Modified macromolecules in the prevention of silica scale
-


Abstract
Silicic acid polycondensation leads to the formation of amorphous silica. This process is of great importance to the survival of certain living organisms, such as diatoms and sponges, but presents a significant problem in various production facilities that use water for heating or cooling. In the latter, amorphous silica can be a recalcitrant deposit that can hamper proper system operation. Hence, inhibition of silicic acid polycondensation by chemical inhibitors is an intensely sought strategy by water system operators. In this manuscript, we report the inhibitory effect of zwitterionic phosphonated analogs (PPEI’s) of the cationic polymeric chemical additive polyethyleneimine (PEI) in mildly supersaturated silica solutions (500 ppm/8.3 mM “Si”) at pH=7. The inhibition efficiency of PPEI’s depends on a variety of parameters, such as concentration and degree of phosphonomethylation of the parent PEI polymer.
Introduction
Water is the most common cooling medium used worldwide in industries because it is cost-effective, easily accessible in large scale and with high heat capacity [1]. Nevertheless, its reuse is essential for both economical and environmental reasons, especially in arid areas. During its reuse in cooling towers, some undesirable precipitates are formed, the identity of which depends on the particular water chemistry.
The most frequently-encountered ones are salts of calcium (carbonates [2], phosphates [3], sulfates [4]), barium/strontium (mostly sulfate [5]), magnesium (mostly silicate [6]), etc. However, there are deposits that are not “salts” and require special attention. Such an example is silicon dioxide or hydrated colloidal/amorphous silica (SiO2·nH2O, n is variable and dependent on hydration), the solubility of which is important to the operation of water-dominated production processes [7].
In arid areas such as Texas, New Mexico, Arizona, parts of California and southern Europe, the water used for industrial applications contains high silica concentrations (50–100 parts per million [ppm], expressed as silicon dioxide [SiO2]). These concentrations result from quartz (crystalline SiO2) and other silicate-based minerals dissolution from rock formations into the groundwater. The potential for silica-scale deposition poses serious problems in water with high dissolved silica content. Silica is an undesirable scale for several reasons including the blockage of heat transfer and the costly (and potentially hazardous) deposit removal techniques [8], [9], [10].
In order to chemically intervene and prevent these undesirable issues, it is important to take a closer look at the intricacies of silica chemistry, particularly its polycondensation processes. In dilute aqueous solutions, “soluble silica” is found as monosilicic acid Si(OH)4. This monomer exists in two forms in an about-neutral pH environment, protonated [major, Si(OH)4] and deprotonated [minor, Si(OH)3O−]. As pH increases, the concentration of the deprotonated form (Si(OH)3O−) increases. Increasing the silicate concentration and adjusting the pH value to ~7 leads to condensation of two monomers, with simultaneous loss of one water molecule. The procedure follows a SN2 – like mechanism (the terminology is borrowed from the organic chemistry substitution process), in which a deprotonated silicic acid molecule attacks a fully protonated one (see Fig. 1) [11].

SN2 – like mechanism of mono-silicic acid condensation to yield di-silicic acid.
The aforementioned step is the most crucial one as far as the kinetics of the polycondensation is concerned and is the rate-determining step in the complex silica polycondensation process [12]. The next steps involve the formation of trimers, tetramers and then higher oligomeric species until amorphous silica nanoparticles (1–2 nm) are formed [13].
It is worth-mentioning that the pKa of the polysilicic acids at these stages of the polymerization is ~6.5, so negatively charged silicate species become predominant in the pH regime 7–8 [14]. In these conditions, silicic acid on the surface of colloidal species exists in equilibrium with dissolved silicic acid molecules and, thus, further particle growth occurs via the Ostwald ripening process (see Fig. 2), leading to stable sols. In contrast, below pH 7, particles are only slightly charged so no electrostatic repulsion prevents them from aggregation, leading to gel formation [15], [16].
![Fig. 2:
Schematic presentation of the “Ostwald ripening” process [15].](/document/doi/10.1515/pac-2016-0807/asset/graphic/j_pac-2016-0807_fig_013.jpg)
Schematic presentation of the “Ostwald ripening” process [15].
The silicic acid polycondensation process and the physicochemical factors that influence it have been part of on-going investigations in our laboratory. The goal is to prevent silica formation, by stabilizing silicic acid, through the discovery and application of chemical scale inhibitors and additives [17], [18], [19], [20], [21], [22], [23], [24], [25], [26], [27], [28], [29], [30], [31], [32], [33].
