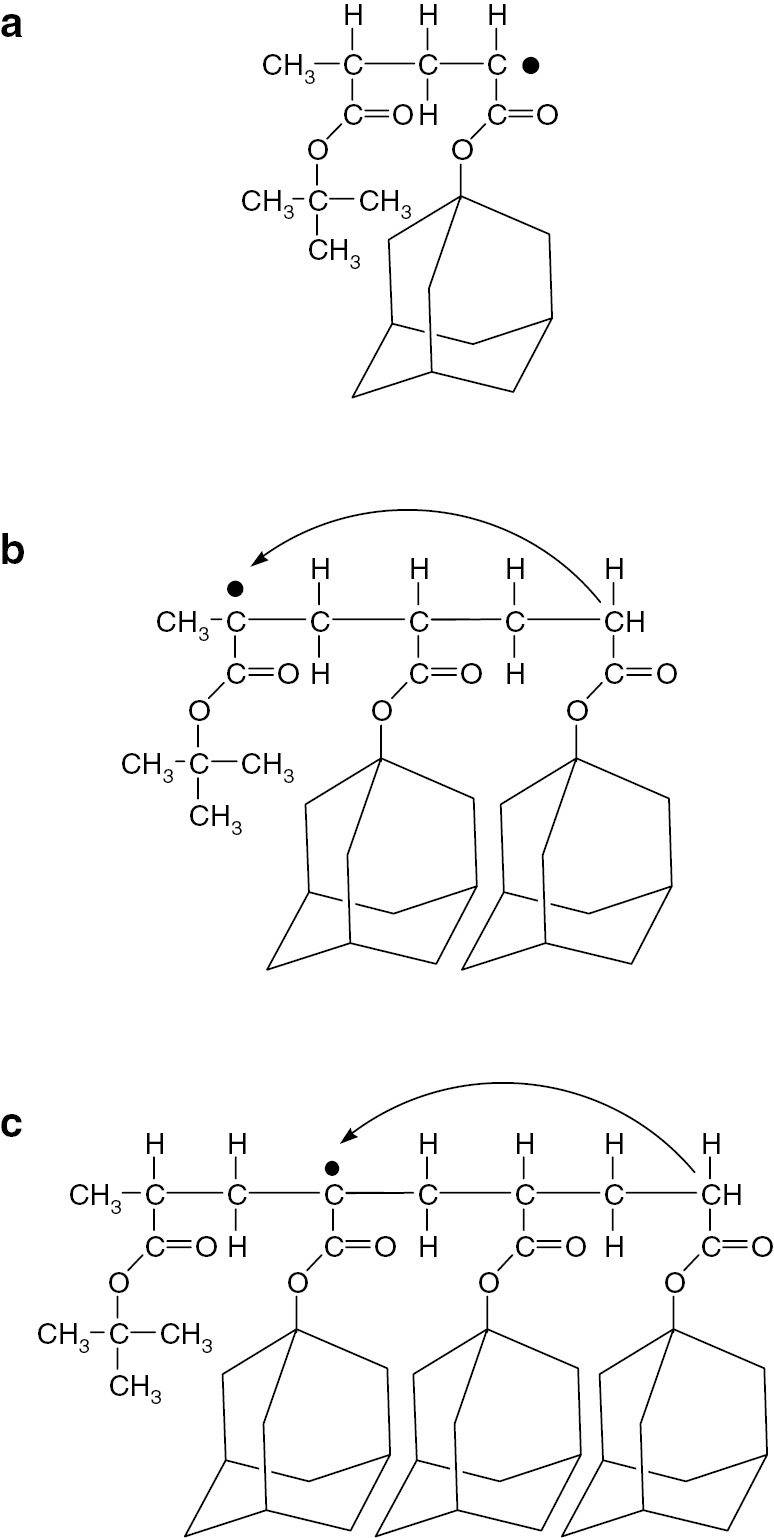
Structures of the observed (a) dimeric, (b) trimeric and (c) tetrameric model propagating radicals.
Different types of radicals were observed for trimeric and tetrameric model radicals at 120°C. These results allow the possibility of intermolecular chain transfer to be discarded. Nothing happened for dimeric model radical and the migrated radicals were located at the unit two units before of the original position strongly indicating the occurrence of a 1,5-hydrogen shift reaction in acrylate radical polymerizations. The original model propagating radical is a secondary radical and the resulting radical formed by transfer reaction is a tertiary radical. This transformation is a thermodynamically favorable process and the reaction through a six-membered ring structure is a kinetically favorable process. Therefore, it is clear that the propagating radical migrated to form mid-chain type radical by a 1,5-hydrogen shift mechanism.
The combination method of ATRP/ESR clearly confirmed the mechanism of the radical transfer reaction of acrylates. On the other hand, there is still no clear evidence for the mechanism of radical transfer reactions that had been discussed in the cases of ethylene [50], vinyl esters [52], [53], [54] and acrylamides [56], [57] polymerizations. Although clear ESR/EPR spectra of mid-chain radicals are shown in the cases of acrylamide polymerizations, the radical migration mechanism is still not clear. Kinetics studies of acrylate radical polymerizations including mid-chain radical formation have been conducted by Buback and co-workers using ESR/EPR techniques [58], [59], [60], [61].
Penultimate unit effect
The combination of ATRP and ESR techniques can also clarify penultimate unit effects (PUE) in radical copolymerizations of (meth)acrylate and styrene. Hetero dimeric model radical precursors were prepared by ATRP and purified. The dimeric model radical precursors are H-tBA-tBA-Br, H-ethyl methacrylate (EMA)-tBA-Br and H-St-tBA-Br. Structures of the generated radicals from these precursors by a reaction with an organo-tin compound under irradiation are shown in Fig. 10.
