Organocatalytic functionalisation through boron chemistry
-


Abstract
Diboron reagents can be activated both, from a metal or free-metal context, and consequently the addition of boryl units to unsaturated substrates proceeds sometimes with complementary selectivity. We highlight here the power of boron chemistry to functionalize molecules and provide new routes towards challenging compounds.
Introduction
Diboron reagents have been used on borylation reactions since Schlesinger et al. [1, 2] found that diboron tetrachloride reacted with one mole of ethylene to form a liquid compound assigned as Cl2BCH2CH2BCl2. The search of diboron reagents that can be easier to handle has provided nowadays the possibility to use commercially available bis(pinacolato)diboron, B2pin2, which has been essentially used in catalytic diboration, β-boration, hydroboration and borylation of unsaturated substrates, by means of transition metals complexes that activates the pinB–Bpin bond to generate M-Bpin units [3–5]. Depending on the metal involved, the Bpin moiety can react as an electrophile or nucleophile motive, opening a wide range of applicability [6, 7]. Recently, another mode of activation of diboron reagents in the absence of transition metal complexes, has pointed out the possibility to run organocatalytic boron addition reactions with quimio-, regio- and enantioselectivity, by the sole use of catalytic amounts of Lewis bases (such as alkoxides or N-heterocyclic carbenes) [8–16]. In our hands, we have observed that the addition of methoxide to B2pin2 can quaternize the Bpin moiety and enhance the nucleophilic character of the sp2 Bpin fragment [10, 16]. At that point, the reactivity of the Lewis acid–base adducts become somewhat unpredictable. In this manuscript we highlight the use of MeO–→Bpin–Bpin and MeO–→Bpin–Bdan (dan = naphthalene-1,8-diaminato) towards the functionalization of activated and non-activated substrates. We also compare here, in some cases, the reactivity of the nucleophilic sp2 Bpin fragments with Cu–Bpin species, taking into consideration the remarkable high nucleophilic character of Bpin moiety when is coordinated to Cu (I).
Catalytic β-boration of α,β-unsaturated carbonyl compounds
We were working on the copper mediated β-boration of α,β-unsaturated carbonyl compounds, following the catalytic cycle established by Yun et al. (Scheme 1a) [17, 18] when we realized that a control experiment (in the absence of Cu(I) salt) allowed the reaction to proceed, albeit with a slightly minor conversion (Scheme 1b) [9].

(a) Catalytic cycle proposed by Yun et al., on copper mediated β-boration of α,β-unsaturated esters and ketones, (b) Catalytic cycle proposed in the absence of copper salts and phosphines, in our control experiment.
Our attempts to improve the yields on the metal-free β-boration of activated olefins required higher temperature (70 °C) and the use of catalytic amounts of basic phosphines, such as PPh3 and PCy3 [9]. Even more remarkably, the use of chiral phosphines as additives, allowed the β-boration to proceed in enantioselective fashion, (Scheme 2).

Enantioselective metal-free β-boration of α,β-unsaturated esters and ketones.
Based on spectroscopic and theoretical studies, we explored the use of PR3 in the reaction and we found that phosphines were essential to interact with the substrate, resulting in the formation of a zwitterionic phosphonium enolate. This species can further deprotonate MeOH when B2pin2 is present, forming eventually the ion pair [α-(H), β-(PR3)-ketone]+-[B2pin2·MeO]– that is responsible for the catalysis (Scheme 3) [19].

Suggested role of PR3 in the formation of zwitterionic phosphinium enolate and its reactivity with a model substrate.
The Lewis acid-base formation with mixed diboron reagents was first established by Santos et al. [20–22] reporting that sp2–sp3 hybridized mixed PDIPA diboron (pinacolato diisopropanolaminato diboron) easily transmetallates to CuX favouring the copper-catalyzed β-boration of α,β-unsaturated conjugated compounds, under mild conditions. We have been interested to find out the activation of the mixed diboron reagent Bpin–Bdan (dan = 1,8-diaminonaphthalene), by alkoxides. With the assistance of DFT calculations and spectroscopic studies it was possible to postulate the exclusive formation of the Lewis acid-base adduct [RO–→B(pin)–B(dan)], which reacts with α,β-unsaturated carbonyl compounds to give exclusively the Cβ-Bdan carbonyl compound with high yields (Scheme 4) [23].
![Scheme 4
Organocatalytic β-boration of α,β-unsaturated ketones and esters with [MeO–→B(pin)–B(dan)].](/document/doi/10.1515/pac-2014-0934/asset/graphic/j_pac-2014-0934_fig_004.jpg)
Organocatalytic β-boration of α,β-unsaturated ketones and esters with [MeO–→B(pin)–B(dan)].
In addition to the unprecedented conjugate Bdan addition to α,β-unsaturated ketones and esters, the presence of chiral diphosphine as additive assisted the asymmetric induction in a more efficient way than in the analogous borylation with B2pin2 (Scheme 5). The new synthetic platform opens a non-existing methodology to prepare selectively Cβ-Bdan carbonyl compounds in a selective straightforward pathway.
![Scheme 5
Enantioselective organocatalytic β-boration of α,β-unsaturated ketones and esters with [MeO–→B(pin)–B(dan)].](/document/doi/10.1515/pac-2014-0934/asset/graphic/j_pac-2014-0934_fig_005.jpg)
Enantioselective organocatalytic β-boration of α,β-unsaturated ketones and esters with [MeO–→B(pin)–B(dan)].
In a similar way, we have conducted the β-boration of α,β-unsaturated imines formed in situ from the corresponding ketone and amine, and once again the presence of base and phospine as additives, were essential to obtain good conversion and asymmetric induction (Scheme 6) [24].

