Arylxanthones and arylacridones: a synthetic overview
-
Clementina M. M. Santos


Abstract
Arylxanthones and arylacridones although not yet found in nature are becoming an important group of heterocyclic compounds due to their promising biological activities. Their central cores, xanthone and acridone, are recognized as interesting motifs for drug development mainly to be used in antitumour chemotherapy. The synthesis of this type of compounds is still scarce but several successful examples were recently published and a large variety of arylated xanthone and acridone derivatives were prepared. A systematic survey of the literature dedicated to their synthesis will be presented and discussed in this review.
Introduction
Xanthone (9H-xanthen-9-one) derivatives (Fig. 1), natural or synthetic, comprise a group of structurally diverse biologically active oxygen heterocyclic compounds. Many derivatives exhibited noticeable biological properties, from which antitumor effects can be highlighted [1], [2]. This family has been studied for over a century and several reviews, covering their natural sources [3], [4], [5], biological activities [6], chemical synthesis [7], [8] and even biosynthesis [1], [9], were published. Nonetheless, the field of xanthone chemistry is growing rapidly, due to several update publications, it is noteworthy that arylxanthones are not the focus of these articles. There are no evidences that arylxanthones occur in nature but some synthetic derivatives already proved their importance in medicinal chemistry research [10], [11], [12].
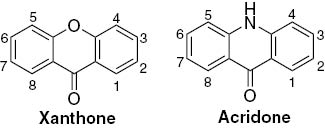
Xanthone and acridone monomer cores with numbering.
Acridone (acridin-9(10H)-one) derivatives (Fig. 1), the xanthones aza-analogues, are also an important group of natural bioactive compounds [13]. Both natural and synthetic derivatives are recognized as possible chemotherapeutic agents [14], [15]. These derivatives are less studied but some synthetic methodologies were developed and recently reviewed [13], [14]. Likewise for the above mentioned xanthones, arylacridones are not found in nature and only a few papers reporting their synthesis can be found in literature.
This review pinpoints the latest progress in the synthesis of arylxanthones and arylacridones, mainly mono- and diaryl derivatives. Developments for the synthesis of more intricate examples will be also disclosed. Through this review the structural numbering will follow the monomeric parent xanthone and acridone cores depicted in Fig. 1.
Synthesis of arylxanthones
The ubiquitous 9H-xanthen-9-one core occurs in a large number of naturally occurring and synthetic compounds and several studies highlight their biological and pharmacological importance [1], [2], [16]. As far as we know, 9H-xanthen-9-ones linked to an aryl group are scarce and only a few synthetic derivatives have been reported. Herein, we will review the synthesis of monoarylated structures at positions 1, 2 or 3 of the 9H-xanthen-9-one scaffold, 2,3-diaryl-9H-xanthen-9-ones and other complex related analogues.
Synthesis of monoaryl-9H-xanthen-9-ones
A series of 1-aryl-9H-xanthen-9-ones have been prepared starting from 2′-hydroxy acetophenones 1, in a multi-step sequence [17], [18] (Scheme 1). It involves acetylation of 2′-hydroxyacetophenones 1, base-catalyzed Baker–Venkataraman rearrangement to afford 1,3-diketones 3 and cyclodehydration using a catalytic amount of p-toluenesulfonic acid (p-TSA) to give the corresponding 2-methyl-4H-chromen-4-ones 4. The condensation of 2-methyl-4H-chromen-4-ones 4 with cinnamaldehydes 5 carried out in the presence of sodium ethoxide (generated in situ) at room temperature provides 2-[(1E,3E)-4-arylbuta-1,3-dien-1-yl]-4H-chromen-4-ones 6 in good yields (62–80%). The parent cinnamaldehyde was available while the 4-substituted cinnamaldehydes 5 were prepared from the reaction of the appropriate halobenzenes 7 with acrolein diacetal using palladium acetate as catalyst in DMF at 90°C [17]. The synthesis of (E)-3-(3,4-dimethoxyphenyl)acrylaldehyde involved a two-step strategy starting with an acid-catalyzed iodination of 1,2-dimethoxybenzene to prepare the corresponding iodobenzene 7 followed by the palladium(II)-catalyzed reaction described before for the synthesis of cinnamaldehydes 5 [18]. 2-[(1E,3E)-4-Arylbuta-1,3-dien-1-yl]-4H-chromen-4-ones 6 undergo subsequent electrocyclization and oxidation reactions in the presence of a catalytic amount of iodine in refluxing 1,2,4-trichlorobenzene (1,2,4-TCB) to obtain the target 1-aryl-9H-xanthen-9-ones 8 (35–70%). From this reaction it was also possible to isolate some semi-oxidized intermediates of the final xanthones, 1-aryl-1,4-dihydro-9H-xanthen-9-ones 9. In order to prepare hydroxylated derivatives, demethylation of methoxyxanthones 8 was attained by their treatment with boron tribromide in anhydrous dichloromethane. Unfortunately, deprotection of 6-OMe xanthone did not occur and to overcome this drawback methoxyethoxymethyl (MEM) was used instead of methyl protecting group, in a similar procedure described in Scheme 1 [18]. Hydroxylated 1-aryl-9H-xanthen-9-ones 10 were shown to be remarkable scavengers of reactive oxygen species (ROS) and reactive nitrogen species (RNS), in non-cellular systems [18].
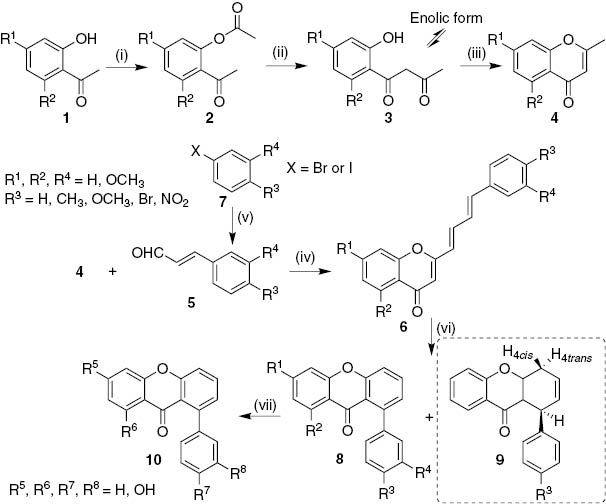
Reagents and conditions: (i) MeCOCl, pyridine, r.t., 2 h; (ii) NaH, THF, reflux, 2 h; (iii) p-TSA, DMSO, 100°C, 2 h; (iv) Na, EtOH, r.t., 12 h; (v) acrolein diethylacetal, Bu4NOAc, K2CO3, Pd(OAc)2, DMF, 90°C, 4 h; (vi) I2, 1,2,4-TCB, reflux, 48 h; (vii) BBr3, dry CH2Cl2, –78°C to r.t., 3–4 h.
A reference to 2-phenylxanthone appeared in a European Patent and later also as US Patent [19] describing a novel crystalline aromatic polyketone copolymer and the process for producing the same. In this invention, aromatic ketones such as xanthone and 2-phenylxanthone were used as polymerization solvents, although no reference to their synthesis was presented.
The synthesis of parnafungin A and C 11, hexacyclic antifungal agents bearing a xanthone unit, involved Suzuki–Miyaura coupling reaction of substituted 2-iodo-9H-xanthen-9-ones 12 with an excess of 3-carbomethoxy-2-nitrophenyl pinacol boronate as the first key step. The resulting 2-(3-carbomethoxy-2-nitrophenyl)-9H-xanthen-9-ones 13 were obtained in good yields (53–60%) using the system Pd(OAc)2/Sphos in a 3:1 mixture of THF/H2O (Scheme 2) [20].
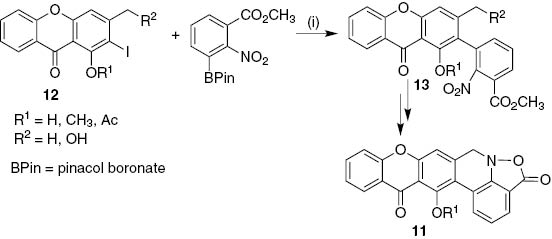
Reagents and conditions: (i) Pd(OAc)2/Sphos, K3PO4, THF/H2O (3:1), 80°C, 1–2 h.
