Asymmetric calixarene derivatives as potential hosts in chiral recognition processes
-
Catalin V. Maftei


Abstract
New chiral derivatives of 15,35,55,75-tetra-tert-butyl-1,3,5,7(1,3)-tetrabenzenacyclooctaphane-12,32,52,72-tetraol [(1); tert-butyl-calix[4]-arene] were synthesized by coupling modified chiral quinuclidines derived from the natural-product-based alkaloids quincorine and quincoridine with the calix[4]arene 1 via either an ester bond or an amide bond. X-ray analyses of two products were performed. Applications of the products in asymmetric catalytic hydrogen transfer reactions are described. A protocol is presented to multi-substitute calix[4]arene at the methylene bridges, resulting in, e.g., 2,6-carboxyl-all-tert-butyl all-methoxy-calix[4]arene.
Introduction
The chemistry of calixarenes has been investigated intensively over the last few decades [1–6], whereby calix[4]arenes have been used especially often. In the past these molecules were modified mostly at the so-called upper and lower rims (Fig. 1). More recently, the methylene bridges have also been focus of research. Various calixarenes differ in the number of benzene cores and the bridge length. The calix[4]arene shown in Fig. 1 is a very common structure in the literature. A major modification to this framework is the replacement of the methylene bridges by 2-oxa-propylene bridges, leading to the family of the homo-oxa-calixarenes [7].
![Fig. 1:
Positions of modifications of calix[4]arenes.](/document/doi/10.1515/pac-2014-1121/asset/graphic/j_pac-2014-1121_fig_001.jpg)
Positions of modifications of calix[4]arenes.
In the description of calixarenes two aspects are generally mentioned; their cavity, which allows them to incorporate smaller molecules or atoms, and their fixed hydroxy groups, which allow them to form stable and rigid complexes. Although calixarenes themselves are quite rigid structures, they do have some conformational flexibility. Figure 2 shows the well-known family of conformers based on the example of 15,35,55,75-tetra-tert-butyl-1,3,5,7(1,3)-tetrabenzenacyclooctaphane-12,32,52,72-tetraol (1, tert-butyl-calix[4]arene).
![Fig. 2:
Conformations of tert-butyl-calix[4]arene 1.](/document/doi/10.1515/pac-2014-1121/asset/graphic/j_pac-2014-1121_fig_002.jpg)
Conformations of tert-butyl-calix[4]arene 1.
Calixarenes have found widespread uses. In general chemistry they are used as surfactants, building blocks for synthesis, and catalysts. An example is the use of ligand A (Fig. 3) as catalyst [8–11]; A was synthesized in a three-step sequence and was used in a rhodium-catalysed hydroformylation reaction (Scheme 1).
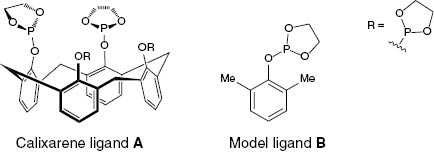
Ligands used for catalytic hydroformylation.
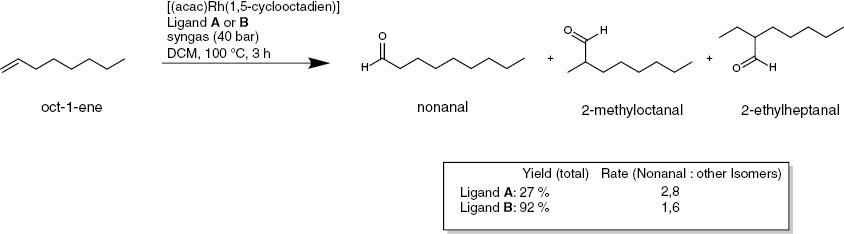
Catalytic hydroformylation of oct-1-ene.
The results were compared to those of catalysis ligand B (4 equivalents based on the rhodium species). The conversion in the case of calixarene ligand A was much slower but the purity of the nonanal reaction product was much better. The ratio of nonanal to all other detected isomeric products of the hydroformylation was 2.8 to 1.
Calixarenes are also widely used in coordination chemistry. Several transition metal complexes have been published [12–14] (see Scheme 2 for examples). In some digold-calix[4]arene complexes the gold(I) ions were shown to be mobile [15]. Other examples with calix[4]resorcinarenes have been presented [16–18], which are structurally very similar to calix[4]arenes. Industrial applications have been described by Perrin et al. [19]. Calixarenes have been employed in electronic noses [20], calcium-selective electrodes [21] and metalloreceptors [22]. They are even used in the cosmetic industry for fixation of hair coloring agents. In photochemistry they find applications in toners and as stabilizers for photographic films.
![Scheme 2:
Synthesis of transition metal complexes [4].](/document/doi/10.1515/pac-2014-1121/asset/graphic/j_pac-2014-1121_fig_005.jpg)
Synthesis of transition metal complexes [4].
An important use of a calix[4]arene in the field of organic chemistry is the β-stereoselective galactosylation of glucose-derivative D (Scheme 3). The activated compound C is used as a galactosyl donor, which is activated by AgClO4. The tetra-O-propylated cone-calix[4]arene 2 has a stabilizing effect on the intermediate galactosyl cation by cation-π-interactions, and a β-stereoselctive galactosylation of the anomerically pure glucose-derivative D to compound E could therefore be conducted [23, 24]. A calix[4]arene has also been introduced into a polymer backbone [25].
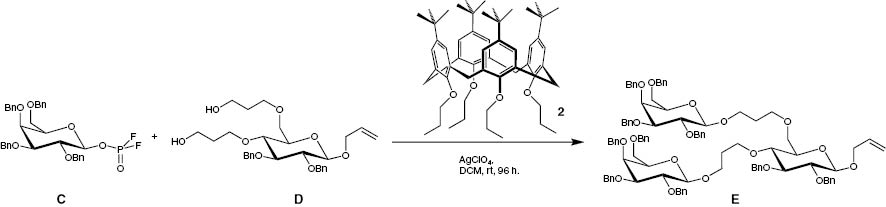
β-stereoselctive galactosylation of 3-(((2R,3R,4S,5R,6R)-6-(allyloxy)-4,5-bis(benzyloxy)-2-((3-hydroxypropoxy)methyl)tetrahydro-2H-pyran-3-yl)oxy)propan-1-ol (D) by cation-π-interactions.
A major theme in the use of calixarenes is based on their ability to bind selectively to structures by utilising their cavity. Thus they have been employed in environmental technology as absorbers of heavy metals such as uranium and mercury [26], and they have found applications as extractants of organic molecules coordinated to precious metals [27, 28] or rare earths [29, 30]. They are capable of separating the Buckminsterfullerenes C60 and C70 [31]. In the field of medicine they are used as capsules [32] or modified as antibody mimetic compounds [33, 34]. The company Synaptec has developed stationary phases based on calixarenes; these “Caltrex®-phases” were able to separate ortho-, meta- and para-toluidine.
A further development has been the coupling of chiral residues to calixarenes; sugar derivatives can be coupled at the lower [35, 36] and the upper rim [37]. Such compounds find applications in the field of asymmetric synthesis.
Maier et al. discovered another combination by coupling derivatives of quinine with a calix[4]arene and immobilizing these on silica gel via a thio-linker (Fig. 4). With this material they tested the separation of racemic mixtures of various protected amino acids, e.g., Z-alanine, by liquid chromatography. For comparison they also used a stationary phase where they replaced the calix[4]arene by a structurally simple tert-butyl rest [38, 39]. The lowest ratio of the retention times (S-enantiomer : R-enantiomer) was 1.57 for Z-alanine using the stationary phase F, where the configuration of the Cinchona alkaloid at C9 was unnatural and the connection was realized via a urea bond. The urethane-bonded material with the same unnatural stereo chemistry as F has a ratio of just 1.14. Interestingly the tert-butyl-variation of F also has a ratio of 1.42. The result is therefore that the main discrimination was associated with the Cinchona alkaloid but the calix[4]arene also gave an improvement of around 10 %. There are further examples where the effect is up to 20 %. The naturally configured materials (an example is G) had the opposite elution order but with smaller ratios.
![Fig. 4:
Tetra-O-ethylated calix[4]arene bonded to Cinchona alkaloids, which are immobilized on silica gel via their former double bond.](/document/doi/10.1515/pac-2014-1121/asset/graphic/j_pac-2014-1121_fig_007.jpg)
Tetra-O-ethylated calix[4]arene bonded to Cinchona alkaloids, which are immobilized on silica gel via their former double bond.
Inspired by this work, we wished to investigate if such an effect could also observed in asymmetric catalytic hydrogen transfer reactions. Arvidsson et al. described in their work the iridium-catalysed reduction of acetophenone using derivatives of quincorine-C9-amine and quincoridine-C9-amine (Fig. 6) as chiral ligands [40, 41]. They achieved an enantiomeric excess of around 80 % and good yields (Scheme 4).
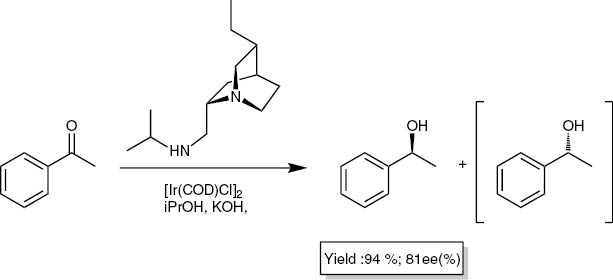
Asymmetric catalytic reduction of acetophenone by Arvidsson et al.
Our plan was to couple derivatives of quincorine and quincoridine with tert-butyl-calix[4]arene 1 (Fig. 5) and test the products in asymmetric catalytic reductions. Here we report the results.
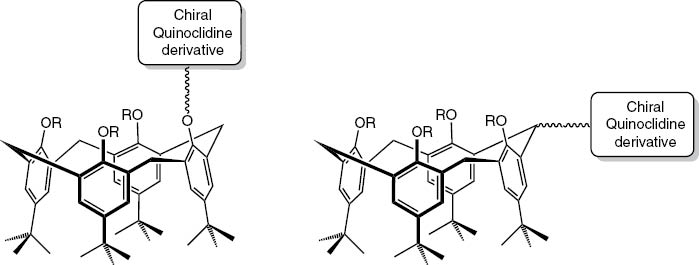
Introduction of a chiral group at the lower rim or at the methylene bridge as ligand partners for metal centers.
Results and discussions
The tert-butyl-calix[4]arene 1 was chosen as the starting point. This was to be combined with modified derivatives of quincorine (QCI) and quincoridine (QCD) as sources of chirality; these are based on the Cinchona alkaloids quinine and quinidine and have in their simplest forms an amine or an alcohol functionality. The coupling of these two moieties should then be realized through an ester or an amide bond, so that the calix[4]arene must be modified by the introduction of a carboxyl functionality.
Lower rim derivatives
Preparation of the acids
The preparation of the acid functionality followed an established protocol. First the calix[4]arene was coupled with ethyl bromoacetate followed by the cleavage of the ethyl ester (Scheme 5). The di-acid 4 was obtained using potassium carbonate [42] and the tetra-acid 6 using NaH as base [43, 44]. In both cases the esters were cleaved using sodium hydroxide in aqueous ethanol.
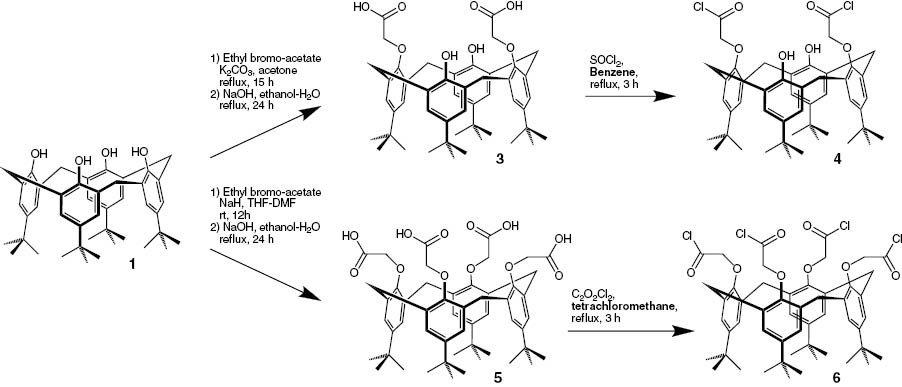
Synthesis of lower rim acids followed by the synthesis of the acid chlorides.
Introduction of the chiral decoration
These acids were now coupled with the C9-amine-quincorine (QCI-C9-NH2) [45], establishing an amide bond. The method of choice was to transform the acids to their corresponding acyl chlorides. The di-acid was synthesized using benzene and thionyl chloride [46, 47] and the tetra-acyl-chloride was generated by a protocol of Zlatuskova et al. using tetrachloromethane as solvent [44]. In both cases alternative attempts using other solvents failed. Having the acyl chlorides in hand, the amides were synthesized successfully in good yields (Scheme 6).
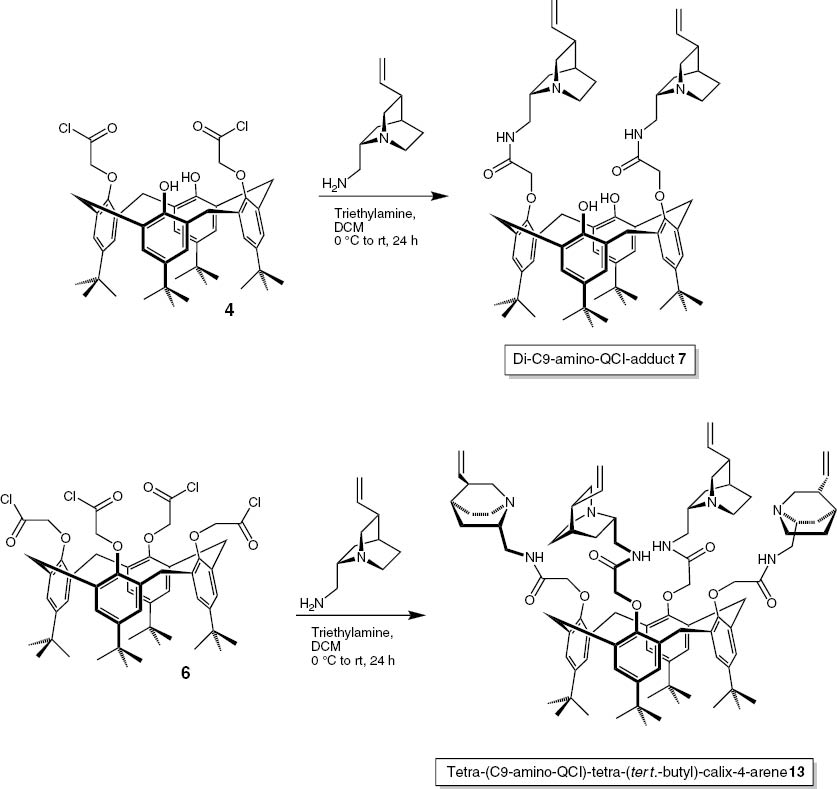
Synthesis of the QCI-C9-NH2 amides
These reactions were also performed successfully for the dihydro- and didehydro-derivatives of QCI-C9-NH2 [48] giving the compounds 8, 9, 14, 15 and the corresponding series of quincoridine derivatives (Fig. 6) giving the compounds 10–12 and 16–18.
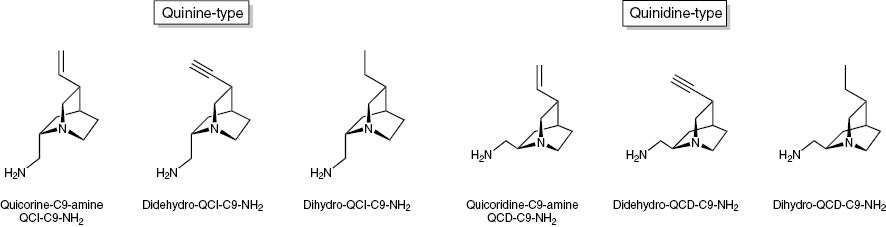
QCI-C9-NH2 and QCD-C9-NH2 and their dihydro-and didehydro-derivatives.
