Hydrogenation of carboxylic acid derivatives with bifunctional ruthenium catalysts
-
Takao Ikariya
 and
Yoshihito Kayaki
and
Yoshihito Kayaki

Abstract
This paper focuses on recent advances in the chemistry of bifunctional Ru catalysts with chelate protic amine ligands effective for hydrogenation of polar carbonyl compounds. The rational design of the cooperating amine ligand that adjusts the balance of the electronic factors on the M/NH units in the bifunctional catalysts is crucial to exploit the characteristic catalytic performance with a wide scope and high practicability. These bifunctional molecular catalysts offer a great opportunity to develop new fundamental processes for the straightforward hydrogenation of carboxylic acid derivatives as a powerful alternative for classical reduction using stoichiometric amounts of metal hydride reagents.
Introduction
Molecular hydrogen (H2), the most atom-efficient reducing chemical agent, is a component of the largest-volume human-made chemical reactions, hydrogenation of organic compounds [1]. Reduction of functional groups with molecular hydrogen is one of the fundamental reactions in organic chemistry. A great breakthrough was made through Noyori’s discovery of bifunctional ruthenium catalysts for direct hydrogenation and transfer hydrogenation of ketones [2]. Significant efforts have been continuously undertaken to design bifunctional molecular catalysts having the combination of two or more active sites working in concert, to attain highly efficient molecular transformation of polar substrates via H+ and H– transfer processes [3]. Detailed experimental and theoretical analyses of the real catalysts revealed that both an amidoruthenium complex having a square-planar geometry and a coordinatively saturated hydrido(amine)ruthenium complex are involved in the catalytic cycle. These reactions proceed via two steps, an enantio-determining hydride transfer and proton transfer, through a contact ion-pair intermediate, as depicted in Fig. 1 [4].
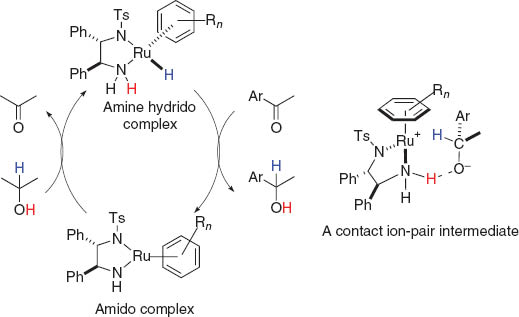
Interconversion between amido and amine(hydrido)Ru complex.
In 2001, we reported an effect of alcoholic solvent facilitating the heterolytic cleavage of H2 bound to the 16-electron [Cp*Ru{L(CH2)2 NH2}]+ fragment as shown in Fig. 2 [5]. Further systematic study of catalytic hydrogenations of polar substrates showed that the replacement of the tertiary amino group in the ligand with a tertiary phosphino group results in the expansion in the scope of the Ru/NH bifunctionality [6]. Whereas the half-sandwich bifunctional precatalysts Cp*RuCl[(CH3)2 N(CH2)2 NH2], 2, are only effective for ketone reductions, Cp*RuCl(P–N) (P–N: (C6 H5)2 P(CH2)2 NH2), 1 combined with a base efficiently promote hydrogenation of either ketones, imides or epoxides [7]. Presumably, the back-bonding properties of the phosphorus atom might enhance the Lewis acidity of the NH2 protons, thus facilitating the hydrogen transfer to various polar functionalities.
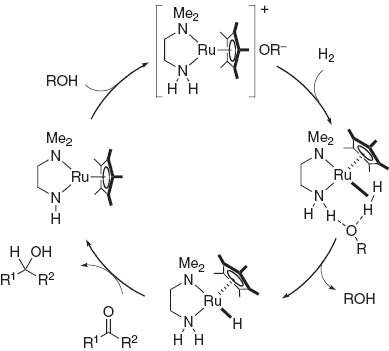
A possible mechanism of H2-activation by a cationic Ru fragment.