[4+2] Cycloaddition reaction of 2-styrylchromone derivatives 14 with 2-pyrrolidinoprop-1-ene 15, generated in situ from acetone and a catalytic amount of pyrrolidine, provided a wide variety of 3-aryl-1-methyl-9H-xanthen-9-ones 16 (56–76% yield, Scheme 3) [21]. It was also possible to isolate a couple of 3-aryl-1-methylidene-1,2,3,4-tetrahydro-9H-xanthen-9-ones 17 as minor products, which after treatment with a mixture of acetic acid/sulfuric acid yielded the corresponding 3-aryl-1-methyl-9H-xanthen-9-ones 16. The mechanism proposed for this [4+2] inverse electron demand cycloaddition reaction involves the formation of tetrahydroxanthones 17 and subsequent migration of the exocyclic double bond and dehydrogenation of the C-ring to afford the fully aromatized 1-methyl-9H-xanthen-9-ones 16. Meanwhile, cycloaddition reaction with the pyrrolidine enamine of butan-2-one 18 led to 3-aryl-2-methyl-1-methylidene-1,2,3,4-tetrahydro-9H-xanthen-9-ones 19 as major products (36–55%) with a small amount of 3-aryl-1-ethyl-9H-xanthen-9-ones 20 (13–16% yield, Scheme 3). These results indicate that two isomers were produced during enamine formation with butan-2-one, 2-pyrrolidinobut-2-ene as major product and 2-pyrrolidinobut-1-ene as minor component. Moreover, the synthesis of 2-methyl-3-aryl- 21 and 2-ethyl-3-heteraryl- 22 -9H-xanthen-9-ones was only possible when the pyrrolidine group is at the end of the alkyl chain as in aldehydes (propanal and butanal, respectively) rather than ketones (Scheme 3). Treating 2-methyl-1-methylidene-3-phenyl-1,2,3,4-tetrahydro-9H-xanthen-9-one with strong acid led to the corresponding 1,2-dimethyl-3-phenyl-9H-xanthen-9-one 23 in almost quantitative yield (Scheme 3) [21].
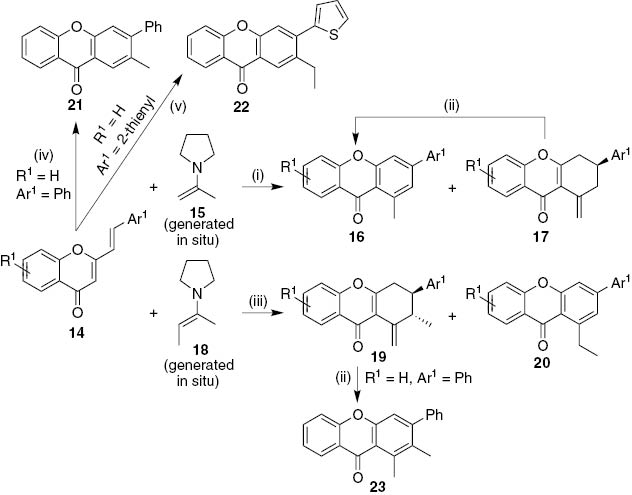
Reagents and conditions: (i) refluxing acetone, 2–28 h; (ii) AcOH/H2SO4, 100°C, 3 h; (iii) refluxing butan-2-one, 70 min-3 days; (iv) refluxing propanal, drop of pyrrolidine, 4 days; (v) refluxing butanal, drop of pyrrolidine, 4 days.
Synthesis of 2,3-diaryl-9H-xanthen-9-ones
Two different approaches were reported for the synthesis of 2,3-diaryl-9H-xanthen-9-ones. The first one uses 3-bromo-2-methyl-4H-chromen-4-one 24 as starting material, obtained through bromination, with bromine in ethanol, and acidic cyclization of 1,3-diketones 3, in a one-pot process (Scheme 4) [22]. Aldol condensation of 3-bromo-2-methyl-4H-chromen-4-one 24 with benzaldehydes 25 (the reaction only occurred using parent benzaldehyde and 4-benzyloxybenzaldehyde) in the presence of sodium in methanol at room temperature afforded 3-bromo-2-styryl-4H-chromen-4-ones 26, which undergo Heck reaction with styrenes 27 to produce 2,3-diaryl-9H-xanthen-9-ones 28 as major products (25–66%) and the semi-oxidized xanthones 29 as minor products (6–27%). The mechanism proposed involves Heck reaction of brominated chromones with styrenes to form 2,3-distyrylchromones followed by in situ electrocyclization, [1,5]-sigmatropic hydrogen migration to give 3,4-dihydroxanthones and oxidation reactions to achieve the corresponding 2,3-diaryl-9H-xanthen-9-ones [22]. On the other hand, Heck reaction of 3-bromo-2-methyl-4H-chromen-4-one 24 with styrenes 27 led to 2-methyl-3-styryl-4H-chromen-4-ones 30 (48–52%). Subsequent aldol condensation was only possible with parent benzaldehyde 25 to form 2,3-distyryl-4H-chromen-4-ones 31 (53–69%), which in refluxing 1,2,4-TCB gave the desired 2,3-diaryl-9H-xanthen-9-ones 28 (80–85%) [23].
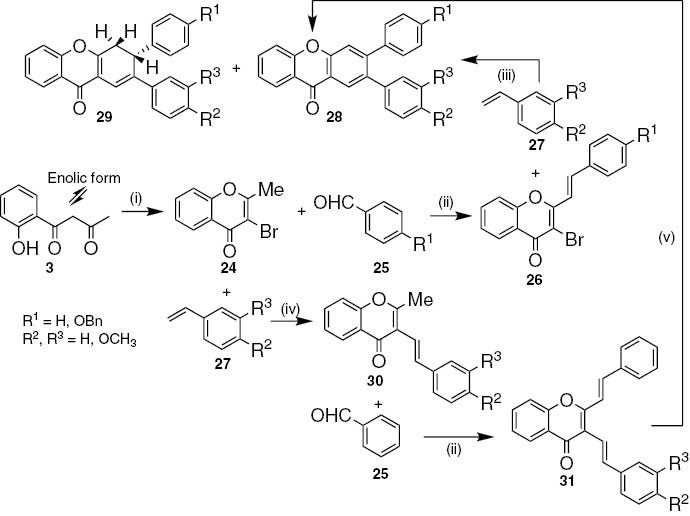
Reagents and conditions: (i) 1. Br2, EtOH, r.t., 2 h; 2. HCl, reflux, 2 h; (ii) Na, MeOH, r.t., 48 h; (iii) Pd(PPh3)4, PPh3, Et3N, NMP, 160°C to reflux; (iv) PdCl2, PPh3, Et3N, NMP, 160°C, 9 h; (v) 1,2,4-TCB, reflux, 18 h.
Due to the lack of reactivity of certain derivatives, a second and more general approach was developed to the synthesis of 2,3-diaryl-9H-xanthen-9-ones, starting from 2′-hydroxy acetophenones 1 and cinnamic acid derivatives (Scheme 5) [24]. The resulting 2-acetylphenyl cinnamates 32 (50–97%) in the presence of potassium hydroxide in DMSO via Baker–Venkataraman rearrangement afforded 5-aryl-3-hydroxy-1-(2-hydroxyaryl)penta-2,4-dien-1-ones 33 in good yields (54–95%). Phenyltrimethylammonium tribromide (PTT) in THF at room temperature promoted bromination and cyclization of these ketones to attain 3-bromo-2-styryl-4H-chromen-4-ones 26 (30–97%) [24]. A great number of derivatives were obtained from the Heck reaction of several 3-bromo-2-styryl-4H-chromen-4-ones 26 with styrenes 27 carried out in the presence of tetrakis(triphenylphosphine)palladium(0), triphenylphosphine, triethylamine and 1-methyl-2-pyrrolidin-1-one (NMP) as solvent: the desired 2,3-diaryl-9H-xanthen-9-ones 28 as major products (13–80%), 2,3-diaryl-3,4-dihydro-9H-xanthen-9-one intermediates 29 (3–32%) and also 6,7-diaryl-1-hydroxy-9H-xanthen-9-ones 34 (1–37%) when 3-bromo-5-methoxy-2-styrylchromones were used as brominated derivatives (Scheme 5) [23].