Furthermore we coupled C10-modified C9-amine derivatives of QCI and QCD as a third mode of providing chiral information. The modification is described in Scheme 7. After a Boc-protection of the QCI-C9-amine the double bond is transformed to an aldehyde functionality. This aldehyde is reduced to the corresponding alcohol, which can be isolated in a good yield of 78 % [49]. The QCD derivative 20 is produced following the same protocol. The structure is illustrated in Fig. 7.
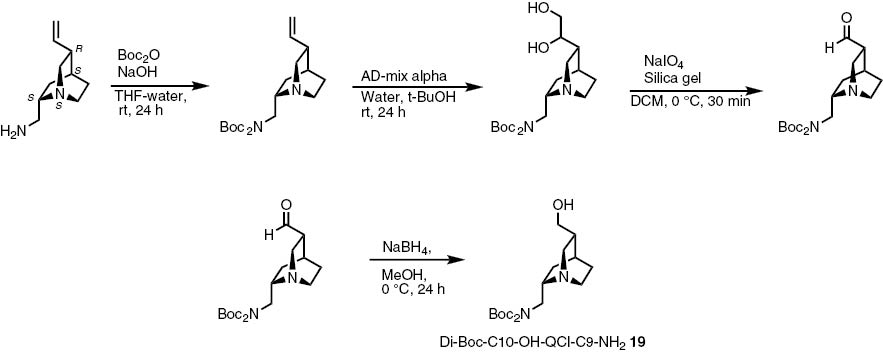
Synthesis of Boc-protected C10-hydroxy-QCI-C9-amine 19.
Structure of Boc-protected C10-hydroxy-QCD-C9-amine 20.
The protected aminoalcohols 19 and 20 were now coupled to the modified tert-butyl-calix[4]arene 3, which has two carboxyl groups, using 1-methyl-pyridinium iodide. After the successful coupling in both cases the Boc-groups were cleaved using an ethereal solution of hydrogen chloride (Scheme 8).
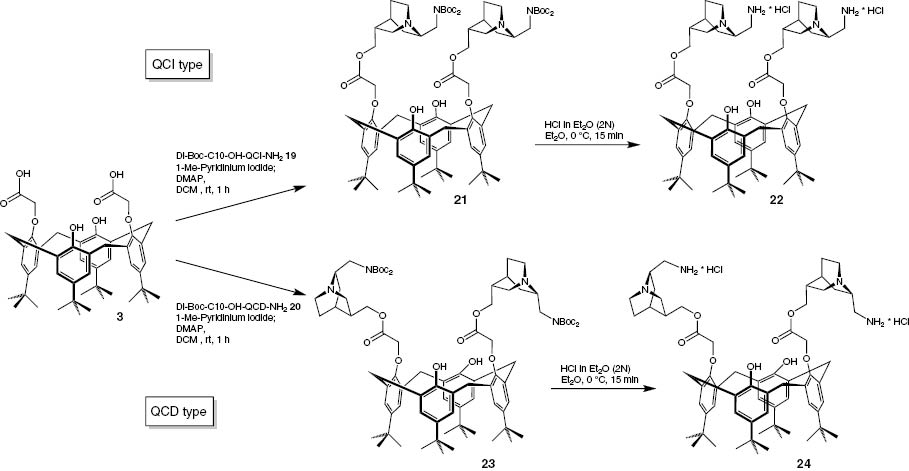
Synthesis of the C10-esters 21 and 23 and their N-deprotected products 22 and 24.
X-ray analysis
Single crystals of di-dihydro-QCI-C9-NH2-calix-4-arene amide 9 and di-dihydro-QCD-C9-NH2-calix[4]arene amide 12 (Fig. 8) were obtained and X-ray determinations carried out for both compounds (Figs. 9 and 10) [50]. Compound 9 crystallizes in the monoclinic space group C2; the asymmetric unit contains two half molecules (each of which corresponds to a molecule with exact twofold symmetry) and two chloroform molecules. For the sake of brevity, only the structure of one independent molecule is discussed in detail (the other molecule is similar, see Supplementary Material for details). Compound 12 crystallizes solvent-free in the hexagonal space group P65 with one molecule in the asymmetric unit.
![Fig. 8:
Di-dihydro-QCI-C9-NH2 calix[4]arene amide 9 and Di-dihydro-QCD-C9-NH2 calix[4]arene amide 12.](/document/doi/10.1515/pac-2014-1121/asset/graphic/j_pac-2014-1121_fig_016.jpg)
Di-dihydro-QCI-C9-NH2 calix[4]arene amide 9 and Di-dihydro-QCD-C9-NH2 calix[4]arene amide 12.
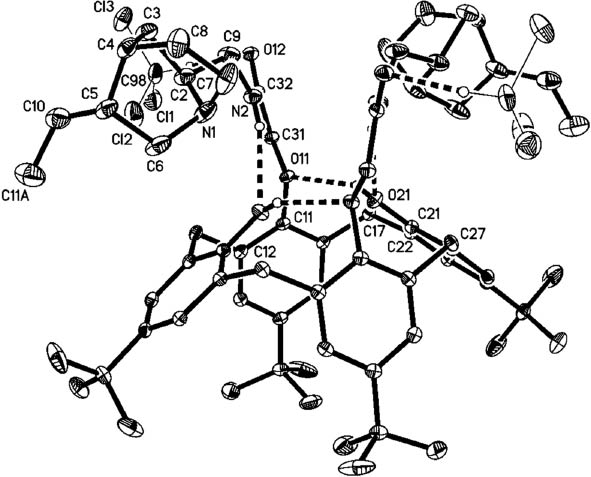
X-ray structure analysis of 9. Only one of the two independent formula units is shown. Ellipsoids correspond to 30 % probability levels. Dashed lines indicate hydrogen bonds. H atoms not involved in H bonding are omitted for clarity. Only one orientation of disordered butyl groups is shown.
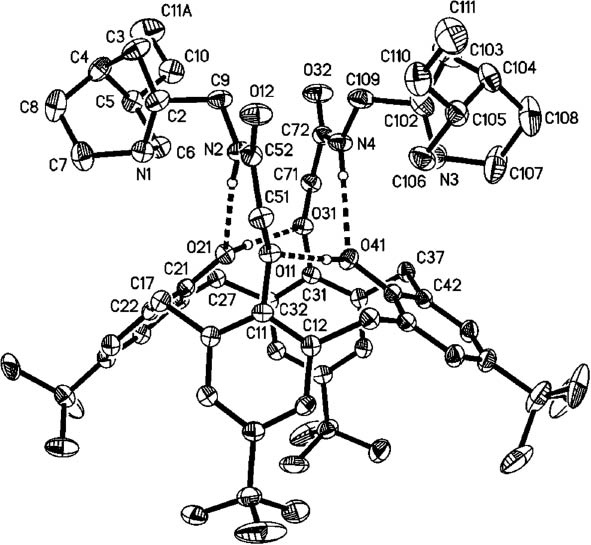
X-ray structure analysis of 12. Ellipsoids correspond to 30 % probability levels. Dashed lines indicate hydrogen bonds. H atoms not involved in H bonding are omitted for clarity. Only one orientation of disordered butyl groups is shown.
The structure analyses confirm that the acids are 1,3-coupled to the calix-4-arene, whereby the bridgehead amines of the quinuclidines are orientated towards the center of the calix-4-arene cavity. The molecules of 9 and 12 both display a distorted cone geometry.
For compound 9, the facing phenolic units (aromatic ring C11–16 and its symmetry-equivalent ring) that bear the substituents are (very) approximately parallel; the interplanar angle is 19° and the angle between the vector C11···C14 and the crystallographic twofold axis is 10°. The intercentroid distance between these two rings is 5.7 Å. The other two rings, C21–C26 and its equivalent ring, are much more strongly tilted with respect to the twofold axis, with a corresponding angle of 51°, an interplanar angle of 102° and an intercentroid distance of 7.5 Å. The interplanar angle between C11–C16 and C21–C26 is 83°. The distance between the two bridgehead nitrogens (N1 and its symmetry-equivalent) is 6.2 Å.
The molecule of 9 displays a system of hydrogen bonds. One intramolecular bond is formed from the amide group to the symmetry-equivalent phenol OH group (N2–H02···O21, with H···O 2.39 Å) and a second such bond from the same phenol group to its substituted counterpart (O21–H21···O11, with H···O 2.14 Å). Furthermore, the chloroform molecule is connected to the carbonyl oxygen of the calixarene by an extremely short interaction C98–H98···O12, with H···O just 2.12 Å); for a review of hydrogen bonding to solvent chloroform and dichloromethane, see [51].
The molecular structure of compound 12 is in many respects closely similar to that of 9. The phenolic units (aromatic rings C11–C16 and C31–C36) that bear the substituents subtend an interplanar angle is 19°; the intercentroid distance between these two rings is 5.7 Å. The other two rings, C21–C26 and C41–C46, subtend an interplanar angle of 104° and have an intercentroid distance of 7.5 Å. The distance between the two bridgehead nitrogens (N1 and N3) is 7.2 Å. The intramolecular hydrogen bonds are: N2–H02···O21 with H···O 2.36 Å; O21–H21···O31 1.86 Å; N4–H04···O41 2.29 Å and O41–H41···O11 1.84 Å.
Methylene-bridge derivatives
Although most modifications of calixarene-type molecules are carried out at the lower or upper rim, Weber et al. have presented a calix[4]arene modified at the methylene bridge.
Preparation of the acids
The synthesis started from the all-methoxy-calix[4]arene 25 [52]. This compound was treated with an excess of n-BuLi at –78 °C and quenched with solid carbon dioxide [53, 54]. The resulting compound contained exactly one carboxyl functionality (Scheme 9, compound 26). No trace of a double addition was found, either by us or by Weber et al.
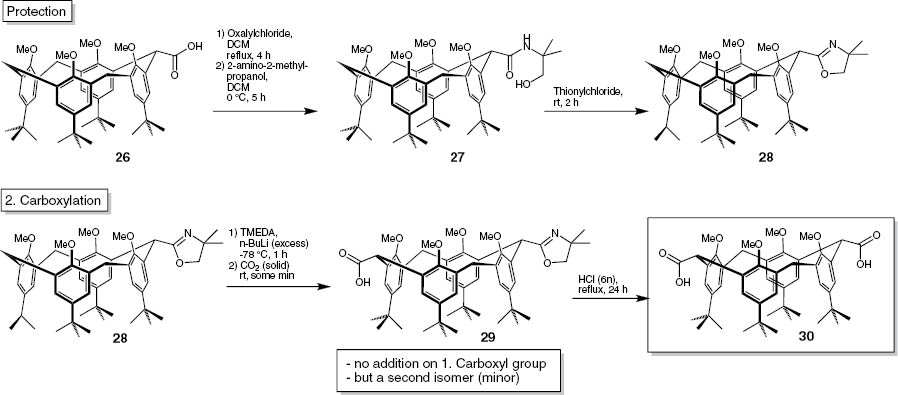
Synthesis of 15,35,55,75-tetra-tert-butyl-12,32,52,72-tetramethoxy-1,3,5,7(1,3)-tetrabenzena-cyclooctaphane-2,6-dicarboxylic acid (30).
In order to introduce a second carboxylic group, the first had to be protected by a group stable to n-BuLi. Attempts to use the free acid 26 failed even for large excesses of a variety of deprotonating agents such as n-BuLi, LDA or NaH. The group of choice was the 2-oxazoline moiety, which was established in a three-step protocol. When the first acid is protected, the second can be introduced using the Weber protocol (Scheme 9). After deprotection under acid conditions the free di-acid 30 was generated. The acid groups were established at the 1,3-positions of the methylene bridges.
Introduction of the chiral groups
With the methylene-bridged mono-acid 26 and methylene-bridged di-acid 30 in hand, it was planned to introduce the chiral derivatives of QCI and QCD. Here we used the corresponding alcohols, which were to be connected via an ester bond, and the C9-amines, which were to be connected via an amide bond. Furthermore we tried to couple C10-modified C9-amine as a third mode of providing chiral information.
The coupling with the Boc-protected C10-hydroxy-QCI-C9-amine and Boc-protected C10-hydroxy-QCD-C9-amine were performed successfully by using 1-methyl-pyrimidinium iodide under standard conditions. The various C9-amines (Fig. 6) were introduced with success via the acyl chloride route. The attempts to couple QCI, QCD and their dihydro and didehydro-derivatives unfortunately showed no conversion to the desired esters, and neither the use of coupling agents such as EDC, DCC, DIC, 1-methyl-pyrimidinium iodide nor the use of the acyl chloride as active intermediate was successful. In Scheme 10 some examples are given.
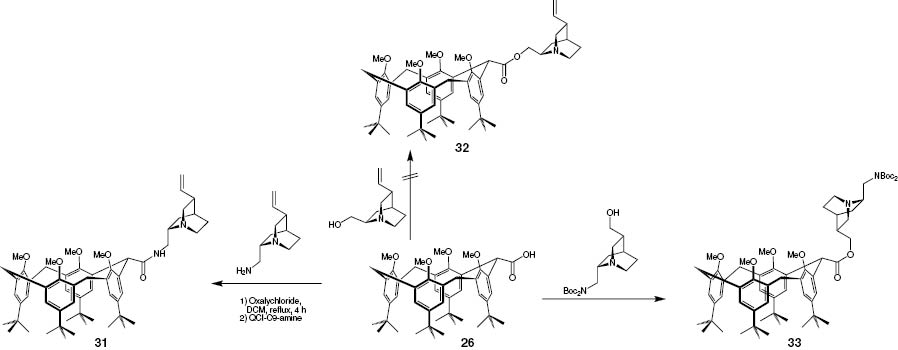
Examples of the synthesis of chiral ester and amides of the methylene-bridged-mono-acid.
Furthermore all products based on the C9-amines are illustrated in Fig. 11. To complete the row the acid 26 was also coupled with the Boc-protected C10-hydroxy-QCD-C9-amine 20 forming the ester 39.
![Fig. 11:
Chiral amides of the methylene bridge modified tert-butyl-calix[4]arene.](/document/doi/10.1515/pac-2014-1121/asset/graphic/j_pac-2014-1121_fig_021.jpg)
Chiral amides of the methylene bridge modified tert-butyl-calix[4]arene.
The Boc-protected C10-esters were cleaved by using ethereal solutions of hydrogen chloride. The products are illustrated in Fig. 12.
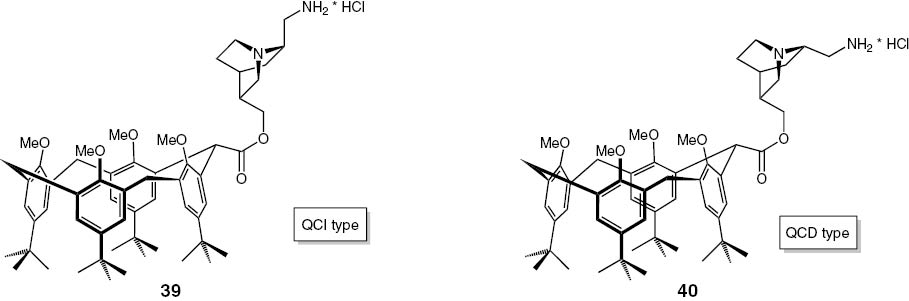
N-deprotected C10-esters 39 and 40.
The methylene-bridged di-acid could also be transformed to the various types of products. As an example, the esterification with Boc-protected C10-hydroxy-QCD-C9-amine 28 is given (Scheme 11).
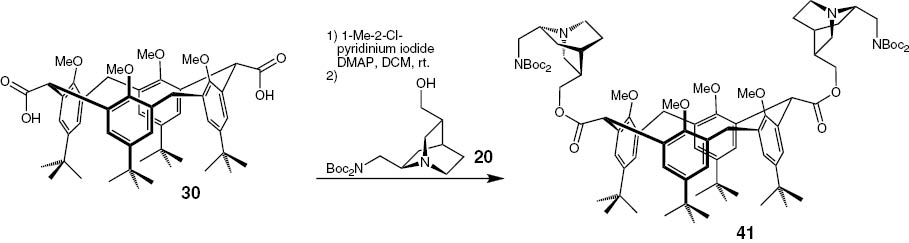
Synthesis of 2-(((1S,3R,4S,6R)-6-((bis(tert-butoxycarbonyl)amino)methyl)quinuclidin-3-yl)methyl) 6-(((3R,4S,6R)-6-((bis(tert-butoxycarbonyl)amino)methyl)quinuclidin-3-yl)methyl) 15,35,55,75-tetra-tert-butyl-12,32,52,72-tetramethoxy-1,3,5,7(1,3)-tetrabenzenacyclooctaphane-2,6-dicarboxylate (41).
