Recent progress in asymmetric synthesis of aminophosphonic acids mediated by chiral sulfinyl auxiliary
-
Marian Mikołajczyk
 and
Piotr Łyżwa
and
Piotr Łyżwa

Abstract
This account outlines the recent results on our strategy of the diastereoselective asymmetric synthesis of aminophosphonic acids (APs) using enantiomeric sulfinimines as chirality inducing reagents. Three important topics are discussed: (a) application of a double asymmetric induction in the synthesis of enantiomerically pure APs; (b) development of a general approach to the synthesis of γ-aminophosphonic acids (γ-APs) and (c) the use enantiomeric N-(p-tolylsulfinyl)cinnamaldimines in the synthesis of diverse α- and β-aminophosphonic acids (AP3, AP4) including the first synthesis of (R)-phosphoemeriamine.
Introduction
Aminoalkylphosphonic acids (APs) are analogs of natural and unnatural amino acids in which the planar carboxylic group is replaced by a tetrahedral phosphonic acid moiety (Scheme 1). Due to a wide spectrum of biological activity, this class of organophosphorus compounds has received considerable attention over the last decades [1]. Aminophosphonic acids (APs) and their conjugates with peptides exhibit antibacterial, anticancer, antiviral and antifungal activity. They are inhibitors of proteolytic enzymes and act as plant growth regulators. Therefore, some of them have found commercial application in agriculture and medicine. In spite of the fact that a lot of experimental material has been accumulated in the field of chemistry and biology of APs [1], [2], [3], [4], [5], [6], [7], [8], there is a continued interest in this class of P-compounds focused particularly on the synthesis and stereostructure-bioactivity studies of enantiomerically pure APs.

General structures of amino acids, aminophosphonic acids (APs) and sulfinimines.
As part of our long-standing program aimed at development of new synthetic methods using organic phosphorus and sulfur reagents, we devised a new asymmetric synthesis of α- and β-aminophosphonic acids using as the first the enantiopure sulfinimines as chiral reagents. A key reaction in this general synthesis of enantiomerically pure α- and β-APs is a highly or fully diastereoselective addition of trivalent phosphorus nucleophiles and α-phosphonate carbanions to sulfinimines. Separation of diastereoisomeric adducts and their subsequent acidic hydrolysis afforded the desired enantiomerically pure α- and β-APs. The synthesis of enantiomeric α-aminobenzylphosphonic acid 2 and β-amino-β-phenylethyl phosphonic acid 3 best illustrates the results of an early stage of our investigations in this field (Scheme 2).