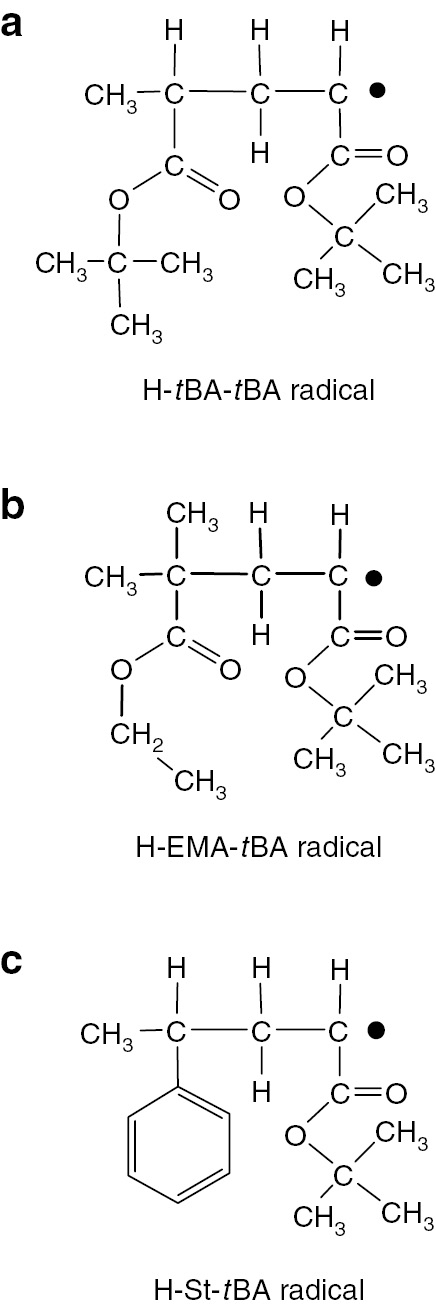
Structures of model dimeric radicals for observation of penultimate unit effects to terminal acrylate radicals. (a) H-tBA-tBA radical, (b) H-EMA-tBA radical and (c) H-St-tBA radical.
In each case the terminal radical structure was the tBA radical but the penultimate units are different. ESR/EPR spectra of H-St-tBA radical observed at +30 and 0°C (at 303 and 273 K) are shown in Fig. 11a and b, respectively. These are typical ESR/EPR spectra for dimeric model propagating radicals of acrylates as discussed above.
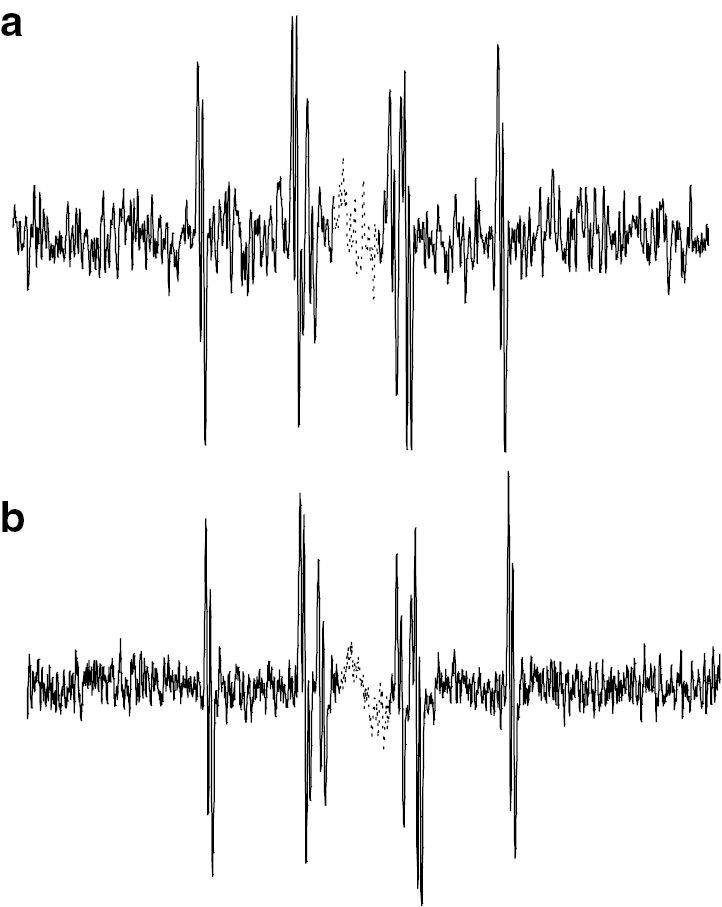
Observed ESR spectra of H-St-tBA radical at (a) 30°C and (b) 0°C.
These spectra are highly resolved and suitable for precise analysis of the hyperfine splitting patterns. The values of hfc’s can be measured precisely from these spectra. Plots in Fig. 12 are the temperature dependent hfc values of the dimeric radicals of H-tBA-tBA, H-EMA-tBA and H-St-tBA, respectively. Although the values of hfc of H-tBA-tBA and H-EMA-tBA radicals did not show clear temperature dependency, characteristic temperature dependent change was observed for the H-St-tBA radical as shown in Fig. 12c.
