When diazo compounds meet with organoboron compounds
-
Jianbo Wang


Abstract
Transition-metal free reactions of diazo compounds with organoboron compounds provide some unique approaches for the formation of C–C, C–B and C–Si bonds. With N-tosylhydrazones as the precursors for non-stabilized diazo compound, this type of reaction becomes practically useful in organic synthesis. Transition-metal-free synthetic methodologies for borylation, gem-diborylation, gem-silylborylation arylation, 2,2,2-trifluoroethylation and gem-difluorovinylation have been successfully developed.
Introduction
Diazo compounds are best known as metal carbene precursors in transition-metal-catalyzed reactions. The classic metal carbene transformations, such as cyclopropanations and C–H bond insertions, have been well-established as useful methods in organic synthesis [1], [2], [3], [4]. More recently, transition-metal-catalyzed cross-coupling with carbene precursors have attarcted considerable attentions [5], [6], [7], [8], [9], [10], [11].
In addition to serve as metal carbene precursors, diazo compounds also function as nucleophiles because of the electron-rich character of the carbon bearing the diazo group [12]. An interesting reaction of diazo compounds as nucleophiles is the interaction with electron-defficient boron, generating a tetra-coordinated boron complex. Subsequently, one of the substituents (the X group) migrates from boron to carbon center, with simutanoues release of dinitrogen. The process can be considered as a formal carbene insertion into the B–X bond of the organoboron reagent (Scheme 1) [13].
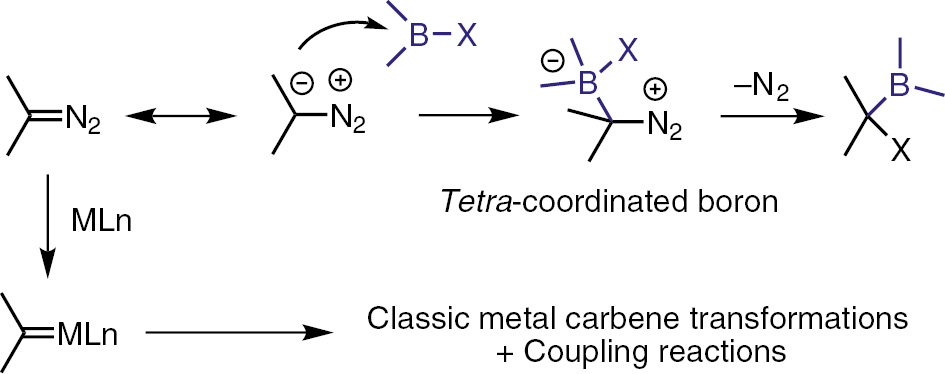
Diazo compounds as nucleophiles vs. as carbene precusors.
Results and discussion
The reaction of α-diazocarbonyl compounds with organoborons was previously reported to form C–C bonds between alkyl or aryl and an α carbon of carbonyl group. Hooz and Linke [14] established the alkylation of diazonitrile, ethyldiazoacetate and diazoketones by means of trialkylboranes (Scheme 2a). The reactions reported by Hooz and Linke were limited by narrow scope and low efficiency. Further improvement was made by Brown, Hooz and coworkers by using dialkylchloroboranes, and alkyl- or aryldichloroboranes, which are more reactive boron reagents toward diazo compounds (Scheme 2b and c) [15], [16].