Synthesis of enantiomerically pure α-aminobenzylphosphonic acid 2 and β-amino-β-phenylethylphosphonic acid 3.
It was found that the addition of dialkyl phosphite anions to (+)-(S)-1 resulted in the predominant formation of α-aminobenzylphosphonic acid (+)-2 with the (R)-configuration of the α-stereogenic carbon atom (Scheme 2a) [9]. With dialkyldiamido phosphite anions the steric course of the addition to (+)-(S)-1 was opposite and (S)-aminobenzylphosphonic acid (−)-2 was formed (Scheme 2b) [9].
The same stereochemistry of the addition of lithiated diaminophosphine borane complex to (+)-(S)-1 was observed and the corresponding adduct formed was directly converted into free α-aminophosphonic acid (−)-(S)-2 (Scheme 2c) [10]. It is interesting to point out that diastereoselectivity of the latter addition reaction was very high or complete and in many cases α-aminophosphonic acids were obtained after hydrolysis as pure enantiomers. Since at the beginning of our work the synthetic approaches to enantiomeric β-aminophosphonic acids (β-APs) were few in number and of limited applicability, their asymmetric synthesis mediated by chiral sulfinimines has been elaborated in the first place and reported as early as 1996 (Scheme 2d) [11]. These early results on the sulfinimine methodology for asymmetric synthesis of α- and β-aminophosphonic acids briefly mentioned above have been reviewed in 2005 [12].
It is worthy to notice that our sulfinimine methodology has been very soon applied by other research groups for the synthesis of diverse structures of enantiomeric α- and β-aminophosphonic acids [5], [6].
The present account will deal with a brief summary of our recent efforts to improve and extend the scope and applicability of our sulfinimine methodology for the synthesis of enantiomerically pure APs. At first we asked ourselves the following questions: (a) how can we achieve a full diastereoselectivity in the phosphite addition to chiral sulfinimines?, (b) can our asymmetric synthesis be extended to γ-amino- phosphonic acids?, (c) can we design a chiral sulfinimine structure suitable for asymmetric synthesis of structurally diverse aminophosphonic acids?
The results obtained in our laboratory aimed at solution to the above questions are reported below.
Double asymmetric induction in asymmetric synthesis of α-aminophosphonic acids
As mentioned above the most important step in asymmetric synthesis of α-APs is nucleophilic addition of phosphite anions to enantiomeric sulfinimines 1. In the majority of cases investigated in our and other laboratories such additions lead to separable mixtures of diastereoisomeric adducts which upon hydrolysis were converted into free enantiomerically pure α-APs (Scheme 2a). With regard to the diastereoselectivity of the addition reaction discussed here, a step forward has been made by the use of N-t-butylsulfinylimines which usually induce higher diastereoselectivities than those observed with N-p-tolylsulfinylimines [13]. Since for the stereostructure-bioactivity studies pure enantiomers of α-APs are required and in many cases the diastereoisomeric adducts could not be separated, we turned our attention to a double asymmetric induction in the hope to obtain directly diastereoisomerically pure adducts. In our case, this approach consists in the synthesis of both enantiomers of a sulfinimine as well as of a phosphite, and subsequent formation of four possible diastereoisomeric adducts. The utility of a double asymmetric induction was demonstrated in the synthesis of α-aminophosphonic acid 2 using (−)- and (+)-O,O -dimenthyl phosphites 4 and (+)- and (−)-N-(p-tolylsulfinyl)benzaldimines 1a as the addition partners (Scheme 3) [14].

Structures of enantiomeric O,O-dimenthyl phosphites 4 and sulfinimines 1a.
The results obtained are shown in Scheme 4. Thus, the addition of the lithium salt of (+)-dimenthyl phosphite 4 to the sulfinimine (−)-(R)-1a yielded the corresponding adduct (−)-5 as a single diastereoisomer with the configuration (RS, SC) as evidenced by its hydrolysis to the well-known α-aminophosphonic acid (−)-(S)-2. On the other hand, condensation of the lithium (−)-dimenthyl phosphite 4 with the sulfinimine (+)-(S)-1a yielded also the diastereoisomerically pure adduct (+)-5 which upon acidic hydrolysis gave aminophosphonic acid (+)-(R)-2. The two remaining addition reactions i.e. addition of (−)-4 to (−)-1a and (+)-4 to (+)-1a afforded the corresponding adducts 5 as diastereoisomeric mixtures. So, it was shown that the enantiomers (+)-4 and (−)-1a as well as (−)-4 and (+)-1a are so-called matched pairs whereas (−)-4 and (−)-1a and (+)-4 and (+)-1a are the mismatched pairs. In this way, we have demonstrated that the method of a double asymmetric induction allows to obtain both enantiomerically pure (−)-(S)- and (+)-(R)-aminobenzylphosphonic acids 2.

Synthesis of enantiomerically pure α-aminophosphonic acid 2 by a double asymmetric induction.
Asymmetric synthesis of γ-aminophosphonic acids
A successful use of chiral sulfinimines for asymmetric synthesis of enantiomerically enriched or pure α- and β-aminophosphonic acids prompted us to apply the same methodology for the synthesis of enantiomeric γ-aminophosphonic acids (γ-APs) [15], [16]. The most representative members of this class of APs are 2-amino-4-phosphonobutanoic acid 6 (AP4) and its phosphinic analog 7 commonly named phosphinotricin (Scheme 5). The first of them is a phosphonic analog of glutamic acid and acts as a modulator for the N-methyl-D-aspartate (NMDA) receptor site and the (S)-enantiomer is 20–24 times more active than the (R)-form in suppression of glutamate mediated neurotransmission. The aminophosphinic acid 7 shows antibacterial and herbicidal activity and is a potent inhibitor of glutamine synthetase.