In situ α,β-unsaturated imine formation and enantioselective organocatalytic β-boration assisted by chiral phosphines.
The last example can be compared with a related Cu-mediated β-boration of in situ formed α,β-unsaturated imines (carried out in collaboration with Whiting’s group), and it can be said that the copper catalyzed reaction favours the formation of the desired β-borated product with higher enantioselectivity, upon optimization of the chiral phosphine additives present in the reaction media. In addition, it has been studied a further reduction/oxidation of the enantioenriched β-borated compound, and depending on the reducing reagent selected, the syn or anti γ-aminoalcohol could be obtained with retention of the asymmetric induction (Scheme 7) [25–30].

Sequential strategic synthesis of chiral syn and anti γ-aminoalcohols throughout copper catalyzed β-boration of α,β-unsaturated imines.
The C=N bond of tosylaldimines has also been susceptible of precise interaction with the Lewis acid–base adduct [MeO–→B(pin)–B(pin)], forming the corresponding α-amino boronate ester, which can be also obtained with high asymmetric induction by the presence of chiral phosphines, as additives. Interestingly, further functionalization of the chiral α-amino boronate ester, following a homologation protocol, delivered easily β-amino alcohols with total retention of the asymmetry in the chiral carbon [16] (Scheme 8).

Asymmetric organocatalytic Bpin addition to tosylaldimines to form chiral α-amino boronate esters, which can be homologated to the corresponding β-aminoalcohols.
Catalytic β-boration-α-halogenation of α,β-unsaturated carbonyl compounds
Haloboration reaction has become an essential route to difunctionalize unsaturated substrates, in an efficient one step process [31, 32]. The nature of the haloborating reagent is a key point on the efficiency of the reaction [33]. In this way, boron trihalides and B-bromo- or B-iodo-9-BBN (BBN=borabicyclo[3.3.1]nonane) have been the most used reagents to haloborate terminal alkynes [31, 32, 34–39], however, none of these haloborating reagents have proved to haloborate internal alkynes. That limitation has been sorted out, by Ingleson and co-workers, increasing the electrophilicity at the boron centre in the haloborating reagents, through an elegant design of boronium and borenium cations. Hence, dichloroborenium cations enable the chloroboration of internal alkynes, in a regio- and stereoselectively way [40]. Further esterification on the boron moiety, provides the corresponding trisubstituted vinyl pinacol boronate esters. In that context we also envisaged that organohaloborated products can be prepared by a sequential borylation/halogenation, in a one pot protocol. Towards this end, we have recently developed a methodology that allows the sequential C–B and C–F bond formation of activated alkenes through a one pot regio-, diastereo- and enantioselective strategy [41]. Therefore, α′-fluoro β-boryl ketones can be obtained by using a sequential organocatalytic β-boration of α,β-unsaturated ketones and a consecutive electrophilic fluorination reaction (with Selectfluor, F-TEDA-BF4) in an acidic media (Scheme 9a). In parallel, α-fluoro β-boryl ketones can also be synthesised, in high yields, as an enriched mixture of the anti diastereomer, by copper mediated β-boration followed by in situ electrophilic fluorination of the boron enolate, in the presence of a base (Scheme 9b).

Sequential boration/fluorination of α,β-unsaturated ketones.
Taking into consideration the importance of introducing two vicinal functionalities in a selective way, and generate tetrasubstituted carbons of potential transformation through the C–B bond [42–46], we report a convenient route for copper mediated β-boration of α,β-unsaturated ketones followed by electrophilic chlorination in α-position (Scheme 10a). We have also carried out the copper β-boration/protonation of α-chloro α,β-unsaturated ketones for comparison (Scheme 10b).