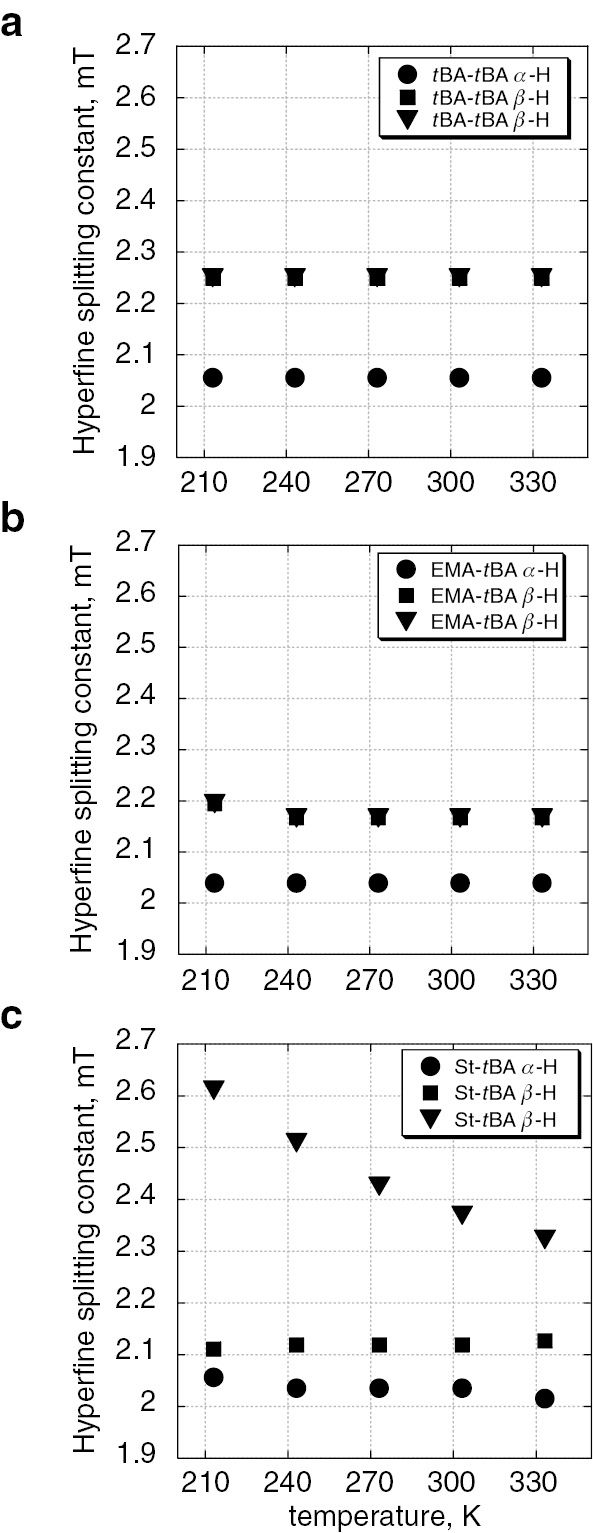
Temperature dependent change of hfc’s of α-methin and β-methylene protons of (a) H-tBA-tBA radical, (b) H-EMA-tBA radical, and (c) H-St-tBA radical.
This characteristic change is considered to be due to an electronic interaction between π-electrons in the phenyl ring and unpaired electron in pπ-orbital. These observations are clear experimental evidence of the electronic effects of the PUE that have been discussed based on kinetic studies [25], [26], [27], [28]. Various combinations of monomer units would provide significant information on the PUE of radical co-polymerization.
Investigation of the initial stage of a radical polymerization using time-resolved ESR spectroscopy
Alternating copolymerization of styrene with maleic anhydride
The first radical addition reaction in the alternating copolymerization of styrene with maleic anhydride has been investigated by TR ESR spectroscopy. Cleavage of a radical initiator was caused by laser pulse irradiation and the generated radial would then add to a monomer to form a chain initiating radical. TR ESR can exclusively observe the formation of the chain initiating radical. The general procedure for the alternating copolymerization of styrene with maleic anhydride is shown in Fig. 13a and the potential first radical addition reaction that can be observed in TR ESR spectroscopy are shown in Fig. 13b.
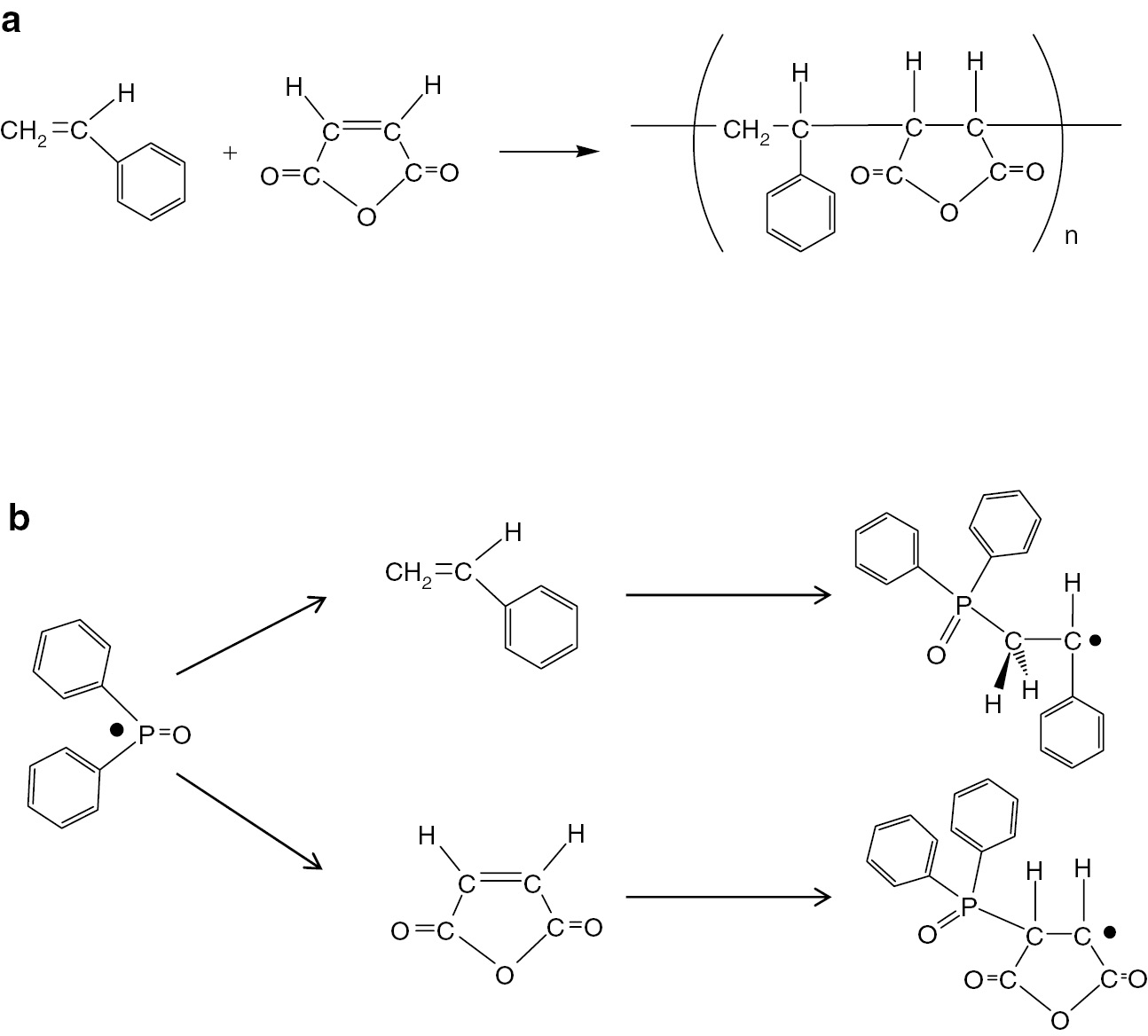
(a) Alternating copolymerization of styrene with maleic anhydride and (b) first radical addition of diphenyl phosphinoyl radical to styrene and maleic anhydride.