Although the original bifunctional amido–Ru catalysts, Ru[Tsdpen](η6-arene) (Tsdpen = N-(p-toluenesulfonyl)-1,2-diphenylethylenediamine), cannot efficiently promote hydrogenation of ketonic substrates, some isolable cationic amine Ru and Ir complexes, Ru(OTf)[Tsdpen](η6-p-cymene) [8] and [Cp*Ir(Tscydn)(CH3 CN)]+SbF6– [9] are highly effective for the asymmetric hydrogenation of either ketones or imines. Similarly to the mechanism in Fig. 2, a key step is the heterolytic H2 cleavage, which is promoted by cationic species [10]. It generates an active hydride complex such as RuH(Tsdpen)(η6-p-cymene) with simultaneous release of HOTf in a polar solvent like methanol. After the hydride/proton transfer from the amine hydrido complex, the regenerated 16-electron amido Ru complex reacts with HOTf to complete the catalytic cycle. These experimental results prompted us to develop new chiral hydrogenation catalysts, [η6:η1-C6 H5(CH2)n NTf]Ru[(S,S)-Tsdpen] and [η5:η1-(CH3)4 C5(CH2)n NTf]M[(S,S)-Msdpen] (M = Rh and Ir) in which the NTf unit is linked to a η6-arene or η5-tetramethylcyclopentadienyl ring [11]. The introduction of a suitable tether unit dramatically facilitates the heterolytic cleavage of H2 suggesting that the hemilabile NTf group on the tether participates in the proton transfer processes as shown in Fig. 3.
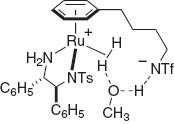
Heterolytic H2-cleavage by NTf tethered-Ru complex.
Significant progress in the field of selective hydrogenation of C–O double bonds of ketones with both chiral and achiral catalysts has been made in the branch of homogeneous transition metal-based molecular catalysis. Until now only limited progress was observed in the hydrogenation of the polar functionalities bearing less electrophilic carbonyl groups, such as carboxylic acids and their derivatives [12]. The recent progress in the hydrogenation of esters and lactones exploits the conceptually new bifunctional molecular catalysts originating from the metal/NH synergy effect or aromatization-dearomatization concept [13]. In addition to the half-sandwich chelate amine complex, several pincer ligands involving a metal/NH group have been developed for powerful hydrogenation systems. For example, Takasago Int. Corp. released an efficient precatalyst, RuHCl(CO)(dpa) (4a; dpa = HN(CH2 CH2 PPh2)2), which has high potential industrial application. Hence, our further efforts were directed towards these synthetically important transformations by using a series of bifunctional Ru catalysts depicted in Fig. 4.
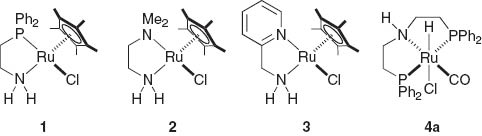
Bifunctional Ru precursors for catalytic hydrogenation.
Hydrogenation of imides and N-acylcarbamates with bifunctional Ru catalysts bearing P–NH2 ligands
The Cp*RuCl(P–N) catalyst allows the straightforward catalytic hydrogenation of carboxyimides and N-acylcarbamates yielding N-protected amines and alcohols selectively [14]. Prochiral cyclic imides such as 4-substitued glutarimides and cyclic meso-imides, such as 3,4-disubstituted succinimides, are important starting compounds in asymmetric synthesis. The known carbonyl-selective reductions of N-substituted cyclic imides affording lactams or carboxamide alcohols, mostly rely on the stoichiometric amount of metal hydride reagents in organic synthesis [15].
We found that a variety of imides are chemoselectively reduced to the corresponding alcohols and carboxamides in 2-propanol containing bifunctional Ru precatalyst, Cp*RuCl(P–N) 1, and the base KOtC4 H9 as the catalytic system under mild conditions, as shown in Scheme 1 [14a]. Only the catalysts with P–N ligands are effective in these reactions, whereas the analogous Cp*RuCl[k2(N,N)-(CH3)2 N(CH2)2 NH2] catalyst 2 exhibits no catalytic activity toward the imide hydrogenation. Notably, the tertiary amine variant (C6 H5)2 P(CH2)2 N(CH3)2 as the ligand is completely ineffective, indicating the crucial importance of the NH group in the ligand for the M/NH bifunctional activation of the carbonyl compounds.

Chemoselective hydrogenation of imides catalyzed by a binary catalyst system composed of Cp*RuCl(P–N) 1 with base.
The hydrogenation method is characterized by its excellent chemoselectivity and a wide substrate scope. A less substituted acyl group in the 3,3-dimethyl substituted glutarimide was exclusively hydrogenated to give the corresponding amide alcohol, as shown in Scheme 1. The orientation of the two carbonyl groups possibly plays a key role in the selectivity. This chemoselective hydrogenation is applicable to the deprotection of primary amines from N-phthaloyl-protected amino acid ester derivatives in Gabriel amino acid synthesis. Thus, N-phthaloyl-l-Phe methyl ester undergoes hydrogenation under neutral conditions to generate N-(o-hydroxymethylbenzoyl)-l-Phe methyl ester without any loss of the ee value after acid hydrolysis, as shown in Scheme 2.