Sequential boration/chlorination of α,β-unsaturated ketones.
Our initial attempts were focused on the α-chlorination of cylohexenone (1), to prepare the corresponding α-chloro-2-cyclohexen-1-one (1-Cl) which could be further β-borated with B2pin2 in the presence of Cu(I). Several transition metals catalyze the β-boration of α,β-unsaturated carbonyl compounds, (Pt, Rh and Ni), however copper has emerged as the most convenient and efficient metal to mediate the chemoselective formation of β-borated esters, ketones, nitriles and amides [47–52]. Towards this end, we based the synthesis of 1-Cl in a previous reported methodology using bisacetoxyiodobenzene (BAIB) and pyridinium chlorochromate (PCC) [53]. The α-chlorination was conducted under mild reaction conditions and allowed the isolation of 1-Cl in 67 % yield. When this compound was exposed to the copper catalyzed β-boration protocol, [54] using CuPF6(CH3CN)4/PCy3 as the precursor of the catalytic system and B2pin2 as the boryl source, the reaction proceed smoothly and the desired α-chloro-β-pinacolboryl-cyclohexan-1-ona (2) was isolated with 65 %, as the syn isomer (Scheme 11, Method A). To the light of this promising result, we were interested to extend the synthesis of α-chloro-cyclohexenones with substituents on β-position to establish a methodology to selectively β-borate them to generate tetrasubstituted carbons with chloride functionality in the vicinal position. Therefore α-chloro-3-methylcyclohexenone (3-Cl) and α-chloro-3-(p-chlorophenyl)cyclohexenone (5-Cl) were prepared from the corresponding cyclohexen-1-ones 3 and 5, and Py·HCl/BAIB [53]. In particular 5-Cl was prepared for the first time in this work with the aim to enhance the electrophilicity on the β-position. Despite the fact that the copper mediated β-boration was carried under identical conditions to the β-boration of 1-Cl, the substrates 3-Cl and 5-Cl were not converted into the desired product, probably as a consequence of the more hindered β-position on 3-Cl and 5-Cl with respect to 1-Cl (Scheme 11).

Sequential α-chlorination followed by copper mediated β-boration/protonation.
In order to circumvent the lack of success on the β-boration of α-chloro-β-substituted-cyclohexenones, we conducted the alternative Method B in which copper(I) (CuPF6(CH3CN)4) initially mediated the β-boration of β-substituted-cyclohexenones 3 and 5, which was followed by the in situ addition of 2 eq of N-chlorosuccinimide (NCS) as the electrophilic chloride source. An advantage of this methodology is the lack of intermediate purification. As illustrated in Scheme 12, substrate 3 could be converted into the desired product 4 (anti-isomer), by a one-pot procedure, although the regioisomer 4α′ was also observed in minor percentage (4/4α′ =2/1), as a mixture of the syn and anti isomer.

Sequential copper mediated β-boration/α-chlorination of 3-methylcyclohexenone (3).
Having demonstrated the possibility to obtain α-chloro-β-pinacolboryl cyclohexenones from β-substituted α,β-unsaturated cyclic ketones, our next goal was to enhance the regioselectivity promoting the α-chlorination versus the α′-chlorination. To this end, we focused our efforts on introducing substituents at the β-position that stabilise the β-boryl enolate towards the α-chlorination. Substrate 5 was prepared with an electronwithdrawing substituent (p-chlorophenyl) on the β-position to support the previous hypothesis. When substrate 5 was β-borated with the Cu(I) catalyst followed by α-chlorination in the presence of N-chlorosuccinimide, in a one pot sequential protocol, only the α-chloro-β-pinacolboryl-3-p-chlorophenylcyclohexanona (6) was observed and isolated (73 %) as a diastereomeric mixture (anti/syn = 8/1) (Table 1, entry 1). Having established the efficient methodology to simultaneously β-borate α-chlorinate, we next extended the reactivity to a representative scope of β-aryl α,β-unsaturated ketones. Substrates 3-phenylcyclohexenona (7), 3-p-fluorophenylcyclohexenona (9), 3-p-methylphenylcyclohexenona (11) and 3-p-napthylcyclohexenona (13) were conveniently converted to the corresponding α-chloro-β-pinacolboryl cyclohexenones 8, 10, 12 and 14 respectively (Table 1, entries 2-5) with a notorious preference towards the anti diastereoisomer. Interestingly, product 14 could be prepared in 50 % isolated yield despite the bulky napthyl substituent on the β-position (Table 1, entry 5).
Cu mediated β-boration/α-chlorination of 3-aryl 2-cyclohexen-1-ones.a
| Entry | Substrate | Product | Conversion (%)b | Isolated yield (%) |
|---|---|---|---|---|
| 1 |
 |
 |
99 % | 73 % |
| 2 |
 |
 |
44 % | 30 % |
| 3 |
 |
 |
73 % | 62 % |
| 4 |
 |
 |
54 % | 37 % |
| 5 |
 |
 |
67 % | 50 % |
(a) Standard conditions: Cu(CH3CN)4PF6 (0.025 mmol), PCy3 (0.025 mmol), substrate (0.25 mmol), B2pin2 (1.1 eq., 0.35 mmol), LiOtBu (0.015 mmol) DMF (2 mL), r.t., 2 h, after that period N-chlorosuccinimide (NCS, 0.5 mmol), rt., 16 h. (b) Conversion calculated by G.C. and NMR spectroscopy.
In order to have a global picture of the potential methodology to β-borate α-halogenate, we have performed on substrate 3 the β-boration/α-bromination and the β-boration/α-fluorination, for comparison with the β-boration/α-chlorination, under the same reaction conditions. When substrate 3 was subjected to the copper borylation with (CuPF6(CH3CN)4), followed by electrophilic fluorination with F-TEDA-BF4, the corresponding α-fluoro β-boryl ketone 4-F was quantitatively obtained (87 % isolated yield) with a similar preferred diastereoselectivity on the anti isomer (anti/syn = 9/1) (Scheme 13). However, the β-boration α-bromination of 3 with N-bromosuccinimide (NBS), as the electrophilic bromide source, only provided 18 % of the corresponding α-bromo β-boryl ketone 4-Br. Electronic and steric factors are likely responsible for the decreasing trend towards the β-boration α-halogenation from F>Cl>Br.
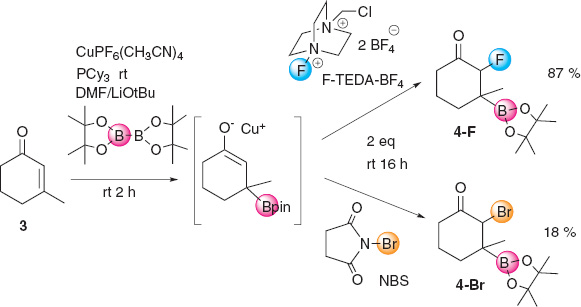
Sequential copper mediated β-boration/α-fluorination and β-boration/α-bromination for comparison.
Another interesting point is the possibility to induce asymmetry in the tetrasubstituted carbon. Therefore we decided to perform the asymmetric copper-catalyzed conjugate boration, with CuPF6(CH3CN)4 modified with QuinoxP* [54], to further react the boron enolate with FTEDA-BF4. Thus, the substrate 3-methylcyclohexenone was β-borated / α-fluorinated providing the desired anti isomer with 72 % enantiomeric excess (Scheme 14). The advantage of this synthetic methodology is due to the simplicity of the construction of two vicinal bonds in the same step, and represents an alternative to the current efforts to develop enantioselective electrophilic fluorination routes [55–67].