In this work, TR ESR spectroscopy has been applied to investigate the initiation mechanism of an alternating copolymerization of styrene and maleic anhydride and the result was compared with that of copolymerization of styrene with 1,3-butadiene. Structures of the chain initiating radicals formed by radical addition of diphenyl phosphinoyl radical to styrene (a), maleic anhydride (b) and 1,3-butadiene (c) are shown in Fig. 14.
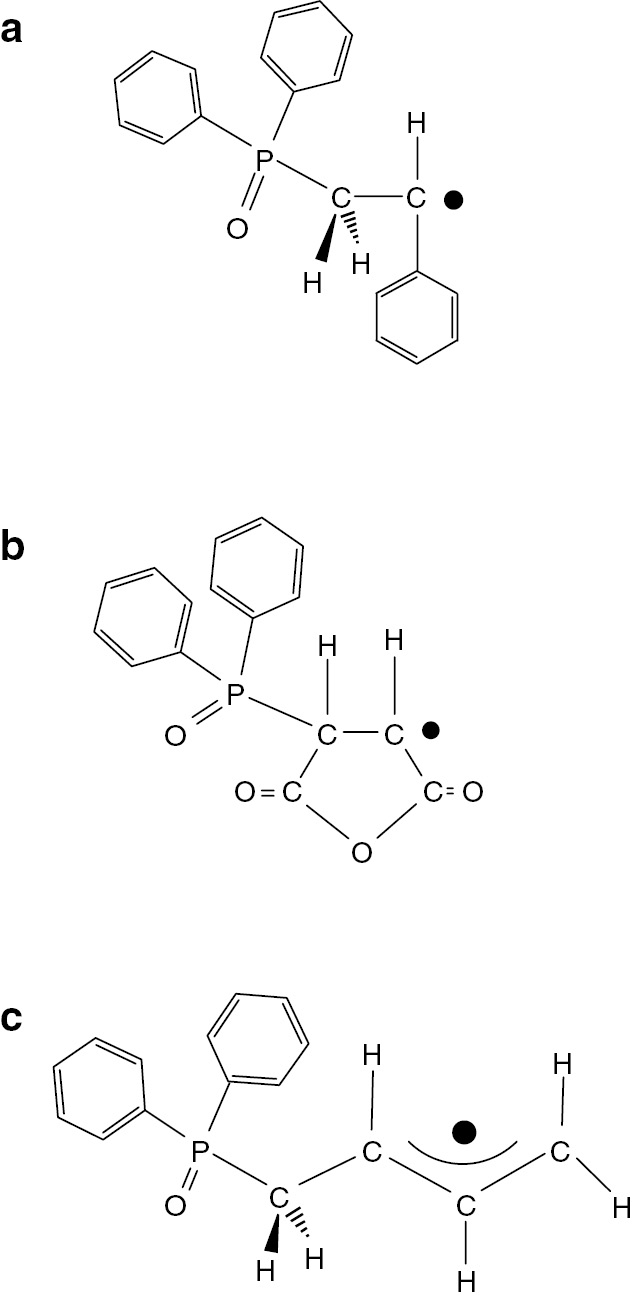
Structures of chain initiating radicals formed by radical addition of diphenyl phosphinoyl radical to (a) styrene, (b) maleic anhydride, and (c) 1,3-butadiene.
In a previous study, SS ESR spectroscopy demonstrated that the maleic anhydride terminal radical was the predominant radical species during the propagation processes using a reversible addition fragmentation chain-transfer (RAFT) agent as a spin trapping agent [24]. In this research work, the initiation step of the alternating copolymerization was examined by TR ESR. TR ESR spectra of styrene only, maleic anhydride only, styrene with maleic anhydride (1:1), and styrene-d8 with maleic anhydride (1:1) are shown in Fig. 15.