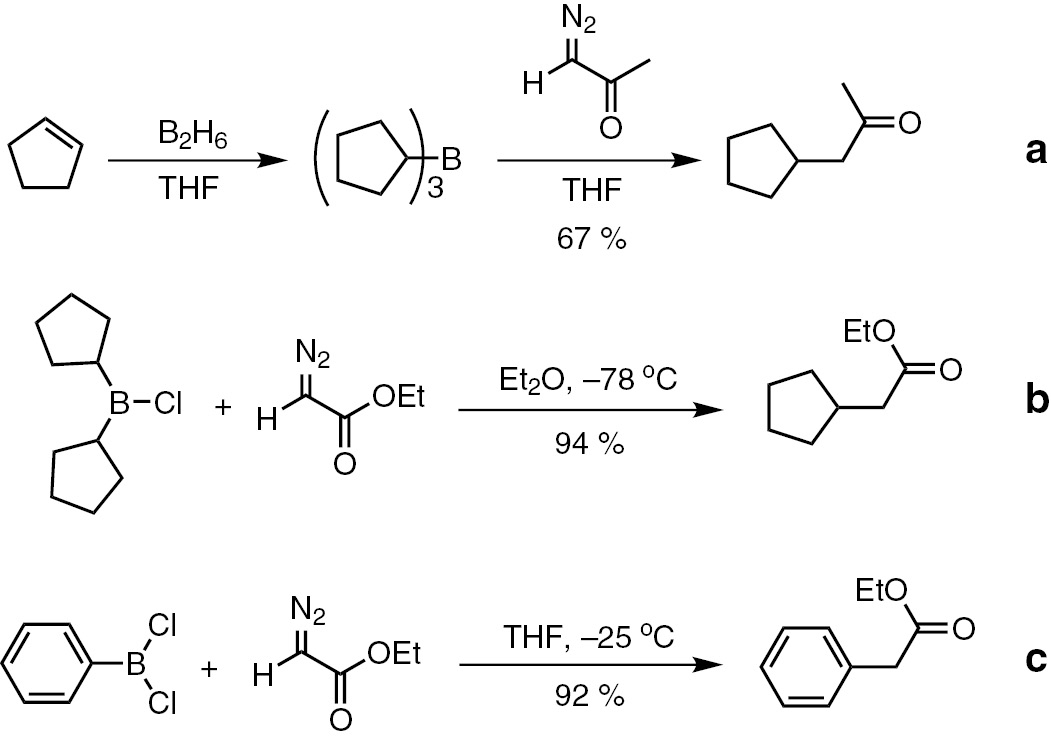
The pioneering work on the reaction between diazo compounds and organoborons.
However, these otherwise very attractive transition-metal-free C–C bond forming reactions have received limited attention following therir publications, presumably because of the toxic and unstable boron compounds being used, and the reactions have to be carried out under stringent conditions. Besides, the scope of diazo substrates is very limited. Mechanistically, these reactions are initiated by the nucleophilic attack of diazo substrate to boron. Thus, increasing the electonpositivity in the boron center and/or increasing the nucleophilicity of diazo substrate should enhance the reaction. In view of the facts that organoboronic acids and their derivatives, which are usually stable, non-toxic and easily available, have gained tremendous popularity among synthetic organic community, it is thus worthwhile to investigate the reaction of diazo compounds with boronic acids and their derivatives.
In the process of studying a Pd-catalyzed reaction of arylboronic acid with diazocarbonyl compounds [17], we have found that the C–C bond forming reaction occurred under transition-metal-free conditions. The reaction with phenylboron pinacolate gave no product, while the reaction with boroxine afforded moderate yield. The reaction is quite general in terms of the structure of diazocarbonyl compounds (Scheme 3) [18].

Metal-free arylation and vinylation of α-diazocarbonyl compounds with boroxines.
Two reaction pathways are proposed to account for the reaction (Scheme 4). In path a, the oxygen anion functions as a nucleophile to coordinate with the boron reagent to generate A, while in path b, the carbon nucleophile interacts with the boron to generate C, both giving boron enolate B as the intermediate.

Proposed reaction mechanism.
In the same year, Barluenga and co-workers reported the transition-metal-free couplings of hydrazones with arylboronic acids. Since N-tosylhydrazone is easily derived from the corresponding ketones or aldehydes, the reaction is highly attractive as a C–C bond forming method [19]. This reaction follows the same mechanism as described in Scheme 4, except that in this case the non-stablized diazo intermediate is generated in situ through Bamford–Stevens reaction [20], [21].
We have conceived that this chemistry may be expanded by empoying other methods of in situ generation of diazo intermediates. As shown in Scheme 5, under almost neutral reaction conditions, the α-aminoesters, α-aminonitriles can be converted into the diazo intermediates through diazotization and deprotonation. The reaction of this diazo intermediate with arylboronic acid forms C–C bond. The entire process is a transition-metal-free deaminative coupling of α-aminoesters, α-aminonitriles with arylboronic acids. The reaction conditions are very mild under almost neutral conditions [22].

Deaminative coupling of α-aminoesters and nitriles with arylboronic acids.
The reaction shows excellent functional group tolerance. In the report 75 examples of the reaction have been demonstrated, and some reactions have been carried out in a gram-scale. Some selected examples have been summarized in Scheme 6. Since the reaction is under tranistion-metal-free conditions, various useful functional groups such as halogen substituents can be tolerated, which is beneficial for the further transformations. This is a highly reliable method for the synthesis of α-aminoesters and α-aminonitriles. These compounds have attracted attentions because of their importance, and much efforts have been devoted to the synthetic method development, mostly by employing transition-metal-catalyzed approaches.