Structures of γ-aminophosphonic acid 6 and γ-aminophosphinic acid 7.
A simple retrosynthetic analysis indicated that sulfinimine 8 would be the best chiral reagent for the synthesis of γ-APs since in this structure the imino-nitrogen atom is bonded to the γ-carbon atom of the phosphonate moiety, securing in this way the γ-aminophosphonic acid framework. The desired enantiomerically pure sulfinimine (+)-(S)-8 was easily prepared by condensation of 3-(diethoxy- phosphoryl)propanal with (+)-(S)-p-toluenesulfinamide in the presence of Ti(OEt)4 (eq. 1). The condensation product isolated in 80% yield was contaminated (up to 5%) with impurities which were difficult to remove by column chromatography. Therefore, as such it was used for addition reactions.
 (1)
(1)
With the main aim to accomplish the synthesis of (S)-6 (AP4), addition of the cyano group to the carbon-nitrogen double bond in (+)-(S)-8 was at first investigated. Among the reagent systems tested [(Et2AlCN/BF3.Et2O, Me3SiCN/CsF, (MeO)2C(Li)CN)] in the addition reaction to (+)-(S)-8, diethylcyanoaluminum in the presence of isopropanol, (Et2AlCN/i-PrOH), turned out to be the most efficient from the viewpoint of the reaction diastereoselectivity. The addition product 9 obtained was a mixture of the two unseparable diastereoisomers in a 9:1 ratio. For this reason it was subjected to hydrolysis under acidic conditions to afford (+)-(S)-2-amino-4-phosphonobutanoic acid 6. This two reaction sequence shown in Scheme 6 represents the shortest approach to our target.

Synthesis of γ-aminophosphonic acid (+)-(S)-6 from sulfinimine (+)-(S)-8.
The sulfinimine (+)-(S)-8 was found to be useful chiral reagent in the first synthesis of optically active aminoalkane-bis-phosphonic acids (Scheme 7). Thus, addition of lithium bis(diethylamino)phosphide-borane complex or α-phosphonate carbanion to (+)-(S)-8 results in the formation of the corresponding addition products 10a and 10b which upon acidic hydrolysis were converted into 1-amino-3-propylphosphonic acid 11a and 2-amino-4-phosphono-butylphosphonic acid 11b, respectively.
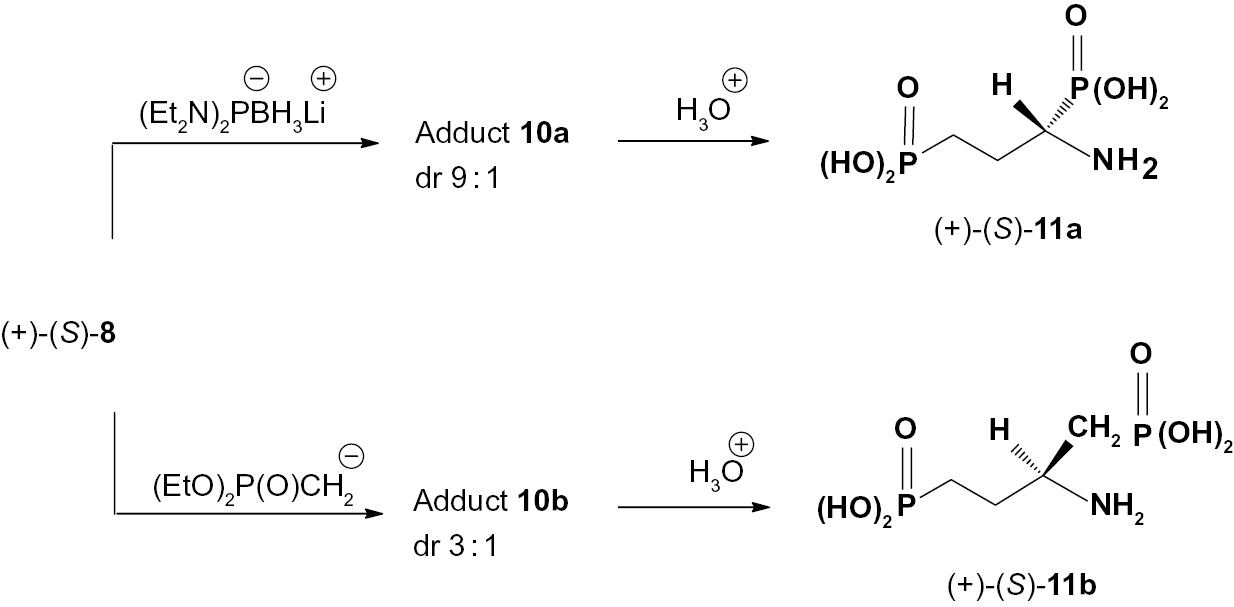
Asymmetric synthesis of amino-bis-(phosphonic acids) 11.
When in the structure of γ-aminophosphonic acid to be prepared there are one or more stereogenic atoms besides the carbon atom bearing the amino group, the synthetic strategy is different. At first, enantiomerically pure phosphono-aldehyde should be prepared, then converted into sulfinimine which after nucleophilic addition and subsequent hydrolysis of the addition product could afford a desired γ-aminophosphonic acid. This strategy has been employed by Midura and her group for asymmetric synthesis of (2S,1′S,2′R)-2-(2′-phosphonocyclopropyl)glycine 12 (Scheme 8) [17].