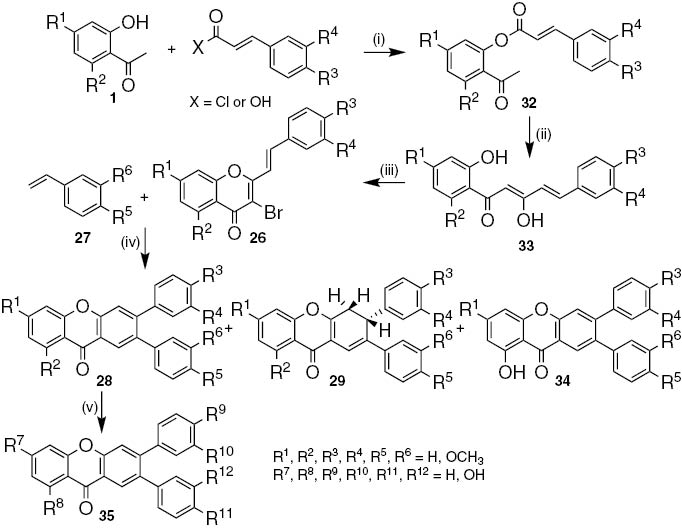
Reagents and conditions: (i) X=Cl, pyridine, r.t., 2 h; X=OH, POCl3, pyridine, 60°C, 2 h; (ii) KOH, DMSO, r.t., 2 h; (iii) PTT, THF, r.t., 12 h; (iv) Pd(PPh3)4, PPh3, Et3N, NMP, 160°C to reflux; (v) BBr3, dry CH2Cl2, –78°C to r.t..
A series of hydroxylated 2,3-diaryl-9H-xanthen-9-ones 35 were prepared in moderated to good yields (37–94%) after demethylation of 2,3-diaryl-9H-xanthen-9-ones 28 with boron tribromide in anhydrous dichloromethane [23], [24]. Some of these hydroxylated 2,3-diaryl-9H-xanthen-9-ones were tested for their ROS and RNS scavenging properties and were considered promising antioxidant agents taking into account the nanomolar to micromolar range of the IC50 values found [23].
Synthesis of arylbenzoxanthones
Benzoxanthones are a small class of xanthones with no significant representation in nature and only a few publications on their synthesis and/or biological activities have been reported in the last two decades. Nevertheless the most recent publications indicate that these tetracyclic ring systems can be potential lead compounds in the development of new antitumour agents [25], [26]. To the best of our knowledge arylbenzoxanthones are not found in natural sources and their synthesis is restricted [27], [28].
Four types of arylbenzoxanthones, 6-arylbenzo[c]xanthones 38, 5-arylbenzo[c]xanthones 39, 6-arylbenzo[b]xanthones 40 and 6-arylbenzo[a]xanthones 41, were obtained as minor products in the oxidation of 3-aryl-1,2,3,4-tetrahydro-2-naphthyl 2-hydroxyphenyl ketones 36 with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) under microwave irradiation (MW) conditions (Scheme 6) [27]. In addition to the low yields obtained in this reaction (1–17%), selectivity is also very low being the nitro substituent selective towards the 6-arylbenzo[c]xanthones 38 whereas the methoxy group was selective towards the 6-arylbenzo[b]xanthones 40. The reaction mechanism [27] seems to indicate that the methoxy group favors a 1,2-aryl shift, which is consistent with its electron-releasing effect.
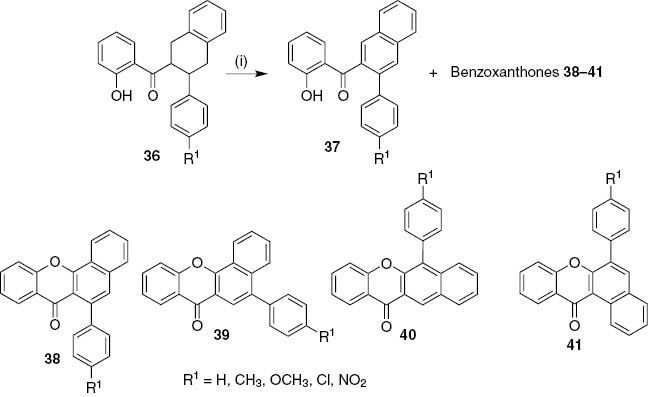
Reagents and conditions: (i) DDQ, 1,2,4-TCB, MW (800 W).
Later our research group has also developed a new approach to obtain 5-arylbenzo[c]xanthones involving the photoinduced electrocyclization and oxidation of (E)-3-styrylflavones 47 (see below Scheme 9) [28]. Two strategies to synthesize the required (E)-3-styrylflavones 47 were established. The first one uses the Heck reaction of 3-bromoflavones 46, obtained through previous established procedures, with styrenes 27 (Scheme 7) [28]. 2′-Hydroxyacetophenones 1, benzoic acids or benzoyl chlorides 42, were used to prepare 2-acetylaryl benzoates 43. When using benzoic acids the catalytic system N,N′-dicyclohexylcarbodiimide (DCC) and 4-pyrrolidinopyridine (4-ppy) was used. In both conditions the benzoates 43 were obtained in very good yields (above 70%). These esters were afterwards treated with base, potassium hydroxide or sodium hydride, to perform the Baker–Venkataraman rearrangement and obtain a tautomeric mixture of 3-aryl-1-(2-hydroxyphenyl)propane-1,3-diones 44/(Z)-3-aryl-3-hydroxy-1-(2-hydroxyphenyl)prop-2-en-1-ones 45 in moderate to good yields (above 40%). The use of sodium hydride was essential to perform the rearrangement in good yields for the polymethoxylated derivatives. Treating these ketones with PTT in THF at room temperature promotes bromination at their carbon C-2 and cyclization into 3-bromoflavones 46. The yields were generally moderate although the presence of several methoxy substituents lowers the yields to values around 30%.
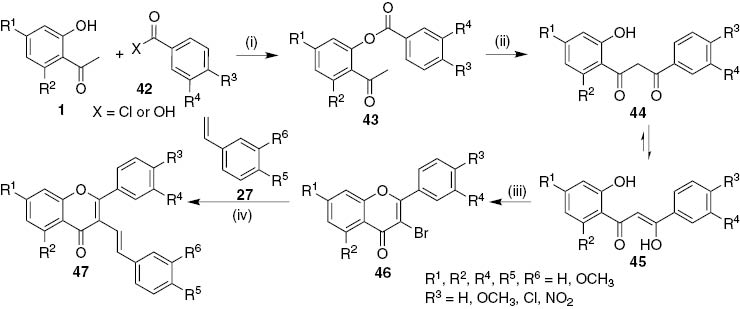
Reagents and conditions: (i) X=Cl, dry pyridine, r.t., 24 h; X=OH, DCC, 4-ppy, CH2Cl2, r.t., 5 days; (ii) KOH, DMSO, r.t., 4 h or NaH, dry THF, reflux, 4 h; (iii) PTT, dry THF, r.t., 24–48 h; (iv) Pd(OAc)2, K2CO3, Bu4NBr, DMF, MW (300 W), 5–10 min.
Diastereoselective synthesis of (E)-3-styrylflavones 47 were obtained by the Heck/Jeffery reaction conditions of 3-bromoflavones 46 with styrenes 27 under MW irradiation, using palladium acetate, potassium carbonate, tetrabutylammonium bromide (TBAB) and DMF as solvent [28]. Although the (E)-3-styrylflavones 47 were the major products in several cases the yields were poor due to the formation of by-products such as the corresponding flavone derivatives. This was most evident in the case of polymethoxylated derivatives.
The second approach involves the synthesis of 3-methylflavones 49 and their transformation into 3-(bromotriphenylphosphoranyl)methylflavones 51 which were afterwards used in the diastereoselective synthesis of (E)-3-styrylflavones 47 through Wittig reactions (Scheme 8) [29]. The key intermediates in this procedure 3-methylflavones 49 were prepared, in very good yields (above 74%) or moderate for the polymethoxylated derivatives (around 30%), through a one-pot tandem reaction of 1-(2-hydroxyphenyl)propan-1-ones 48 with benzoyl chlorides 42, using lithium bis(trimethylsilyl)amide (LiHMDS) as base [29], [30]. The selective bromination of 3-methylflavones 49 occurred with N-bromosuccinimide (NBS), benzoyl peroxide and using CCl4 as solvent. The presence of other methyl groups or several methoxy substituents can interfere and lower the yield of the desired 3-bromomethylflavones 50. The subsequent transformation of 50 into 3-(bromotriphenylphosphoranyl)methylflavones 51 was accomplished by their reaction with triphenylphosphine in toluene. Finally, Wittig reaction was successfully employed in the synthesis of (E)-3-styrylflavones 47 using 3-(bromotriphenylphosphoranyl)methylflavones 51 and benzaldehydes 52 (Scheme 8).