We previously reported some preliminary results on the inhibitory activity of cationic polyethyleneimine (PEI, see Fig. 3) for silica control [27], [29]. The PEI polymeric system contains an excess of amine moieties as surface groups, from which ~25% are primary, ~50% are secondary and ~25% are tertiary amines. PEI showed a peculiar inhibition behavior. Specifically, PEI demonstrates inhibition at low (10 ppm) concentrations, but its inhibitory activity drops as its concentration increases. This phenomenon was very interesting and deserved further study. The most probable explanation is that because of its excessive cationic charge, the formed amorphous silica matrix entraps the PEI inhibitor and forms aggregates and clusters that precipitate out of solution. As a result, the inhibitor can no longer be available in the working solution, and is thus no longer available to perform polycondensation inhibition. PEI’s inhibitory capability was previously tested in various blends with anionic polymer additives in an effort to ease the entrapment phenomenon [34]. The working scenario was that the anionic polyelectrolyte would “neutralize” some of the excessive cationic charge on the PEI backbone, and would prevent, to a certain extent, inhibitor entrapment and deactivation.

Derivatization of cationic polyethyleneimine (PEI, left) to zwitterionic phosphonated polyethyleneimine (PPEI, right). Cationic (amine) moieties are highlighted in red, whereas anionic (phosphonate) ones in blue.
Inspired by our previous approaches, we pursued a PEI derivative that would combine both cationic and anionic moieties on the same polymer backbone, instead of the use of blends of PEI with anionic polyelectrolytes. Hence, in this paper we present the inhibitory effects of the zwitterionic polymeric additive phosphonomethylated polyethyleneimine (PPEI) (see Fig. 3), on the formation of amorphous silica. PPEI is the phosphonomethylation derivative of PEI and can be synthetically accessed via the well-known Mannich-type (also known as Irani) process [35], [36], [37], [38], [39], [40]. By varying the reagent ratios, various phosphonomethylation degrees can be achieved, from as low as 8% (mole percent) to the fully substituted analog. The Mannich-type reaction has been extensively utilized to synthesize numerous amino-methylenephosphonic acids that have been used in the synthesis of metal-organic framework and hybrid materials and coordination networks [41].
As shown in Fig. 3, every primary amine is converted to an amino-bis(methylenephosphonic acid) moiety, every secondary amine is converted to an amino-methylenephosphonic acid group, however, the tertiary amines remain unreactive. These chemical modifications on the PEI backbone result in the formation of zwitterionic polymers with various degrees of anionic groups and are ideal to evaluate them in structure/activity studies, related to silica scale inhibition. Similar zwitterionic polymers, e.g. phosphonomethylated chitosan, have been used in our group as silica scale inhibitors [42].
Experimental section
Reagents and chemicals
Sodium silicate pentahydrate Na2SiO3·5H2O was purchased from Sigma-Aldrich. Ammonium molybdate ((NH4)6Mo7O24·4H2O) was obtained from Alfa-Aesar and oxalic acid (H2C2O4·2H2O) from EM Science (Merck). Sodium hydroxide (NaOH) and hydrochloric acid (HCl) 37% were purchased from Sigma-Aldrich. Deionized water from an ion-exchange resin was used for all experiments and stock preparations. This water was tested for molybdate-reactive silica (interference tests) and was found to contain negligible amounts.
Solution preparation
Sodium silicate stock solution: A solution containing silicate (500 ppm as SiO2) was prepared by dissolving 4.08 g of Na2SiO3·5H2O in 2 L of deionized water (a non-glass container must be used to avoid silicate leaching from glass), followed by overnight rigorous stirring.
Inhibitor stock solutions: Stock solutions of the additives in water (PPEI in various phosphonated forms) were 1% w/v (10 000 ppm).
Reagent solutions for the silicomolybdate method. The following solutions were prepared for the silicate spectrophotometric detection test: (a) An ammonium molybdate solution: 10 g of ammonium molybdate was dissolved in 100 mL of water, and its pH was adjusted between 7 and 8 with NaOH to avoid precipitation of ammonium molybdate. (b) A hydrochloric acid solution: one volume 37% HCl was mixed with equal volume water. (c) An oxalic acid solution: 8.75 g of oxalic acid was dissolved in 100 mL of water. All solutions were kept in polyethylene containers. HCl and ammonium molybdate were kept in the fridge, while silica and oxalic acid solution were kept at room temperature.
Silicic acid polycondensation protocol (“control” experiment)
Hundred milliliter from the 500 ppm sodium silicate stock solution (see above) was placed in a polyethylene beaker and the initial pH was found 11.8. The pH is then adjusted to 7.00±0.1 by addition of HCl and/or NaOH, as needed (the change in the resulting volume was minor and did not affect any of the calculations). Then, the beaker was covered with plastic membrane and set aside without stirring. The solutions were checked for molybdate-reactive silica by the silicomolybdate method every 1 h for the first 8 h or after 24, 48, and 72 h after the pH adjustment (see below).