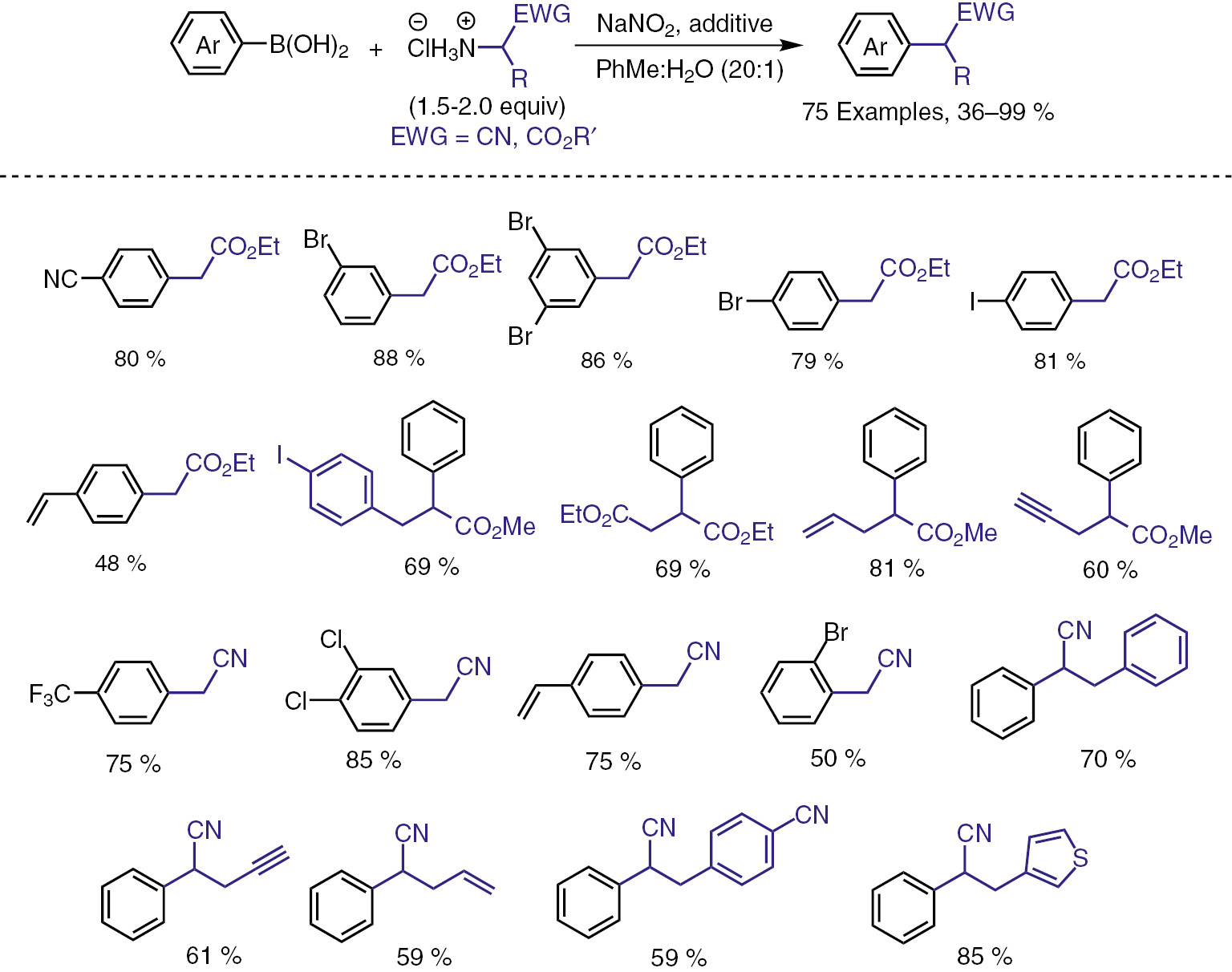
Selected examples of the transition-metal-free deaminative coupling.