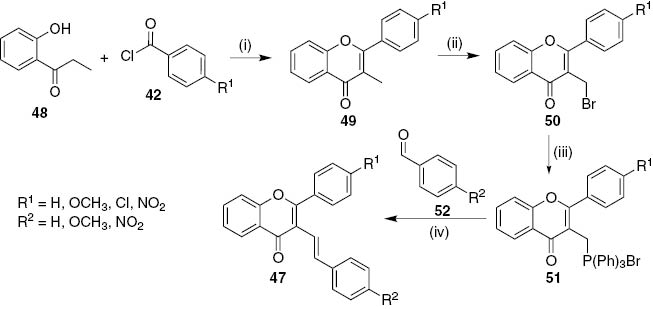
Reagents and conditions: (i) LiHMDS, dry toluene, r.t., 37 h; (ii) NBS, benzoyl peroxide, CCl4, reflux, 7 h; (iii) PPh3, dry toluene, reflux, 24–43 h; (iv) NaH, dry THF, r.t., 2.5 h.
The one-pot photoinduced electrocyclization and oxidation of (E)-3-styrylflavones 47 gave 5-aryl-7H-benzo[c]xanthen-7-ones 39 (Scheme 9) [28]. The procedure presents few drawbacks such as long reaction times and, when several methoxy substituents are present, yields are lower than 30%. Long reaction times are essential to obtain the xanthone derivatives, otherwise the oxidation step into 5-aryl-7H-benzo[c]xanthen-7-ones 39 did not occur and 5-aryl-5,6-dihydro-7H-benzo[c]xanthen-7-ones 53 were obtained (Scheme 9). Furthermore the presence of the nitro electron-withdrawing group leads to the formation of the 6-hydroxy-3-nitro-5-phenyl-7H-benzo[c]xanthen-7-one 54, as the main product (Scheme 9). Considering the proposed reaction mechanism [28], the presence of the nitro group should improve the acidic character of the α-protons allowing the formation of a diene which undergoes cycloaddition reaction with singlet oxygen. This cycloadduct can successively, rearrange, dehydrate and, through a keto-enol tautomerism, aromatize to give 6-hydroxy-3-nitro-5-phenyl-7H-benzo[c]xanthen-7-one 54.
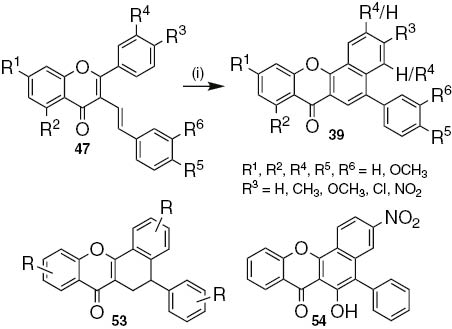
Reagents and conditions: (i) 1,2,4-TCB, I2 (cat.), hυ (high pressure mercury lamp), 3–7 days.
Synthesis of miscellaneous arylxanthones
3-(1,6-Diynyl)-4H-chromen-4-ones 55 underwent microwave-assisted tandem reaction using potassium tert-butoxide, tetrabutylammonium chloride and phenylacetonitrile as anion reagent transfer in DMSO to deliver tetracyclic 2-aryl-9H-xanthen-9-ones 56 (68–80% yield, Scheme 10) [31]. This transformation involves ring-opening and ring-closing tandem process, [4+2] cyclization and aromatization reactions. A single example of 1,2-diphenyl-9H-xanthen-9-one derivative was obtained in 28% yield when chromone possesses a 2-phenyl substituent.
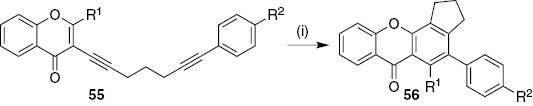
Reagents and conditions: (i) Bu4NCl, t-BuOK, phenylacetonitrile, DMSO, MW, 110°C, 20 min.
Although 2-styrylchromones 14 were first used as dienes in Diels–Alder (DA) reactions, the reactivity in [4+2] cycloaddition reactions of their isomers (Z)-3-styrylchromones 57 was also studied. Dienophiles such as N-methylmaleimide (58, X=NCH3), N-phenylmaleimide (58, X=NPh) and 2-(2-nitrovinyl)thiophene 61 were used and their DA reaction with (Z)-3-styrylchromones 57 gave several xanthone type compounds (Scheme 11) [32], [33]. The (Z)-3-styrylchromone derivatives 57 attained in previous works [34] undergo MW-assisted solvent-free DA reaction with maleimides 58 affording the respective cycloadducts, rel-(3aR,4S,11aR,11bR)-4-aryl-2-(methyl or phenyl)-3a,4,11a,11b-tetrahydrochromeno[2,3-e]isoindole-1,3,6(2H)-triones 59 in very good yields (>75%, Scheme 11). The authors tried the same transformation using classic heating conditions, but the results were not improved (59 X=NCH3, up to 51%) and other by-products such as 4-aryl-2-methylchromeno[2,3-e]isoindole-1,3,6(2H)-triones 60 (40–47%), were formed [32], [33]. These xanthone derivatives 60 were also attained in very good yields using DDQ as oxidant in 1,2,4-TCB under MW irradiation. The use of 2-(2-nitrovinyl)thiophene 61 as dienophile allowed the direct synthesis of 2-aryl-3-(thiophen-2-yl)-9H-xanthen-9-one 62, which results from the DA reaction followed by in situ oxidation due to the favoured HNO2 elimination (Scheme 11) [32]. Moreover, the reaction is regioselective as only 2-aryl-3-(thiophen-2-yl)-9H-xanthen-9-ones 62 were obtained in very good yields (68–75%). This regioselectivity is explained by orbital coefficients calculation that revealed the partially positively charged carbon atom in the dienophile and the most negatively charged carbon in the diene.
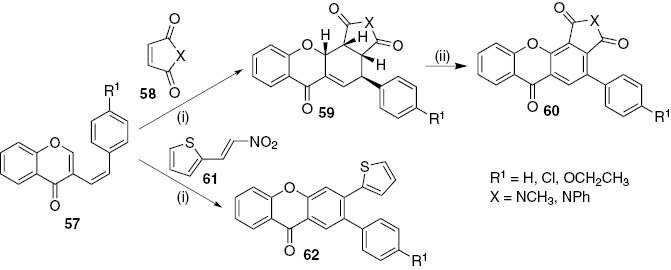
Reagents and conditions: (i) MW (270 W), 30 min; (ii) DDQ, 1,2,4-TCB, MW (300 W), 45 min.
The stereochemistry observed in the obtained cycloadducts 59 confirms the DA reaction stereoselectivity towards the endo isomer. Theoretical calculations regarding the regio- and stereoselectivity of the reaction explain the experimental results [32]. The extension of the cycloaddition reaction using N-methylmaleimide (58, X=NCH3) and N-phenylmaleimide (58, X=NPh) as dienophiles with (E)-3-styrylchromone derivatives 63 afforded in a stereoselective manner, the exo cycloadducts rel-(3aR,4S,11aS,11bR)-4-aryl-2-(methyl/phenyl)-3a,4,11a,11b-tetrahydrochromeno[2,3-e]isoindole-1,3,6(2H)-triones 64 (Scheme 12) [32], [33].
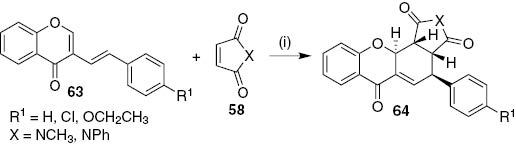
Reagents and conditions: (i) MW (270 W), 30 min.
Various examples of 3-aryl-1,2,3,9a-tetrahydro-9H-xanthen-9-ones are known since 1954 when 2-styrylchromone derivatives 14 undergo DA reaction with maleic anhydride (58, X=O) in boiling xylene (Scheme 13) [35]. More derivatives arise when 2 years later the same group tested in the same reaction conditions the reactivity with N-arylmaleimides (58, X=NAr2) as dienophiles [36]. Cycloaddition reaction of 2-styrylchromones with dibenzoylethylenes [37] and 1,4-benzoquinones [38], [39] to give 1,2,3,9a-tetrahydro-9H-xanthen-9-ones were also reported. In 1992, Letcher and Yue revised the cycloadduct structures 65 to the isomeric 1,2,3,4-tetrahydro-9H-xanthen-9-ones 66 [40], [41]. They also studied the reactivity of 2-styrylchromone derivatives 14 with electron rich dienophile enamine 1-pyrrolidinylcyclopentene 67, in equimolar amounts, in refluxing 95% ethanol (Scheme 13) [42]. The trans-stereochemistry of the cycloadducts obtained indicated that the DA reaction occurred via an exo-addition and the corresponding 1,2,3,4-tetrahydro-9H-xanthen-9-one derivatives 68 were obtained in 50–60% yield.