Deprotection of N-phthaloyl amino acid derivative.
The chiral version of the Cp*RuCl(P–N) catalyst bearing the chiral P–N ligand derived from l-proline promoted the enantioselective hydrogenation of prochiral 4-arylglutarimides to provide the corresponding hydroxyamides with excellent enantiomeric excesses and in high yields, as shown in Scheme 3. The resulting chiral hydroxyamides are readily converted by a ring-closing followed by deprotection to chiral piperidinone derivatives, which serve as important synthetic intermediates for a number of physiologically active chiral compounds, including the antidepressant paroxetine. Enantioselective hydrogenation of readily accessible bicyclic imides with the 3,4-(OCH2 O)C6 H3 group derived from cyclic dicarboxylic acids gave the corresponding hydroxyamides with excellent enantiomeric excesses as shown in Scheme 4 [14b]. Thus, the cyclic dicarboxylic acids can be transformed to chiral cyclic compounds bearing two different functional groups on two chiral centers, which would otherwise require tedious multistep synthesis.

Enantioselective hydrogenation of prochiral cyclic and bicyclic imides with bifunctional chiral Cp* RuCl(P–N) catalysts.

Catalytic hydrogenation of N-acylcarbamates and related compounds with Cp*Ru(P–N) catalyst 1.
Similarly to the hydrogenation of imides with the catalyst 1, N-acylcarbamates, N-acyloxazolidinone, and N-acylsulfonamides can be hydrogenated, as shown in Scheme 4 [14c]. tert-Butyl alcohol is the preferable solvent for the hydrogenation of these substrates because other alcohols can cause undesired alcoholysis of the substrates. The reaction of N-alkoxycarbonyl protected cyclic carboxamides (lactams) gave preferentially N-protected aminoalcohols in almost quantitative yield. The rates of the reaction strongly rely on the electron-withdrawing nature of substituents on nitrogen in the substrates, the rate increasing in the order of Cbz<Boc<CO2 CH3<Ts.
Notably, the hydrogenation was applicable to the reductive transformation of chiral N-acyloxazolidiones, which are useful synthetic intermediates in the asymmetric synthesis. The resulting chiral alcohol and the original chiral auxiliary can be obtained without any loss in the optical purity as shown in Scheme 4. This method may be an environmentally benign catalytic alternative to the method using LiAlH4, which sometimes causes difficulty in the recovery of the chiral auxiliaries.
Hydrogenation of esters and lactones with bifunctional Ru catalysts
Bifunctional catalyst 1, Cp*RuCl(P–N), in the presence of a large amount of base promotes the hydrogenation of lactones and simple esters, leading to the corresponding diols and alcohols, respectively [16, 17]. For example, phthalide was cleanly hydrogenated to oxylyleneglycol under 50 atm of H2 at 100 °C in the presence of catalyst 1, as shown in Scheme 5. A variety of aprotic solvents, including THF, dioxane, and toluene, can be equally used as alcoholic solvents under similar conditions. The amount of base has a significant influence on the reaction rate, and the addition of more than 25 equiv of NaOCH3 as the base to the Cp*RuCl(P–N) complex resulted in an improvement of its catalytic performance, although the reason for this is still unclear. Structural modification of the protic chelating amine ligand of the Cp*Ru catalyst system caused marked improvement in the catalytic performance and provided efficient access to diols from lactones, as shown in Scheme 5 [18]. Thus, Cp*RuCl(N–N) (N–N: 2-C5 H4 NCH2 NH2) 3 bearing a pyridyl amine chelating ligand is more effective for hydrogenation of lactones than Cp*RuCl(P–N) catalyst. The precatalyst 3 combined with 25 mol% of NaOCH3 was efficient for hydrogenation of several kinds of lactones at 100 °C under 50 atm of H2 in 2-propanol to give the corresponding diols in reasonably good to excellent yields. The catalytic activity of Cp*RuCl(N–N) catalyst is comparable with Milstein’s RuH(NNP)(CO) pincer complex, although the latter works without any base additive and under lower H2 pressure [19]. A variety of esters and lactones can be reduced to the corresponding alcohols and diols, respectively.