Silicic acid polycondensation in the presence of PPEI inhibitors
Hundred milliliter portions of the 500 ppm sodium silicate stock solution (see above) were placed in polyethylene containers. In each container, different volumes of PPEI (from the prepared 10 000 ppm stock solutions) were added to achieve desirable concentration. After that, the same procedure as the “control” protocol was followed.
Determination of molybdate-reactive silica (silicomolybdate method)
Molybdate-reactive silica (mainly mono- and some disilicic acid) was quantified using the well-established silicomolybdate spectrophotometric method [43]. As in our previous studies, we used the “yellow molybdate” method (using Spectrophotometer HACH DR/890,) as follows: 2 mL from the working solution is filtered through a 0.45 μm syringe filter and then diluted to 25 mL in a special cylindrical cell of 1 cm path length, made of quartz. Next, 1 mL of ammonium molybdate stock solution and 0.5 mL of HCl (1:1 dilution of the concentrated solution) are added to the sample cell, the solution is shaken well and left standing for 10 min. Afterward 1 mL of oxalic acid solution is added and the cell contents are mixed well. The solution is set aside for 2 min. The photometer is now set to “zero absorbance” using a sample of deionized water (“blank”). Finally, the sample absorbance is measured (at 452 nm) and is expressed as “ppm SiO2”. The detectable concentration range for this specific protocol is 6–75 ppm. To calculate the concentration in the original solution, an appropriate dilution factor (×27.5/2) is applied. The basic working principal of the silicomolybdate test is that ammonium molybdate reacts only with mono- and disilicic acid and any phosphate present and forms yellow-colored complexes (see Fig. 4).
![Fig. 4:
Structure of the yellow silicomolybdic acid cage-like cluster. The silicon atom (gray sphere at the center) is encaged within 12 MoO6 octahedra (oxygen atoms in white) [44].](/document/doi/10.1515/pac-2016-0807/asset/graphic/j_pac-2016-0807_fig_015.jpg)
Structure of the yellow silicomolybdic acid cage-like cluster. The silicon atom (gray sphere at the center) is encaged within 12 MoO6 octahedra (oxygen atoms in white) [44].
This reaction requires acidic environment in order to take place, and this is why the hydrochloric acid is added to the samples. It should be noted that colloidal silica does not participate in the reaction and thus does not affect the intensity of yellow color, which is proportional to the concentration of the reactive silica present in the sample experiment. Oxalic acid is added to destroy any molybdophosphoric acid formed, leaving the silicomolybdate complex intact, and thus eliminating any color interference from phosphates.
Results and discussion
Inhibitory activity of PPEI derivatives
PPEI was tested in “long-term” (up to 3 days) and “short term” (8 h) experiments. Long term experiments are an important time frame for scale inhibitors, as it evaluates the ability of a certain inhibitor to prevent scale formation for a prolonged time period. Short term experiments examine the silica polycondensation in its early stages.
At first, PPEI (full, i.e. 100% phosphonomethylation degree, in mole percent) was tested as a silica scale inhibitor in long term (3 days) experiments at various concentrations (20–80 ppm), see Table 1 and Fig. 5. Results obtained with PEI as the inhibitor are also presented for comparison in Table 1 and Fig. 5 (upper). PEI, due to its high positive charge density is not active at concentrations above 40 ppm, whereas, at 20 ppm dosage, it enhances silicic acid solubility by only 53 ppm above the control. By introducing the phosphonate moieties, inhibitory activity is dramatically increased in PPEI, see Fig. 5, lower. This is particularly evident at inhibitor dosages >40 ppm. Specifically, at 60 ppm dosage PPI (full) is able to enhance silicic acid solubility more than 200 ppm above the control after 24 h. This behavior of PPEI may be explained by the “relief” the anionic phosphonate groups exert on the cationic backbone. By balancing the excess cationic charge, they most likely prevent inhibitor entrapment in the forming silica matrix.
Silica inhibition measurements involving the PPEI (full) and, for comparison, the parent PEI in long-term (3-day) experiments.
| Time (h) | Control | PPEI (full) |
|||
|---|---|---|---|---|---|
| 20 ppm | 40 ppm | 60 ppm | 80 ppm | ||
| 24 | 171 | 242 | 363 | 385 | 373 |
| 48 | 143 | 166 | 230 | 264 | 314 |
| 72 | 136 | 152 | 184 | 220 | 263 |
|
PEI
|
|||||
| 24 | 171 | 224 | 216 | 182 | 186 |
| 48 | 143 | 193 | 179 | 164 | 164 |
| 72 | 136 | 186 | 173 | 165 | 157 |
The numbers are expressed in “ppm as SiO2”.