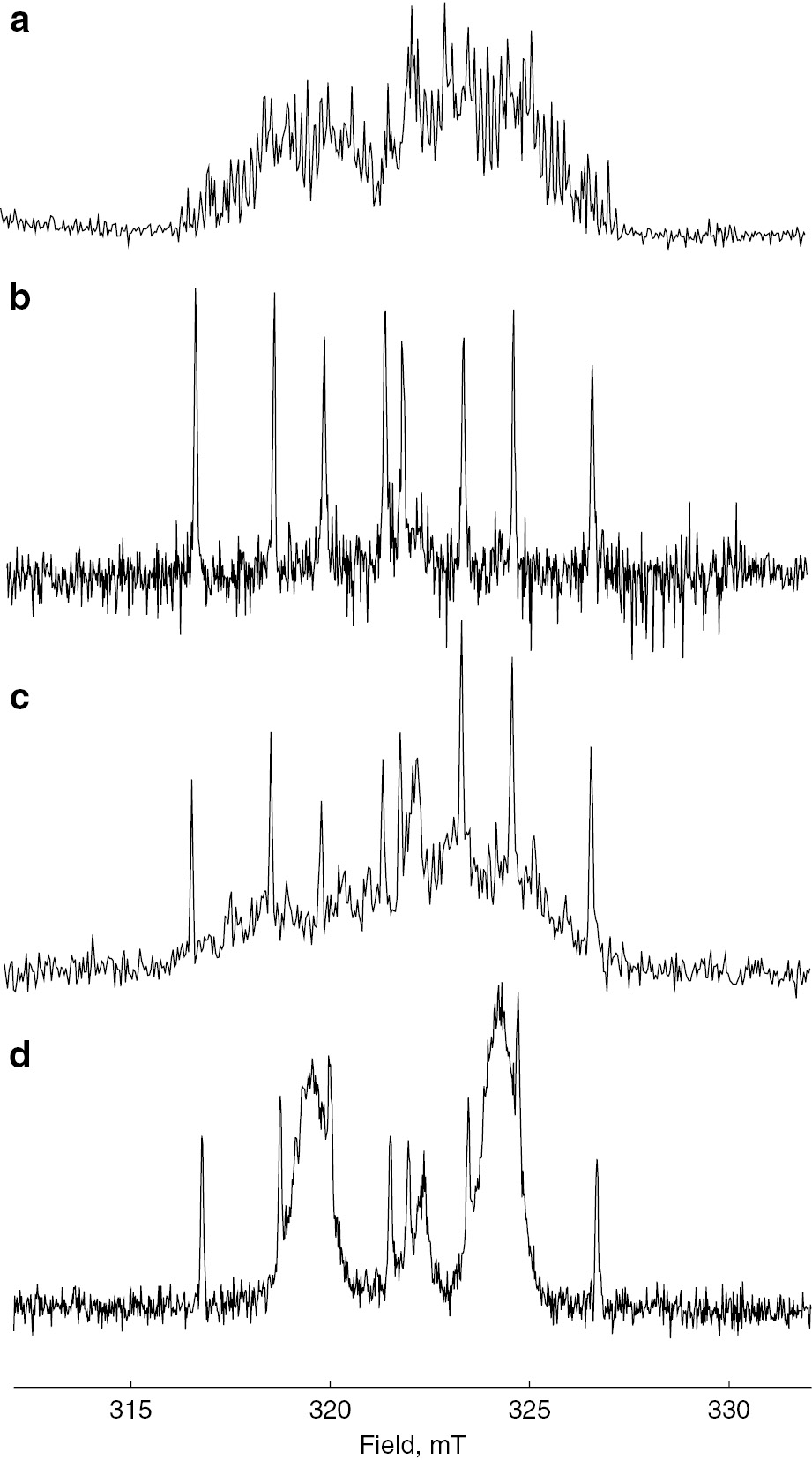
Time-resolved ESR spectra of (a) styrene only, (b) maleic anhydride only, (c) styrene with maleic anhydride and (d) styrene-d8 with maleic anhydride.
When copolymerization systems, with both styrene and maleic anhydride present, were examined overlapped signals of those styrene and maleic anhydride were observed as shown in Fig. 15c indicating that P-centered radical added to the each monomer just as in a homo-polymerization even in a co-polymerization system. Although, in principle, TR ESR spectroscopy does not provide quantitative results, we can state that first radical addition of the P-centered radical to each of the monomers was done without any selectivity. The reactivity of the radical addition reaction of each monomer seemed to be determined by the individual reactivity of the each monomer. TR ESR spectrum of the chain initiating radical of styrene is somehow complicated due to many splitting lines of phenyl protons. In order to simplify the spectrum, fully deuterated styrene (St-d8) was employed. The resulting TRESR spectrum of St-d8 with maleic anhydride is shown in Fig. 15d. The TR ESR spectrum of the chain initiating radical of St-d8 is appeared as two broad peaks and the peaks can be distinguished clearly from those of maleic anhydride. There was no clear observation of a radical species due to some kinds of interaction between styrene and maleic anhydride with the potential formation of a charge transfer complex that had been discussed in such alternating copolymerization systems [62]. Kinetics of fully deuterated styrene polymerization were also reported by Kattner and Buback [63].
Next, copolymerization of styrene with 1,3-butadiene was examined by TR ESR. Monomer reactivity ratios of this copolymerization are r1 (styrene)=0.58 and r2 (1,3-butadiene)=1.35. In this case, the reactivity of 1,3-butadiene is higher than that of styrene for both styrene and 1,3-butadiene radicals. TR ESR spectra of styrene only, 1,3-butadiene only, and styrene with 1,3-butadiene are shown in Fig. 16. Figure 16c is the TR ESR of styrene with 1,3-butadiene (1:1). The spectrum of 1,3-butadiene looks predominant in the figure which means that radical addition reaction of P-centered radical to the 1,3-butadiene is more favorable than that to styrene. This result is reasonable from the values of the monomer reactivity ratios.