Similarly, we can use 2,2,2-trifluorodiazoethane, generated from the corresponding amine, to develop a method for trifluoroethylation and gem-difluorovinylation of organoboronic acids, depending on the reaction conditions (Scheme 7) [23]. The reactions follow similar mechanism, except the final terminating step. Under weak acidic conditions, the reaction was terminated by protonation to afford trifluoroethylation products. Under week basic conditions with LiOH as the additive, the reaction is terminated by elimination of fluoride rather than protonation, giving gem-difluorovinylation product. Both reactions shows wide substrate scope.

2,2,2-Trifluoroethylation and gem-difluorovinylation of organoboronic acids with 2,2,2-trifluorodiazoethane.
Furthermore, with trimethylsilyldiazomethane (TMSCHN2), we could develop a method for homologating the arylboronic acids, by employing very similar method. The transformation is shown in Scheme 8. First, the coordination of the diazo substrate to boronic acid, followed by 1,2-shift, esterification with pinacol, and finally protodesilylation to afford the pinacol benzylboronates. The reaction gave the homologation products in moderately good yields [24].

Homologation of arylboronic acid.
To further develop the chemistry, we can consider varying the migrating group X, as shown in Scheme 1. An interesting development along this line is the use of diboron or borane reagent, in which two C–B bonds can be formed. Thus, with N-tosylhydrazone as the precusor of diazo compounds, we have developed a transition-metal-free C–B bond forming transformations. The reaction represents an alternative access toward pinacol alkylboronates. The reaction works well over a wide range of N-tosylhydrazone substrates, with both diboron and also borane reagents (Scheme 9) [25].
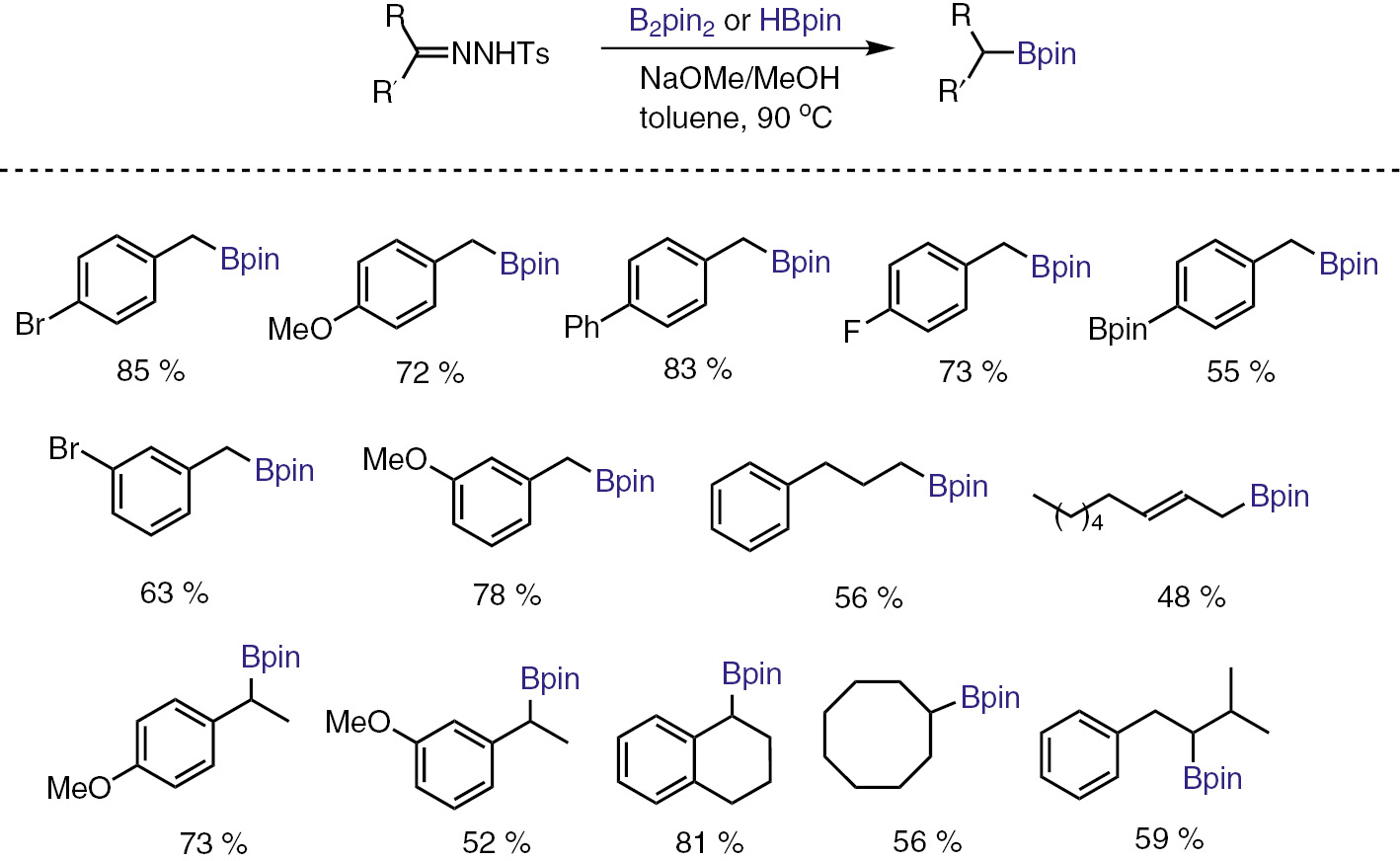
Transition metal-free synthesis of pinacol alkylboronates from tosylhydrazones.