Silica inhibition by PEI (upper) and PPEI (full, lower) in long-term (3-day) experiments.
The results presented in Table 1 and Fig. 5 establish that by grafting anionic phosphonate moieties onto the PEI backbone, thus creating the zwitterionic PPEI polymer, profoundly increases four-fold the inhibitory activity. However, this is true for the fully grafted PPEI (100% phosphonomethylation). Hence, in order to evaluate the effect of the extent of phosphonometylation on inhibitory activity, a series of partially grafted PPEI’s were studied. For direct comparison reasons, the 60 ppm dosage was studied for all inhibitors. These results are shown in Table 2 and Fig. 6.
Silica inhibition measurements at a constant PPEI concentration (60 ppm) in all phosphonated forms (0, 40, 60, 80, 100%) in long-term experiments.
| Time (h) | Control | PPEI phosphonomethylation degree (%) |
||||
|---|---|---|---|---|---|---|
| 0 | 40 | 60 | 80 | 100 | ||
| 24 | 171 | 182 | 229 | 245 | 283 | 373 |
| 48 | 143 | 164 | 158 | 169 | 176 | 314 |
| 72 | 136 | 165 | 147 | 141 | 139 | 263 |
The numbers are expressed in “ppm as SiO2”.

Effect of PPEI% phosphonomethylation degree on silicic acid inhibitory activity of all PPEI forms synthesized, at 60 ppm PPEI concentration, in long-term (3 days) experiments. Upper: 3-day results. Lower: linear dependence of the 24-h measurements.
There appears to be an almost linear dependence of inhibitory activity on % grafting degree. The maximum inhibition performance is achieved when PPEI is fully (100%) substituted. Specifically, it stabilizes 202 ppm of silicic acid higher than the control after 24 h. Its inhibitory activity drops after 48 and 72 h, as is expected from our past experience of silica inhibitors, but still PPEI 100% is able to stabilize 171 ppm and 127 ppm, respectively above the control (Fig. 6 upper).
The concentration dependence of the PPEI (40, 60, 80, 100%) inhibitors was also studied, and the results are shown in Fig. 7. In all grafting degrees inhibitory activity increases as the PPEI concentration increases. For the grafting degrees 40, 60, and 80%, the enhancement is very mild as concentration increases from 0 to 20, to 40 ppm. In contrast, the same concentration increase for PPEI 100% causes a drastic enhancement in silicic acid stabilization. All PPEI inhibitors reach a plateau after the 60 ppm concentration.

Dependence of inhibitory activity on concentration. Measurements include those after the first 24 h.
Based on the above-mentioned “long-term” (24 h) results, we decided to further study the silica inhibition event at its earlier stages, i.e. during the first 8 h. For this purpose, we selected all phosphonated forms of PPEI (40, 60, 80 and 100%), which were tested in short – term measurements at 60 ppm concentration. These results are presented in Table 3 and plotted in Fig. 8. The same dependence of inhibitory performance on % grafting was noted, as with the long-term experiments. Specifically, as the grafting degree increases, silicic acid stabilization ability increases. Again the fully substituted PPEI is the most efficient inhibitor. It can stabilize 400 ppm (246 ppm above the control) silicic acid after 8 h.
Silica inhibition measurements with PPEI 40%, 60%, 80% and 100% (full) at a constant concentration 60 ppm in short-term experiments (8 h).
| Time (h) | Control | PPEI (40%) | PPEI (60%) | PPEI (80%) | PPEI (100%) |
|---|---|---|---|---|---|
| 0 | 500 | 500 | 500 | 500 | 500 |
| 1 | 349 | 369 | 381 | 403 | 455 |
| 2 | 271 | 297 | 315 | 368 | 432 |
| 3 | 227 | 249 | 269 | 341 | 416 |
| 4 | 204 | 231 | 254 | 324 | 411 |
| 5 | 184 | 209 | 235 | 301 | 410 |
| 6 | 171 | 201 | 227 | 281 | 406 |
| 7 | 160 | 193 | 220 | 268 | 404 |
| 8 | 157 | 181 | 218 | 259 | 403 |
The numbers are expressed in “ppm as SiO2”.