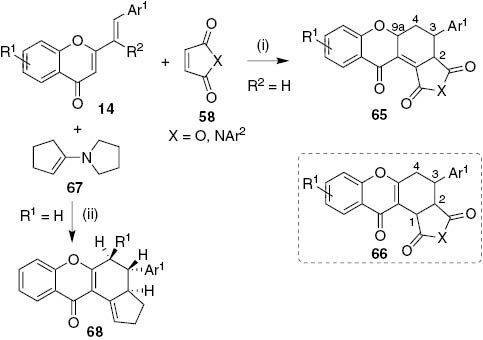
Reagents and conditions: (i) boiling xylene; (ii) refluxing 95% EtOH.
During a study of methyl groups cleavage of 3-cinnamoyl-2-styrylchromones 69 with boron tribromide very interesting (E)-3-aryl-4-arylidene-8-hydroxy-3,4-dihydro-1H-xanthene-1,9(2H)-dione derivatives 70 were obtained in moderate to very good yields (37–89%, Scheme 14) [43]. The attempts to oxidize xanthenediones 70 were not fruitful due to the formation of several xanthone and tetracyclic xanthone derivatives, the yields were not improved and the reaction was not reproducible [44]. From this work one can also highlight the high antioxidant and the potent acetylcholinesterase inhibition demonstrated for (E)-3-aryl-4-arylidene-8-hydroxy-3,4-dihydro-1H-xanthene-1,9(2H)-dione (70, R1=H, R2=R3=OH) [44].
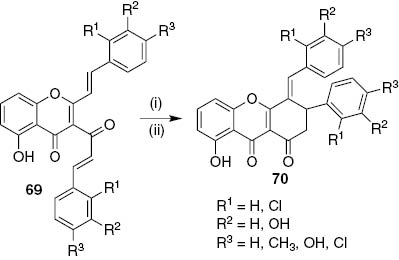
Reagents and conditions: (i) BBr3, dry CH2Cl2, r.t.; (ii) H2O, r.t..
Synthesis of arylacridones
Acridin-9(10H)-ones, also called dibenzo-γ-pyridones, are tricyclic compounds presenting a γ-pyridone with two ortho-fused benzene rings and are the aza-analogues of 9H-xanthen-9-ones. Several naturally occurring and synthetic acridin-9(10H)-ones are known, the natural ones are found predominantly in plants of Rutaceae family [45], and their importance is recognized due to their biomedical potential mainly as antiviral [46], antimalarial [47], antitumoral and anticancer agents [13]. Moreover acridin-9(10H)-ones have important applications in host-guest interactions, as fluorescence probes in chemical, biochemical and environmental analysis and as analytical tools in biomimetic chemistry [48], [49], [50]. To the best of our knowledge, synthetic acridin-9(10H)-ones linked to one or more aryl rings are scarce and natural derivatives were not yet reported. However, there are synthetic acridin-9(10H)-ones unrelated to the natural products now emerging as promising bioactive compounds. Herein we report the recent studies on the synthesis of 1-arylacridin-9(10H)-ones and 2,3-diarylacridin-9(10H)-ones and other related analogues.
Synthesis of 1-arylacridones
Palladium(0)-mediated Suzuki–Miyaura cross-coupling reaction of 3,6-disubstituted-10-methylacridin-9(10H)-ones 71, bearing a 1-bromo or 1-trifluoromethylsulfonyloxy substituent, with 2-(pivaloylamino)benzeneboronic acids 72 afforded 1-arylacridin-9(10H)-ones 73 in very good yields (Scheme 15) [51]. The reaction conditions depend on the acridin-9(10H)-one used as coupling partner. Thus, the reaction of bromoacridin-9(10H)-one 71 (X=Br) with 2-(pivaloylamino)benzeneboronic acid 72 (R3=H), using Pd(PPh3)4 as catalyst in aqueous DME with sodium bicarbonate (3 equiv) as base gave 1-arylacridin-9(10H)-one 73 (R1=R2=R3=H) in very good yield (88%). Treating 73 with HCl (6 mol·dm−3) in THF, or hot POCl3, led to pentacycle 74 in yields higher than 90%. The coupling of triflates 71 (X=SO2CF3) with boronic acids is more challenging to avoid triflate hydrolysis. In the best conditions involving Pd(PPh3)4, NaHCO3 in DME with minimal water, 1-arylacridin-9(10H)-ones 73 (R1=OCH3, R2=R3=H, Cl) were obtained but cyclize directly to pentacyclic quinoacridines 74. Other conditions such as Pd2(dba)3–P(t-Bu)3–KF or Pd(OAc)2–PCy3–KF, either gave no coupling, very limited conversions, or hydrolysis. To facilitate purification, a resin bound catalyst (Deloxan resin) under aqueous conditions was used but once again the hydrolysis of triflate groups occurred [51].
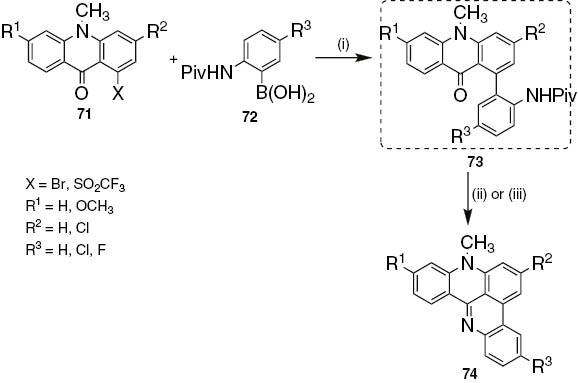
Reagents and conditions: (i) Pd(PPh3)4, NaHCO3, DME, H2O, 100°C, 5 h; (ii) 6 mol⋅dm−3 HCl in THF (1:1 or 1:1.5), reflux, 18 h to 5 days; (iii) POCl3, 100°C, 20 min.
Later, it was reported the Suzuki–Miyaura cross-coupling reaction of bromoacridin-9(10H)-one 75 with the dioxaborolane 76 as an alternative coupling partner giving the 1-arylacridin-9(10H)-one 77 in 47% yield with dichloro[1,1-bis(diphenylphosphino)ferrocene]palladium(II) and Na2CO3 in 1,4-dioxane (Scheme 16) [52].
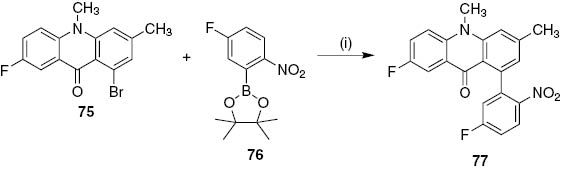
Reagents and conditions: (i) PdCl2(dppf), Na2CO3, 1,4-dioxane, 80°C, 24 h.
Another synthetic approach towards new 1-arylacridin-9(10H)-ones is being developed in our research group and until now two new derivatives were synthesized (Scheme 17). This approach involves the condensation of 2′-aminoacetophenone 78 with (2E,4E)-5-arylpenta-2,4-dienoic acids 81, which were obtained from the reaction of cinnamaldehydes 79 with malonic acid 80 in dry pyridine at 120°C, affording the (2E,4E)-N-(2-acetylphenyl)-5-arylpenta-2,4-dienamides 82. Then, the base-induced cyclodehydration of 82 gave new 2-[(1E,3E)-4-arylbuta-1,3-dienyl]quinolin-4(1H)-ones 83 in good yields (80–91%). Attempts to cyclize compound 83 (R=H) into the desired 1-phenylacridin-9(10H)-one were unsuccessful, so an alternative approach involved methylation of the NH group prior to cyclization. Thus, direct methylation of compound 83 (R=H) with methyl iodide and sodium hydride in dry THF gave a mixture of 1-methy-2-[(1E,3E)-4-phenylbuta-1,3- dienyl]quinolin-4(1H)-ones 85 as main compound (59%), and the isomeric 4-methoxy-2-[(1E,3E)-4-phenylbuta-1,3-dienyl]quinoline 84 (10%), due to the non-regioselective nature of this reaction [53]. A significant amount of starting material was also recovered (30%) (Scheme 17). In a modification of this synthetic route, the methylation and in situ base-induced cyclodehydration of the (2E,4E)-N-(2-acetylphenyl)-5-arylpenta-2,4-dienamides 82 was carried out affording 2-[(1E,3E)-4-arylbuta-1,3-dienyl]-1-methylquinolin-4(1H)-ones 85 as the sole reaction product. Finally, electrocyclization of 2-[(1E,3E)-4-arylbuta-1,3-dienyl]-1-methylquinolin-4(1H)-ones 85 in the presence of a catalytic amount of iodine in refluxing 1,2,4-TCB afforded the desired 1-aryl-10-methylacridin-9(10H)-ones 86 in good yields (53–65%) (Scheme 17). The main limitation of this synthetic approach is the lack of commercially available substituted cinnamaldehydes to allow the preparation of diversely substituted cinnamic acids 81 and the low yields obtained in their synthesis.