Asymmetric synthesis of conformationally constrained γ-amino- phosphonic acid 12.
Asymmetric synthesis of structurally diverse aminophosphonic acids using enantiomeric n-(p-tolylsulfinyl)cinnamaldimines as reagents
In continuation of our work on extension of the sulfinimine methodology for asymmetric synthesis of APs we turned our attention to chiral N-(p-tolylsulfinyl)cinnamaldimine 13 (Scheme 9). The presence of cinnamylidene moiety in the structure of 13 creates a nice possibility for its elaboration into various functional groups [18].

Structure of sulfinimine 13.
At the outset of this part of work the synthesis of enantiomerically pure unsaturated α-amino- and β-aminophosphonic acids was elaborated. To the best of our knowledge, this group of enantiopure APs was unknown. The synthesis of enantiomeric α-amino-β,γ-unsaturated phosphonic acids 14 is depicted in Scheme 10 and briefly discussed below.

Synthesis of enantiopure α-amino-β,γ-unsaturated phosphonic acids 14.
Addition of diethyl phosphite anion to sulfinimine (+)-(S)-13 gave the addition product as a mixture of two diastereoisomers in a 16:1 ratio. The major diastereoisomer was then isolated and hydrolyzed under acidic conditions to afford enantiomerically pure α-aminophosphonic acid (+)-(R)-14. With lithium bis(diethylamino)phosphide-borane complex as a nucleophile, the addition reaction to (+)-(S)-13 occurred with complete diastereoselectivity but with opposite stereochemical course affording (−)-(S)-14.
In a similar way, both enantiomers of β-amino-γ,δ-unsaturated phosphonic acids 15 were synthesized (Scheme 11). In this case, addition of diethyl lithiummethanephosphonate to both enantiomers of sulfinimine 13 was performed. The addition products were obtained as mixtures of diastereoisomers from which the major diastereoisomers were isolated and easily converted into enantiomeric aminophosphonic acids 15.