Sequential asymmetric copper mediated β-boration/α-fluorination of 3-methylclyclohexenone with CuPF6(CH3CN)4 /(R,R)-QuinoxP*.
Borylative ring-opening of vinyl epoxides and aziridines
A rational approach towards the borylative ring-opening of vinylepoxides and vinylaziridines, by the in situ formed [MeO–→B(pin)–B(pin)] adduct, has also been developed. The enhanced nucleophilic character of the Bpin (sp2) moiety from the reagent favours the SN2′ conjugated boron addition with the concomitant opening of the epoxide or aziridine rings. The reaction proceeds with total chemoselectivity towards the polyfunctionalised (–OH or –NHTs) allyl boronate (Scheme 15). Theoretical calculations have determined the transition states that come from the reaction of the vinylic substrates with the activated [MeO–→B(pin)–B(pin)] adduct, and a plausible mechanism for the organocatalytic borylative ring opening reaction has been suggested [68].
![Scheme 15
Organocatalytic borylative ring-opening of vinyl epoxides and vinyl aziridines with [MeO-→B(pin)–B(pin)] adduct.](/document/doi/10.1515/pac-2014-0934/asset/graphic/j_pac-2014-0934_fig_025.jpg)
Organocatalytic borylative ring-opening of vinyl epoxides and vinyl aziridines with [MeO-→B(pin)–B(pin)] adduct.
Interestingly, in a comparative study carried out using CuCl to activate the B2pin2 we found that the transition metal catalyzed reaction afforded the 1,2-cyclohexenyl hydroxyboronate via SN2 addition (Scheme 16). This result nicely highlights the complementariety of the organocatalytic and copper mediated borylative ring opening reactions.

Copper mediated chemoselective nucleophilic Bpin attack at the epoxide functional group of 3,4-epoxy-1-cyclohexene. X-ray structure of 4-hydroxy-cyclohex-2-enyl-phenyl-methanol.
Organocatalytic diboration
Very recently, we have reported a reaction that represents a very uncommon reactivity pattern: a reaction between a nucleophilic reagent and an otherwise nucleophilic substrate. The boron nucleophile is easily generated from the [MeO–→B(pin)–B(pin)] adduct, which also provide the electrophilic counterpart B(sp3) of the nucleophilic boron moiety B(sp2). The net result is a new, Lewis base catalyzed diboration method, which, because of the simple reagents and catalysts, the complete atom economy, and the high synthetic value of the products, represents a great step towards a future industrial organoborane synthesis (Scheme 17) [16, 69]. The catalytic system for the nucleophilic diboration of non-activated olefins is a combination of base and alcohol. Both additives are crucial to achieve high activity. After the screening of various bases and alcohols, we have concluded that, in THF solutions, a combination of Cs2CO3 and MeOH provides synthetically useful conversions and chemoselectivities towards the diborated product. It is worth noting that various other bases, such as methoxides (Li+, Na+, K+), or NaOtBu give comparable results. Despite the fact that for optimal activities MeOH is added in excess with respect to the substrate, the formation of the “hydroborated” by-product rarely exceeds 5 mol %.