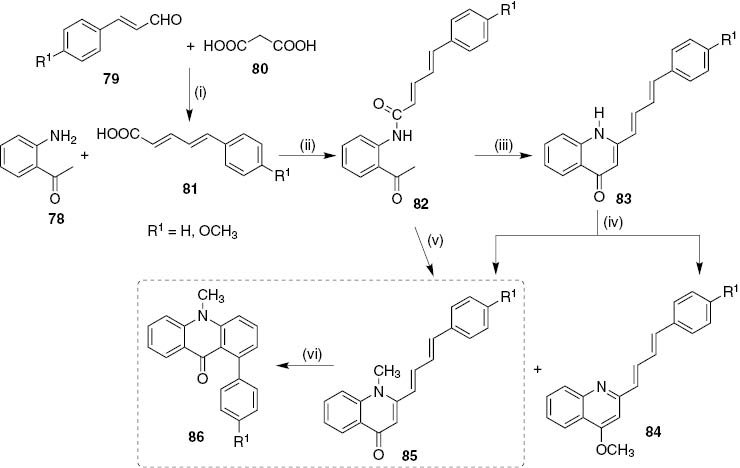
Reagents and conditions: (i) dry pyridine, 120°C, overnight; (ii) DCC, 4-ppy, CH2Cl2, r.t., 6–8 days; (iii) t-BuOK, t-BuOH, 90°C, 6 h; (iv) CH3I (20 equiv), NaH (1.0 equiv), dry THF, r.t., 24 h; (v) CH3I (1.5 equiv after 30 min.), NaH (1.5 equiv+1.0 equiv after 18 h), dry THF, r.t., 24 h; (vi) I2 (cat.), 1,2,4-TCB, reflux, 2 h.
Synthesis of 2,3-diarylacridones
As far as we know, no natural 2,3-diarylacridin-9(10H)-ones have been described to date. Recently, our group reported two different approaches for the synthesis of 2,3-diarylacridin-9(10H)-ones 93 and 97 [54], [55]. The first one (Scheme 18) involves the condensation of 2′-amino acetophenone 78 with cinnamic acids 87 in the presence of DCC and 4-ppy to afford (E)-N-(2-acetylphenyl)-3-arylacrylamides 88 which after base-induced cyclodehydration into 89 followed by C-3 iodination, using iodine and sodium carbonate in dry THF, gave (E)-3-iodo-2-styrylquinolin-4(1H)-ones 90 in very good yields (82–95%). Heck reaction of these (E)-3-iodo-2-styrylquinolin-4(1H)-ones 90 with styrenes 27, using Pd(PPh3)4 as catalyst, gave (E,E)-2,3-distyrylquinolin-4(1H)-ones 91 in moderate to good yields (36–81%), which when heated at high temperatures cyclize in two different ways: i) electrocyclization and further in situ oxidation in the presence of a catalytic amount of iodine and p-TSA in refluxing 1,2,4-TCB leads to 2,3-diarylacridin-9(10H)-ones 93 (2–40%), whereas ii) tautomerization, cyclization by nucleophilic addition followed by in situ oxidation produces (E)-2-aryl-4-styrylfuro[3,2-c]quinolines 92 as the main compounds (36–60%) [54], [55]. When the electrocyclization and in situ oxidation reactions were performed in 1,2,4-TCB without addition of iodine and p-TSA, in general higher amounts of (E)-2-aryl-4-styrylfuro[3,2-c]quinolines 92 were obtained (35–66%) together with 2,3-diarylacridin-9(10H)-ones 93 (16–37%), after a longer reaction time [54].
![Scheme 18: Reagents and conditions: (i) DCC, 4-ppy, CH2Cl2, r.t., 6–8 days; (ii) Classical heating: t-BuOK, dry THF, 80°C, 4 h. Microwave: t-BuOH, NaOH, 120°C, 18 min.; (iii) I2, Na2CO3, dry THF, r.t., 4–5 h; (iv) Pd(PPh3)4, Et3N, with ligand [PPh3, or tris(o-tolyl)phosphine] or without ligand, NMP or CH3CN, 80–100°C, 3–6.5 h; (v) 1,2,4-TCB, reflux, 24 h or 1,2,4-TCB, I2 (10%), p-TSA, reflux, 2–3 h.](/document/doi/10.1515/pac-2016-0407/asset/graphic/j_pac-2016-0407_scheme_018.jpg)
Reagents and conditions: (i) DCC, 4-ppy, CH2Cl2, r.t., 6–8 days; (ii) Classical heating: t-BuOK, dry THF, 80°C, 4 h. Microwave: t-BuOH, NaOH, 120°C, 18 min.; (iii) I2, Na2CO3, dry THF, r.t., 4–5 h; (iv) Pd(PPh3)4, Et3N, with ligand [PPh3, or tris(o-tolyl)phosphine] or without ligand, NMP or CH3CN, 80–100°C, 3–6.5 h; (v) 1,2,4-TCB, reflux, 24 h or 1,2,4-TCB, I2 (10%), p-TSA, reflux, 2–3 h.
The second approach that gives only 2,3-diaryl-10-methylacridin-9(10H)-ones 97 (Scheme 19) [55], involves the methylation and in situ cyclodehydration of (E)-N-(2-acetylphenyl)-3-arylacrylamides 88 with methyl iodide and sodium hydride in dry THF to afford the (E)-1-methyl-2-styrylquinolin-4(1H)-ones 94 (66–86%), which after C-3 iodination with iodine and cerium(IV) ammonium nitrate in acetonitrile at 80°C were converted into the corresponding (E)-3-iodo-1-methyl-2-styrylquinolin-4(1H)-ones 95 in good yields (66–83%). The Heck coupling reaction of (E)-3-iodo-1-methyl-2-styrylquinolin-4(1H)-ones 95 with styrenes 27 using Pd(PPh3)4 as catalyst, led to the expected (E,E)-1-methyl-2,3-distyrylquinolin-4(1H)-ones 96 in moderate to good yields (36–77%), which when heated at high temperatures in refluxing 1,2,4-TCB in the presence of a catalytic amount of iodine and p-TSA underwent electrocyclization and oxidation processes affording only the expected 2,3-diaryl-10-methylacridin-9(10H)-ones 97 in good yields (60–79%) [55].
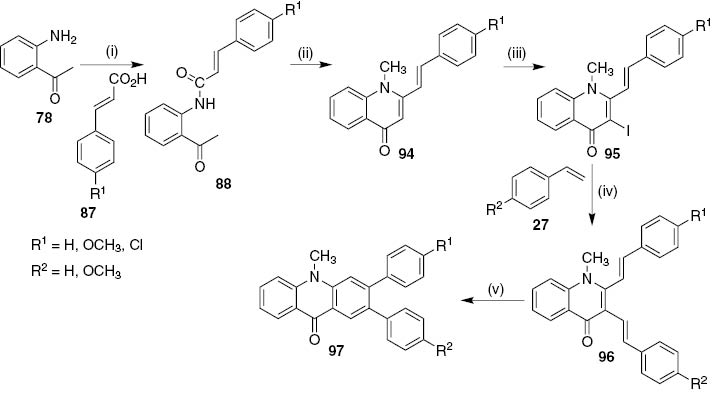
Reagents and conditions: (i) DCC, 4-ppy, CH2Cl2, r.t., 6–8 days; (ii) CH3I (1.5 equiv after 30 min.), NaH (1.5 equiv+1.0 equiv after 18 h), dry THF, r.t., 24 h; (iii) I2 (10%), (NH4)2Ce(NO3)6, CH3CN, 80°C, 3–4 h; (iv) Pd(PPh3)4, Et3N, with tris(o-tolyl)phosphine or without ligand, NMP or CH3CN, 80–100°C, 3–7 h; (v) 1,2,4-TCB, I2 (10%), p-TSA, reflux, 2–3 h.