Catalytic hydrogenation of esters and lactones with bifunctional Cp*Ru catalysts.
Since the discovery of Milstein’s pincer catalyst for the hydrogenation of esters, modified pincer catalysts have been designed by their group and others [13]; namely, a pincer-type bifunctional complex, RuHCl(CO)(dpa) 4a (dpa = [(C6 H5)2 P(CH2)2]2 NH) serves as more efficient catalyst for ester hydrogenation [20]. In particular, the commercially available catalyst 4a effects hydrogenation of α-fluorinated esters with S/C = 4000–5000 in methanol containing NaOCH3 under 10 atm of H2 at 40 °C giving fluorinated alcohols in almost quantitative yields as shown in Scheme 6 [21]. Notably, non-fluorinated methyl acetate and methyl trifluoropropionate bearing fluorine atoms at the β-position were hydrogenated inefficiently. Hence, the introduction of the fluorine atom at α-position of esters caused a marked increase in the rate of the reaction.

Selective hydrogenation of α-fluorinated esters with bifunctional pincer-type Ru (II) catalysts.
This carbonyl selective hydrogenation is applicable to the synthesis of α-fluorinated unsaturated alcohols. For example, the reduction of ethyl 2,2-difluoropentenoate gave unsaturated alcohol with 99.6 % selectivity, in which the E/Z ratio (94/6) of the substrate was invariable during the reaction. Noticeably, the hydrogenation of chiral (R)-2-fluoropropionate gave the monofluorinated alcohol in almost quantitative yield although with a low ee due to racemization under basic conditions. To minimize racemization of the chiral ester, the drop-wise slow addition of the chiral ester provided the corresponding chiral monofluorinated alcohol without any serious decrease of the ee value.
The catalyst precursor, RuHCl(CO)(dpa) 4a, was found to be readily converted to the real catalyst, trans-RuH2(CO)(dpa) 4b by treatment of 4a with 1 equiv of KOtC4 H9 in THF-d8 under a hydrogen atmosphere. In addition to the dihydride complex 4b, trans-RuCl2(CO)(dpa) 4c also worked well for the hydrogenation of methyl trifluoroacetate albeit under longer reaction times, as listed in Table 1. Fluorinated esters, including CF2 HCO2 CH3, CF3 CO2 CH3, CClF2 CO2 CH3, and C2 F5 CO2 CH3, were selectively hydrogenated with 4a or 4b in the reaction with S/C = 5000 to hemiacetal intermediates, which are among the most important synthons for organic synthesis. Notably, the outcome of the reaction was delicately influenced by the reaction conditions. A decrease of the reaction temperature to 15 °C in the hydrogenation of CF2 HCO2 CH3 caused a drastic change in the product ratio of alcohol/hemiacetal, giving preferentially the corresponding hemiacetal CF2 HCH(OH)OCH3. When the H2 pressure was decreased under less than 10 atm at 40 °C, the hemiacetals (B) were obtainable preferentially. These results suggest that the highly fluorinated hemiacetal is stable enough to resist further conversion to alcohols (A) under the reaction conditions. After optimization of hydrogenation conditions, a preparative scale reaction of methyl trifluoroacetate, 128 g with S/C = 20 000 using the catalyst 4a gave fluoral hemiacetal in 89 % yield and with 96 % selectivity.
Hydrogenative transformation of α-fluorinated esters into alcohols A and hemiacetals B.
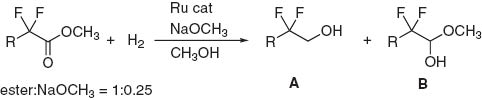 | |||||||
|---|---|---|---|---|---|---|---|
| Cat | R | S/C | Time, h | Temperature, °C | H2, atm | % Yield | A:B |
| 4a | F | 5000 | 6 | 40 | 10 | 92 | 9:91 |
| 4ba | F | 5000 | 8 | 40 | 10 | 89 | 8:92 |
| 4c | F | 3000 | 21 | 50 | 10 | 84 | 4:96 |
| 4a | H | 5000 | 6 | 40 | 10 | >99 | 100:0 |
| 4a | H | 5000 | 6 | 15 | 10 | 58 | 8:92 |
| 4a | F | 2000 | 6 | 40 | 20 | 99 | 81:15 |
aEster:NaOCH3 = 1:0.1
Continuum methanol reaction field DFT analysis with the SMD (solvation model based on density) of the selective hydrogenation of CF3 C(O)OCH3 to the hemiacetal intermediate suggests a very facile outer-sphere hydride transfer (ΔG‡298 K = 7.8 kcal·mol–1) from the trans-RuH2(CO)(dpa) 4b to CF3 C(O)OCH3 through a H-bonded complex Int1 via TS1 via a two-step process to afford the inner-sphere ion pair intermediate Int2, as shown in Fig. 5. A similar step-wise hydride and proton transfer mechanism from the bifunctional Ru hydride complex to carbonyl compound through the contact ion-pair intermediate was observed as discussed in Fig. 1 [4].