The Boc-groups were removed using the standard method with hydrogen chloride, resulting in the deprotected compound 42.
Asymmetric hydrogenations
The synthesized chiral compounds were tested in the asymmetric hydrogenation of acetophenone. A common protocol of the catalytic hydrogen transfer reaction was used. As metal sources {[(η6-p-cymene)Ru(μ-Cl)Cl}2] [55, 56], Cp*RuCl(isoprene) [57] and [IrCl(COD)]2 [43] were used (Scheme 12).
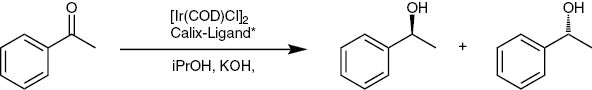
Hydrogen transfer with iridium and a chiral calix-ligand.
Calix[4]arene C9 amides – lower rim and methylene bridged
The reductions were performed in the presence of the amides 7–18, 31 and 34–38 using {[(η6-p-cymene)Ru(μ-Cl)Cl}2] and [IrCl(COD)]2 as catalyst. The reductions were faster in the presence of the amides than in their absence. Unfortunately no chiral influence of the co-catalyst on the product 1-phenylethanol was observable.
Calix[4]arene C10 esters – lower rim and methylene bridged
The reductions were performed in the presence of the esters 22, 24, 39, 40 and 42 using {[(η6-p-cymene)Ru(μ-Cl)Cl}2], Cp*RuCl(isoprene) and [IrCl(COD)]2 as catalyst. The reductions were complete in less than half an hour, but again no chiral influence of the co-catalysts was observed.
Conclusions
The syntheses of serveral chiral derivatives of tert-Butyl-calix[4]arene 1 and all-methoxy-tert-butyl-calix[4]arene 25 are described. The modification route in the case of 1 followed the established protocols while in the case of 25 with the di-acid 30 also an unprecedented derivative was synthesized. Several amides based on the chiral compounds QCI-C9-amine and QCD-C9-amine and their derivatives were generated. The corresponding esters using QCI and QCD could not be synthesized. The esters formed by the various calix[4]arenes and the di-Boc-C10-hydroxy-QCI-C9-amine 19 or the di-Boc-C10-hydroxy-QCD-C9-amine 20 were accessible.
Unfortunately the use of these new chiral compounds provided no enantioselective effect in the hydrogen transfer reaction of acetophenone, although the reaction time was shortened.
Furthermore we present for compounds 9 and 12 X-ray analyses that confirm the cone conformation of these derivatives.
Experimental
Compound 7: A mixture of the 1,3-calix[4]arene acid chloride 4 (300 mg, 0.374 mmol) was dissolved in dry DCM (10 mL) and dropped to a solution of ethenyl-QCI-NH2 (136 mg, 0.822 mmol) and Et3N (95 mg, 0.935 mmol) in dry DCM (10 mL). The reaction mixture was stirred at r.t. overnight. The solution was concentrated to dryness and the crude product, was purified by flash chromatography. It remained a white solid. Yield: 30 % (126 mg, 0.115 mmol).
IR (ATR): 1/λ = 3347, 3048, 2953, 2864, 1755, 1677, 1599, 1527, 1483, 1392, 1362, 1300, 1194, 1125, 1100, 1041, 992, 910, 873, 820, 802, 777, 756, 724, 669, 628, 573, 555. UV/Vis (DCM): λmax. (lg ε) = 228 (0.690), 282 (0.166), 289 (0.164). 1H NMR (300.1 MHz, CDCl3): δ = 8.82 (bs, 2H, NH), 7.49 (s, 2H, OH), 7.11–7.01 (m, 4 H, Ar-H), 6.88–6.79 (m, 4 H, Ar-H), 5.82–5.65 (m, 2 H, H-10), 4.93–4.82 (m, 4 H, H-11, H-11), 4.72–4.38 (m, 4 H, OCH2), 4.25–4.07 (d, J = 13.4, 4 H, Ar-CH2-Ar), 3.74–3.58 (m, 2 H, H-2), 3.42–3.35 (d, J = 13.4, 4 H, Ar-CH2-Ar), 3.34–3.26 (m, 2 H, H-9), 3.09–2.86 (m, 6H, H-9, H-6, H-7), 2.61–2.43 (m, 4 H, H-6, H-7), 2.26–2.11 (m, 2 H, H-5), 1.95–1.81 (m, 2 H, H-3), 1.74–1.65 (m, 2 H, H-4), 1.58–1.37 (m, 4 H, H-8, H-8), 1.28 (s, 18 H, t-Bu), 1.03–0.92 (m, 20 H, t-Bu, H-3). 13C NMR (75.47 MHz, CDCl3): δ = 168.64, 149.72, 149.33, 147.92, 142.68, 141.81, 132.34, 132.11, 127.78, 127.19, 126.27, 125.73, 125.42, 125.23, 114.09, 78.44, 74.71, 55.53, 55.46, 42.43, 40.49, 39.54, 34.00, 33.86, 32.05, 31.90, 31.61, 30.91, 27.82, 27.52, 26.96, 26.59. MS (ESI): 1061.75 [M + H+], 1083.78 [M + Na+].
Compound 8: A mixture of the 1,3-calix[4]arene acid chloride 4 (300 mg, 0.374 mmol) was dissolved in dry DCM (10 mL) and dropped to a solution of ethynyl-QCI-NH2 (134 mg, 0.822 mmol) and Et3N (95 mg, 0.935 mmol) in dry DCM (10 mL). The reaction mixture was stirred at room temperature overnight. The solution was concentrated to dryness and the crude product, was purified by flash chromatography. It remained a white solid. Yield: 28 % (115 mg, 0.108 mmol).
IR (ATR): 1/λ = 3346, 3311, 2953, 2867, 2110, 1667, 1598, 1536, 1482, 1392, 1362, 1299, 1193, 1125, 1105, 1040, 946, 912, 872, 818, 778, 750, 627, 588, 544. UV/Vis (DCM): λmax. (lg ε) = 228 (0.781), 282 (0.198), 289 (0.196). 1H NMR (300.1 MHz, CDCl3): δ = 8.84 (bs, 2H, NH), 7.48 (s, 2H, OH), 7.10–6.99 (m, 4 H, Ar-H), 6.91–6.82 (m, 4 H, Ar-H), 4.74–4.62 (m, 4 H, OCH2), 4.28–4.03 (d, J = 13.4, 4 H, Ar-CH2-Ar), 3.78–3.63 (m, 2 H, H-2), 3.47–3.31 (d, J = 13.4, 4 H, Ar-CH2-Ar), 3.32–3.05 (m, 2 H, H-9), 3.03–2.78 (m, 4 H, H-6, H-7), 2.72–2.64 (m, 2 H, 9-H), 2.46–2.33 (m, 4 H, H-6, H-7), 2.22–2.19 (m, 4 H, H-5, H-3), 1.87 (d, J = 2.4, 2 H, H-11), 1.59–1.48 (m, 2 H, H-4), 1.46–1.30 (m, 4 H, H-8, H-8), 1.27 (s, 18 H, t-Bu), 1.03–0.95 (m, 20 H, t-Bu, H-3). 13C NMR (75.47 MHz, CDCl3): δ = 237.74, 236.88, 168.52, 149.75, 149.19, 148.08, 142.63, 132.46, 132.13, 127.76, 126.89, 126.43, 125.75, 125.50, 125.18, 87.92, 74.71, 68.49, 56.87, 55.01, 41.94, 39.83, 34.04, 33.85, 32.18, 31.96, 31.62, 30.94, 27.44, 26.73, 26.68, 26.33. MS (ESI): 1057.76 [M + H+], 1079.73 [M + Na+].
Compound 9: A mixture of the 1,3-calix[4]arene acid chloride 4 (300 mg, 0.374 mmol) was dissolved in dry DCM (10 mL) and dropped to a solution of Ethyl-QCI-NH2 (138 mg, 0.822 mmol) and Et3N (95 mg, 0.935 mmol) in dry DCM (10 mL). The reaction mixture was stirred at room temperature overnight. The solution was concentrated to dryness and the crude product, was purified by flash chromatography. It remained a white solid. Yield: 35 % (142 mg, 0.133 mmol).
IR (ATR): 1/λ = 3441, 3345, 3047, 2955, 2927, 2863, 1679, 1524, 1483, 1362, 1300, 1195, 1125, 1103, 1044, 981, 945, 911, 873, 821, 802, 773, 760, 687, 657, 629, 574. UV/Vis (DCM): λmax. (lg ε) = 229 (1.246), 282 (0.322), 289 (0.320). 1H NMR (300.1 MHz, CDCl3): δ = 8.83 (bs, 2H, NH), 7.51 (s, 2H, OH), 7.09–7.02 (m, 4 H, Ar-H), 6.86–6.78 (m, 4 H, Ar-H), 4.72–4.37 (m, 4 H, OCH2), 4.25–4.06 (d, J = 13.4, 4 H, Ar-CH2-Ar), 3.73–3.59 (m, 2 H, H-2), 3.44–3.35 (d, J = 13.4, 4 H, Ar-CH2-Ar), 3.34–3.24 (m, 2 H, H-9), 3.07–2.81 (m, 6H, H-9, H-6, H-7), 2.62–2.42 (m, 2 H, H-6), 2.24–2.13 (m, 2 H, H-7), 1.88–1.74 (m, 2 H, H-5), 1.66–1.58 (m, 2 H, H-4), 1.56–1.43 (m, 4 H, H-8, H-8), 1.36–1.15 (m, 24 H, t-Bu, H-3, H-10, H-10), 0.98 (m, 20 H, t-Bu, H-3), 0.78–0.69 (m, 6 H, CH3). 13C NMR (75.47 MHz, CDCl3): δ = 168.61, 149.75, 149.35, 147.80, 142.62, 132.31, 132.06, 127.89, 127.35, 126.23, 125.65, 125.36, 125.16, 74.67, 57.30, 55.31, 42.55, 40.58, 37.22, 33.96, 33.84, 31.97, 31.85, 31.60, 30.90, 28.52, 27.43, 26.95, 26.41, 25.19, 11.96. MS (ESI): 1065.75 [M + H+].
Compound 10: A mixture of the 1,3-calix[4]arene acid chloride 4 (300 mg, 0.374 mmol) was dissolved in dry DCM (10 mL) and dropped to a solution of ethenyl-QCD-NH2 (136 mg, 0.822 mmol) and Et3N (95 mg, 0.935 mmol) in dry DCM (10 mL). The reaction mixture was stirred at room temperature overnight. The solution was concentrated to dryness and the crude product, was purified by flash chromatography. It remained a white solid. Yield: 37 % (147 mg, 0.138 mmol).
IR (ATR): 1/λ = 3342, 3048, 2951, 2867, 1679, 1533, 1482, 1392, 1362, 1299, 1195, 1125, 1099, 1040, 994, 911, 872, 821, 756, 660, 629, 574, 553. UV/Vis (DCM): λmax. (lg ε) = 229 (1.105), 282 (0.276), 289 (0.275). 1H NMR (300.1 MHz, CDCl3): δ = 8.78 (bs, 2H, NH), 7.48 (s, 2H, OH), 7.12–7.02 (m, 4 H, Ar-H), 6.87–6.79 (m, 4 H, Ar-H), 5.94–5.78 (m, 2 H, H-10), 5.09–4.96 (m, 4 H, H-11, H-11), 4.71–4.36 (m, 4 H, OCH2), 4.24–4.06 (d, J = 13.4, 4 H, Ar-CH2-Ar), 3.62–3.51 (m, 2 H, H-2), 3.43–3.34 (d, J = 13.4, 4 H, Ar-CH2-Ar), 3.33–3.27 (m, 2 H, H-6), 2.98–3.84 (m, 2 H, H-9), 2.79–2.58 (m, 8 H, H-6, H-7, H-7, H-9), 2.27–2.11 (m, 2 H, H-5), 1.73–1.64 (m, 2 H, H-4), 1.59–1.45 (m, 6 H, H-3, H-8, H-8), 1.36–1.31 (m, 2 H, H-3), 1.28 (s, 18 H, t-Bu), 0.98 (s, 18 H, t-Bu). 13C NMR (75.47 MHz, CDCl3): δ = 237.66, 237.52, 168.57, 149.79, 149.34, 147.86, 142.60, 140.51, 132.33, 132.11, 127.88, 127.31, 126.24, 125.69, 125.36, 125.20, 114.54, 74.72, 55.27, 48.92, 46.76, 41.48, 39.78, 33.98, 33.86, 31.88, 31.61, 30.91, 27.60, 26.96, 26.71, 25.79. MS (ESI): 1061.94 [M + H+], 1083.67 [M + Na+].
Compound 11: A mixture of the 1,3-calix[4]arene acid chloride 4 (300 mg, 0.374 mmol) was dissolved in dry DCM (10 mL) and dropped to a solution of ethynyl-QCI-NH2 (134 mg, 0.822 mmol) and Et3N (95 mg, 0.935 mmol) in dry DCM (10 mL). The reaction mixture was stirred at room temperature overnight. The solution was concentrated to dryness and the crude product, was purified by flash chromatography. It remained a white solid. Yield: 26 % (105 mg, 0.099 mmol).
IR (ATR): 1/λ = 3409, 3347, 3286, 2945, 2868, 2111, 1680, 1599, 2525, 1483, 1439, 1392, 1362, 1298, 1194, 1124, 1100, 1040, 979, 945, 911, 874, 819, 802, 774, 751, 653, 628, 589, 571. UV/Vis (DCM): λmax. (lg ε) = 229 (0.971), 282 (0.250), 289 (0.248). 1H NMR (300.1 MHz, CDCl3): δ = 8.81 (bs, 2H, NH), 7.41 (s, 2H, OH), 7.09–7.01 (m, 4 H, Ar-H), 6.87–6.78 (m, 4 H, Ar-H), 4.76–4.37 (m, 4 H, OCH2), 4.26–4.05 (d, J = 13.4, 4 H, Ar-CH2-Ar), 3.68–3.56 (m, 2 H, H-2), 3.54–3.47 (m, 2 H, H-6), 3.45–3.28 (d, J = 13.4, 4 H, Ar-CH2-Ar), 3.04–2.54 (m, 8 H, H-6, H-7, H-9, H-9), 2.48–2.37 (m, 2 H, H-7), 2.29–2.17 (m, 2 H, H-5), 2.12 (d, J = 2.4, 2H, H-11), 1.91–1.84 (m, 2 H, H-4), 1.66–1.58 (m, 4 H, H-8, H-8), 1.56–1.36 (m, 4 H, H-3, H-3), 1.28 (s, 18 H, t-Bu), 0.98 (s, 18 H, t-Bu). 13C NMR (75.47 MHz, CDCl3): δ = 168.58, 149.76, 149.37, 147.98, 142.64, 132.36, 132.06, 127.84, 127.05, 126.36, 125.69, 125.45, 125.21, 74.79, 69.47, 55.02, 48.50, 48.03, 41.54, 34.02, 33.89, 31.93, 31.63, 30.92, 28.22, 27.36, 26.04. MS (ESI): 1057.99 [M + H+], 1079.71 [M + Na+], 1095.60 [M + K+].
Compound 12: A mixture of the 1,3-calix[4]arene acid chloride 4 (300 mg, 0.374 mmol) was dissolved in dry DCM (10 mL) and dropped to a solution of ethyl-QCI-NH2 (138 mg, 0.822 mmol) and Et3N (95 mg, 0.935 mmol) in dry DCM (10 mL). The reaction mixture was stirred at room temperature overnight. The solution was concentrated to dryness and the crude product, was purified by flash chromatography. It remained a white solid. Yield: 28 % (112 mg, 0.105 mmol).