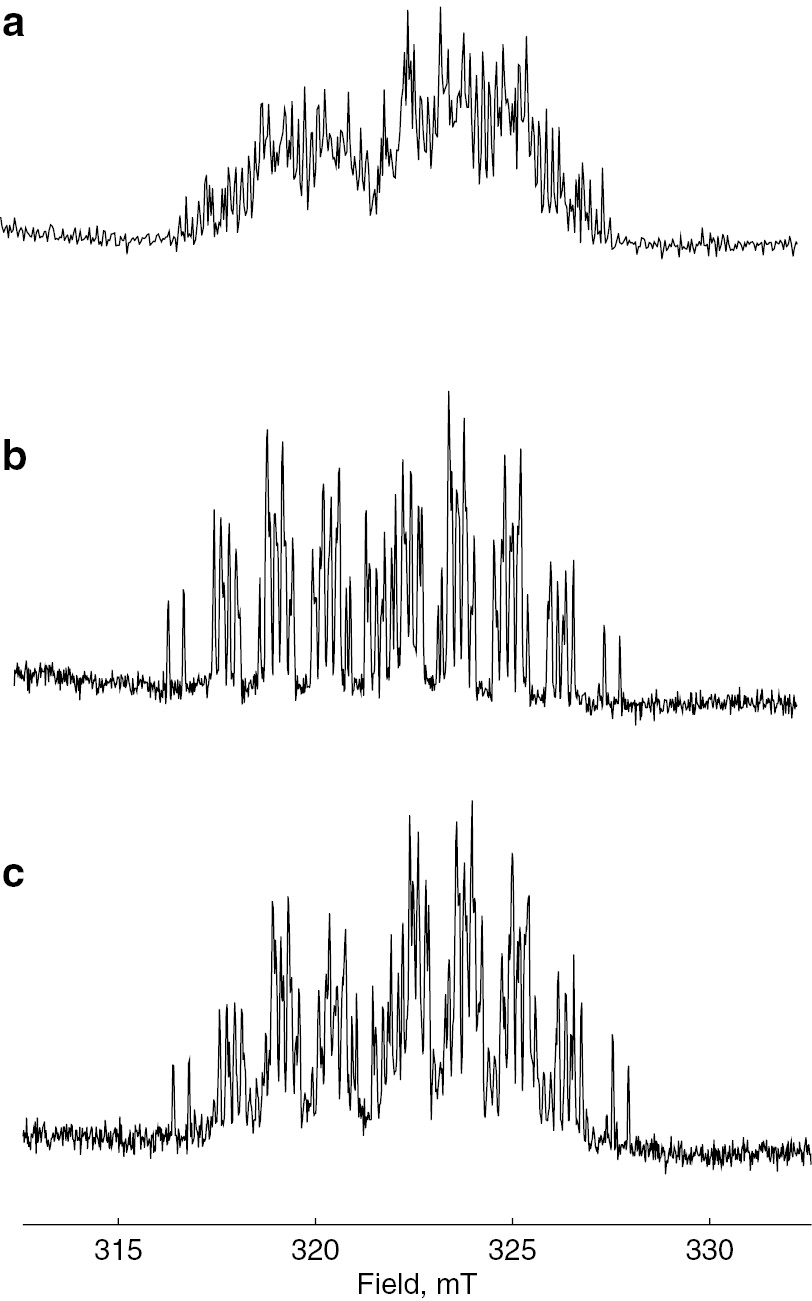
Time-resolved ESR spectra of (a) styrene only, (b) 1,3-butadiene only, and (c) styrene with 1,3-butadiene.
Judging from the results of both previous studies and the present study, an alternating copolymerization of styrene with maleic anhydride is initiated with no selectivity but is propagated in alternating manner probably from second monomer addition, and the maleic anhydride radical is predominant during propagation steps. In the case of copolymerization of styrene with 1,3-butadiene, radical addition reaction to 1,3-butadiene looked predominant, although addition reactions to both styrene and 1,3-butadiene occurred.
Conclusion
Several unsolved problems regarding the propagating radicals in conventional radical polymerization processes have been clarified using a combination of ATRP and SS ESR and TR ESR. Direct ESR observation of actual radical polymerizations of (meth)acrylates, styrene, and 1,3-butadiene clearly showed the structures of the propagating radicals. A combination of ATRP and ESR provided information on the chain lengths of the observed radicals in actual polymerization systems, the mechanism of the chain transfer reaction in acrylate polymerizations, and origin of PUE of copolymerization. TR ESR spectroscopy gave information on initial radical addition reaction of alternating copolymerization of styrene with maleic anhydride and copolymerization of styrene with 1,3-butadiene. Characterization of the radicals in radical polymerizations using various ESR techniques would provide interesting and useful information on conventional radical polymerizations.
Article note:
A collection of invited papers based on presentations at the 25th POLYCHAR 2017 World Forum on Advanced Materials, Kuala Lumpur, Malaysia, 9–13 October 2017.
References
[1] H. Fischer. Adv. Polym. Sci.5, 463 (1968).Suche in Google Scholar
[2] H. Fischer. “Electron spin resonance”, in Polymer Spectroscopy, D. O. Hummel (Ed.), Chapter 4, pp. 289–354, Verlag Chemie GmbH, Weinheim, Germany (1974).Suche in Google Scholar
[3] M. Kamachi. Adv. Polym. Sci.82, 207 (1987).Suche in Google Scholar
[4] B. Yamada, D. G. Westmoreland, S. Kobatake, O. Konosu. Prog. Polym. Sci.24, 565 (1999).10.1016/S0079-6700(99)00012-XSuche in Google Scholar
[5] M. Kamachi. J. Polym. Sci. Part A Polym. Chem.40, 269 (2002).10.1002/pola.10092.absSuche in Google Scholar
[6] J. A. Weil, J. R. Bolton. Electron Paramagnetic Resonance, 2nd ed. Wiley, Hoboken, NJ, USA (2007).10.1002/0470084987Suche in Google Scholar
[7] M. Kamachi, A. Kajiwara. Macromol. Symp.179, 53 (2002).10.1002/1521-3900(200203)179:1<53::AID-MASY53>3.0.CO;2-RSuche in Google Scholar
[8] M. Kamachi, A. Kajiwara. Macromolecules29, 2378 (1996).10.1021/ma951280jSuche in Google Scholar
[9] M. Kamachi, A. Kajiwara. Macromol. Chem. Phys.198, 787 (1997).10.1002/macp.1997.021980310Suche in Google Scholar
[10] M. Kamachi, A. Kajiwara. Macromol. Chem. Phys.201, 2165 (2000).10.1002/1521-3935(20001101)201:16<2165::AID-MACP2165>3.0.CO;2-DSuche in Google Scholar
[11] A. Kajiwara, M. Kamachi. Macromol. Chem. Phys.201, 2160 (2000).10.1002/1521-3935(20001101)201:16<2160::AID-MACP2160>3.0.CO;2-6Suche in Google Scholar
[12] A. Kajiwara, K. Maeda, N. Kubo, M. Kamachi. Macromolecules36, 526 (2003).10.1021/ma021205qSuche in Google Scholar
[13] A. Kajiwara. Macromol. Symp.248, 50 (2007).10.1002/masy.200790010Suche in Google Scholar
[14] A. Kajiwara. JEOL News 43, 39 (2008).10.1016/S0029-7437(08)70515-9Suche in Google Scholar
[15] M. D. Saban, M. K. Georges, R. P. N. Veregin, G. K. Hamer, P. M. Kazmaier. Macromolecules28, 7032 (1995).10.1021/ma00124a049Suche in Google Scholar
[16] J.-S. Wang, K. Matyjaszewski. Macromolecules28, 7901 (1995).10.1021/ma00127a042Suche in Google Scholar
[17] M. Kato, M. Kamigaito, M. Sawamoto, T. Higashimura. Macromolecules28, 1721 (1995).10.1021/ma00109a056Suche in Google Scholar
[18] J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo, S. H. Thang. Macromolecules31, 5559 (1998).10.1021/ma9804951Suche in Google Scholar