Synthesis of enantiopure β-amino-γ,δ-unsaturated phosphonic acids 15.
The synthesis of enantiomeric unsaturated α- and β-aminophosphonic acids 14 and 15 is of great importance because they or their precursors can be used as chiral building blocks for the synthesis of other functionalized aminophosphonic acids. This idea was put into practice and exemplified by the stereoselective synthesis of enantiomerically pure 2-amino-3-phosphonopropanoic acid 16 (AP3) and its 3-amino regioisomer 17. As shown in Scheme 12, the synthesis of (+)-(R)-16 was accomplished in three simple steps from the diastereoisomerically pure adduct 18 – a precursor of aminophosphonic acid (+)-(R)-15.

Synthesis of (+)-(R)-2-amino-3-phosphonopropanoic acid 16.
Similarly, starting from the diastereoisomerically pure adduct (+)-(SS,RC)-21 with the amino function at the α-carbon atom the synthesis of α-aminophosphonic acid 17 was accomplished in two steps only as shown in Scheme 13.

Synthesis of (−)-(R)-3-amino-3-phosphonopropanoic acid 17.
Looking for the new phosphonic analogs of biologically active amino acids, we found that the phosphonic analog of emeriamine is unknown. Emeriamine, called also aminocarnitine, inhibits fatty-acid oxidation and reduces hyperglycemia and ketosis. Therefore, in an extension of our earlier work on the synthesis phosphocarnitine [19], we decided to synthesize enantiomerically pure phosphoemeriamine 23. Retrosynthetic analysis for the synthesis of (R)-23 indicated that (R)-β-amino-γ-hydroxypropanephosphonate 19 would be the best starting material. It has been already obtained as intermediate product in the synthesis of (+)-(R)-16 (see Scheme 12). The first total synthesis of (−)-(R)-phosphoemeriamine 23 from (+)-19 is depicted in Scheme 14. It was accomplished in four simple steps (mesylation, amination, methylation and acidic hydrolysis) in 24% overall yield.