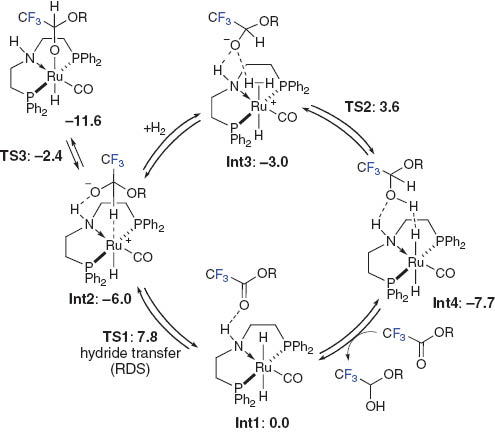
A possible catalytic cycle for the formation of trifluoroacetaldehyde methyl hemiacetal. All the free energies (ΔG°298 K, kcal·mol–1) are calibrated relative to Int1.
Hydrogenation of carboxamides with bifunctional Ru catalysts bearing N–NH2 ligands
Carboxamides with a less electrophilic carbonyl group are still one of the challenging targets in the hydrogenation of carboxylic acid derivatives [18, 22, 23]. Possible reaction pathways in the hydrogenation of carboxamides or lactams are presented in Scheme 7. Path A, which represents reductive cleavage of the C–O bond along with production of water, is a typical pathway for heterogeneous systems or traditional homogeneous catalysts [22], whereas path B, which represents CO reduction followed by C–N bond cleavage, is a typical pathway for bifunctional catalysts [18, 23].

Chemoselectivity in hydrogenation of carboxamides or lactams.
The Cp*RuCl(N–N) catalyst (N–N: 2-C5 H4 NCH2 NH2) 3 combined with base catalyzes selective and direct hydrogenation of lactams and carboxamides to form amino alcohols and a mixture of amines and alcohols, respectively (path B in Scheme 7). For example, N-phenylpyrrolidinone was smoothly hydrogenated in 2-propanol containing 10 mol% of 3 to give 4-(phenylamino)butan-1-ol in 73 % isolated yield, although N-benzylpyrrolidinone was reluctant under similar conditions. Nevertheless, a wide variety of carboxamides were susceptible to the present hydrogenation as long as they had an aryl group on their nitrogen (Scheme 8). The inertness of carboxamides lacking any aryl group on the nitrogen allowed a chemoselective hydrogenation of a certain type of amide esters bearing an ester unit in the same molecule, which cleanly gave the hydroxyalkyl-substituted lactams under similar conditions. Notably, in the hydrogenation of lactams bearing no substituent on the nitrogen, the corresponding cyclic amines can be obtained via selective dehydration (path A in Scheme 6) [24].

Catalytic hydrogenation of carboxamides and lactams with Cp*Ru(N–N) catalyst 3.
Conclusions
We demonstrated that a family of Ru catalysts with the metal/NH bifunctional moiety, such as Cp*Ru(P–N) 1 and Cp*Ru(N–N) 3, and RuHCl(CO)(dpa) 4a complexes, serves as efficient catalysts for hydrogenation of carbonyl compounds including carboxylic acid derivatives, esters and carboxamides. The amido complexes formed from chloro(amine) complexes and KOC(CH3)3, promoted heterolytic cleavage of dihydrogen in alcoholic solvents. Significant and rapid progress, particularly for the ester hydrogenation, has been achieved for the past decade by utilization of conceptually new bifunctional molecular catalysts originating from the metal–ligand cooperation effects. However, most molecular catalysts still need relatively forced reaction conditions in terms of the temperature and pressure. From a practical point of view, development of base-free catalysts for hydrogenation is also highly desirable. Therefore, the rational design of new molecular catalyst systems is crucially important to explore unprecedented catalytic performance for hydrogenation of polar functionalities. The industrial outlook for the hydrogenation with the sophisticated catalysts should be bright because of their excellent catalytic performance, operational simplicity, and economic viability as well as their suitability for the green chemistry.