IR (ATR): 1/λ = 3346, 3047, 2954, 2867, 1678, 1535, 1482, 1459, 1391, 1361, 1298, 1197, 1125, 1099, 1040, 946, 812, 872, 821, 755, 661, 627, 573, 551. UV/Vis (DCM): λmax. (lg ε) = 229 (1.254), 282 (0.326), 289 (0.323). 1H NMR (300.1 MHz, CDCl3): δ = 8.76 (bs, 2H, NH), 7.52 (s, 2H, OH), 7.11–7.01 (m, 4 H, Ar-H), 6.87–6.76 (m, 4 H, Ar-H), 4.71–4.36 (m, 4 H, OCH2), 4.22–4.08 (d, J = 13.4, 4 H, Ar-CH2-Ar), 3.65–3.53 (m, 2 H, H-2), 3.44–3.26 (m, 6 H, Ar-CH2-Ar, H-6), 2.97–2.39 (m, 10 H, H-6, H-7, H-7, H-9, H-9), 2.28–2.14 (m, 2 H, H-5), 1.62–1.19 (m, 32 H, H-4, H-3, H-8, H-8, H-3, H-10, H-10, t-Bu), 0.97 (s, 18 H, t-Bu), 0.88–0.81 (m, 6 H, H-11). 13C NMR (75.47 MHz, CDCl3): δ = 169.62, 149.86, 149.42, 147.74, 142.58, 132.34, 132.13, 128.08, 127.56, 126.20, 125.65, 125.32, 125.17, 74.71, 55.42, 49.45, 49.05, 48.61, 41.49, 37.56, 33.98, 33.87, 31.87, 31.63, 30.93, 27.47, 26.97, 25.69, 25.55, 11.98. MS (ESI): 1065.73 [M + H+].
Compound 13: A mixture of the calix[4]arene acid 5 (180 mg, 0.2 mmol) and oxalyl chloride (2 mL) in dry CCl4 (20 mL) was refluxed for 3 h. After cooling, the solution was evaporated to dryness and the residue was dried under vacuum. The resulting yellow solid was dissolved in dry DCM (5 mL) and evaporated again to dryness under vacuum. The remaining acid chloride was dissolved in dry DCM (5 mL) and dropped to a solution of ethenyl-QCI-NH2 (198 mg, 1.2 mmol) and Et3N (161.9 mg, 1.6 mmol) in dry DCM (10 mL). The reaction mixture was stirred at room temperature overnight. The solution was concentrated to dryness and the crude product, was purified by flash chromatography. It remained a white solid. Yield: 29 % (87 mg, 0.059 mmol).
IR (ATR): 1/λ = 3310, 3073, 2950, 2927, 2863, 1659, 1538, 1477, 1391, 1361, 1298, 1238, 1194, 1126, 1043, 992, 909, 870, 817, 749, 665, 586, 556. UV/Vis (DCM): λmax. (lg ε) = 229 (0.706), 276 (0.054). 1H NMR (300.1 MHz, CDCl3): δ = 7.79 (bs, 4H, NH), 6.74 (s, 8 H, Ar-H), 5.93–5.78 (m, 4 H, H-10), 5.09–4.94 (m, 8 H, H-11, H-11), 4.62–4.41 (m, 12 H, OCH2, Ar-CH2-Ar), 3.73–3.48 (m, 8 H, H-2, H-9), 3.21–2.89 (m, 16H, Ar-CH2-Ar, H-9, H-6, H-7), 2.69–2.51 (m, 8 H, H-6, H-7), 2.33–2.14 (m, 4 H, H-5), 1.88–1.76 (m, 4 H, H-3), 1.73–1.64 (m, 4 H, H-4), 1.54–1.42 (m, 8 H, H-8, H-8), 1.06 (s, 36 H, t-Bu), 0.91–0.79 (m, 4 H, H-3). 13C NMR (75.47 MHz, CDCl3): δ = 169.79, 152.90, 145.26, 141.72, 132.82, 132.76, 125.62, 125.49, 114.40, 74.39, 55.73, 55.21, 42.12, 40.45, 39.78, 33.79, 31.65, 31.30, 27.68, 27.51, 26.28. MS (ESI): 1473.99 [M + H+], 1496.94 [M + Na+].
Compound 14: A mixture of the calix[4]arene acid 5 (180 mg, 0.2 mmol) and oxalyl chloride (2 mL) in dry CCl4 (20 mL) was refluxed for 3 h. After cooling, the solution was evaporated to dryness and the residue was dried under vacuum. The resulting yellow solid was dissolved in dry DCM (5 mL) and evaporated again to dryness under vacuum. The remaining acid chloride was dissolved in dry DCM (5 mL) and dropped to a solution of ethynyl-QCI-NH2 (195.6 mg, 1.2 mmol) and Et3N (161.9 mg, 1.6 mmol) in dry DCM (10 mL). The reaction mixture was stirred at overnight. The solution was concentrated to dryness and the crude product, was purified by flash chromatography. It remained a white solid. Yield: 32 % (93.5 mg, 0.064 mmol).
IR (ATR): 1/λ = 3289, 3061, 2950, 2865, 2110, 1650, 1542, 1477, 1391, 1361, 1299, 1238, 1194, 1126, 1049, 941, 870, 816, 777, 744, 628. UV/Vis (DCM): λmax. (lg ε) = 229 (0.776), 276 (0.065), 281 (0.065). 1H NMR (300.1 MHz, CDCl3): δ = 7.75 (bs, 4H, NH), 6.74 (s, 8 H, Ar-H), 4.63–4.39 (m, 12 H, OCH2, Ar-CH2-Ar), 3.65–3.52 (m, 4 H, H-2), 3.34–2.73 (m, 24H, Ar-CH2-Ar, H-9, H-9, H-6, H-7, H-7), 2.64–2.45 (m, 8 H, H-5, H-6), 2.24–2.12 (m, 4 H, H-3), 2.08 (d, 4 H, J = 2.01, H-11), 1.97–1.88 (m, 4 H, H-4), 1.60–1.39 (m, 8 H, H-8, H-8), 1.07 (s, 36 H, t-Bu), 1.02–0.99 (m, 4 H, H-3). 13C NMR (75.47 MHz, CDCl3): δ = 236.32, 169.84, 153.03, 145.34, 132.77, 125.70, 125.54, 74.51, 68.87, 56.80, 54.92, 41.77, 40.02, 33.83, 31.66, 31.34, 27.47, 26.75, 26.49, 26.17. MS (ESI): 1467.13 [M + H+], 1488.20 [M + Na+].
Compound 15: A mixture of the calix[4]arene acid 5 (180 mg, 0.2 mmol) and oxalyl chloride (2 mL) in dry CCl4 (20 mL) was refluxed for 3 h. After cooling, the solution was evaporated to dryness and the residue was dried under vacuum. The resulting yellow solid was dissolved in dry DCM (5 mL) and evaporated again to dryness under vacuum. The remaining acid chloride was dissolved in dry DCM (5 mL) and dropped to a solution of ethyl-QCI-NH2 (200 mg, 1.2 mmol) and Et3N (161.9 mg, 1.6 mmol) in dry DCM (10 mL). The reaction mixture was stirred at room temperature overnight. The solution was concentrated to dryness and the crude product, was purified by flash chromatography. It remained a white solid. Yield: 32 % (93.5 mg, 0.064 mmol).
IR (ATR): 1/λ = 3310, 3047, 2953, 2926, 2862, 1665, 1536, 1477, 1459, 1361, 1298, 1237, 1194, 1126, 1044, 948, 911, 870, 816, 736, 556. UV/Vis (DCM): λmax. (lg ε) = 230 (1.316), 275 (0.117), 281 (0.115). 1H NMR (300.1 MHz, CDCl3): δ = 7.76 (bs, 4H, NH), 6.73 (s, 8 H, Ar-H), 4.61–4.39 (m, 12 H, OCH2, Ar-CH2-Ar), 3.62–3.43 (m, 4 H, H-2), 3.31–2.82 (m, 24H, Ar-CH2-Ar, H-9, H-9, H-6, H-7, H-7), 2.63–2.45 (m, 4 H, H-6,), 2.39–2.24 (m, 4 H, H-5), 1.89–1.75 (m, 4 H, H-3), 1.72–1.64 (m, 4 H, H-4), 1.59–1.29 (m, 16 H, H-8, H-8, H-10), 1.07 (s, 36 H, t-Bu), 0.99–0.79 (m, 16 H, H-3, H-11). 13C NMR (75.47 MHz, CDCl3): δ = 169.72, 153.06, 145.19, 132.85, 132.74, 125.62, 125.46, 74.46, 57.43, 54.94, 42.26, 40.58, 37.39, 33.79, 31.67, 31.32, 28.55, 27.52, 26.19, 25.31, 12.12. MS (ESI): 1483.16 [M + H+], 1505.13 [M + Na+].
Compound 16: A mixture of the calix[4]arene acid 5 (180 mg, 0.2 mmol) and oxalyl chloride (2 mL) in dry CCl4 (20 mL) was refluxed for 3 h. After cooling, the solution was evaporated to dryness and the residue was dried under vacuum. The resulting yellow solid was dissolved in dry DCM (5 mL) and evaporated again to dryness under vacuum. The remaining acid chloride was dissolved in dry DCM (5 mL) and dropped to a solution of ethenyl-QCD-NH2 (198 mg, 1.2 mmol) and Et3N (161.9 mg, 1.6 mmol) in dry DCM (10 mL). The reaction mixture was stirred at room temperature overnight. The solution was concentrated to dryness and the crude product, was purified by flash chromatography. It remained a white solid. Yield: 27 % (80 mg, 0.054 mmol).
IR (ATR): 1/λ = 3305, 3073, 2935, 2867, 1657, 1537, 1477, 1458, 1391, 1361, 1299, 1239, 1195, 1126, 1048, 994, 909, 870, 822, 755, 661, 582, 554. UV/Vis (DCM): λmax. (lg ε) = 229 (0.986), 275 (0.079). 1H NMR (300.1 MHz, CDCl3): δ = 7.92 (bs, 4H, NH), 6.74 (s, 8 H, Ar-H), 5.99–5.76 (m, 4 H, H-10), 5.12–4.96 (m, 8 H, H-11, H-11), 4.68–4.41 (m, 12 H, OCH2, Ar-CH2-Ar), 3.63–3.41 (m, 4 H, H-2), 3.34–3.09 (m, 8 H, Ar-CH2-Ar, H-6), 3.08–2.62 (m, 20 H, H-6, H-7, H-7, H-9, H-9), 2.35–2.14 (m, 4 H, H-5), 1.78–1.69 (m, 4 H, H-4), 1.63–1.49 (m, 12 H, H-3, H-8, H-8), 1.37–1.24 (m, 4 H, H-3), 1.07 (s, 36 H, t-Bu). 13C NMR (75.47 MHz, CDCl3): δ = 169.86, 153.19, 145.22, 140.23, 132.82, 125.64, 125.53, 114.75, 74.47, 55.31, 48.94, 46.87, 41.18, 39.91, 33.81, 31.73, 31.33, 27.63, 26.55, 25.51. MS (ESI): 1473.98 [M + H+], 1496.93 [M + Na+].
Compound 17: A mixture of the calix[4]arene acid 5 (180 mg, 0.2 mmol) and oxalyl chloride (2 mL) in dry CCl4 (20 mL) was refluxed for 3 h. After cooling, the solution was evaporated to dryness and the residue was dried under vacuum. The resulting yellow solid was dissolved in dry DCM (5 mL) and evaporated again to dryness under vacuum. The remaining acid chloride was dissolved in dry DCM (5 mL) and dropped to a solution of ethynyl-QCD-NH2 (195.6 mg, 1.2 mmol) and Et3N (161.9 mg, 1.6 mmol) in dry DCM (10 mL). The reaction mixture was stirred at room temperature overnight. The solution was concentrated to dryness and the crude product, was purified by flash chromatography. It remained a white solid. Yield: 33 % (98mg, 0.066 mmol).
IR (ATR): 1/λ = 3304, 2948, 2868, 2110, 1660, 1534, 1477, 1459, 1391, 1361, 1299, 1239, 1194, 1126, 1104, 1037, 1001, 870, 817, 751, 624, 559. UV/Vis (DCM): λmax. (lg ε) = 229 (0.96), 275 (0.083). 1H NMR (300.1 MHz, CDCl3): δ = 7.73 (bs, 4H, NH), 6.74 (s, 8 H, Ar-H), 4.67–4.41 (m, 12 H, OCH2, Ar-CH2-Ar), 3.61–3.48 (m, 4 H, H-2), 3.39–3.27 (m, 4 H, H-6), 3.20 (d, J = 13.7, 4 H, Ar-CH2-Ar), 3.01–2.58 (m, 16 H, H-6, H-7, H-9, H-9), 2.56–2.43 (m, 4 H, H-7), 2.28–2.19 (m, 4 H, H-5), 2.12 (d, J = 2.37, 4H, H-11), 1.98–1.89 (m, 4 H, H-4), 1.63–1.44 (m, 16 H, H-3, H-8, H-8, H-3), 1.07 (s, 36 H, t-Bu). 13C NMR (75.47 MHz, CDCl3): δ = 169.78, 153.12, 145.29, 132.83, 132.76, 125.66, 125.55, 77.20, 74.48, 69.45, 55.11, 48.49, 47.99, 41.21, 33.83, 31.70, 31.35, 28.23, 27.49, 25.93, 25.32. MS (ESI): 1467.10 [M + H+], 1488.9 [M + Na+].
Compound 18: A mixture of the calix[4]arene acid 5 (180 mg, 0.2 mmol) and oxalyl chloride (2 mL) in dry CCl4 (20 mL) was refluxed for 3 h. After cooling, the solution was evaporated to dryness and the residue was dried under vacuum. The resulting yellow solid was dissolved in dry DCM (5 mL) and evaporated again to dryness under vacuum. The remaining acid chloride was dissolved in dry DCM (5 mL) and dropped to a solution of ethyl-QCD-NH2 (200 mg, 1.2 mmol) and Et3N (161.9 mg, 1.6 mmol) in dry DCM (10 mL). The reaction mixture was stirred at room temperature overnight. The solution was concentrated to dryness and the crude product, was purified by flash chromatography. It remained a white solid. Yield: 30 % (89.78 mg, 0.060 mmol).
IR (ATR): 1/λ = 3307, 3058, 2931, 2865, 1660, 1536, 1477, 1459, 1361, 1298, 1239, 1195, 1126, 1053, 947, 870, 821, 752, 555. UV/Vis (DCM): λmax. (lg ε) = 229 (1.164), 276 (0.098). 1H NMR (300.1 MHz, CDCl3): δ = 8.01 (bs, 4H, NH), 6.73 (s, 8 H, Ar-H), 4.65–4.39 (m, 12 H, OCH2, Ar-CH2-Ar), 3.57–3.42 (m, 4 H, H-2), 3.29–3.08 (m, 8 H, Ar-CH2-Ar, H-6), 3.02–2.61 (m, 20 H, H-6, H-7, H-7, H-9, H-9), 2.52–2.37 (m, 4 H, H-5), 1.68–1.19 (m, 28 H, H-4, H-3, H-8, H-8, H-3, H-10, H-10), 1.07 (s, 36 H, t-Bu), 0.91–0.83 (m, 12 H, H-11). 13C NMR (75.47 MHz, CDCl3): δ = 169.86, 153.42, 145.13, 132.83, 132.69, 125.63, 125.51, 77.2, 74.49, 55.42, 49.09, 48.75, 41.36, 37.65, 33.81, 31.79, 31.35, 27.46, 25.90, 25.57, 25.43, 12.01. MS (ESI): 1482.96 [M + H+], 1504.06 [M + Na+].
Compound 21: A mixture of 0.1 g (0.13 mmol, 1 eq.) of diacid 3, was dissolved in 15 mL of dry DCM. To this mixture was added 0.145 g of Di-Boc-QCD-C10-OH 20 (0.39 mmol, 3.0 eq.). Then 127 mg of DMAP (1.04 mmol, 8 eq.) and 166 mg of 1-methylpyridinium iodite (0.65 mmol, 5 eq.) were added. The reaction was stirred at room temperature for 1 h (TLC shows complete conversion diethylether : MeOH NH3 20:2:4 drops, rf = 0.8). After the reaction was finished, water was added to the solution, the phases were separated. The organic phase was dried and evaporated, and the residue was purified by flash chromatography (20 g silica gel, eluent Et2O:MeOH 500:50:5 mL NH3). Yield: 53 % (101.3 mg, 0.069 mmol).