Synthesis of miscellaneous acridones
Cycloaddition reaction of 1-methyl-2-styrylquinolin-4(1H)-ones 94 with N-methylmaleimide 58 in refluxing toluene afforded 4-aryl-2,6-dimethyl-4,5,6,11b-tetrahydro-1H-pyrrolo[3,4-a]acridine-1,3,11(2H,3aH)-triones 99 (34–52%), unreacted starting material 94 (13–15%) and traces of the oxidized cycloadducts 100 (Scheme 20) [56]. However, 4-aryl-2,6-dimethyl-1H-pyrrolo[3,4-a]acridine-1,3,11(2H,6H)-triones 100 were obtained directly carrying out the DA reaction in 1,2,4-TCB at 180°C. Under these reaction conditions the in situ oxidation of cycloadducts 99 occurred leading to acridin-9(10H)-ones 100 in moderate yields (45–56%) after 42–48 h [56]. When the reaction was performed in 1,2,4-TCB at 150°C under MW heating under sealed vessels the cycloadducts 99 were obtained in a shorter reaction time (1 h of irradiation), however, the yields were disappointingly lower (13–34%) and only traces of the oxidized cycloadducts 100 were obtained. Thus, the one-pot DA and oxidation reaction proved to be a valuable methodology to prepare acridin-9(10H)-ones 100 and led to the investigation of the oxidation of the cycloadducts 99 with chloranil in dry 1,4-dioxane at 80°C. Under these conditions acridin-9(10H)-ones 100 were obtained in very good yields (87–90%) (Scheme 20) [56].
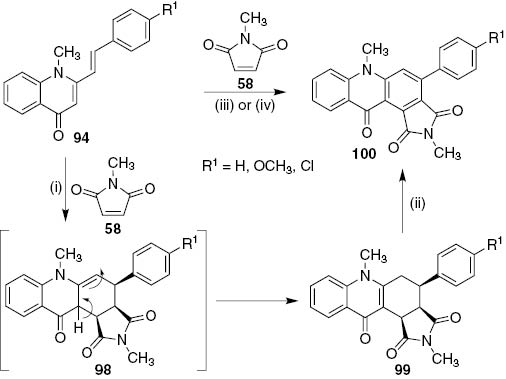
Reagents and conditions: (i) 58 (3 equiv+3 equiv after 48 h), toluene, 120°C, 72 h; (ii) chloranil, dry 1,4-dioxane, 80°C, 3 h. (iii) Classical heating: 1,2,4-TCB, 180°C, 42–48 h. Microwave: sealed vessels, 1,2,4-TCB, 150°C, 1 h. (iv) 58 (3 equiv+3 equiv after 7 h), 1,2,4-TCB, Sc(OTf)3, 80°C (for 4a and 4b) or reflux (for 4c), 24–27 h.
The formation of the acridin-9(10H)-ones 100 from the reaction of 1-methyl-2-styrylquinolin-4(1H)-ones 94 with N-methylmaleimide 58 can be explained by a 1,3-hydrogen shift of cycloadducts 98 leading to the formation of the more stabilized quinolin-4(1H)-one nucleus (Scheme 20).
The Lewis acid catalyzed DA reaction of 1-methyl-2-styrylquinolin-4(1H)-ones 94 with N-methylmaleimide 58 also led to the acridin-9(10H)-ones 100 with moderate yields (42–51%) in a shorter reaction time (24–27 h) and Sc(OTf)3 (20 mol%) proved to be more efficient than AlCl3 (Scheme 20) [56].
Conclusions
The most recent methodologies towards the synthesis of arylxanthone and arylacridone derivatives were covered in this review. The efficiency and scope of the described methods are largely corroborated by the wide range of structures prepared with classical and/or alternative heating conditions; however, efforts dedicated to reactions with higher performance and less environmental impact would be much appreciated. It should be highlighted that some chemical transformations were equally applied to both ring systems. Thus, it is expected that the already established chemistry, reactivity and biological properties for these compounds can boost the development of new methodologies and the search for novel decorated bioactive arylxanthone and arylacridone derivatives.
Article note:
A collection of invited papers based on presentations at the Trans Mediterranean Colloquium on Heterocylic Chemistry (TRAMECH VIII) in Anatalya, Turkey, 11–15 November 2015.
Acknowledgments
Thanks are due to the University of Aveiro, Polytechnic Institute of Bragança, Fundação para a Ciência e a Tecnologia (FCT) FCT/MEC for the financial support of the QOPNA research Unit (FCT UID/QUI/00062/2013) through national founds and, where applicable, co-financed by the FEDER, within the PT2020 Partnership Agreement. V.L.M. Silva also acknowledges FCT for the financial support of her post-doctoral grant (SFRH/BPD/108807/2015), in the ambit of “QREN e POCH e Tipologia 4.1 e Formação Avançada”, co-sponsored by FSE and by national funds of MCTES.
References
[1] H. R. El-Seedi, M. A. El-Barbary, D. M. H. El-Ghorab, L. Bohlin, A. K. Borg-Karlson, U. Goransson, R. Verpoorte. Curr. Med. Chem.17, 854 (2010).10.2174/092986710790712147Suche in Google Scholar
[2] M. M. M. Pinto, M. E. Sousa, M. S. J. Nascimento. Curr. Med. Chem.12, 2517 (2005).10.2174/092986705774370691Suche in Google Scholar
[3] F. Imperato. Curr. Top. Phytochem.6, 63 (2004).Suche in Google Scholar
[4] S. Bräse, A. Encinas, J. Keck, C. F. Nising. Chem. Rev.109, 3903 (2009).10.1021/cr050001fSuche in Google Scholar
[5] K.-S. Masters, S. Bräse. Chem. Rev.112, 3717 (2012).10.1021/cr100446hSuche in Google Scholar
[6] M. M. M. Pinto, R. A. P. Castanheiro. in Natural Products: Chemistry, Bio-chemistry and Pharmacology, G. Brahmachari (Ed.), pp. 520–675, Narosa Publishing House Pvt, Ltd, New Delhi (2009).Suche in Google Scholar
[7] M. E. Sousa, M. M. M. Pinto. Curr. Med. Chem.12, 2447 (2005).10.2174/092986705774370736Suche in Google Scholar
[8] C. M. G. Azevedo, C. M. M. Afonso, M. M. M. Pinto. Curr. Org. Chem.16, 2818 (2012).10.2174/138527212804546921Suche in Google Scholar
[9] C.-H. Yang, L. Ma, Z.-P. Wei, F. Han, J. Gao. Chin. Herb. Med.4, 87 (2012).Suche in Google Scholar
[10] C. M. M. Santos, M. Freitas, D. Ribeiro, A. Gomes, A. M. S. Silva, J. A. S. Cavaleiro, E. Fernandes. Bioorg. Med. Chem.18, 6776 (2010).10.1016/j.bmc.2010.07.044Suche in Google Scholar
[11] C. M. M. Santos, A. M. S. Silva, P. Filipe, R. Santus, L. K. Patterson, J.-C. Mazière, J. A. S. Cavaleiro, P. Morlière. Org. Biomol. Chem.9, 3965 (2011).10.1039/c0ob00841aSuche in Google Scholar
[12] P. Morlière, L. K. Patterson, C. M. M. Santos, A. M. S. Silva, J.-C. Mazière, P. Filipe, A. Gomes, E. Fernandes, M. B. Garcia, R. Santus. Org. Biomol. Chem.10, 2068 (2012).10.1039/c2ob06612bSuche in Google Scholar
[13] G. Cholewiński, K. Dzierzbicka, A. M. Kołodziejczyk. Pharmacol. Rep.63, 305 (2011).10.1016/S1734-1140(11)70499-6Suche in Google Scholar
[14] R. Kumar, M. Kumari. J. Chem. Pharm. Res.3, 217 (2011).Suche in Google Scholar
[15] C. S. Sepúlveda, M. L. Fascio, C. C. Garcia, N. B. D’Accorso, E. B. Damonte. Curr. Med.Chem. 20, 2402 (2013).10.2174/0929867311320190002Suche in Google Scholar PubMed
[16] L. M. M. Vieira, A. Kijjoa. Curr. Med. Chem.12, 2413 (2005).10.2174/092986705774370682Suche in Google Scholar PubMed
[17] C. I. C. Esteves, C. M. M. Santos, C. M. Brito, A. M. S. Silva, J. A. S. Cavaleiro. Synlett 1403 (2011).10.1055/s-0030-1260567Suche in Google Scholar
[18] C. Proença, H. M. T. Albuquerque, D. Ribeiro, M. Freitas, C. M. M. Santos, A. M. S. Silva, E. Fernandes. Eur. J. Med. Chem.115, 381 (2016).10.1016/j.ejmech.2016.03.043Suche in Google Scholar PubMed