Comparative perspective of metal mediated diboration reaction and organocataytic diboration procedure.
This simple catalytic system is capable of mediating the addition of different diboron reagents to various non-activated unsaturated substrates. Interestingly enough, the unsymmetrical diboron Bpin–Bdan can be added regioselectively to terminal and internal alkenes, by meand of the methoxide activation of the diboron, representing the first mixed diboration of alkenes [70]. At this point is worthy to note that, even in the presence of transition metal complexes, the diboron Bpin–Bdan could only be added to alkynes (Scheme 18) [71].

Comparative perspective of metal mediated diboration reaction and organocataytic diboration procedure.
The organocatalytic diboration is only in its early stages, but since 2011 there have been significat input in this new methodology. Morken and coworkers have recently used this concept for the directed diboration of alkenyl alcohols [72]. The reaction occurs in a stereoselective fashion and is demonstrated with cyclic and acyclic homoallylic and bishomoallylic alcohol substrates. After oxidation, the reaction generates 1,2-diols such that the process represents a method for the stereoselective directed dihydroxylation of alkenes. Another appealing application of the organocatalytic diboration is the one formulated by Hirano and Uchiyama, because it represents the first trans-selective diboration of alkynes [73]. The authors designed a pseudo-intramolecular reaction of diboron, propargyl alcohol and a base, to facilitate the B-B activation and C-B formation with high efficiency. This approach provides synthetically versatile and densely functionalized 4-borylated 1,2-oxaborol-2(5H)-oles (vinyldiboronates) in a straightforward manner. Detailed computational analysis showed that the directing alkoxide functionality markedly lowers the activation energy of B–C bond formation.
Carbonyl functionality activates phenylselenium boranes
Activated olefins directly react with phenylselenium boranes, at room temperature, without the need of metal or organocatalytic assistance. A simple mechanism that involves the interaction of the electron pair of carbonyl functional group in α,β-unsaturated ketones and aldehydes, with the empty p orbital of the boron atom, justifies the efficient reaction towards the kinetically and thermodynamically most stable 1,4-addition product (Scheme 19). Up to 12 examples of β-(phenylseleno) substituted ketones and aldehydes have been prepared with moderate to high yield (Scheme 20). The simplicity and efficiency of this new reactivity generates new strategic platforms towards the C–Se bond formation and opens non existing pathways to create C-heteroatom bonds, as a general tool [74].

Synthesis of β-(phenylseleno) substituted carbonyl compounds by simple interaction of a,b-unsaturated carbonyl compounds and phenylselenium boranes.