The mechanistic rationale is shown in Scheme 10. The diazo intermediate is first generated in situ in the presence of base, reacting with borone, then 1,2-hydrogen shift occurs to give the product directly. While in the case of B2pin2, 1,2-Bpin shift occurs to afford gem-diboron intermediate, which is then followed by protodeborylation to give the product. In the absence of proton source, we could isolate the gem-diboron product in 73% yield, which was a strong evidence to support the proposed reaction mechanism.

The mechanistic rationale and control experiments.
On the other hand, the mechanistic experiment demonstrates the potential of developing a method for the synthesis of gem-diboron compounds. Indeed, under the conditions that proton source is absent, the gem-diboron compounds could be obtained in moderately high yields. Very similarly, gem-silylborylation compounds could be obtained by using silylborane (pinBSiMe2Ph). The reaction shows wide substrate scope for both cases (Scheme 11) [26].

gem-Diborylation and gem-silylborylation of N-tosylhydrazone.
Conlusions
From the reactions presented in this lecture, it can be concluded that the reaction between nucleophilic diazo carbon and electron-deficient boron center is a general process and can be utilized in various transformations. In particular, the 1,2-shift of the substituent from boron to the diazo carbon generates C–C, C–B and C–Si bonds. The interemdirates can be further applied to various transformations. By using tosylhydrazone as the non-stablized diazo compound precursors, this type of reaction becomes practically useful in organic synthesis [27], [28], [29], [30], [31], [32], [33].
Article note
A collection of invited papers based on presentations at the 16th International Meeting on Boron Chemistry (IMEBORON-16), Hong Kong, 9–13 July 2017.
Acknowledgments
The project is supported by National Basic Research Program of China (973 Program, No. 2015CB856600) and NSFC (Grant No. 21332002).
References
[1] T. Ye, M. A. McKervey. Chem. Rev.94, 1091 (1994).10.1021/cr00028a010Search in Google Scholar
[2] M. P. Doyle, M. A. McKervey, T. Ye. Modern Catalytic Methods for Organic Synthesis with Diazo Compounds, Wiley, New York, NY (1998).Search in Google Scholar
[3] Z. Zhang, J. Wang. Tetrahedron64, 6577 (2008).10.1016/j.tet.2008.04.074Search in Google Scholar
[4] A. Ford, H. Miel, A. Ring, C. N. Slattery, A. R. Maguire, M. A. McKervey. Chem. Rev.115, 9981 (2015).10.1021/acs.chemrev.5b00121Search in Google Scholar PubMed
[5] Y. Zhang, J. Wang. Eur. J. Org. Chem. 1015 (2011).10.1002/ejoc.201001588Search in Google Scholar
[6] J. Barluenga, C. Valdés. Angew. Chem. Int. Ed.50, 7486 (2011).10.1002/anie.201007961Search in Google Scholar PubMed
[7] Z. Shao, H. Zhang. Chem. Soc. Rev.41, 560 (2012).10.1039/C1CS15127DSearch in Google Scholar PubMed
[8] Q. Xiao, Y. Zhang, J. Wang. Acc. Chem. Res.46, 236 (2013).10.1021/ar300101kSearch in Google Scholar PubMed
[9] Z. Liu, J. Wang. J. Org. Chem.78, 10024 (2013).10.1021/jo401850qSearch in Google Scholar PubMed
[10] Y. Xia, Y. Zhang, J. Wang. ACS Catal.3, 2586 (2013).10.1021/cs4006666Search in Google Scholar
[11] F. Hu, Y. Xia, C. Ma, Y. Zhang, J. Wang. Chem. Commun.51, 7986 (2015).10.1039/C5CC00497GSearch in Google Scholar PubMed