Stereoselective synthesis of (−)-(R)-phosphoemeriamine 23.
Further studies on the application of chiral sulfinimine 13 in the synthesis of other phosphonic analogs of biologically active amino acids are in progress in this laboratory.
Article note:
A collection of invited papers based on presentations at the 21st International Conference on Phosphorous Chemistry (ICPC-21) held in Kazan, Russia, 5–10 June 2016.
References
[1] V. P. Kukhar, H. Hudson. Aminophosphonic and Aminophosphinic Acids: Chemistry and Biological Activity. John Wiley, New York (2000).Search in Google Scholar
[2] M. Ordonez, J. L. Viveros-Ceballos, C. Cativiela, F. J. Sayago. Tetrahedron71, 1745 (2015).10.1016/j.tet.2015.01.029Search in Google Scholar
[3] A. Mucha. Molecules17, 13530 (2012).10.3390/molecules171113530Search in Google Scholar
[4] Z. H. Kudzin, M. H. Kudzin, J. Drabowicz, C. V. Stevens. Curr. Org. Chem.15, 2015 (2011).10.2174/138527211795703612Search in Google Scholar
[5] M. Ordonez, H. Rojas-Cabrera, C. Cativiela. Tetrahedron65, 17 (2009).10.1016/j.tet.2008.09.083Search in Google Scholar
[6] F. Palacios, C. Alonso, J. M. de los Santos. Chem. Rev.105, 899 (2005).10.1021/cr040672ySearch in Google Scholar
[7] F. Orsini, G. Sello, M. Sisti. Curr. Med. Chem.17, 264 (2010).10.2174/092986710790149729Search in Google Scholar
[8] V. D. Romanenko, M. Shevchuk, V. P. Kukhar. Curr. Org. Chem.15, 2774 (2011).10.2174/138527211796378505Search in Google Scholar
[9] M. Mikołajczyk, P. Łyżwa, J. Drabowicz. Tetrahedron: Asymmetry8, 3991 (1997).10.1016/S0957-4166(97)00606-XSearch in Google Scholar
[10] M. Mikołajczyk, P. Łyżwa, J. Drabowicz. Tetrahedron: Asymmetry13, 2571 (2002).10.1016/S0957-4166(02)00684-5Search in Google Scholar
[11] M. Mikołajczyk, P. Łyżwa, J. Drabowicz, M. W. Wieczorek, J. Błaszczyk. Chem. Commun. 1503 (1996).10.1039/CC9960001503Search in Google Scholar
[12] M. Mikołajczyk. J. Organomet. Chem.690, 2488 (2005).10.1016/j.jorganchem.2004.10.045Search in Google Scholar
[13] H. A. Khan, J. A. Ellman. Synthesis45, 3147 (2013) and references therein.10.1055/s-0033-1339712Search in Google Scholar
[14] P. Łyżwa. Heteroatom Chem.25, 15, (2014).10.1002/hc.21130Search in Google Scholar
[15] P. Łyżwa, M. Mikołajczyk. Pure Appl. Chem.82, 577 (2010).10.1351/PAC-CON-09-11-06Search in Google Scholar
[16] P. Łyżwa, M. Mikołajczyk. Heteroatom Chem.22, 594 (2011).10.1002/hc.20710Search in Google Scholar
[17] W. H Midura, J. Krysiak, A. Rzewnicka, A. Supeł, P. Łyżwa, A. M. Ewas. Tetrahedron69, 730 (2013).10.1016/j.tet.2012.10.085Search in Google Scholar
[18] P. Łyżwa, J. Błaszczyk, L. Sieroń, M. Mikołajczyk. Eur. J. Org. Chem. 2106 (2013).10.1002/ejoc.201201589Search in Google Scholar
[19] M. Mikołajczyk, J. Łuczak, P. Kiełbasiński. J. Org. Chem.67, 7872 (2002).10.1021/jo020330xSearch in Google Scholar PubMed
©2017 IUPAC & De Gruyter. This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License. For more information, please visit: http://creativecommons.org/licenses/by-nc-nd/4.0/
Articles in the same Issue
- Frontmatter
- In this issue
- Guest Editorial
- Research Papers from the 21st International Conference on Phosphorus Chemistry (ICPC-21)
- Conference papers
- Sterically protected organophosphorus compounds of unusual structures
- Estimation of the phosphorus loading with consideration for the planetary boundaries (for the Russian Federation as an example)
- Cyclic aminomethylphosphines as ligands. Rational design and unpredicted findings
- Eco-efficient electrocatalytic C–P bond formation
- Macrocyclic tetrakis-phosphines and their copper(I) complexes
- Strategies toward phosphorus-containing PAHs and the effect of P-substitution on the electronic properties
- Recent progress in asymmetric synthesis of aminophosphonic acids mediated by chiral sulfinyl auxiliary
- Exploring allene chemistry using phosphorus-based allenes as scaffolds
- Hydroxylated phosphines as ligands for chalcogenide clusters: self assembly, transformations and stabilization
- Polarity and structure of derivatives of bis(2-phenylethyl)selenophosphinic acid
Articles in the same Issue
- Frontmatter
- In this issue
- Guest Editorial
- Research Papers from the 21st International Conference on Phosphorus Chemistry (ICPC-21)
- Conference papers
- Sterically protected organophosphorus compounds of unusual structures
- Estimation of the phosphorus loading with consideration for the planetary boundaries (for the Russian Federation as an example)
- Cyclic aminomethylphosphines as ligands. Rational design and unpredicted findings
- Eco-efficient electrocatalytic C–P bond formation
- Macrocyclic tetrakis-phosphines and their copper(I) complexes
- Strategies toward phosphorus-containing PAHs and the effect of P-substitution on the electronic properties
- Recent progress in asymmetric synthesis of aminophosphonic acids mediated by chiral sulfinyl auxiliary
- Exploring allene chemistry using phosphorus-based allenes as scaffolds
- Hydroxylated phosphines as ligands for chalcogenide clusters: self assembly, transformations and stabilization
- Polarity and structure of derivatives of bis(2-phenylethyl)selenophosphinic acid