Scope of organocatalytic synthesis of β-(phenylseleno) substituted carbonyl compounds.
Conclusions
We conclude that both activation of diboron reagents (copper or methoxide) provides a nucleophilic boryl unit that can behave in a similar way or complementary. This circumstance is an advantage towards the functionalisation of organic compounds through an easy and efficient way. In addition, enantioselectivity can be achieved when copper is modified with a chiral ligand, but also the organocatalylic pathway can also induce asymmetry when chiral additives are involved in the reaction media. The Lewis acidity of the boron atom as well as the facility to be transformed into a quaternized system, are the key roles of this potential methodology for synthesis.
Article note
A collection of invited papers based on presentations at the 15th International Meeting on Boron Chemistry (IMEBORON-XV), Prague, Czech Republic, 24–28 August 2014.
References
[1] G. Urry, J. Kerrigan, T. D. Parsons, H. I. Schlesinger. J. Am. Chem. Soc.76, 5299 (1954).10.1021/ja01650a011Search in Google Scholar
[2] W. B. Fox, T. Wartik. J. Am. Chem. Soc.83, 498 (1961).10.1021/ja01463a066Search in Google Scholar
[3] I. Beletskaya, Ch. Moberg. Chem Rev. 106, 2320 (2006).10.1021/cr050530jSearch in Google Scholar
[4] T. B. Marder, N. C. Norman. Top. Catal. 5, 63 (1998).10.1023/A:1019145818515Search in Google Scholar
[5] T. Ishiyama, N. Miyaura. Chem. Rec. 3, 271 (2004).10.1002/tcr.10068Search in Google Scholar
[6] J. Cid, H. Gulyás, J. J. Carbó, E. Fernández. Chem. Soc. Rev. 41, 3558 (2012).10.1039/c2cs15291fSearch in Google Scholar
[7] J. Cid, J. J. Carbó, E. Fernández. Chem. Eur. J.18, 12794 (2012).10.1002/chem.201200987Search in Google Scholar
[8] K. Lee, A. R. Zhugralin, A. H. Hoveyda. J. Am. Chem. Soc. 131, 7253 (2009).10.1021/ja902889sSearch in Google Scholar
[9] A. Bonet, H. Gulyás, E. Fernández. Angew. Chem. Int Ed.49, 5130 (2010).10.1002/anie.201001198Search in Google Scholar
[10] C. Pubill-Ulldemolins, A. Bonet, C. Bo, H. Gulyás, E. Fernández. Chem. Eur. J.18, 1121 (2012).10.1002/chem.201102209Search in Google Scholar
[11] C. Solé, E. Fernández. Angew.Chem. Int. Ed. 52, 11351 (2013).10.1002/anie.201305098Search in Google Scholar
[12] H. Ito, Y. Horita, E. Yamamoto. Chem. Commun. 48, 8006 (2012).10.1039/c2cc32778cSearch in Google Scholar
[13] I. Ibrahem, P. Breistein, A. Córdova. Chem. Eur. J. 18, 5175 (2012).10.1002/chem.201103572Search in Google Scholar
[14] H. Wu, S. Radomkit, J. M. O’Brien, A. H. Hoveyda. J. Am. Chem. Soc. 134, 8277 (2012).10.1021/ja302929dSearch in Google Scholar
[15] C. Kleeberg, A. G. Crawford, A. S. Batsanov, P. Hodgkinson, D. C. Apperley, M. S. Cheung, Z. Y. Lin, T. B. Marder. J. Org. Chem. 77, 785 (2012).10.1021/jo202127cSearch in Google Scholar
[16] A. Bonet, C. Pubill-Ulldemolins, C. Bo, H. Gulyás, E. Fernández. Angew. Chem. Int. Ed.50, 7158 (2011).10.1002/anie.201101941Search in Google Scholar
[17] S. Mun, J. E. Lee, J. Yun. Org. Lett. 8, 4887 (2006).10.1021/ol061955aSearch in Google Scholar
[18] J. E. Lee, J. Kwon, J. Yun. Chem. Commun. 733 (2008).10.1039/B716697DSearch in Google Scholar
[19] C. Pubill-Ulldemolins, A. Bonet, H. Gulyás, C. Bo, E. Fernández. Org. Biomol. Chem.10, 9677 (2012).10.1039/c2ob26899jSearch in Google Scholar
[20] M. Gao, S. B. Thorpe, W. L. Santos. Org. Lett. 11, 3478 (2009).10.1021/ol901359nSearch in Google Scholar
[21] M. Gao, S. B. Thorpe, Ch. Kleeberg, C. Slebodnick, T. B. Marder, W. L. Santos. J. Org. Chem. 76, 3997 (2011).10.1021/jo2003488Search in Google Scholar
[22] S. B. Thorpe, X. Guo, W. L. Santos. Chem. Commun. 424 (2011).10.1039/C0CC02270ESearch in Google Scholar
[23] J. Cid, J. J. Carbó, E. Fernández. Chem. Eur J. 20, 3616 (2014).10.1002/chem.201304615Search in Google Scholar
[24] E. La Cascia, X. Sanz, C. Bo, A. Whiting, E. Fernández. Org. Biomol. Chem. (2014).Search in Google Scholar
[25] C. Sole, A. Whiting, H. Gulyás, E. Fernández. Adv. Synth. Catal.353, 376 (2011).10.1002/adsc.201000842Search in Google Scholar