[19] A. Kajiwara. in Controlled/Living Radical Polymerization: Progress in ATRP, K. Matyjaszewski (Ed.), ACS Symposium Series 1023, pp. 49–59, ch. 4, American Chemical Society, Washington, DC (2009).10.1021/bk-2009-1023.ch004Suche in Google Scholar
[20] A. Kajiwara, M. Kamachi. in Advances in Controlled/Living Radical Polymerization, K. Matyjaszewski (Ed.), pp. 86–100, ACS Symposium series 854, ch. 7, American Chemical Society, Washington, DC (2003).10.1021/bk-2003-0854.ch007Suche in Google Scholar
[21] A. Kajiwara. in Progress in Controlled Radical Polymerization: Mechanisms and Techniques, K. Matyjaszewski (Ed.), pp. 33–46, ACS Symposium Series, American Chemical Society, Washington, DC (2012).Suche in Google Scholar
[22] A. Kajiwara, K. Matyjaszewski, M. Kamachi. in Controlled/Living Radical Polymerization, K. Matyjaszewski (Ed.), pp. 68–81, ACS Symposium series 768, ch. 5, American Chemical Society, Washington, DC (2000).10.1021/bk-2000-0768.ch005Suche in Google Scholar
[23] A. Kajiwara. ACS Symposium Series 1187, pp. 73–85, Washington, DC, San Francisco (2015).10.1021/bk-2015-1187.ch004Suche in Google Scholar
[24] F. S. Du, M. Q. Zhu, H. Q. Guo, Z. C. Li, F. M. Li, M. Kamachi, A. Kajiwara. Macromolecules35, 6739 (2002).10.1021/ma0202179Suche in Google Scholar
[25] T. Fukuda, A. Goto, Y. Kwak, C. Yoshikawa, Y. D. Ma. Macromolecular Symp.182, 53 (2001).10.1002/1521-3900(200206)182:1<53::AID-MASY53>3.0.CO;2-SSuche in Google Scholar
[26] Y. D. Ma, K.-S. Sung, Y. Tsujii, T. Fukuda. Macromolecules34, 4749 (2001).10.1021/ma001337oSuche in Google Scholar
[27] T. Fukuda, K. Kubo, Y. D. Ma. Prog. Polym. Sci.17, 875 (1992).10.1016/0079-6700(92)90012-NSuche in Google Scholar
[28] T. Fukuda, Y. D. Ma, H. Inagaki, K. Kubo. Macromolecules24, 370 (1991).10.1021/ma00002a005Suche in Google Scholar
[29] A. Kajiwara, A. K. Nanda, K. Matyjaszewski. Macromolecules.37, 1378 (2004).10.1021/ma035516sSuche in Google Scholar
[30] A. Kajiwara. Macromol. Rapid Comm.30, 1975 (2009).10.1002/marc.200900606Suche in Google Scholar
[31] T. Sumiyoshi, A. Henne, P. Lechtken, W. Schnabel. Z. Naturforsch. A39, 434 (1984).10.1515/zna-1984-0503Suche in Google Scholar
[32] M. Kamachi, K. Kuwata, T. Sumiyoshi, W. Schnabel. J. Chem. Soc. Perkin Trans.2, 961 (1988).10.1039/p29880000961Suche in Google Scholar
[33] A. Kajiwara, Y. Konishi, Y. Morishima, W. Schanbel, K. Kuwata, M. Kamachi. Macromolecules26, 1656 (1993).10.1021/ma00059a025Suche in Google Scholar
[34] Y. Mizuta, N. Morishita, K. Kuwata. Chem. Lett. 311 (1999).10.1246/cl.1999.311Suche in Google Scholar
[35] M. Weber, I. V. Khudyakov, N. J. Turro. J. Phys. Chem. A106, 1938 (2002).10.1021/jp011813sSuche in Google Scholar
[36] M. D. E. Forbes, H. Yashiro. Macromolecules40, 1460 (2007).10.1021/ma062438aSuche in Google Scholar
[37] B. Giese, W. Damm, F. Wetterich, H.-G. Zeitz. Tetrahedron Lett.33, 1863 (1992).10.1016/S0040-4039(00)74162-1Suche in Google Scholar
[38] A. Kajiwara. in Controlled/Living Radical Polymerization, K. Matyjaszewski (Ed.), pp. 111–124, ACS Symposium Series 944, ch. 9, American Chemical Society, Washington, DC (2006).Suche in Google Scholar
[39] H. Yoshida, B. Rånby. J. Polym. Sci. Part C16, 1333 (1967).10.1002/polc.5070160311Suche in Google Scholar
[40] H. Fischer, G. Giacometti. J. Polym. Sci. Part C16, 2763 (1967).10.1002/polc.5070160529Suche in Google Scholar
[41] M. E. Best, P. H. Kasai. Macromolecules22, 2622 (1989).10.1021/ma00196a014Suche in Google Scholar
[42] B. C. Gilbert, J. R. Lindsay Smith, E. C. Milne, A. C. Whitwood, P. Taylor. J. Chem. Soc. Perkin Trans.2, 2025 (1993).10.1039/P29930002025Suche in Google Scholar
[43] B. C. Gilbert, J. R. Lindsay Smith, E. C. Milne, A. C. Whitwood, P. Taylor. J. Chem. Soc. Perkin Trans.2, 1759 (1994).10.1039/P29940001759Suche in Google Scholar
[44] D. C. Doetschman, R. C. Mehlenbacher, D. Cywar. Macromolecules29, 1807 (1996).10.1021/ma951444wSuche in Google Scholar