IR (ATR): 1/λ = 3458, 2954, 2871, 1740, 1694, 1482, 1391, 1363, 1299, 1257, 1174, 1155, 1125, 1062, 985, 921, 859, 780, 754, 597, 552. UV/Vis (DCM): λmax. (lg ε) = 228 (0.550), 284 (0.138). 1H NMR (400 MHz, CDCl3): δ = 7.02 (bs, 4 H, Ar-H), 6.96 (s, 2H, OH), 6.79 (bs, 4 H, Ar-H), 4.71 (s, 4 H, OCH2), 4.43 (dd, J = 13.2, J = 1.6, 4 H, Ar-CH2-Ar), 4.24–4.13 (m, 4 H, H-10, H-10), 3.69 (dd, J = 14.3, J = 9.3, 2 H, H-9), 3.50 (dd, J = 14.3, J = 6.1, 2 H, H-9), 3.32–3.21 (m, 2 H, H-6), 3.31 (d, J = 13.2, 4 H, Ar-CH2-Ar), 3.17–3.05 (m, 2 H, H-2), 2.90–2.70 (m, 4 H, H-7, H-7), 2.29–2.14 (m, 2 H, H-6), 2.00–1.88 (m, 2 H, H-5), 1.87–1.80 (m, 2 H, H-4), 1.78–1.69 (m, 2 H, H-3), 1.69–1.58 (m, 2 H, H-8), 1.49 (s, 36 H, t-BuO), 1.35–1.25 (m, 2 H, H-8), 1.27 (s, 18 H, t-Bu), 1.15–1.05 (m, 2 H, H-3), 0.96 (s, 18 H, t-Bu). 13C NMR (100 MHz, CDCl3): δ = 169.23, 153.11, 150.65, 150.26, 147.11, 141.48, 132.36, 132.33, 127.88, 127.86, 125.74, 125.08, 82.11, 77.21, 72.25, 67.24, 54.60, 49.92, 48.07, 45.25, 35.16, 33.89, 33.79, 31.81, 31.64, 31.40, 30.99, 28.05, 27.92, 23.05, 20.32. HR-MS (ESI): calculated for C86H124N4O16 + H+ (M + H+): 1469.9085, found 1469.9091 [M + H+], 735.4592 ½*[M + 2 * H+].
Compound 22: A solution of 0.05 g of compound 21 (0.034 mmol) in Et2O (10 mL) was cooled to 0 °C and 0.7 mL of a 2N solution of HCl (0.136 mmol) in Et2O was added dropwise. The solid thus formed was filtered off, washed with Et2O, dried and suspended in H2O. The pH was adjusted to basic value (pH-10) by addition of NaHCO3 and the mixture was extracted with ethyl acetate. After the separation of the two phases the organic one was dried over Na2SO4 and the solvent was removed under reduced pressure. Yield: 96 % (0.035 g, 0.032 mmol).
1H NMR (400 MHz, CDCl3): δ = 7.01 (bs, 4 H, Ar-H), 6.99 (s, 2H, OH), 6.80 (bs, 4 H, Ar-H), 4.79–4.69 (m, 4 H, OCH2), 4.45 (d, J = 13.1, 4 H, Ar-CH2-Ar), 4.27–4.14 (m, 4 H, H-10, H-10), 3.71 (dd, J = 14.1, J = 8.9, 2 H, H-9), 3.61 (dd, J = 14.1, J = 6.1, 2 H, H-9), 3.31 (d, J = 13.1, 4 H, Ar-CH2-Ar), 3.16–2.91 (m, 6 H, H-2, H-6, H-7), 2.65–2.51 (m, 2 H, H-7), 2.42–2.31 (m, 2 H, H-6), 2.40–2.32 (m, 2 H, H-3), 1.80–1.74 (m, 2 H, H-4), 1.69–1.53 (m, 6 H, H-5, H-8, H-8), 1.49 (s, 36 H, t-BuO), 1.26 (s, 18 H, t-Bu), 1.17–1.11 (m, 2 H, H-3), 0.96 (s, 18 H, t-Bu). 13C NMR (100 MHz, CDCl3): δ = 169.13, 152.89, 150.69, 150.33, 147.11, 141.36, 132.38, 127.83, 127.73, 125.76, 124.98, 82.00, 77.20, 72.21, 71.46, 54.69, 53.18, 47.82, 42.62, 34.16, 34.03, 33.86, 33.74, 31.82, 31.59, 30.85, 30.83, 28.03, 23.44, 21.55. HR-MS (ESI): calculated for C66H92N4O8 + H+ (M + H+): 1069.6988, found 1069.6965 [M + H+], further signal: 1091.6801 [M + Na+].
Compound 23: A mixture of 0.1 g (0.13 mmol, 1 eq.) of diacid 3, was dissolved in 15 mL of dry DCM. To this mixture was added 0.145 g of Di-Boc-QCI-C10-OH 19 (0.39 mmol, 3.0 eq.). Then 127 mg of DMAP (1.04 mmol, 8 eq.) and 166 mg of 1-methylpyridinium iodite (0.65 mmol, 5 eq.) were added. The reaction was stirred at room temperature for 1 hour (TLC shows complete conversion diethylether : MeOH NH3 20:2:4 drops, rf = 0.8). After the reaction was finished, water was added to the solution, the phases were separated. The organic phase was dried and evaporated, and the residue was purified by flash chromatography (20 g silicagel, eluent Et2O:MeOH:NH3 500:50:5). Yield: 57 % (108.8 mg, 0.074 mmol).
IR (ATR): 1/λ = 3459, 2955, 2868, 1740, 1695, 1482, 1392, 1363, 1300, 1259, 1174, 1155, 1125, 1056, 979, 889, 859, 821, 780, 754, 552. 1H NMR (400 MHz, CDCl3): δ = 7.02 (bs, 4 H, Ar-H), 7.01 (s, 2H, OH), 6.81 (bs, 4 H, Ar-H), 4.77–4.66 (m, 4 H, OCH2), 4.43 (d, J = 13.2, 4 H, Ar-CH2-Ar), 4.27–4.14 (m, 4 H, H-10, H-10), 3.68 (dd, J = 14.2, J = 9.0, 2 H, H-9), 3.56 (dd, J = 14.2, J = 6.1, 2 H, H-9), 3.31 (d, J = 13.2, 4 H, Ar-CH2-Ar), 3.18–2.93 (m, 6 H, H-2, H-6, H-7), 2.65–2.51 (m, 2 H, H-7), 2.42–2.31 (m, 2 H, H-6), 2.40–2.32 (m, 2 H, H-3), 1.80–1.74 (m, 2 H, H-4), 1.69–1.53 (m, 6 H, H-5, H-8, H-8), 1.49 (s, 36 H, t-BuO), 1.27 (s, 18 H, t-Bu), 1.17–1.11 (m, 2 H, H-3), 0.97 (s, 18 H, t-Bu). 13C NMR (100 MHz, CDCl3): δ = 169.25, 153.02, 150.59, 150.23, 147.11, 141.46, 132.33, 127.81, 127.79, 125.73, 125.06, 82.00, 77.21, 72.24, 66.40, 54.69, 53.18, 47.79, 41.52, 34.18, 33.98, 33.87, 33.76, 31.80, 31.61, 30.97, 30.85, 28.02, 23.40, 21.52. HR-MS (ESI): calculated for C86H124N4O16 + H+ (M + H+): 1469.9085, found 1469.9098 [M + H+], further signals: 1491.8907 [M + Na+], 735.4593 ½*[M + 2*H+], 746.4498 ½*[M + H+ + Na+], 735.4593 ½*[M + 2*Na+].
Compound 24: A solution of 0.05 g of compound 23 (0.034 mmol) in Et2O (10 mL) was cooled to 0 °C and 0.7 mL of a 2N solution of HCl (0.136 mmol) in Et2O was added dropwise. The solid thus formed was filtered off, washed with Et2O, dried and suspended in H2O. The pH was adjusted to basic value (pH-10) by addition of NaHCO3 and the mixture was extracted with ethyl acetate. After the separation of the two phases the organic one was dried over Na2SO4 and the solvent was removed under reduced pressure. Yield: 94 % (0.034 g, 0.031 mmol).
1H NMR (400 MHz, CDCl3): δ = 7.01 (bs, 4 H, Ar-H), 6.95 (s, 2H, OH), 6.78 (bs, 4 H, Ar-H), 4.71 (s, 4 H, OCH2), 4.44 (dd, J = 13.2, J = 1.5, 4 H, Ar-CH2-Ar), 4.28–4.15 (m, 4 H, H-10, H-10), 3.68 (dd, J = 14.1, J = 9.4, 2 H, H-9), 3.548 (dd, J = 14.1, J = 6.0, 2 H, H-9), 3.33–3.21 (m, 2 H, H-6), 3.31 (d, J = 13.2, 4 H, Ar-CH2-Ar), 3.17–3.05 (m, 2 H, H-2), 2.90–2.70 (m, 4 H, H-7, H-7), 2.29–2.14 (m, 2 H, H-6), 2.00–1.88 (m, 2 H, H-5), 1.87–1.80 (m, 2 H, H-4), 1.78–1.69 (m, 2 H, H-3), 1.71–1.59 (m, 2 H, H-8), 1.35–1.25 (m, 2 H, H-8), 1.27 (s, 18 H, t-Bu), 1.13–1.03 (m, 2 H, H-3), 0.97 (s, 18 H, t-Bu). 13C NMR (100 MHz, CDCl3): δ = 169.33, 153.12, 150.64, 150.21, 147.21, 141.58, 132.32, 132.30, 127.91, 127.89, 125.75, 125.09, 82.13, 77.31, 72.25, 71.24, 54.50, 49.98, 48.01, 45.35, 35.16, 33.91, 33.79, 31.81, 31.64, 31.03, 28.06, 27.93, 23.09, 20.42. HR-MS (ESI): calculated for C66H92N4O8 + H+ (M + H+): 1069.6988, found 1069.6971 [M + H+], further signal: 1091.6798 [M + Na+].
Compound 26: A solution of 2.8 mL (18.3 mmol) TMEDA in solved in 100 mL dry THF was cooled down to –78 °C. At this temperature 5.7 mL (13.8 mmol) n-BuLi (2.4 M in n-hexane) was added. After 30 min a solution of 2.0 g (2.8 mmol) 25,26,27,28-tetramethoxycalix[4]arene in 50 mL dry THF was added by syringe. The resulting dark red solution was allowed to warm up to room temperature. After 1 h, solid CO2 was added in excess, immediately changing the color of the solution to yellow. Removal of all volatiles resulted in a yellow residue, which was dissolved in a small amount of methanol. By addition of water, a white precipitate was formed giving after recrystallization from methanol as a microcrystalline solid. Yield: 42 % (0.88 g, 1.17 mmol)
IR (ATR): 1/λ = 3459, 2955, 2868, 1740, 1695, 1482, 1392, 1363, 1300, 1259, 1174, 1155, 1125, 1056, 979, 889, 859, 821, 780, 754, 552. 1H NMR (300 MHz, CDCl3): δ = 12.63 (bs, 1 H, COOH), 7.50–6.70 (m, 8 H, Ar-H), 5.03 (m, 1 H, Ar-CH-Ar), 4.40–3.05 (m, 18 H, Ar-CH2-Ar, OMe), 1.48–1.02 (m, 36 H, t-Bu).
13C NMR (100 MHz, CDCl3): δ = 169.25, 153.02, 150.59, 150.23, 147.11, 141.46, 132.33, 127.81, 127.79, 125.73, 125.06, 82.00, 77.21, 72.24, 66.40, 54.69, 53.18, 47.79, 41.52, 34.18, 33.98, 33.87, 33.76, 31.80, 31.61, 30.97, 30.85, 28.02, 23.40, 21.52. HR-MS (ESI): calculated for C86H124N4O16 + H+ (M + H+): 1469.9085, found 1469.9098 [M + H+], further signals: 1491.8907 [M + Na+], 735.4593 ½*[M + 2*H+], 746.4498 ½*[M + H+ + Na+], 735.4593 ½*[M + 2*Na+].
Compound 27: A mixture of 1.6 g of acid 26 (2.1 mmol), 1 mL of SOCl2 (1.64 g, 13.7 mmol) and one drop of DMF in dry DCM was stirred at room temperature under an argon atmosphere for 3 h. The excess SOCl2 was removed by distillation and the residue, was dissolved in DCM (5 mL) and added dropwise to a solution containing 0.5 g of 2-amino-2-methyl-1-propanol (5.0 mmol) in DCM (5 mL) at 0 °C. After the addition was complete, the reaction was stirred for 12 h. The precipitate was removed by filtration was purified by flash chromatography (30g silica gel, eluent ethyl-acetate:hexane 1:2). Yield: 68 % (1.1 g, 1.42 mmol).
IR (ATR): 1/λ = 3390, 2957, 2870, 2822, 1739, 1650, 1519, 1481, 1461, 1392, 1361, 1245, 1204, 1174, 1120, 1066, 1019, 949, 871, 799, 754, 665, 643, 552. UV/Vis (DCM): λmax. (lg ε) = 229 (0.610), 274 (0.062), 280 (0.065). 1H NMR (300 MHz, CDCl3): δ = 7.97 (bs, 1 H, NH), 7.40–6.55 (m, 8 H, Ar-H), 6.26 (bs, 1 H, OH), 5.35 (m, 1 H, Ar-CH-Ar), 4.35–2.75 (m, 20 H, Ar-CH2-Ar, OMe, CH2), 1.45–0.95 (m, 42 H, t-Bu, C(Me)2). 13C NMR (75 MHz, CDCl3): δ = 169.25, 153.02, 150.59, 150.23, 147.11, 141.46, 132.33, 127.81, 127.79, 125.73, 125.06, 82.00, 77.21, 72.24, 66.40, 54.69, 53.18, 47.79, 41.52, 34.18, 33.98, 33.87, 33.76, 31.80, 31.61, 30.97, 30.85, 28.02, 23.40, 21.52. HR-MS (ESI): calculated for C53H73N1O6 + Na+ (M + Na+): 842.5330, found 842.5347 [M + Na+], further signals: 843.54, 844.54.
Compound 28: One milliliter of SOCl2 was added dropwise to 1 g of the amide 27 (1.2 mmol) under argon atmosphere. After the solution was stirred for 2 h, the reaction mixture was poured into 20 mL iced water. The pH was adjusted to basic value with a solution of KOH (4 N) and the mixture twice extracted with ethyl-acetate (2 × 50 mL). The organic layer was separated, dried over Na2SO4 and evaporated. The rezidue was purified by flash chromatography (20 g silica gel, eluent Ethyl-Acetate:Hexane 1:2). Yield: 72 % (0.69 g, 0.864 mmol).
IR (ATR): 1/λ = 2959, 2870, 2822, 1660, 1603, 1482, 1462, 1392, 1361, 1286, 1246, 1204, 1120, 1020, 871, 798, 754, 665, 643, 608, 557. UV/Vis (DCM): λmax. (lg ε) = 230 (1.446), 274 (0.171), 280 (0.179). 1H NMR (300 MHz, CDCl3): δ = 7.46–6.67 (m, 8 H, Ar-H), 5.53 (bs, 1 H, Ar-CH-Ar), 4.42–2.35 (m, 20 H, Ar-CH2-Ar, OMe, CH2), 1.55–0.85 (m, 42 H, t-Bu, C(Me)2). 13C NMR (75 MHz, CDCl3): δ = 166.17, 155.04, 154.63, 153.88, 144.61, 144.42, 143.99, 134.83, 133.87, 132.81, 132.58, 127.17, 126.83, 126.17, 125.49, 125.03, 124.57, 123.81, 79.17, 77.21, 66.95, 61.11, 60.71, 60.00, 37.35, 37.01, 36.57, 36.02, 33.99, 33.85, 31.44, 31.37, 30.12, 28.39. HR-MS (ESI): calculated for C53H71N1O4 + Na+ (M + Na+): 824.5225, found 842.5248 [M + Na+], further signals: 825.53, 826.54.