[26] C. Sole, A. Tatla, J. A. Mata, A. Whiting, H. Gulyás, E. Fernández. Chem, Eur J.17, 14248 (2011).10.1002/chem.201102081Search in Google Scholar
[27] A. D. J. Calow, A. S. Batsanov, E. Fernández, C. Sole, A. Whiting. Chem Commun.48, 11401 (2012).10.1039/c2cc36129aSearch in Google Scholar
[28] A. D. J. Calow, C. Sole, A. Whiting, E. Fernández. Chem Cat Chem. 8, 2233 (2013).10.1002/cctc.201300113Search in Google Scholar
[29] A. D. J. Calow, A. S. Batsanov, A. Pujol, C. Solé, E. Fernández, A. Whiting. Org. Lett.15, 4810 (2013).10.1021/ol4022029Search in Google Scholar
[30] A. D. J. Calow, E. Fernández, A. Whiting. Org. Biomol. Chem, 12, 6121 (2014).10.1039/C4OB01142BSearch in Google Scholar
[31] S. Hara, H. Dojo, S. Takinami, A. Suzuki. Tetrahedron Lett.24, 731 (1983).10.1016/S0040-4039(00)81511-7Search in Google Scholar
[32] Y. Satoh, H. Serizawa, S. Hara, A. Suzuki. J. Am. Chem. Soc.107, 5225 (1985).10.1021/ja00304a032Search in Google Scholar
[33] C. Wang, M. Uchiyama. Eur. J. Org. Chem. 6548 (2012).10.1002/ejoc.201200975Search in Google Scholar
[34] A. Suzuki. Pure Appli. Chem. 58, 629 (1986).10.1351/pac198658040629Search in Google Scholar
[35] Z. Wang, A. Wang, P. Tarlí, K. K. Gannett. J. Am. Chem. Soc. 118, 10783 (1996).10.1021/ja9622620Search in Google Scholar
[36] C. Wang, T. Tobrman, Z. Xu, E. -i Negishi. Org. Lett. 11, 4092 (2009).10.1021/ol901566eSearch in Google Scholar
[37] M.-L. Yao, M. S. Reddy, W. Zeng, K. Hall, I. Walfish, G. W. Kabalka. J. Org. Chem. 74, 1385 (2009).10.1021/jo802207ySearch in Google Scholar
[38] M. F. Lappert, B. Prokai. J. Organomet. Chem. 1, 384 (1964).10.1016/S0022-328X(00)80030-3Search in Google Scholar
[39] B. Wrackmeyer. Polyhedron5, 1709 (1986).10.1016/S0277-5387(00)84848-2Search in Google Scholar
[40] J. R. Lawson, E. R. Clark, I. A. Cade, S. A. Solomon, M. J. Ingleson. Angew. Chem. Int Ed. 52, 7518 (2013).10.1002/anie.201302609Search in Google Scholar
[41] G. Palau-Lluch, E. Fernández. Adv. Synth. Catal. 355, 1464, (2013).10.1002/adsc.201300282Search in Google Scholar
[42] F. C. Pigge. Synthesis42, 1745 (2010).10.1055/s-0029-1218756Search in Google Scholar
[43] P. Zhang, H. Le, R. E. Kyne, J. P. Morken. J. Am. Chem. Soc.133, 9716 (2011).10.1021/ja2039248Search in Google Scholar
[44] R. Shintani, K. Takatsu, M. Takeda, T. Hayashi. Angew. Chem.50, 8656 (2011).10.1002/anie.201103581Search in Google Scholar
[45] B. Jung, A. H. Hoveyda. J. Am. Chem. Soc.134, 1490 (2012).10.1021/ja211269wSearch in Google Scholar
[46] F. Gao, J. L. Carr, A. H. Hoveyda. Angew. Chem. Int. Ed.51, 6613 (2012).10.1002/anie.201202856Search in Google Scholar
[47] J. A. Schiffner, K. Müther, M. Oestreich. Angew. Chem. Int. Ed. 49, 1194 (2010).10.1002/anie.200906521Search in Google Scholar
[48] E. Hartmann, D. J. Vyas, M. Oestreich. Chem. Commun. 7917 (2011).10.1039/c1cc10528kSearch in Google Scholar PubMed
[49] V. Lillo, A. Bonet, E. Fernández. Dalton Trans. 2899 (2009).10.1039/b819237eSearch in Google Scholar PubMed
[50] L. Dang, Z. Lin, T. B. Marder. Chem. Commun. 3987 (2009).10.1039/b903098kSearch in Google Scholar PubMed
[51] L. Mantilli, C. Mazet. ChemCatChem2, 501 (2010).10.1002/cctc.201000008Search in Google Scholar
[52] A. D. J. Calow, A. Whiting. Org. Biomol. Chem.29, 5485 (2012).10.1039/c2ob25908gSearch in Google Scholar
[53] M. Ngatimin, Ch. J. Gartshore, J. P. Kindler, S. Naidu, D. W. Lupton. Tetrahedron Lett.50, 6008 (2009).10.1016/j.tetlet.2009.08.038Search in Google Scholar
[54] I.-H. Chen, L. Yin, W. Itano, M. Kanai, M. Shibasaki. J. Am. Chem. Soc.131, 11664 (2009).10.1021/ja9045839Search in Google Scholar
[55] T. Furuka, A. S. Kamlet, T. Ritter. Nature473, 470 (2010).10.1038/nature10108Search in Google Scholar PubMed PubMed Central
[56] M. Oestreich. Angew. Chem. Int. Ed. 44, 2324 (2005).10.1002/anie.200500478Search in Google Scholar PubMed
[57] J.-A. Ma, D. Cahard. Chem. Rev. 108, PR1–PR43 (2008).10.1021/cr800221vSearch in Google Scholar PubMed
[58] S. Lectard, Y. Hamashima, M. Sodeoka. Adv. Synth. Catal. 352, 2708 (2010).10.1002/adsc.201000624Search in Google Scholar