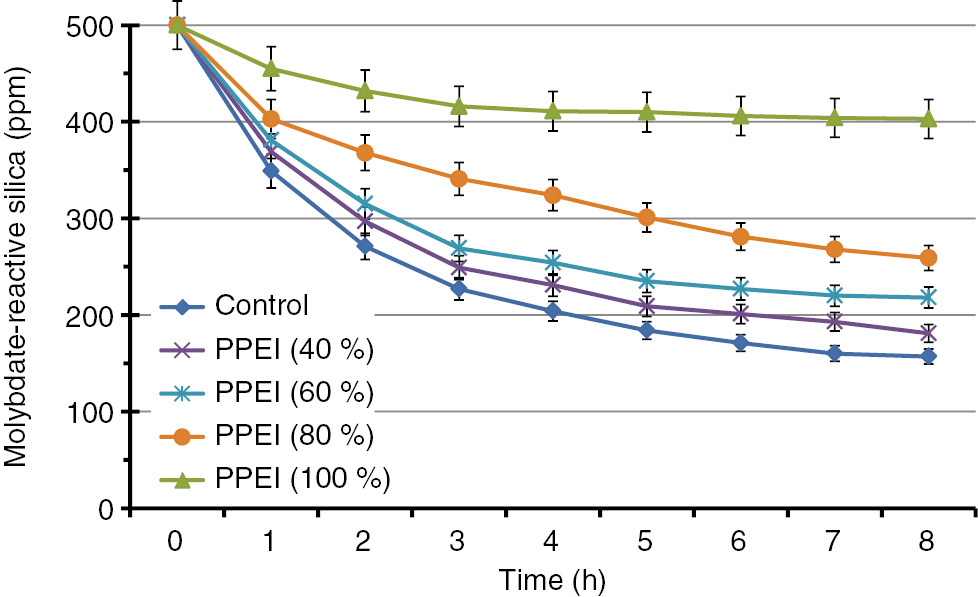
Silica inhibition measurements with PPEI 8%, 40%, 60%, 80% and 100% (full) at a constant concentration 60 ppm in short-term experiments (8 h).
Characterization of precipitated silica
Although PPEI derivatives are efficient inhibitors of silica polycondensation, there is always some colloidal silica formed. This appears as a white flocculant precipitate in the working solutions. Its appearance and severity is variable and depends on the actual PPEI form and is a result of the competition of two concomitant processes: the silica condensation and the inhibition by PPEI. As shown in Fig. 9, as the phosphonomethylation% degree increases, the solution turbidity decreases. This is reasonably rationalized by the excessive cationic charge of the PPEI at lower substitution degrees, that causes flocculation of the formed silica particles at pH=7. It is well established that the pKa of surface silanols decreases as the silica polyconcensation degree increases [45]. In other words, the silanol groups on a silica particle are much more acidic (pKa~4) than silicic acid (pKa~9.3) [46]. Hence, cationically charged macromolecules at appropriate concentrations can act as flocculants in silica containing solutions, and, thus, create silica-inhibitor precipitates through electrostatic interactions and hydrogen bonding. This has been observed before [19], [21], [29]. However, when the negatively-charged phosphonate groups are introduced, the cationic charge density is lowered, hence, the tendency to generate silica-inhibitor precipitates is eased.

Visual demonstration of the turbidity results at 60 ppm inhibitor concentration at pH=7.0, after 8 h, at 25°C. 1: PPEI 8%, 2: PPEI 40%, 3: PPEI 80%, 4: 100%.
The visual observations presented in Fig. 9 were quantified by turbidity measurements taken over the course of 8 h at pH 7, shown on Fig. 10. Increase of the phosphonomethylation degree on the PPEI backbone has two significant effects: (a) the PPEI inhibitor becomes more efficient (see Figs. 5 and 6), and (b) the tendency of formation of silica-PPEI precipitates is substantially reduced.

Evolution of turbidity over time in solutions containing PPEI inhibitors with various phosphonomethylation degrees. All PPEI inhibitors are at 60 ppm concentration.
Selected silica precipitates were studied by scanning electron micrpscopy (SEM), in order to evaluate morphological effects of the PPEI inhibitors on the silica particle morphology and texture. The SEM images are shown in Fig. 11. SEM images were recorded on precipitates formed at pH 7.0, after 8 h and after 3 days of polycondensation time, in the presence of various concentrations of fully phosphonomethylated (100%) PPEI, as indicated. The solids appear as aggregates of smaller, nearly spherical primary particles of submicron size.

SEM images of silica precipitates in the absence (control) and presence of PPEI inhibitors.
Conclusion
In this paper we discussed the use of zwitterionic phosphonomethylated polyethyleneimine (PPEI) in the inhibition of silicic acid polycondensation. The use of PPEI was inspired from previous research from our group that demonstrated that the combination of cationic, amine-based polymeric inhibitors and anionic polyelectrolytes are beneficial in silica inhibition. However, PPEI combines these two attributes on the same polymeric backbone.