Compound 29: A solution of 0.43 mL (3.9 mmol) TMEDA in solved in 10 mL dry THF was cooled down to –78 °C. At this temperature 1.16 mL (2.9 mmol) n-BuLi (2.4 M in n-hexane) was added. After 30 min a solution of 0.5 g (0.6 mmol) compound 28 in 20 mL dry THF was added by syringe. The resulting dark red solution was allowed to warm up to room temperature. After 1 h, solid CO2 was added in excess, immediately changing the color of the solution to yellow. Removal of all volatiles resulted in a yellow residue, which was dissolved in a small amount of methanol. By addition of water, a white precipitate was formed. Yield: 48 % (0.244 g, 0.288 mmol).
IR (ATR): 1/λ = 2959, 2870, 2822, 1660, 1603, 1482, 1462, 1392, 1361, 1286, 1246, 1204, 1120, 1020, 871, 798, 754, 665, 643, 608, 557. UV/Vis (DCM): λmax. (lg ε) = 230 (1.446), 274 (0.171), 280 (0.179). 1H NMR (300 MHz, CDCl3): δ = 12.55 (bs, 1 H, COOH), 7.54–6.73 (m, 8 H, Ar-H), 5.56 (bs, 1 H, Ar-CH-Ar), 5.12 (bs, 1 H, Ar-CH-Ar), 4.52–2.55 (m, 18 H, Ar-CH2-Ar, OMe, CH2), 1.75–0.96 (m, 42 H, t-Bu, C(Me)2). 13C NMR (75 MHz, CDCl3): δ = 176.93, 166.27, 155.14, 154.73, 153.91, 144.73, 144.62, 144.03, 134.82, 133.82, 132.88, 132.58, 127.27, 126.91, 126.19, 125.53, 125.11, 124.62, 123.83, 79.18, 77.25, 67.07, 61.17, 60.74, 60.13, 40.57, 37.36, 37.03, 36.04, 34.01, 33.86, 31.44, 31.39, 30.13, 28.42. HR-MS (ESI): calculated for C54H71N1O5 + Na+ (M + Na+): 868.5123 and C54H71N1O5 + H+ (M + H+): 846.5303, found 868.5131 [M + Na+] and 846.5311 [M + H+], further signals: 869.52, 870.53, 847.54, 848.54.
Compound 30: A solution of 0.2 g (0.236 mmol) compound 29 was suspended in an 10N NaOH solution. The mixture was refluxed for 48 h, cooled to room temperature and acidified to pH = 2 with HCl. The solid thus formed was collected by filtration, washed with water and dried. Yield: 57 % (0.106 g, 0.134 mmol).
IR (ATR): 1/λ = 2959, 2870, 2822, 1660, 1603, 1482, 1462, 1392, 1361, 1286, 1246, 1204, 1120, 1020, 871, 798, 754, 665, 643, 608, 557. UV/Vis (DCM): λmax. (lg ε) = 230 (1.446), 274 (0.171), 280 (0.179). 1H NMR (300 MHz, CDCl3): δ = 12.23 (bs, 2 H, COOH), 7.68–6.78 (m, 8 H, Ar-H), 5.78–5.52 (m, 2 H, Ar-CH-Ar), 4.75–2.58 (m, 16 H, Ar-CH2-Ar, OMe), 1.75–0.96 (m, 36 H, t-Bu). 13C NMR (75 MHz, CDCl3): δ = 176.93, 176.83, 155.14, 154.73, 153.91, 144.73, 144.62, 144.03, 134.82, 133.82, 132.88, 132.58, 127.27, 126.91, 126.19, 125.53, 125.11, 124.62, 123.83, 67.07, 61.17, 60.74, 60.13, 41.23, 40.57, 37.36, 37.03, 34.01, 33.86, 31.44, 31.39, 30.13. HR-MS (ESI): calculated for C50H64O6 + Na+ (M + Na+): 815.4493 and C50H64O6 + H+ (M + H+): 793.4674, found 815.4503 [M + Na+] and 793.4682 [M + H+], further signals: 816.45, 817.45, 794.46, 795.47.
Compound 31: A mixture of 25,26,27,28-tetramethoxycalix[4]arene-2-carboxylic acid (cone) 26 (100 mg, 0.133 mmol) and oxalyl chloride (3 mL) in dry CCl4 (10 mL) was refluxed for 3 h. After cooling, the solution was evaporated to dryness and the residue was dried under vacuum. The resulting yellow solid was dissolved in dry DCM (5 mL) and evaporated again to dryness under vacuum. The remaining acid chloride was dissolved in dry DCM (5 mL) and dropped to a solution of ethenyl-QCI-NH2 (24.26 mg, 0.146 mmol) and Et3N (16.15 mg, 0.156 mmol) in dry DCM (10 mL). The reaction mixture was stirred at r.t. overnight. The solution was concentrated to dryness and the crude product, was purified by flash chromatography. It remained a white solid. Yield: 29 % (35 mg, 0.039 mmol).
IR (ATR): 1/λ = 3415, 2952, 2867, 2822, 1669, 1481, 1462, 1392, 1361, 1245, 1204, 1174, 1120, 1020, 911, 871, 812, 796, 748, 666, 644, 554. UV/Vis (DCM): λmax. (lg ε) = 230 (1.487), 274 (0.163), 281 (0.169). 1H NMR (300.1 MHz, CDCl3): δ = 7.31 (bs, 1 H, NH), 7.26–6.52 (m, 8 H, Ar-H), 5.89–5.78 (m, 1 H, H-10), 5.41 (s, 1 H, CH-CONH), 4.97–5.03 (m, 2 H, H-11, H-11), 4.44–2.34 (m, 25 H, O-CH3, Ar-CH2-Ar, H-2, H-9, H-9, H-6, H-6, H-7, H-7), 2.32–2.27 (m, 1 H, H-5), 1.98–1.81 (m, 1 H, H-3), 1.72–1.61 (m, 1 H, H-4), 1.51–0.82 (m, 39 H, t-Bu, H-3, H-8, H-8). 13C NMR (75.47 MHz, CDCl3): δ = 172.49, 155.06, 154.95, 154.63, 154.2, 145.03, 144.42, 141.74, 134.75, 134.11, 133.47, 132.95, 132.68, 132.42, 127.21, 126.79, 126.06, 125.48, 124.95, 124.14, 123.08, 114.18, 77.21, 61.09, 60.59, 59.93, 55.75, 55.20, 45.65, 42.25, 40.15, 39.76, 37.01, 34.02, 33.92, 33.86, 33.78, 31.39, 31.28, 31.26, 29.89, 27.81, 27.39, 26.35. MS (ESI): 897.82 [M + H+], 919.75 [M + Na+].
Compound 33: A mixture of 0.1 g (0.133 mmol, 1 eq.) of acid 26, was dissolved in 15 mL of dry DCM. To this mixture was added 0.074 g of Di-Boc-QCI-C10-OH 19 (0.2 mmol, 1.5 eq.). Then 63.5 mg of DMAP (0.52 mmol, 4 eq.) and 83 mg of 1-methyl-pyridinium iodide (0.33 mmol, 2.5 eq.) were added. The reaction was stirred at room temperature for 1 h (TLC shows complete conversion diethylether : MeOH : NH3 20:2:4 drops, rf = 0.8). After the reaction was finished, water was added to the solution, the phases were separated. The organic phase was dried and evaporated, and the residue was purified by flash chromatography (20 g silica gel, eluent Et2O:MeOH 500:50:1 mL NH3).
IR (ATR): 1/λ = 2955, 2868, 2823, 1735, 1696, 1481, 1391, 1362, 1251, 1204, 1174, 1152, 1126, 1021, 949, 869, 795, 755, 642, 555. 1H NMR (400 MHz, CDCl3): δ = 7.56–6.55 (m, 8 H, Ar-H), 5.52 (bs, 1 H, Ar-CH-Ar), 4.22–4.12 (m, 2 H, H-10, H-10), 4.05–2.80 (m, 21 H, Ar-CH2-Ar, OMe, H-2, H-6, H-7), 3.67 (dd, J = 14.2, J = 9.1, 1 H, H-9), 3.54 (dd, J = 14.2, J = 6.1, 1 H, H-9), 2.59–2.45 (m, 1 H, H-7), 2.38–2.28 (m, 1 H, H-6), 2.05–1.88 (m, 1 H, H-5), 1.73–1.67 (m, 1 H, H-4), 1.65–1.49 (m, 1 H, H-3), 1.48 (s, 18 H, t-BuO), 1.46–1.05 (m, 39 H, t-Bu, H-3, H-8, H-8). 13C NMR (100 MHz, CDCl3): δ = 173.41, 155.01, 154.86, 154.64, 153.79, 153.05, 144.97, 144.29, 134.93, 134.80, 133.97, 132.73, 132.51, 131.86, 127.66, 127.41, 126.73, 125.85, 125.57, 125.49, 125.06, 124.04, 123.22, 82.02, 77.21, 66.07, 60.73, 60.28 59.93, 54.73 53.44, 47.81, 43.19, 41.55, 37.10, 34.14, 34.09, 34.01, 33.93, 33.86, 31.91, 31.71, 31.48, 31.45, 31.38, 31.00, 30.31, 29.68, 29.34, 28.22, 28.04, 23.51, 23.41, 22.67, 21.60, 14.09. HR-MS (ESI): calculated for C68H96N2O10 + H+ (M + H+): 1101.7138, found 1101.7149 [M + H+], further signals: 1102.71, 1103.72, 1104.72, 1123.69, 1124.70.
Compound 34: A mixture of 25,26,27,28-tetramethoxycalix[4]arene-2-carboxylic acid (cone) 26 (100 mg, 0.133 mmol) and oxalyl chloride (3 mL) in dry CCl4 (10 mL) was refluxed for 3 h. After cooling, the solution was evaporated to dryness and the residue was dried under vacuum. The resulting yellow solid was dissolved in dry DCM (5 mL) and evaporated again to dryness under vacuum. The remaining acid chloride was dissolved in dry DCM (5 mL) and dropped to a solution of ethyl-QCI-NH2 (24.55 mg, 0.146 mmol) and Et3N (16.15 mg, 0.156 mmol) in dry DCM (10 mL). The reaction mixture was stirred at r.t. overnight. The solution was concentrated to dryness and the crude product, was purified by flash chromatography. It remained a white solid. Yield: 29 % (35 mg, 0.039 mmol).
IR (ATR): 1/λ = 3414, 2953, 2866, 2822, 1669, 1481, 1462, 1392, 1361, 1246, 1204, 1174, 1120, 1020, 949, 871, 796, 644, 553. UV/Vis (DCM): λmax. (lg ε) = 230 (1.301), 274 (0.152), 280 (0.156). 1H NMR (300.1 MHz, CDCl3): δ = 7.31 (bs, 1 H, NH), 7.28–6.53 (m, 8 H, Ar-H), 5.41 (s, 1 H, CH-CONH), 4.45–2.08 (m, 25 H, O-CH3, Ar-CH2-Ar, H-2, H-9, H-9, H-6, H-6, H-7, H-7), 1.92–1.75 (m, 1 H, H-5), 1.73–1.62 (m, 1 H, H-4), 1.58–0.78 (m, 45 H, t-Bu, H-3, H-3, H-8, H-8, H-10, H-10, H-11). 13C NMR (75.47 MHz, CDCl3): δ = 155.00, 145.06, 132.99, 125.53, 59.99, 57.37, 42.23, 40.31, 37.36, 33.97, 33.84, 31.44, 31.33, 31.30, 30.29, 28.44, 27.47, 26.14, 25.14, 12.02. MS (ESI): 899.66 [M + H+], 921.67 [M + Na+].
Compound 35: A mixture of 25,26,27,28-tetramethoxycalix[4]arene-2-carboxylic acid (cone) 26 (100 mg, 0.133 mmol) and oxalyl chloride (3 mL) in dry CCl4 (10 mL) was refluxed for 3 h. After cooling, the solution was evaporated to dryness and the residue was dried under vacuum. The resulting yellow solid was dissolved in dry DCM (5 mL) and evaporated again to dryness under vacuum. The remaining acid chloride was dissolved in dry DCM (5 mL) and dropped to a solution of ethynyl-QCI-NH2 (23.95 mg, 0.146 mmol) and Et3N (16.15 mg, 0.156 mmol) in dry DCM (10 mL). The reaction mixture was stirred at r.t. overnight. The solution was concentrated to dryness and the crude product, was purified by flash chromatography. It remained a white solid. Yield: 33 % (41 mg, 0.045 mmol).
IR (ATR): 1/λ = 3414, 3310, 2953, 2869, 2822, 1669, 1481, 1462, 1392, 1361, 1298, 1246, 1204, 1174, 1120, 1020, 945, 871, 812, 796, 746, 625, 554. UV/Vis (DCM): λmax. (lg ε) = 230 (1.452), 274 (0.169), 281 (0.174). 1H NMR (300.1 MHz, CDCl3): δ = 7.31 (bs, 1 H, NH), 7.27–6.49 (m, 8 H, Ar-H), 5.42 (s, 1 H, CH-CONH), 4.38–2.41 (m, 25 H, O-CH3, Ar-CH2-Ar, H-2, H-9, H-9, H-6, H-6, H-7, H-7), 2.23–2.19 (m, 2 H, H-5, H-3), 2.04 (d, J = 2.4, 1 H, H-11), 1.94–1.86 (m, 1 H, H-4), 1.58–0.81 (m, 39 H, t-Bu, H-3, H-8, H-8). 13C NMR (75.47 MHz, CDCl3): δ = 155.00, 145.12, 133.01, 125.80, 87.78, 77.21, 68.59, 57.00, 41.89, 39.70, 33.97, 33.84, 31.44, 31.35, 31.31, 27.55, 26.65, 26.51, 26.25. MS (ESI): 895.84 [M + H+], 917.58 [M + Na+].
Compound 36: A mixture of 25,26,27,28-tetramethoxycalix[4]arene-2-carboxylic acid (cone) 26 (100 mg, 0.133 mmol) and oxalyl chloride (3 mL) in dry CCl4 (10 mL) was refluxed for 3 h. After cooling, the solution was evaporated to dryness and the residue was dried under vacuum. The resulting yellow solid was dissolved in dry DCM (5 mL) and evaporated again to dryness under vacuum. The remaining acid chloride was dissolved in dry DCM (5 mL) and dropped to a solution of ethenyl-QCD-NH2 (24.26 mg, 0.146 mmol) and Et3N (16.15 mg, 0.156 mmol) in dry DCM (10 mL). The reaction mixture was stirred at r.t. overnight. The solution was concentrated to dryness and the crude product, was purified by flash chromatography. It remained a white solid. Yield: 32 % (39 mg, 0.043 mmol).
IR (ATR): 1/λ = 3412, 2952, 2869, 2821, 1667, 1480, 1461, 1392, 1361, 1246, 1204, 1174, 1120, 1020, 912, 871, 797, 734, 643, 555. UV/Vis (DCM): λmax. (lg ε) = 230 (1.288), 274 (0.168), 280 (0.172). 1H NMR (300.1 MHz, CDCl3): δ = 7.32 (bs, 1 H, NH), 7.25–6.62 (m, 8 H, Ar-H), 5.92–5.73 (m, 1 H, H-10), 5.42 (s, 1 H, CH-CONH), 5.09–4.94 (m, 2 H, H-11, H-11), 4.36–2.35 (m, 25 H, O-CH3, Ar-CH2-Ar, H-2, H-9, H-9, H-6, H-6, H-7, H-7), 2.31–2.17 (m, 1 H, H-5), 1.78–1.67 (m, 1 H, H-4), 1.51–0.82 (m, 40 H, t-Bu, H-3, H-3, H-8, H-8). 13C NMR (75.47 MHz, CDCl3): δ = 172.67, 155.01, 154.68, 145.05, 139.93, 132.85, 125.54, 125.01, 123.14, 114.85, 77.21, 60.65, 48.93, 46.47, 41.09, 39.63, 33.97, 33.85, 31.45, 31.35, 31.32, 29.91, 27.51, 26.56, 25.55. MS (ESI): 897.65 [M + H+], 919.73 [M + Na+].
Compound 37: A mixture of 25,26,27,28-tetramethoxycalix[4]arene-2-carboxylic acid (cone) 26 (100 mg, 0.133 mmol) and oxalyl chloride (3 mL) in dry CCl4 (10 mL) was refluxed for 3 h. After cooling, the solution was evaporated to dryness and the residue was dried under vacuum. The resulting yellow solid was dissolved in dry DCM (5 mL) and evaporated again to dryness under vacuum. The remaining acid chloride was dissolved in dry DCM (5 mL) and dropped to a solution of ethyl-QCD-NH2 (24.55 mg, 0.146 mmol) and Et3N (16.15 mg, 0.156 mmol) in dry DCM (10 mL). The reaction mixture was stirred at r.t. overnight. The solution was concentrated to dryness and the crude product, was purified by flash chromatography. It remained a white solid. Yield: 32 % (39 mg, 0.043 mmol).