Article note: A collection of invited papers based on presentations at the 44th IUPAC Congress, Istanbul, Turkey, 11–16 August 2013.
Acknowledgments
This work was financially supported by a Grant-in-Aid for Scientific Research (S) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (No. 22225004) and partly supported by the GCOE Program.
References
[1] G. J. Kubas. Chem. Rev.107, 4152 (2007).10.1021/cr050197jSearch in Google Scholar PubMed
[2] (a) T. Ohkuma, H. Ooka, S. Hashiguchi, T. Ikariya, R. Noyori. J. Am. Chem. Soc.117, 2675 (1995). (b) S. Hashiguchi, A. Fujii, J. Takehara, T. Ikariya, R. Noyori. J. Am. Chem. Soc.117, 7562 (1995).Search in Google Scholar
[3] (a) T. Ikariya. Bull. Chem. Soc. Jpn.84, 1 (2011). (b) T. Ikariya, A. J. Blacker. Acc. Chem. Res.40, 1300 (2007). (c) T. Ikariya, K. Murata, R. Noyori. Org. Biomol. Chem.4, 393 (2006). (d) K. Muñiz. Angew. Chem. Int. Ed.44, 6622 (2005).Search in Google Scholar
[4] P. A. Dub, T. Ikariya. J. Am. Chem. Soc.135, 2604 (2013).Search in Google Scholar
[5] M. Ito, M. Hirakawa, K. Murata, T. Ikariya. Organometallics20, 379 (2001).10.1021/om000912+Search in Google Scholar
[6] M. Ito, T. Ikariya. Chem. Commun.43, 5134 (2007).Search in Google Scholar
[7] M. Ito, M. Hirakawa, A. Osaku, T. Ikariya. Organometallics22, 4190 (2003).10.1021/om034006jSearch in Google Scholar
[8] (a) T. Ohkuma, N. Utsumi, K. Tsutsumi, K. Murata, C. Sandoval, R. Noyori. J. Am. Chem. Soc.128, 8724 (2006). (b) C. A. Sandoval, T. Ohkuma, N. Utsumi, K. Tsutsumi, K. Murata, R. Noyori. Chem. Asian J.1–2, 102 (2006). (c) T. Ohkuma, K. Tsutsumi, N. Utsumi, N. Arai, R. Noyori, K. Murata. Org. Lett.9, 255 (2007).Search in Google Scholar
[9] S. Shirai, H. Nara, Y. Kayaki, T. Ikariya. Organometallics28, 802 (2009).10.1021/om800926pSearch in Google Scholar
[10] (a) R. M. Bullock. Chem. Eur. J.10, 2366 (2004). (b) S. E. Clapham, A. Hadzovic, R. H. Morris. Coord. Chem. Rev.248, 2201 (2004).Search in Google Scholar
[11] (a) M. Ito, Y. Endo, T. Ikariya. Organometallics27, 6053 (2008). (b) M. Ito, Y. Endo, N. Tejima, T. Ikariya. Organometallics29, 2397 (2010).Search in Google Scholar
[12] (a) P. A. Dub, T. Ikariya. ACS Catal.2, 1718 (2012). (b) W. W. N. O, R. H. Morris, ACS Catal.3, 32 (2013). (c) T. P. Brewster, A. J. M. Miller, D. M. Heinekey, K. I. Goldberg. J. Am. Chem. Soc.135, 16022 (2013). (d) D. Spasyk, S. Smith, D. G. Gusev. Angew. Chem. Int. Ed.52, 2538 (2013). (e) X.-H. Yang, J.-H. Xie, W.-P. Liu, Q.-L. Zhou. Angew. Chem. Int. Ed.52, 7833 (2013).Search in Google Scholar
[13] C. Gunanathan, D. Milstein. Acc. Chem. Res.44, 588 (2011).Search in Google Scholar
[14] (a) M. Ito, A. Sakaguchi, C. Kobayashi, T. Ikariya. J. Am. Chem. Soc. 129, 290 (2007). (b) M. Ito, C. Kobayashi, A. Himizu, T. Ikariya. J. Am. Chem. Soc. 132, 11414 (2010). (c) M. Ito, L. W. Koo, A. Himizu, C. Kobayashi, A. Sakaguchi, T. Ikariya. Angew. Chem. Int. Ed. 48, 1324 (2009).Search in Google Scholar
[15] J. Seyden-Penne. Reductions by the Alumino and Borohydrides in Organic Synthesis, 2nd Ed., Wiley-VCH, Weinheim (1997).Search in Google Scholar
[16] M. Ito, T. Ikariya. J. Synth. Org. Chem. Jpn.66, 1042 (2008).Search in Google Scholar