[19] I. Fukawa, H. Yoneda, A. K. K. K. Kaisha. EP 0237004, US Patent 4804735, Sep, 1987.Suche in Google Scholar
[20] Q. Zhou, B. B. Snider. J. Org. Chem.75, 8224 (2010).10.1021/jo101826pSuche in Google Scholar PubMed PubMed Central
[21] A. S. Kelkar, R. M. Letcher, K.-K. Cheung, K.-F. Chiu, G. D. Brown. J. Chem. Soc., Perkin Trans I 3732 (2000).10.1039/b004422iSuche in Google Scholar
[22] C. M. M. Santos, A. M. S. Silva, J. A. S. Cavaleiro. Synlett 3095 (2005).10.1055/s-2005-921928Suche in Google Scholar
[23] C. M. M. Santos, A. M. S. Silva, J. A. S. Cavaleiro. Eur. J. Org. Chem. 2642 (2009).10.1002/ejoc.200900026Suche in Google Scholar
[24] C. M. M. Santos, A. M. S. Silva, J. A. S. Cavaleiro. Synlett 3113 (2007).10.1055/s-2007-990900Suche in Google Scholar
[25] H.-J. Cho, M.-J. Jung, S. Woo, J. Kim, E.-S. Lee, Y. Kwon, Y. Na. Bioorg. Med. Chem.18, 1010 (2010).10.1016/j.bmc.2009.12.069Suche in Google Scholar PubMed
[26] Q.-G. Su, Y. Liu, Y.-C. Cai, Y.-L. Sun, Bo. Wang, L.-J. Xian. Invest. New Drugs29, 1230 (2011).10.1007/s10637-010-9468-5Suche in Google Scholar PubMed
[27] C. M. Brito, D. C. G. A. Pinto, A. M. S. Silva, A. M. G. Silva, A. C. Tomé, J. A. S. Cavaleiro. Eur. J. Org. Chem.11, 2558 (2006).10.1002/ejoc.200500872Suche in Google Scholar
[28] D. H. A. Rocha, D. C. G. A. Pinto, A. M. S. Silva, T. Patonay, J. A. S. Cavaleiro. Synlett23, 559 (2012).10.1055/s-0031-1290355Suche in Google Scholar
[29] D. H. A. Rocha, D. C. G. A. Pinto, A. M. S. Silva. Synlett24, 2683 (2013).10.1055/s-0033-1340013Suche in Google Scholar
[30] D. H. A. Rocha, D. C. G. A. Pinto, R. S. G. R. Seixas, A. M. S. Silva. Magn. Reson. Chem.52, 47 (2014).10.1002/mrc.4026Suche in Google Scholar PubMed
[31] Y. Liu, S. Jin, L. Huang, Y. Hu. Org. Lett.17, 2134 (2015).10.1021/acs.orglett.5b00721Suche in Google Scholar PubMed
[32] D. C. G. A. Pinto, A. M. S. Silva, L. M. P. M. Almeida, J. R. Carrillo, A. Díaz-Ortiz, A. de la Hoz, J. A. S. Cavaleiro. Synlett 1415 (2003).10.1055/s-2003-40826Suche in Google Scholar
[33] D. C. G. A. Pinto, A. M. S. Silva, C. M. Brito, A. Sandulache, J. R. Carrillo, P. Prieto, A. Díaz-Ortiz, A. de la Hoz, J. A. S. Cavaleiro. Eur. J. Org. Chem. 2973 (2005).10.1002/ejoc.200500073Suche in Google Scholar
[34] A. Sandulache, A. M. S. Silva, D. C. G. A. Pinto, L. M. P. M. Almeida, J. A. S. Cavaleiro. New J. Chem.27, 1592 (2003).10.1039/b303554aSuche in Google Scholar
[35] A. Schonberg, A. Mustafa, G. Aziz. J. Am. Chem. Soc.76, 4576 (1954).10.1021/ja01647a020Suche in Google Scholar
[36] A. Mustafa, M. I. Ali. J. Org. Chem.21, 849 (1956).10.1021/jo01114a006Suche in Google Scholar
[37] G. Aziz. J. Org. Chem.27, 2954 (1962).10.1021/jo01055a529Suche in Google Scholar
[38] M. A.-F. Elkashef, F. M. E. Abdel-Megeid, K.-E. M. Mokhart, F. A. Gad. Acta Chim. Acad. Sci. Hung.84, 319 (1975).Suche in Google Scholar
[39] H. Marona. Pol. J. Chem.53, 1877 (1979).Suche in Google Scholar
[40] R. M. Letcher, T.-Y. Yue. J. Chem. Res. (S) 248 (1992).Suche in Google Scholar
[41] R. M. Letcher, T.-Y. Yue. J. Chem. Res. (M) 2078 (1992).Suche in Google Scholar
[42] R. M. Letcher, T.-Y. Yue. J. Chem. Soc. Chem. Commun. 1310 (1992).10.1039/C39920001310Suche in Google Scholar
[43] D. C. G. A. Pinto, A. M. L. Seca, S. B. Leal, A. M. S. Silva, J. A. S. Cavaleiro. Synlett 2005 (2011).10.1055/s-0030-1261172Suche in Google Scholar
[44] A. M. L. Seca, S. B. Leal, D. C. G. A. Pinto, M. C. Barreto, A. M. S. Silva. Molecules19, 8317 (2014).10.3390/molecules19068317Suche in Google Scholar PubMed PubMed Central
[45] F. Tillequin. Phytochem. Rev.6, 65 (2007).10.1007/s11101-006-9010-8Suche in Google Scholar
[46] K. F. Bastow. Curr. Drug Targets Infect. Disord.4, 323 (2004).10.2174/1568005043340533Suche in Google Scholar PubMed
[47] V. Barton, N. Fisher, G. A. Biagini, S. A. Ward, P. M. O’Neill. Curr. Opin. Chem. Biol.14, 440 (2010).10.1016/j.cbpa.2010.05.005Suche in Google Scholar PubMed
[48] J. A. Smith, R. M. West, M. Allen. J. Fluoresc.14, 151 (2004).10.1023/B:JOFL.0000016287.56322.ebSuche in Google Scholar
[49] B. Wang, L. Bouffier, M. Demeunynck, P. Mailley, A. Roget, T. Livache, P. Dumy. Bioelectrochemistry, 63, 233 (2004).10.1016/j.bioelechem.2003.10.020Suche in Google Scholar PubMed
[50] C. Huang, S.-J. Yan, Y.-M. Li, R. Huang, J. Lin. Bioorg. Med. Chem. Lett.20, 4665 (2010).10.1016/j.bmcl.2010.05.101Suche in Google Scholar PubMed
[51] R. A. Heald, M. F. G. Stevens. Org. Biomol. Chem.1, 3377 (2003).10.1039/B305177NSuche in Google Scholar PubMed
[52] I. Hutchinson, M. F. G. Stevens. Org. Biomol. Chem.5, 114 (2007).10.1039/B613580NSuche in Google Scholar PubMed
[53] A. I. S. Almeida, V. L. M. Silva, A. M. S. Silva, D. C. G. A. Pinto, J. A. S. Cavaleiro. Synlett 2593 (2008).10.1055/s-0028-1083530Suche in Google Scholar
[54] V. L. M. Silva, A. M. S. Silva, J. A. S. Cavaleiro. Synlett 2565 (2010).10.1055/s-0030-1258579Suche in Google Scholar
[55] V. L. M. Silva, A. M. S. Silva. Tetrahedron70, 5310 (2014).10.1016/j.tet.2014.05.040Suche in Google Scholar
[56] A. I. S. Almeida, V. L. M. Silva, A. M. S. Silva, D. C. G. A. Pinto, J. A. S. Cavaleiro. Synlett 889 (2012).10.1055/s-0031-1290600Suche in Google Scholar
©2016 IUPAC & De Gruyter. This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License. For more information, please visit: http://creativecommons.org/licenses/by-nc-nd/4.0/
Artikel in diesem Heft
- Frontmatter
- In this issue
- Editorial
- VIII Trans-Mediterranean Colloquium on Heterocyclic Chemistry (TRAMECH VIII)
- Conference papers
- Heterocycle-based bifunctional organocatalysts in asymmetric synthesis
- Arylxanthones and arylacridones: a synthetic overview
- Esterification of chitosan with L-alanine and a study on their effect in removing the heavy metals and total organic carbon (TOC) from wastewater
- Nanofibers based on chitin: a new functional food
- Dissolution, gelation, functionalization, and material preparation of chitin using ionic liquids
Artikel in diesem Heft
- Frontmatter
- In this issue
- Editorial
- VIII Trans-Mediterranean Colloquium on Heterocyclic Chemistry (TRAMECH VIII)
- Conference papers
- Heterocycle-based bifunctional organocatalysts in asymmetric synthesis
- Arylxanthones and arylacridones: a synthetic overview
- Esterification of chitosan with L-alanine and a study on their effect in removing the heavy metals and total organic carbon (TOC) from wastewater
- Nanofibers based on chitin: a new functional food
- Dissolution, gelation, functionalization, and material preparation of chitin using ionic liquids