IR (ATR): 1/λ = 3390, 2952, 2868, 2821, 1669, 1480, 1461, 1392, 1361, 1245, 1204, 1174, 1120, 1061, 1020, 946, 870, 796, 644, 554. UV/Vis (DCM): λmax. (lg ε) = 230 (1.378), 274 (0.168), 280 (0.173). 1H NMR (300.1 MHz, CDCl3): δ = 7.32 (bs, 1 H, NH), 7.28–6.63 (m, 8 H, Ar-H), 5.42 (s, 1 H, CH-CONH), 4.38–2.23 (m, 25 H, O-CH3, Ar-CH2-Ar, H-2, H-9, H-9, H-6, H-6, H-7, H-7), 2.38–2.24 (m, 1 H, H-5), 1.68–1.88 (m, 43 H, t-Bu, H-3, H-3, H-4, H-8, H-8, H-10, H-10), 0.85–0.77 (m, 3 H, H-11). 13C NMR (75.47 MHz, CDCl3): δ = 155.10, 145.05, 132.95, 125.55, 55.30, 49.13, 48.26, 41.37, 37.58, 33.97, 33.84, 31.44, 31.33, 30.29, 27.60, 25.81, 25.55, 11.90. MS (ESI): 899.65 [M + H+], 919.67 [M + Na+].
Compound 38: A mixture of 25,26,27,28-tetramethoxycalix[4]arene-2-carboxylic acid (cone) 26 (100 mg, 0.133 mmol) and oxalyl chloride (3 mL) in dry CCl4 (10 mL) was refluxed for 3 h. After cooling, the solution was evaporated to dryness and the residue was dried under vacuum. The resulting yellow solid was dissolved in dry DCM (5 mL) and evaporated again to dryness under vacuum. The remaining acid chloride was dissolved in dry DCM (5 mL) and dropped to a solution of ethynyl-QCD-C9-NH2 (24.55 mg, 0.146 mmol) and Et3N (16.15 mg, 0.156 mmol) in dry DCM (10 mL). The reaction mixture was stirred at r.t. overnight. The solution was concentrated to dryness and the crude product, was purified by flash chromatography. It remained a white solid. Yield: 39 % (48 mg, 0.053 mmol).
IR (ATR): 1/λ = 3417, 3310, 2952, 2869, 2822, 1668, 1481, 1462, 1392, 1361, 1246, 1204, 1174, 1120, 947, 917, 871, 813, 797, 750, 626, 554. UV/Vis (DCM): λmax. (lg ε) = 230 (1.334), 274 (0.163), 280 (0.169). 1H NMR (300.1 MHz, CDCl3): δ = 7.31 (bs, 1 H, NH), 7.27–6.48 (m, 8 H, Ar-H), 5.42 (s, 1 H, CH-CONH), 4.38–2.36 (m, 26 H, O-CH3, Ar-CH2-Ar, H-2, H-9, H-9, H-6, H-6, H-7, H-7, H-5), 2.06 (d, J = 2.4, 1 H, H-11), 1.96–1.83 (m, 1 H, H-4), 1.69–0.68 (m, 40 H, t-Bu, H-3, H-3, H-8, H-8). 13C NMR (75.47 MHz, CDCl3): δ = 155.13, 155.01, 145.09, 144.43, 133.12, 126.95, 125.54, 125.02, 124.03, 87.04, 77.21, 69.35, 65.81, 60.63, 60.64, 59.99, 55.16, 48.59, 47.85, 41.29, 34.08, 33.97, 33.85, 31.44, 31.34, 31.30, 28.19, 27.28, 25.89, 25.42, 15.24. MS (ESI): 895.74 [M + H+], 917.57 [M + Na+].
Compound 39: A mixture of 0.1 g (0.133 mmol, 1 eq.) of acid 26, was dissolved in 15 mL of dry DCM. To this mixture was added 0.074 g of Di-Boc-QCD-C10-OH 20 (0.2 mmol, 1.5 eq.). Then 63.5 mg of DMAP (0.52 mmol, 4 eq.) and 83 mg of 1-Methy-pyridinium iodide (0.33 mmol, 2.5 eq.) were added. The reaction was stirred at room temperature for 1 h (TLC shows complete conversion diethylether : MeOH : NH3 20:2:4 drops, rf = 0.8). After the reaction was finished, water was added to the solution, the phases were separated. The organic phase was dried and evaporated, and the residue was purified by flash chromatography (20 g silica gel, eluent Et2O:MeOH 500:50:1 mL NH3).
IR (ATR): 1/λ = 2954, 2871, 2823, 1735, 1696, 1482, 1462, 1392, 1362, 1250, 1205, 1152, 1124, 1021, 869, 795, 771, 643, 555. 1H NMR (400 MHz, CDCl3): δ = 7.54–6.56 (m, 8 H, Ar-H), 5.53 (bs, 1 H, Ar-CH-Ar), 4.24–4.06 (m, 2 H, H-10, H-10), 4.02–2.85 (m, 21 H, Ar-CH2-Ar, OMe, H-2, H-6, H-7), 3.65 (dd, J = 14.2, J = 9.2, 1 H, H-9), 3.49 (dd, J = 14.2, J = 6.0, 1 H, H-9), 2.83–2.65 (m, 1 H, H-7), 2.20–2.10 (m, 1 H, H-6), 1.96–1.82 (m, 1 H, H-5), 1.80–1.75 (m, 1 H, H-4), 1.75–1.60 (m, 1 H, H-3), 1.50–0.96 (m, 39 H, t-Bu, H-3, H-8, H-8), 1.48 (s, 18 H, t-BuO). 13C NMR (100 MHz, CDCl3): δ = 173.40, 155.01, 154.85, 154.64, 153.10, 145.01, 134.01, 133.25, 132.70, 132.51, 131.85, 127.06, 126.53, 126.03, 125.87, 125.58, 125.06, 124.97, 124.21, 82.04, 77.73, 77.21, 66.83, 60.70, 59.94, 54.56, 49.94, 48.10, 45.30, 37.81, 37.08, 35.34, 34.09, 34.00, 33.93, 33.86, 31.45, 31.38, 30.98, 30.31, 29.68, 28.04, 25.65, 23.12, 20.44, 17.97. HR-MS (ESI): calculated for C68H96N2O10 + H+ (M + H+): 1101.7138, found 1101.7147 [M + H+], further signals: 1102.72, 1103.72, 1104.72, 1123.69, 1124.70.
Compound 40: A solution of 0.05 g of compound 33 (0.045 mmol) in Et2O (10 mL) was cooled to 0 °C and 0.45 mL of a 2N solution of HCl (0.09 mmol) in Et2O was added dropwise. The solid thus formed was filtered off, washed with Et2O, dried and suspended in H2O. The pH was adjusted to basic value (pH-10) by addition of NaHCO3 and the mixture was extracted with ethyl acetate. After the separation of the two phases the organic one was dried over Na2SO4 and the solvent was removed under reduced pressure. Yield: 95 % (0.038 g, 0.042 mmol).
1H NMR (400 MHz, CDCl3): δ = 7.58–6.53 (m, 8 H, Ar-H), 5.50 (bs, 1 H, Ar-CH-Ar), 4.26–4.08 (m, 2 H, H-10, H-10), 4.07–2.70 (m, 21 H, Ar-CH2-Ar, OMe, H-2, H-6, H-7), 3.65 (dd, J = 14.1, J = 9.0, 1 H, H-9), 3.51 (dd, J = 14.1, J = 6.1, 1 H, H-9), 2.59–2.45 (m, 1 H, H-7), 2.38–2.28 (m, 1 H, H-6), 2.05–1.88 (m, 1 H, H-5), 1.73–1.67 (m, 1 H, H-4), 1.65–1.49 (m, 1 H, H-3), 1.49 (s, 18 H, t-BuO), 1.46–1.04 (m, 39 H, t-Bu, H-3, H-8, H-8). 13C NMR (100 MHz, CDCl3): δ = 173.51, 155.11, 154.96, 154.84, 153.79, 152.69, 144.91, 144.21, 134.87, 134.89, 134.03, 132.83, 132.58, 131.89, 127.76, 127.45, 126.73, 125.82, 125.67, 125.42, 125.16, 124.13, 123.33, 82.12, 77.31, 71.16, 60.76, 60.36, 59.91, 54.73, 53.44, 47.81, 43.19, 42.55, 37.10, 34.14, 34.03, 33.86, 33.59, 33.46, 31.81, 31.69, 31.43, 31.41, 31.19, 31.00, 30.31, 29.68, 29.34, 28.46, 28.09, 23.53, 23.43, 22.69, 21.60, 14.13. HR-MS (ESI): calculated for C58H80N2O6 + H+ (M + H+): 901.6089, found 901.6095 [M + H+], further signal: 923.5912 [M + Na+].
Compound 41: A solution of 0.05 g of compound 39 (0.045 mmol) in Et2O (10 mL) was cooled to 0 °C and 0.45 mL of a 2N solution of HCl (0.09 mmol) in Et2O was added dropwise. The solid thus formed was filtered off, washed with Et2O, dried and suspended in H2O. The pH was adjusted to basic value (pH-10) by addition of NaHCO3 and the mixture was extracted with ethyl acetate. After the separation of the two phases the organic one was dried over Na2SO4 and the solvent was removed under reduced pressure. Yield: 96 % (0.039 g, 0.043 mmol).
1H NMR (400 MHz, CDCl3): δ = 7.58–6.58 (m, 8 H, Ar-H), 5.49 (bs, 1 H, Ar-CH-Ar), 4.26–4.06 (m, 2 H, H-10, H-10), 4.02–2.85 (m, 21 H, Ar-CH2-Ar, OMe, H-2, H-6, H-7), 3.68 (dd, J = 14.2, J = 9.1, 1 H, H-9), 3.47 (dd, J = 14.2, J = 6.2, 1 H, H-9), 2.86–2.64 (m, 1 H, H-7), 2.20–2.10 (m, 1 H, H-6), 1.96–1.82 (m, 1 H, H-5), 1.80–1.75 (m, 1 H, H-4), 1.75–1.60 (m, 1 H, H-3), 1.50–0.96 (m, 39 H, t-Bu, H-3, H-8, H-8). 13C NMR (100 MHz, CDCl3): δ = 173.41, 154.81, 154.73, 154.64, 153.10, 145.01, 134.01, 133.25, 132.70, 132.51, 131.85, 127.06, 126.53, 126.03, 125.87, 125.58, 125.16, 124.97, 124.21, 82.04, 77.73, 77.21, 71.83, 60.70, 59.94, 54.56, 49.94, 48.10, 45.43, 42.81, 37.08, 35.34, 34.09, 34.00, 33.93, 33.86, 31.45, 31.48, 31.08, 30.41, 29.71, 28.04, 25.65, 23.12, 20.54, 17.98. HR-MS (ESI): calculated for C58H80N2O6 + H+ (M + H+): 901.6089, found 901.6098 [M + H+], further signal: 923.5910 [M + Na+].
Compound 42: A mixture of 0.2 g (0.252 mmol, 1 eq.) of di-acid 30, was dissolved in 15 mL of dry DCM. To this mixture was added 0.189 g of Di-Boc-QCD-C10-OH 20 (0.511 mmol, 4.5 eq.). Then 214 mg of DMAP (1.04 mmol, 16 eq.) and 332 mg of 1-methyl-pyridinium iodide (1.1 mmol, 10 eq.) were added. The reaction was stirred at room temperature for 1 h (TLC shows complete conversion diethylether : MeOH : NH3 20:2:4 drops, rf = 0.8). After the reaction was finished, water was added to the solution, the phases were separated. The organic phase was dried and evaporated, and the residue was purified by flash chromatography (20 g silica gel, eluent Et2O:MeOH:NH3 500:50:5).
IR (ATR): 1/λ = 3523, 3398, 2955, 2871, 1806, 1735, 1694, 1481, 1392, 1363, 1292, 1251, 1202, 1127, 1082, 1017, 870, 778, 599. 1H NMR (400 MHz, CDCl3): δ = 7.80–6.26 (m, 8 H, Ar-H), 5.65–5.45 (m, 2 H, Ar-CH-Ar), 5.30–2.45 (m, 34 H, Ar-CH2-Ar, OMe, H-2, H-6, H-7, H-9, H-10), 2.20–0.75 (m, 84 H, H-3, H-4, H-5, H-8, t-Bu, t-BuO). 13C NMR (100 MHz, CDCl3): δ = 173.42, 155.36, 155.04, 153.11, 149.84, 146.25, 144.69, 135.76, 132.79, 131.45, 130.89, 130.81, 130.57, 129.06, 128.50, 128.34, 128.25, 128.08, 127.53, 127.19, 127.07, 126.19, 125.82, 125.56, 125.31, 125.00, 124.32, 119.20, 119.09, 82.06, 81.01, 77.72, 77.21, 61.80, 60.68, 59.98, 58.90, 54.72, 54.55, 49.90, 49.29, 49.17, 48.09, 47.25, 37.67, 34.57, 34.50, 34.20, 34.01, 33.95, 33.84, 31.62, 31.55, 31.53, 31.50, 31.41, 31.36, 31.11, 31.06, 31.03, 30.96, 29.69, 29.50, 28.05, 27.89, 27.69, 26.64, 24.26, 23.06, 14.10. HR-MS (ESI): calculated for C88H128N4O16 + H+ (M + H+): 1497.9398, found 1497.9409 [M + H+], further signals: 1497.94, 1498.94, 1499.95.
Compound 43: A solution of 0.05 g of compound 42 (0.033 mmol) in Et2O (10 mL) was cooled to 0 °C and 0.667 mL of a 2N solution of HCl (0.133 mmol) in Et2O was added dropwise. The solid thus formed was filtered off, washed with Et2O, dried and suspended in H2O. The pH was adjusted to basic value (pH-10) by addition of NaHCO3 and the mixture was extracted with ethyl acetate. After the separation of the two phases the organic one was dried over Na2SO4 and the solvent was removed under reduced pressure. Yield: 92 % (0.033 g, 0.030 mmol).
1H NMR (400 MHz, CDCl3): δ = 7.80–6.26 (m, 8 H, Ar-H), 5.65–5.45 (m, 2 H, Ar-CH-Ar), 5.30–2.45 (m, 34 H, Ar-CH2-Ar, OMe, H-2, H-6, H-7, H-9, H-10), 2.20–0.75 (m, 48 H, H-3, H-4, H-5, H-8, t-Bu). 13C NMR (100 MHz, CDCl3): δ = 173.42, 155.36, 155.04, 153.11, 149.84, 146.25, 144.69, 135.76, 132.79, 131.45, 130.89, 130.81, 130.57, 129.06, 128.50, 128.34, 128.25, 128.08, 127.53, 127.19, 127.07, 126.19, 125.82, 125.56, 125.31, 125.00, 124.32, 119.20, 119.09, 82.06, 81.01, 77.72, 77.21, 61.80, 60.68, 59.98, 58.90, 54.72, 54.55, 49.90, 49.29, 49.17, 48.09, 47.25, 37.67, 34.57, 34.50, 34.20, 34.01, 33.95, 33.84, 31.62, 31.55, 31.53, 31.50, 31.41, 31.36, 31.11, 31.06, 31.03, 30.96, 29.69, 29.50, 28.05, 27.89, 27.69, 26.64, 24.26, 23.06, 14.10. HR-MS (ESI): calculated for C68H92N4O8 + H+ (M + H+): 1097.7301, found 1097.7268 [M + H+], further signal: 1119.73.