[45] H. R. Chang, W. Lau, H.-Y. Parker, D. G. Westmoreland. Macromol. Symp.111, 253 (1996).10.1002/masy.19961110123Suche in Google Scholar
[46] Y. Sugiyama. Bull. Chem. Soc. Jpn.70, 1827 (1997).10.1246/bcsj.70.1827Suche in Google Scholar
[47] M. Azukizawa, B. Yamada, D. J. T. Hill, P. J. Pomery. Macromol. Chem. Phys.201, 774 (2000).10.1002/(SICI)1521-3935(20000401)201:7<774::AID-MACP774>3.0.CO;2-KSuche in Google Scholar
[48] B. Yamada, M. Azukizawa, H. Yamazoe, D. J. T. Hill, P. J. Pomery. Polymer41, 5611 (2000).10.1016/S0032-3861(99)00794-6Suche in Google Scholar
[49] A. Kajiwara, K. Matyjaszewski. in Advanced ESR Methods in Polymer Research, ch. 5, pp. 101–132, Wiley Interscience, NJ (2006).Suche in Google Scholar
[50] M. J. Roedel. J. Am. Chem. Soc.75, 6110 (1953).10.1021/ja01120a005Suche in Google Scholar
[51] K. Tanaka, B. Yamada, R. Willemse, A. M. van Herk. Polym. J.34, 692 (2002).10.1295/polymj.34.692Suche in Google Scholar
[52] J. T. Clarke, R. O. Howard, W. H. Stockmayer. Makromol. Chem.44–46, 427 (1961).10.1002/macp.1961.020440136Suche in Google Scholar
[53] R. L. Adelman, R. C. Ferguson. J. Polym. Sci. Polym. Chem. Ed.13, 891 (1975).10.1002/pol.1975.170130409Suche in Google Scholar
[54] Y. Morishima, S.-I. Nozakura. J. Polym. Sci. Polym. Chem. Ed.14, 1277 (1976).10.1002/pol.1976.170140527Suche in Google Scholar
[55] S.-I. Nozakura, Y. Morishima, H. Iimura, Y. Irie. J. Polym. Sci. Polym. Chem. Ed.14, 759 (1976).10.1002/pol.1976.170140323Suche in Google Scholar
[56] H. Kattner, M. Buback. Macromol. Rapid Commun.36, 2186 (2015).10.1002/marc.201500479Suche in Google Scholar
[57] H. Kattner, M. Buback. Macromolecules48, 7410 (2015).10.1021/acs.macromol.5b01921Suche in Google Scholar
[58] R. X. E. Willemse, A. M. van Heck, E. Panchenko, T. Junkers, M. Buback. Macromolecules38, 5098 (2005).10.1021/ma050198dSuche in Google Scholar
[59] M. Buback, P. Hesse, I. Lacik. Macromol. Rapid Commun.28, 2049 (2007).10.1002/marc.200700396Suche in Google Scholar
[60] J. Barth, M. Buback, P. Hesse, T. Sergeeva. Macromol. Rapid Commun.30, 1969 (2009).10.1002/marc.200900531Suche in Google Scholar PubMed
[61] J. Barth, M. Buback, P. Hesse, T. Sergeeva. Macromolecules43, 4023 (2010).10.1021/ma1006039Suche in Google Scholar
[62] E. Tsuchida, T. Tomono. Makromol. Chem.141, 265–298 (1971).10.1002/macp.1971.021410122Suche in Google Scholar
[63] H. Kattner, M. Buback. Macromolecules48, 309 (2015).10.1021/ma5022665Suche in Google Scholar
©2018 IUPAC & De Gruyter. This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License. For more information, please visit: http://creativecommons.org/licenses/by-nc-nd/4.0/
Artikel in diesem Heft
- Frontmatter
- In this issue
- Conference papers
- Core-shell Fe-SiO2-polyamine magnetic nanoparticles for metal recovery using a continuous flow pipeline reactor
- Characterizations of radicals formed in radical polymerizations and transfer reactions by electron spin resonance spectroscopy
- IUPAC Technical Reports
- The ongoing challenge of novel psychoactive drugs of abuse. Part I. Synthetic cannabinoids (IUPAC Technical Report)
- Engineered nanomaterials and human health: Part 1. Preparation, functionalization and characterization (IUPAC Technical Report)
- Engineered nanomaterials and human health: Part 2. Applications and nanotoxicology (IUPAC Technical Report)
Artikel in diesem Heft
- Frontmatter
- In this issue
- Conference papers
- Core-shell Fe-SiO2-polyamine magnetic nanoparticles for metal recovery using a continuous flow pipeline reactor
- Characterizations of radicals formed in radical polymerizations and transfer reactions by electron spin resonance spectroscopy
- IUPAC Technical Reports
- The ongoing challenge of novel psychoactive drugs of abuse. Part I. Synthetic cannabinoids (IUPAC Technical Report)
- Engineered nanomaterials and human health: Part 1. Preparation, functionalization and characterization (IUPAC Technical Report)
- Engineered nanomaterials and human health: Part 2. Applications and nanotoxicology (IUPAC Technical Report)