The results showed that phosphonate grafting on the initial cationic PEI polymeric chain causes enhancement of inhibitory activity by “relieving” the cationic charge of the parent polymer PEI. Hence, the polymeric inhibitor is not entrapped in the forming colloidal silica matrix, a problem severely exhibited by the parent PEI. Furthermore, it appears that the more extensive the phosphonate grafting is on PEI, the higher the inhibitory activity is of the PPEI modified polymer.
Research on the effect of polyamines [30], [47] and related macromolecules such as polysaccharides [48], [49], [50] on (bio)silica formation and control is an active area of research, due to the fact that these biomacromolecules are intimately involved in biosilicification processes in diatoms, sponges and plants.
Article note
A collection of invited papers based on presentations at the 16th International Conference on Polymers and Organic Chemistry (POC-16), Hersonissos (near Heraklion), Crete, Greece, 13–16 June 2016.
Acknowledgments
KDD thanks the EU for funding the Research Program SILICAMPS-153, under the ERA.NET-RUS Pilot Joint Call for Collaborative S&T projects.
References
[1] K. D. Demadis. in Compact Heat Exchangers and Enhancement Technology for the Process Industries, R. K. Shah (Ed.), p. 483, Begell House Inc., New York (2003).Search in Google Scholar
[2] K. D. Demadis, S. D. Katarachia. Phosphorus Sulfur Silicon179, 627 (2004).10.1080/10426500490441514Search in Google Scholar
[3] K. D. Demadis, B. Yang, P. R. Young, D. L. Kouznetsov, D. G. Kelley. in Advances in Crystal Growth Inhibition Technologies, Z. Amjad (Ed.), p. 215, Ch. 16, Plenum Press, New York (2000).Search in Google Scholar
[4] E. Akyol, M. Öner, E. Barouda, K. D. Demadis. Cryst. Growth Des.9, 5145 (2009).10.1021/cg9005423Search in Google Scholar
[5] E. Barouda, K. D. Demadis, S. Freeman, F. Jones, M. I. Ogden. Cryst. Growth Des.7, 321 (2007).10.1021/cg0604172Search in Google Scholar
[6] K. D. Demadis, A. Ketsetzi, E.-M. Sarigiannidou. Ind. Eng. Chem. Res.51, 9032 (2012).10.1021/ie3010836Search in Google Scholar
[7] H. Ehrlich, K. D. Demadis, P. G. Koutsoukos, O. Pokrovsky. Chem. Rev.110, 4656 (2010).10.1021/cr900334ySearch in Google Scholar PubMed
[8] K. D. Demadis, E. Mavredaki, M. Somara. Ind. Eng. Chem. Res.50, 12587 (2011).10.1021/ie201703bSearch in Google Scholar
[9] K. D. Demadis, E. Mavredaki, M. Somara. Ind. Eng. Chem. Res.50, 13866 (2011).10.1021/ie201798eSearch in Google Scholar
[10] K. D. Demadis, M. Somara, E. Mavredaki. Ind. Eng. Chem. Res.51, 2952 (2012).10.1021/ie202806mSearch in Google Scholar
[11] K. D. Demadis, J. Chem. Technol. Biotechnol.80, 630 (2005).10.1002/jctb.1242Search in Google Scholar
[12] T. Tarutani. Anal. Sci.5, 245 (1989).10.2116/analsci.5.245Search in Google Scholar
[13] K. Spinde, K. Pachis, I. Antonakaki, E. Brunner, K. D. Demadis. Chem. Mater.23, 4676 (2011).10.1021/cm201988gSearch in Google Scholar
[14] M. Preari, K. Spinde, J. Lazic, E. Brunner, K. D. Demadis. J. Am. Chem. Soc.136, 4236 (2014).10.1021/ja411822sSearch in Google Scholar PubMed
[15] C. C. Perry, T. Keeling-Tucker. J. Inorg. Biochem.69, 181, (1998).10.1016/S0162-0134(97)10017-4Search in Google Scholar
[16] B. A. Fleming. J. Coll. Interf. Sci.110, 40 (1986).10.1016/0021-9797(86)90351-6Search in Google Scholar
[17] K. D. Demadis, M. Öner. in Green Chemistry Research Trends, J. T. Pearlman (Ed.), p. 265, Ch. 8, Nova Science Publishers, New York (2009).Search in Google Scholar
[18] K. D. Demadis, E. Neofotistou, E. Mavredaki, M. Tsiknakis, E.-M. Sarigiannidou, S. D. Katarachia. Desalination179, 281 (2005).10.1016/j.desal.2004.11.074Search in Google Scholar
[19] K. D. Demadis, E. Neofotistou. Chem. Mater.19, 581 (2007).10.1021/cm062370dSearch in Google Scholar