[59] L. Hintermann, A. Togni. Angew. Chem. Int. Ed.39, 4359 (2000).10.1002/1521-3773(20001201)39:23<4359::AID-ANIE4359>3.0.CO;2-PSearch in Google Scholar
[60] Y. Hamashima, K. Yagi, H. Takano, L. Tamás, M. Sodeoka. J. Am. Chem. Soc. 124, 14530 (2002).10.1021/ja028464fSearch in Google Scholar
[61] D. Enders, M. R. M. Hottl. Synlett 991 (2005).10.1055/s-2005-864813Search in Google Scholar
[62] M. Marigo, D. Fielenbach, A. Braunton, A. Kjaersgaard, K. A. Jørgensen. Angew. Chem. Int. Ed. 44, 3703 (2005).10.1002/anie.200500395Search in Google Scholar
[63] D. D. Steiner, N. Mase, C. F. Barbas III. Angew. Chem. Int. Ed. 44, 3706 (2005).10.1002/anie.200500571Search in Google Scholar
[64] T. D. Beeson, D. W. C. Mac-Millan. J. Am. Chem. Soc. 127, 8826 (2005).10.1021/ja051805fSearch in Google Scholar
[65] T. Ishimaru, N. Shibata, T. Horikawa, N. Yasuda, S. Nakamura, T. Toru, M. Shiro. Angew. Chem. Int. Ed. 47, 4157 (2008).10.1002/anie.200800717Search in Google Scholar
[66] T. D. Kwiatkowsli, J. C. Beeson, D. W. C. Conrad, Mac-Millan. J. Am. Chem. Soc. 133, 1738 (2011).10.1021/ja111163uSearch in Google Scholar
[67] H. Ibrahim, A. Togni. Chem. Commun. 1147 (2004).10.1039/b317004gSearch in Google Scholar
[68] X. Sanz, G. M. Lee, C. Pubill-Ulldemolins, A. Bonet, H. Gulyás, S. A. Westcott, C. Bo, E. Fernández. Org Biomol. Chem.11, 7004 (2013).10.1039/c3ob41328dSearch in Google Scholar
[69] A. Bonet, C. Solé, H. Gulyás, E. Fernández. Org. Biomol. Chem.10, 6621 (2012).10.1039/c2ob26079dSearch in Google Scholar
[70] N. Miralles, J. Cid, A. B. Cuenca, J. J. Carbó, E. Fernández, Chem. Commun. (2014). DOI: 10.1039/C4CC08743G.10.1039/C4CC08743GSearch in Google Scholar PubMed
[71] N. Iwadate, M. Suginome. J. Am. Chem. Soc. 132, 2548 (2010).10.1021/ja1000642Search in Google Scholar
[72] Th. P. Blaisdell, Th. C. Caya, L. Zhang, A. Sanz-Marco, J. P. Morken. J. Am. Chem. Soc. 136, 9264 (2014).10.1021/ja504228pSearch in Google Scholar
[73] Y. Nagashima, K. Hirano, R. Takita, M. Uchiyama. J. Am. Chem. Soc. 136, 8532 (2014).10.1021/ja5036754Search in Google Scholar
[74] X. Sanz, Ch. M. Vogels, A. Decken, C. Bo, S. A. Westcott, E. Fernández. Chem. Commun. 50, 8420 (2014).10.1039/c4cc02098gSearch in Google Scholar
©2014 IUPAC & De Gruyter
Articles in the same Issue
- Frontmatter
- Preface
- 15th International Conference on Boron Chemistry (IMEBORON XV)
- Conference papers
- Nanomaterials for boron and gadolinium neutron capture therapy for cancer treatment
- Acyl chloride carbon insertion into dicarbaborane cages – new route to tricarbollide cages
- Diaryl-substituted carboranes as inhibitors of hypoxia inducible factor-1 transcriptional activity
- Boric acid: a simple molecule of physiologic, therapeutic and prebiotic significance
- Carbaboranes – more than just phenyl mimetics
- Boron clusters in medicinal chemistry: perspectives and problems
- Organocatalytic functionalisation through boron chemistry
- Chemistry of early and late transition metallaboranes: synthesis and structural characterization of periodinated dimolybdaborane [(Cp*Mo)2B4H3I5]
- Invited paper
- Template-directed nonenzymatic oligonucleotide synthesis: lessons from synthetic chemistry
Articles in the same Issue
- Frontmatter
- Preface
- 15th International Conference on Boron Chemistry (IMEBORON XV)
- Conference papers
- Nanomaterials for boron and gadolinium neutron capture therapy for cancer treatment
- Acyl chloride carbon insertion into dicarbaborane cages – new route to tricarbollide cages
- Diaryl-substituted carboranes as inhibitors of hypoxia inducible factor-1 transcriptional activity
- Boric acid: a simple molecule of physiologic, therapeutic and prebiotic significance
- Carbaboranes – more than just phenyl mimetics
- Boron clusters in medicinal chemistry: perspectives and problems
- Organocatalytic functionalisation through boron chemistry
- Chemistry of early and late transition metallaboranes: synthesis and structural characterization of periodinated dimolybdaborane [(Cp*Mo)2B4H3I5]
- Invited paper
- Template-directed nonenzymatic oligonucleotide synthesis: lessons from synthetic chemistry