[12] Y. Zhang, J. Wang. Chem. Commun. 5350 (2009).10.1039/b908378bSearch in Google Scholar PubMed
[13] H. Li, Y. Zhang, J. Wang. Synthesis45, 3090 (2013).10.1055/s-0033-1340041Search in Google Scholar
[14] J. Hooz, S. Linke. J. Am. Chem. Soc.90, 5936 (1968).Search in Google Scholar
[15] H. C. Brown, M. M. Midland, A. B. Levy. J. Am. Chem. Soc.90, 5936 (1968).10.1021/ja01023a070Search in Google Scholar
[16] J. Hooz, J. N. Bridson, J. G. Calzada, H. C. Brown, M. M. Midland, A. B. Levy. J. Org. Chem.38, 2574 (1973).10.1021/jo00954a044Search in Google Scholar
[17] C. Peng, Y. Wang, J. Wang. J. Am. Chem. Soc.130, 1566 (2008).10.1021/ja0782293Search in Google Scholar PubMed
[18] C. Peng, W. Zhang, G. Yan, J. Wang. Org. Lett.11, 1667 (2009).10.1021/ol900362dSearch in Google Scholar PubMed
[19] J. Barluenga, M. Tomása-Gamasa, F. Aznar, C. Valdés. Nat. Chem.1, 494 (2009).10.1038/nchem.328Search in Google Scholar PubMed
[20] W. R. Bamford, T. S. Stevens. J. Chem. Soc. 4735 (1952).10.1039/jr9520004735Search in Google Scholar
[21] J. R. Fulton, V. K. Aggarwal, J. de Vicente. Eur. J. Org. Chem. 1479 (2005).10.1002/ejoc.200400700Search in Google Scholar
[22] G. Wu, Y. Deng, C. Wu, Y. Zhang, J. Wang. Angew. Chem. Int. Ed.53, 10510 (2014).10.1002/anie.201406765Search in Google Scholar PubMed
[23] X. Wang, Y. Zhou, G. Ji, G. Wu, M. Li, Y. Zhang, J. Wang. Eur. J. Org. Chem. 3093 (2014).10.1002/ejoc.201402105Search in Google Scholar
[24] C. Wu, G. Wu, Y. Zhang, J. Wang. Org. Chem. Front.3, 817 (2016).10.1039/C6QO00141FSearch in Google Scholar
[25] H. Li, L. Wang, Y. Zhang, J. Wang. Angew. Chem. Int. Ed.51, 2943 (2012).10.1002/anie.201108139Search in Google Scholar PubMed
[26] H. Li, X. Shangguan, Z. Zhang, S. Huang, Y. Zhang, J. Wang. Org. Lett.16, 448 (2014).10.1021/ol403338sSearch in Google Scholar PubMed
[27] P. K. Elkin, V. V. Levin, A. D. Dilman, M. I. Struchkova, P. A. Belyakov, D. E. Arkhipov, A. A. Korlyukov, V. A. Tartakovsky. Tetrahedron Lett.52, 5259 (2011).10.1016/j.tetlet.2011.07.141Search in Google Scholar
[28] P. K. Elkin, V. V. Levin, A. D. Dilman, M. I. Struchkova, D. E. Arkhipov, A. A. Korlyukov. Tetrahedron Lett.53, 6216 (2012).10.1016/j.tetlet.2012.08.153Search in Google Scholar
[29] O. A. Argintaru, D. Ryu, I. Aron, G. A. Molander. Angew. Chem. Int. Ed.52, 13656 (2013).10.1002/anie.201308036Search in Google Scholar PubMed PubMed Central
[30] Y. Luan, J. Yu, X. Zhang, S. E. Schaus, G. Wang. J. Org. Chem.79, 4694 (2014).10.1021/jo5003505Search in Google Scholar PubMed PubMed Central
[31] A. B. Cuenca, J. Cid, Di. García-López, J. J. Carbó, E. Fernández. Org. Biomol. Chem.13, 9659 (2015).10.1039/C5OB01523ESearch in Google Scholar
[32] D. N. Tran, C. Battilocchio, S.-B. Lou, J. M. Hawkins, S. V. Ley. Chem. Sci.6, 1120 (2015).10.1039/C4SC03072ASearch in Google Scholar
[33] J.-S. Poh, S.-H. Lau, I. G. Dykes, D. N. Tran, C. Battilocchio, S. V. Ley. Chem. Sci.7, 6803 (2016).10.1039/C6SC02581ASearch in Google Scholar
©2018 IUPAC & De Gruyter. This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License. For more information, please visit: http://creativecommons.org/licenses/by-nc-nd/4.0/
Articles in the same Issue
- Frontmatter
- In this issue
- Preface
- 16th International Meeting on Boron Chemistry (IMEBORON XVI)
- Conference papers
- Palladium-promoted sulfur atom migration on carboranes: facile B(4)−S bond formation from mononuclear Pd-B(4) complexes