X-Ray structure determinations
Compound 9 crystallized as a chloroform disolvate and compound 12 solvent-free. Crystals were mounted in inert oil on glass fibers and transferred to the cold gas stream of the diffractometer (Oxford Diffraction Nova A for 9 and Nonius KappaCCD APEX-II for 12). Intensity data were recorded at low temperature using mirror-focussed (9) or monochromated (12) Cu Kα radiation (λ = 1.54184 Å). Absorption corrections were based on multi-scans. The structures were refined anisotropically on F2 using the program SHELXL-97 [30]. Hydrogens of NH and OH groups were located in diference syntheses and refined freely but with X-H distance restraints. Other hydrogens were refined using rigid methyl groups allowed to rotate but not tip, or a riding model starting from calculated positions. Special features: For compound 9, the solvent chlorine provided adequate anomalous scattering to confirm the absolute configuration, with a Flack parameter of 0.008(8). The lower crystal quality of 12 and its lack of heavier atoms prevented the unambiguous determination of the Flack parameter and Friedel opposite reflections were therefore merged. A space group and configuration consistent with the known QCD confguration were chosen. For both structures, some t-butyl groups were disordered over two positions. Appropriate restraints were used to improve refinement stability, but dimensions of disordered groups should be interpreted with caution. Disordered groups in 12 were refined isotropically.
Crystallographic data (Table 1) have been deposited with the Cambridge Crystallographic Data Centre as supplementary publications no. CCDC-1036066 (9), CCDC-1036067 (12). Copies of the data can be obtained free of charge from www.ccdc.cam.ac.uk/data_request/cif.
| Compound | 9 · 2CHCl3 | 12 |
|---|---|---|
| Formula | C70H98Cl6N4O6 | C68H96N4O6 |
| M r | 1304.22 | 1065.49 |
| Habit | colorless block | colorless block |
| Cryst. size (mm) | 0.35 × 0.25 × 0.18 | 0.15 × 0.15 × 0.13 |
| Crystal system | monoclinic | hexagonal |
| Space group | C2 | P65 |
| Temperature (°C) | –173 | –50 |
| Cell constants: | ||
| a (Å) | 27.7917(8) | 13.1682(1) |
| b (Å) | 12.9360(4) | 13.1682(1) |
| c (Å) | 20.8979(6) | 63.2115(12) |
| α (°) | 90 | 90 |
| β (°) | 108.379(3) | 90 |
| γ (°) | 90 | 120 |
| V (Å3) | 7129.8 | 9492.5 |
| Z | 4 | 6 |
| Dx (Mg m–3) | 1.215 | 1.118 |
| μ (mm–1) | 2.6 | 0.55 |
| F(000) | 2784 | 3480 |
| 2θmax | 152 | 131 |
| Refl. measured | 93 573 | 73 133 |
| Refl. indep. | 14 007 | 9852 |
| R int | 0.030 | 0.055 |
| Parameters | 830 | 833 |
| Restraints | 72 | 1006 |
| wR(F2, all refl.) | 0.111 | 0.093 |
| R(F, >4σ(F)) | 0.040 | 0.037 |
| S | 1.03 | 1.06 |
| max. Δρ (e Å–3) | 0.58 | 0.16 |
Article note
Invited paper based on presentation at the 15th International Conference on Polymers and Organic Chemistry (POC-2014), Timisoara, Romania, 10 – 13 June 2014.
Acknowledgments
This work was supported by the Romanian National Authority for Scientific Research through the EXPLORATORY RESEARCH PROGRAM IDEI-PCE-PROJECT NR. 341-/05.10.2011 – Immuno-modulatory Fluoroglycopeptide Molecular Architectures (I. N.).
References
[1] V. Böhmer. Angew. Chem. 107, 785 (1995).10.1002/ange.19951070704Search in Google Scholar
[2] V. Böhmer. Angew. Chem. Int. Ed. 34, 713 (1995).10.1002/anie.199507131Search in Google Scholar
[3] M. Roundhill. Prog. Inorg. Chem. 43, 533 (1995).Search in Google Scholar
[4] A. Arduini, A. Casnati. Macrocycle Synth. 145 (1996).10.1093/oso/9780198558415.003.0007Search in Google Scholar
[5] I. Neda, T. Kaukorat, R. Schmutzler. Main Group Chem. News 6, 4 (1998).Search in Google Scholar
[6] C. D. Gutsche. Calixarenes: An Introduction; Monographs in Supramolecular Chemistry, 2nd ed., Royal Society of Chemistry, Cambridge, UK (2008).Search in Google Scholar
[7] K. Cottet, P. M. Marcos, P. J. Cragg. Beilstein J. Org. Chem. 8, 201 (2008).10.3762/bjoc.8.22Search in Google Scholar
[8] C. Kunze, D. Selent, I. Neda, M. Freytag, P. G. Jones, R. Schmutzler, W. Baumann, A. Börner. Z. Anorg. Allg. Chem. 628, 779 (2002).Search in Google Scholar
[9] C. Kunze, D. Selent, I. Neda, R. Schmutzler, A. Spannenberg, A. Börner. Het. Chem. 12, 577 (2006).10.1002/hc.1088Search in Google Scholar
[10] R. Schmutzler, I. Neda, C. Kunze, A. Börner, D. Selent, C. Borgmann, D. Hess, K.-D. Wiese. EP Patent 1 417 212, Filed 7 August 2002, issued 12 May 2004.Search in Google Scholar
[11] R. Schmutzler, I. Neda, C. Kunze, A. Börner, D. Selent, C. Borgmann, D. Hess, K.-D. Wiese. US Patent 7009 068, Filed 7 August 2002, issued 7 March 2006.Search in Google Scholar
[12] I. Neda, H. J. Plinta, R. Sonnenberg, A. Fischer, P. G. Jones, R. Schmutzler. Chem. Berichte 128, 267 (1995).10.1002/cber.19951280310Search in Google Scholar
[13] I. Neda, H. J. Plinta, R. Sonnenberg, A. Fischer, P. G. Jones, R. Schmutzler. Chem. Berichte 128, 545 (1995).10.1002/cber.19951280310Search in Google Scholar
[14] C. Kunze, I. Neda, M. Freytag, P. G. Jones, R. Schmutzler. Z. Anorg. Allg. Chem. 628, 545 (2002).10.1002/1521-3749(200205)628:4<779::AID-ZAAC779>3.0.CO;2-VSearch in Google Scholar
[15] C. D. Dielemann, D. Matt, I. Neda, R. Schmutzler, H. Thönnessen, P. G. Jones, A, Harriman. J. Chem. Soc., Dalton Trans. 2115 (1998).10.1039/a801586dSearch in Google Scholar
[16] A. Vollbrecht, I. Neda, H. Thönnessen, P. G. Jones, R. K. Harris, L. A. Crowe, R. Schmutzler. Chem. Ber. 130, 1715 (1997).10.1002/cber.19971301124Search in Google Scholar
[17] I. Neda, T. Siedentop, A. Vollbrecht, H. Thönnessen, P. G. Jones, R. Schmutzler. Z. Naturforschung. 53b, 841 (1998).10.1515/znb-1998-0811Search in Google Scholar
[18] P. Sakhaii, I. Neda, M. Freytag, H. Thönnessen, P.G. Jones, R. Schmutzler. Z. Anorg. Allg. Chem. 626, 1246 (2000).10.1002/(SICI)1521-3749(200005)626:5<1246::AID-ZAAC1246>3.0.CO;2-FSearch in Google Scholar
[19] R. Perrin, R. Lamartine, M. Perrin. Pure & Appl. Chem. 65, 1549 (1993).10.1351/pac199365071549Search in Google Scholar
[20] Y Kubo, S. Maeda, S. Tokita, M. Kubo. Nature 382, 522 (1996).10.1038/382522a0Search in Google Scholar
[21] T. McKittrick, D. Diamond, D. J. Marrs, P. O’Hagan, M. A. McKervey. Talanta 43, 1145 (1996).10.1016/0039-9140(96)01888-7Search in Google Scholar
[22] B. R. Cameron, S. J. Loeb, G. P. A. Yap. Inorg. Chem. 36, 5498 (1997).10.1021/ic9705913Search in Google Scholar
[23] A. Ikeda, T. Nagasaki, K. Araki, S. Shinkai. Tetrahedron 48, 1059 (1992).10.1016/S0040-4020(01)88202-8Search in Google Scholar
[24] I. Neda, P. Sakhaii, A, Waßmann, U. Niemeyer, E. Günther, J. Engel. Synthesis 1625 (1999).Search in Google Scholar
[25] M. T. Blanda, E. Adou. Chem. Commun. 139 (1998).10.1039/a704122eSearch in Google Scholar
[26] A. Cecal, K. Popa, I. I. Craciun, I. Neda. Czech. J. Phys. 53, 557 (2003).10.1007/s10582-003-0072-1Search in Google Scholar
[27] T. Oshima, M. Goto, S. Furusaki. J. Incl. Phenom. Macro. Chem. 43, 77 (2002).10.1023/A:1020451421666Search in Google Scholar
[28] S. Moerkerke, S. Le Gac, F. Topic, K. Rissanen, I. Jabin. Eur. J. Org. Chem. 5315 (2013).10.1002/ejoc.201300639Search in Google Scholar
[29] E. M. Georgiev, J. Clymire, G. L. McPherson, D. M. Roundhill. Inorg. Chim. Acta 227, 293 (1994).10.1016/S0020-1693(00)84023-2Search in Google Scholar
[30] M. P. O. Wolbers, F. C. J. M. van Veggel, R. H. M. Heeringa, J. W. Hofstraat, F. A. J. Geurts, G. J. van Hummel, S. Harkema, D. N. Reinhoudt. Liebigs Ann. 2587 (1997).10.1002/jlac.199719971226Search in Google Scholar
[31] M. Kawaguchi, A. Ikeda, S. Shinkai, I. Neda. J. Incl. Phenom. Macro. Chem. 37, 253 (2000).10.1023/A:1008056229653Search in Google Scholar
[32] R. K. Castellano, B. H. Kim, J. Rebek, Jr. J. Am. Chem. Soc. 119, 12671 (1997).10.1021/ja973132+Search in Google Scholar
[33] Y. Hamuro, M. C. Calama, H. S. Park, A. D. Hamilton. Angew. Chem. 109, 2797 (1997).10.1002/ange.19971092342Search in Google Scholar
[34] Y. Hamuro, M. C. Calama, H. S. Park, A. D. Hamilton. Angew. Chem. Int. Ed. 36, 2680 (1997).10.1002/anie.199726801Search in Google Scholar
[35] R. Roy, J. M. Kim. Angew. Chem. 111, 380 (1999).10.1002/(SICI)1521-3757(19990201)111:3<380::AID-ANGE380>3.0.CO;2-USearch in Google Scholar
[36] R. Roy, J. M. Kim. Angew. Chem. Int. Ed. 38, 369 (1999).10.1002/(SICI)1521-3773(19990201)38:3<369::AID-ANIE369>3.0.CO;2-1Search in Google Scholar
[37] A. Marra, A. Dondoni, F.Sansone. J.Org. Chem. 61, 5155 (1996).10.1021/jo960417gSearch in Google Scholar
[38] K. H. Krawinkler, N. M. Maier, R. Ungaro, F. Sansone, A. Casnati, W. Lindner. Chirality 15, 17 (2003).10.1002/chir.10257Search in Google Scholar
[39] K. H. Krawinkler, N. M. Maier, E. Sajovic, W. Lindner. J. Chromatogr. A 1053, 119 (2004).10.1016/S0021-9673(04)01206-3Search in Google Scholar
[40] A. Hartikka, S. A. Modin, P. G. Andersson, P. I. Arvidsson. Org. Biomol. Chem. 1, 2522 (2003).10.1039/b304060gSearch in Google Scholar
[41] C. Hedberg, K. Källström, P. I. Arvidsson, P. Brandt, P. G. Andersson. J. Am. Chem. Soc. 127, 15083 (2005).10.1021/ja051920qSearch in Google Scholar
[42] E. A. Kunze, M. A. McKervey, E. Madigan, M. B. Moran, M. Owens, George Fergusen, S. J. Harris. J. Chem. Soc. Perkin I 1991, 3137 (1991).Search in Google Scholar
[43] A. Arduini, A. Pochini, S. Reverberi, R. Ungaro. J. Chem. Soc. Chem. Com. 981 (1984).10.1039/c39840000981Search in Google Scholar
[44] P. Zlatuskova, I. Stibor, M. Tkadlecova, P. Lhotak. Tetrahedron 60, 11383 (2004).10.1016/j.tet.2004.09.088Search in Google Scholar
[45] I. Neda, T. Kaukorat, A. K. Fischer. Eur. J. Org. Chem. 3784 (2003).10.1002/ejoc.200300211Search in Google Scholar
[46] P. Bandyopadhyay, P. K. Bharadwaj. Synlett 1331 (1998).10.1055/s-1998-1961Search in Google Scholar
[47] J. Dessingou, K. Tabbasum, A. Mitra, V. K. Hinge, C. P. Rao. J. Org. Chem. 77, 1406 (2012).10.1021/jo2022372Search in Google Scholar
[48] I. Neda, T. Kaukorat, C.-G. Hrib. Tetrahedron: Asymmetry 13, 1327 (2002).10.1016/S0957-4166(02)00320-8Search in Google Scholar
[49] I. Neda, E. Fodor, C.V. Maftei, M. Mihorianu, H. D. Ambrosi, M. H. Franz. Eur. J. Org. Chem. 7876 (2013).10.1002/ejoc.201301286Search in Google Scholar
[50] G. M. Sheldrick. Acta Cryst. A64, 112 (2008).10.1107/S0108767307043930Search in Google Scholar
[51] F. H. Allen, P. A. Wood, P. T. A. Galek. Acta Cryst. B69, 379 (2013).10.1107/S2052519213015078Search in Google Scholar
[52] C. David Gutsche, B. Dhawan, J. A. Levine, K. H. No, L. J. Bauer. Tetrahedron 39, 409 (1983).10.1016/S0040-4020(01)88541-0Search in Google Scholar
[53] T. Gruber, M. Gruner, C. Fischer, W. Seichter, P. Bomnicz, E. Weber. New. J. Chem. 34, 250 (2010).10.1039/b904489bSearch in Google Scholar
[54] C. Fischer, W. Seichter, E. Weber, Beilstein J. Org. Chem. 7, 1602 (2011).10.3762/bjoc.7.188Search in Google Scholar
[55] H. Türkmen, I. Kani, B. Cetinkaya. Eur. J. Org. Chem. 4494 (2012).10.1002/ejic.201200638Search in Google Scholar
[56] S. Zhang, W. Baratta. Organometallics 32, 3339 (2013).10.1021/om400280mSearch in Google Scholar
[57] M. Ito, T. Ootsuka, R. Watari, A. Shiibashi, A. Himizu, T. Ikariya. J. Am. Chem. Soc. 133, 4240 (2011).10.1021/ja1117254Search in Google Scholar
©2015 IUPAC & De Gruyter
Articles in the same Issue
- Frontmatter
- Preface
- 22nd IUPAC International Conference on Physical Organic Chemistry (ICPOC-22)
- Conference papers
- Benzhydrylium and tritylium ions: complementary probes for examining ambident nucleophiles
- Catalyzing decarboxylation by taming carbon dioxide
- Mechanistic insights from mass spectrometry: examination of the elementary steps of catalytic reactions in the gas phase
- Nitrosyl–heme and anion–arene complexes: structure, reactivity and spectroscopy
- Gas-phase studies of metal catalyzed decarboxylative cross-coupling reactions of esters
- Wettability of organically coated tridymite surface – molecular dynamics study
- Asymmetric calixarene derivatives as potential hosts in chiral recognition processes
- Erratum
- Erratum to: Nomenclature and graphic representations for chemically modified polymers (IUPAC Recommendations 2014)
Articles in the same Issue
- Frontmatter
- Preface
- 22nd IUPAC International Conference on Physical Organic Chemistry (ICPOC-22)
- Conference papers
- Benzhydrylium and tritylium ions: complementary probes for examining ambident nucleophiles
- Catalyzing decarboxylation by taming carbon dioxide
- Mechanistic insights from mass spectrometry: examination of the elementary steps of catalytic reactions in the gas phase
- Nitrosyl–heme and anion–arene complexes: structure, reactivity and spectroscopy
- Gas-phase studies of metal catalyzed decarboxylative cross-coupling reactions of esters
- Wettability of organically coated tridymite surface – molecular dynamics study
- Asymmetric calixarene derivatives as potential hosts in chiral recognition processes
- Erratum
- Erratum to: Nomenclature and graphic representations for chemically modified polymers (IUPAC Recommendations 2014)