[20] K. D. Demadis, E. Neofotistou. Int. J. Corros. Scale Inhib. 4, 95 (2015).Search in Google Scholar
[21] K. D. Demadis, E. Neofotistou. Mater. Perform.43, 38 (2004).Search in Google Scholar
[22] E. Neofotistou, K. D. Demadis. Coll. Surf. A: Physicochem. Eng. Asp.242, 213 (2004).10.1016/j.colsurfa.2004.04.067Search in Google Scholar
[23] E. Neofotistou, K. D. Demadis. Desalination167, 257 (2004).10.1016/j.desal.2004.06.135Search in Google Scholar
[24] K. D. Demadis. Power148(6), 19 (2004).Search in Google Scholar
[25] E. Mavredaki, E. Neofotistou, K. D. Demadis. Ind. Eng. Chem. Res.44, 7019 (2005).10.1021/ie0501982Search in Google Scholar
[26] K. D. Demadis, E. Mavredaki. Env. Chem. Lett.3, 127 (2005).10.1007/s10311-005-0015-0Search in Google Scholar
[27] K. D. Demadis, A. Stathoulopoulou. Mater. Perform.45, 40 (2005).Search in Google Scholar
[28] K. D. Demadis, E. Mavredaki, A. Stathoulopoulou, E. Neofotistou, C. Mantzaridis. Desalination213, 38 (2007).10.1016/j.desal.2006.01.042Search in Google Scholar
[29] K. D. Demadis, A. Stathoulopoulou. Ind. Eng. Chem. Res.45, 4436 (2006).10.1021/ie0602254Search in Google Scholar
[30] K. D. Demadis, S. Brückner, E. Brunner, S. Paasch, I. Antonakaki, M. Casolaro. Chem. Mater.27, 6827 (2015).10.1021/acs.chemmater.5b03100Search in Google Scholar
[31] K. D. Demadis, M. Preari, I. Antonakaki. Pure Appl. Chem.86, 1663 (2014).10.1515/pac-2014-0705Search in Google Scholar
[32] K. D. Demadis, M. Preari. Des. Wat. Treat.55, 749 (2015).10.1080/19443994.2014.927803Search in Google Scholar
[33] A. Spinthaki, A. Stathoulopoulou, K. D. Demadis. Int. J. Corros. Scale Inhib. 4, 125 (2015).10.17675/2305-6894-2015-4-1-125-138Search in Google Scholar
[34] K. D. Demadis, K. Pachis, A. Ketsetzi, A. Stathoulopoulou. Adv. Coll. Interf. Sci.151, 33 (2009).10.1016/j.cis.2009.07.005Search in Google Scholar PubMed
[35] O. Abderrahim, M. A. Didi, B. Moreau, D. Villemin. Solv. Extr. Ion Exch.24, 943 (2006).10.1080/07366290600952519Search in Google Scholar
[36] O. Abderrahim, M. A. Didi, D. Villemin. Anal. Lett.42, 1233 (2009).10.1080/00032710902901723Search in Google Scholar
[37] O. Abderrahim, M. A. Didi, D. Villemin. J. Radioanal. Nucl. Chem.279, 237 (2009).10.1007/s10967-007-7270-zSearch in Google Scholar
[38] R. R. Navarro, K. Tatsumi. Sep. Sci. Technol.37, 203 (2002).10.1081/SS-120000329Search in Google Scholar
[39] R. R. Navarro, S. Wada, K. Tatsumi. J. Hazard. Mater.123, 203 (2005).10.1016/j.jhazmat.2005.03.048Search in Google Scholar PubMed
[40] R. R. Navarro, S. Wada, K. Tatsumi. Sep. Sci. Technol.38, 2327 (2003).10.1081/SS-120021627Search in Google Scholar
[41] A. Clearfield, K. D. Demadis. Metal phosphonate chemistry: From synthesis to applications. Royal Society of Chemistry, London (2012).10.1039/9781849733571Search in Google Scholar
[42] K. D. Demadis, A. Ketsetzi, K. Pachis, V. M. Ramos. Biomacromolecules9, 3288 (2008).10.1021/bm800872nSearch in Google Scholar PubMed
[43] A. D. Eaton, L. S. Clesceri, E. W. Rice, S. E. Greenberg, M. H. Franson. Standard Methods for Examination of Water and Wastewater, American Public Health Association: Washington, DC (2005).Search in Google Scholar
[44] M. Feist, V. N. Molchanov, L. P. Kazanskii, E. A. Torchenkova, V. I. Spitsyn. Zh. Neorg. Khim.25, 733 (1980).Search in Google Scholar
[45] L. T. Zhuravlev. Coll. Surf. A: Physicochem. Eng. Asp.173, 1 (2000).Search in Google Scholar
[46] T. Coradin, D. Eglin, J. Livage. Spectroscopy18, 567 (2004).10.1155/2004/356207Search in Google Scholar
[47] A. Jantschke, K. Spinde, E. Brunner. Beilstein J. Nanotechnol.5, 2026 (2014).10.3762/bjnano.5.211Search in Google Scholar PubMed PubMed Central