- When diazo compounds meet with organoboron compounds
- Transition-metal complexes with oxidoborates. Synthesis and XRD characterization of [(H3NCH2CH2NH2)Zn{κ3O,O′,O′′-B12O18(OH)6-κ1O′′′}Zn(en)(NH2CH2CH2NH3)]·8H2O (en=1,2-diaminoethane): a neutral bimetallic zwiterionic polyborate system containing the ‘isolated’ dodecaborate(6−) anion
- Novel sulfur containing derivatives of carboranes and metallacarboranes
- Metal–metal bonding in deltahedral dimetallaboranes and trimetallaboranes: a density functional theory study
- Nanostructured boron compounds for cancer therapy
- Heterometallic boride clusters: synthesis and characterization of butterfly and square pyramidal boride clusters*
- Influence of fluorine substituents on the properties of phenylboronic compounds
- Copper-catalyzed asymmetric dearomative borylation: new pathway to optically active heterocyclic compounds
- Borenium and boronium ions of 5,6-dihydro-dibenzo[c,e][1,2]azaborinine and the reaction with non-nucleophilic base: trapping of a dimer and a trimer of BN-phenanthryne by 4,4′-di-tert-butyl-2,2′-bipyridine
- The electrophilic aromatic substitution approach to C–H silylation and C–H borylation
- Recent advances in B–H functionalization of icosahedral carboranes and boranes by transition metal catalysis
- closo-Dodecaborate-conjugated human serum albumins: preparation and in vivo selective boron delivery to tumor
- IUPAC Technical Report
- Risk assessment of effects of cadmium on human health (IUPAC Technical Report)
Articles in the same Issue
- Frontmatter
- In this issue
- Preface
- 16th International Meeting on Boron Chemistry (IMEBORON XVI)
- Conference papers
- Palladium-promoted sulfur atom migration on carboranes: facile B(4)−S bond formation from mononuclear Pd-B(4) complexes
- When diazo compounds meet with organoboron compounds
- Transition-metal complexes with oxidoborates. Synthesis and XRD characterization of [(H3NCH2CH2NH2)Zn{κ3O,O′,O′′-B12O18(OH)6-κ1O′′′}Zn(en)(NH2CH2CH2NH3)]·8H2O (en=1,2-diaminoethane): a neutral bimetallic zwiterionic polyborate system containing the ‘isolated’ dodecaborate(6−) anion
- Novel sulfur containing derivatives of carboranes and metallacarboranes
- Metal–metal bonding in deltahedral dimetallaboranes and trimetallaboranes: a density functional theory study
- Nanostructured boron compounds for cancer therapy
- Heterometallic boride clusters: synthesis and characterization of butterfly and square pyramidal boride clusters*
- Influence of fluorine substituents on the properties of phenylboronic compounds
- Copper-catalyzed asymmetric dearomative borylation: new pathway to optically active heterocyclic compounds
- Borenium and boronium ions of 5,6-dihydro-dibenzo[c,e][1,2]azaborinine and the reaction with non-nucleophilic base: trapping of a dimer and a trimer of BN-phenanthryne by 4,4′-di-tert-butyl-2,2′-bipyridine
- The electrophilic aromatic substitution approach to C–H silylation and C–H borylation
- Recent advances in B–H functionalization of icosahedral carboranes and boranes by transition metal catalysis
- closo-Dodecaborate-conjugated human serum albumins: preparation and in vivo selective boron delivery to tumor
- IUPAC Technical Report
- Risk assessment of effects of cadmium on human health (IUPAC Technical Report)