[48] H. Ehrlich. in Encyclopedia of Geobiology, J. Reitner, V. Thiel (Eds.), p. 796, Springer Verlag, Heidelberg (2011).Search in Google Scholar
[49] H. C. W. Skinner, H. Ehrlich. in Treatise on Geochemistry, Vol. 10: Biogeochemistry, 2nd ed., K. K. Turekian, H. D. Holland (Eds.), p. 105, Elsevier Science, Amsterdam (2013).10.1016/B978-0-08-095975-7.00804-4Search in Google Scholar
[50] E. Brunner, P. Richthammer, H. Ehrlich, S. Paasch, P. Simon, S. Ueberlein, K.-H. van Pee. Angew. Chem. Int. Ed.48, 9724 (2009).10.1002/anie.200905028Search in Google Scholar PubMed
©2016 IUPAC & De Gruyter. This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License. For more information, please visit: http://creativecommons.org/licenses/by-nc-nd/4.0/
Articles in the same Issue
- Frontmatter
- In this issue
- Editorial
- The 2016 Nobel Prize in Chemistry
- Conference papers
- Cyclotriphosphazene, an old compound applied to the synthesis of smart dendrimers with tailored properties
- Milestones in microwave-assisted organophosphorus chemistry
- Synthetic routes to polyphosphoesters as solid polymer electrolytes for lithium ion batteries
- Soluble polymer supports for homogeneous catalysis in flow reactions
- Box-Behnken experimental design for the production of precipitated calcium carbonate
- Synthesis and characterisation of lignin-like oligomers as a bio-inspired consolidant for waterlogged archaeological wood
- Synthesis, characterizations and Pb(II) sorption properties of cobalt phosphonate materials
- Performance of poly(styrene-co-divinylbenzene) functionalized with different aminophosphonate pendant groups, in the removal of phenolic compounds from aqueous solutions
- Synthesis of new dithia[3.3]parapara- and metapara-cyclophane based tectons: toward an universal surface-confined 2D/3D molecular binding motif
- Effects of surface modification and ultrasonic agitation on the properties of PHBV/ZnO nanocomposites
- Modified macromolecules in the prevention of silica scale
- Ionizing radiation: a versatile tool for nanostructuring of polymers
- Non-classical effects in proton or hydrogen transfer
- IUPAC Recommendations
- Source-based nomenclature for single-strand homopolymers and copolymers (IUPAC Recommendations 2016)
Articles in the same Issue
- Frontmatter
- In this issue
- Editorial
- The 2016 Nobel Prize in Chemistry
- Conference papers
- Cyclotriphosphazene, an old compound applied to the synthesis of smart dendrimers with tailored properties
- Milestones in microwave-assisted organophosphorus chemistry
- Synthetic routes to polyphosphoesters as solid polymer electrolytes for lithium ion batteries
- Soluble polymer supports for homogeneous catalysis in flow reactions
- Box-Behnken experimental design for the production of precipitated calcium carbonate
- Synthesis and characterisation of lignin-like oligomers as a bio-inspired consolidant for waterlogged archaeological wood
- Synthesis, characterizations and Pb(II) sorption properties of cobalt phosphonate materials
- Performance of poly(styrene-co-divinylbenzene) functionalized with different aminophosphonate pendant groups, in the removal of phenolic compounds from aqueous solutions
- Synthesis of new dithia[3.3]parapara- and metapara-cyclophane based tectons: toward an universal surface-confined 2D/3D molecular binding motif
- Effects of surface modification and ultrasonic agitation on the properties of PHBV/ZnO nanocomposites
- Modified macromolecules in the prevention of silica scale
- Ionizing radiation: a versatile tool for nanostructuring of polymers
- Non-classical effects in proton or hydrogen transfer
- IUPAC Recommendations
- Source-based nomenclature for single-strand homopolymers and copolymers (IUPAC Recommendations 2016)