[17] T. Ikariya, M. Ito, A. Shiibashi, T. Ootsuka, Tokyo Institute of Technology, Japan; Central Glass Company, Limited; Patent WO2010004883A1, p 31 (2010).Search in Google Scholar
[18] M. Ito, T. Ootsuka, R. Watari, A. Shiibashi, A. Himizu, T. Ikariya. J. Am. Chem. Soc. 133, 4240 (2011).Search in Google Scholar
[19] J. Zhang, G. Leitus, Y. Ben-David, D. Milstein. Angew. Chem. Int. Ed. 45, 1113 (2006).Search in Google Scholar
[20] W. Kuriyama, T. Matsumoto, O. Ogata, Y. Ino, K. Aoki, S. Tanaka, K. Ishida, T. Kobayashi, N. Sayo, T. Saito. Org. Process Res. Dev.16, 166 (2012).Search in Google Scholar
[21] T. Otsuka, A. Ishii, P. A. Dub, T. Ikariya. J. Am. Chem. Soc.135, 9600 (2013).Search in Google Scholar
[22] (a) A. A. N. Magro, G. R. Eastham, D. Cole-Hamilton. Chem. Commun.43, 3154 (2007). (b) M. Stein, B. Breit. Angew. Chem. Int. Ed.52, 2231 (2013). (c) J. Coetzee, D. L. Dodds, J. Klankermayer, S. Brosinski, W. Leitner, A. M. Z. Slawin, D. J. Cole-Hamilton. Chem. Eur. J.19, 11039 (2013).Search in Google Scholar
[23] (a) E. Balaraman, B. Gnanaprakasam, L. J. W. Shimon, D. Milstein. J. Am. Chem. Soc.132, 16756 (2010). (b) J. M. John, S. H. Bergens, Angew. Chem. Int. Ed.50, 10377 (2011). (c) T. Miura, I. E. Held, S. Oishi, M. Naruto, S. Saito. Tetrahedron Lett. 54, 2674 (2013).Search in Google Scholar
[24] R. Watari, T. Otsuka, M. Ito, T. Ikariya, In 5th Kanto Branch Conference, The Chemical Society of Japan, 1B529: Tokyo, Japan, August 30–31, (2011).Search in Google Scholar
©2014 IUPAC & De Gruyter Berlin/Boston
Articles in the same Issue
- Frontmatter
- Preface
- 44th IUPAC World Chemistry Congress: Clean Energy Through Chemistry
- Conference papers
- The energy landscape concept and its implications for synthesis planning
- Near-IR absorbing Bodipy functionalized SPIONs: a potential magnetic nanoplatform for diagnosis and therapy
- Glycerol acetals with antioxidant properties
- Lithiated oxazolinyloxiranes and oxazolinylaziridines: key players in organic synthesis
- Access to pyrrole-based heterocyclic compounds via addition of pyrrole to C=C and C=N bonds
- Hydrogenation of carboxylic acid derivatives with bifunctional ruthenium catalysts
- Generation of singlet oxygen (1O2) from hydrogen peroxide decomposition by in situ generated hypervalent iodoarene reagents
- Organometallic macrocycles and cages based on bis(amidinate) ligands
- Anisotropic core-shell Fe3 O4 @Au magnetic nanoparticles and the effect of the immunomagnetic separation volume on the capture efficiency
- Recent investigations of bioactive natural products from endophytic, marine-derived, insect pathogenic fungi and Thai medicinal plants
- Chemoecological studies on marine natural products: terpene chemistry from marine mollusks
- IUPAC Recommendations
- Abbreviations of polymer names and guidelines for abbreviating polymer names (IUPAC Recommendations 2014)
- IUPAC Technical Report
- Toward a comprehensive definition of oxidation state (IUPAC Technical Report)
Articles in the same Issue
- Frontmatter
- Preface
- 44th IUPAC World Chemistry Congress: Clean Energy Through Chemistry
- Conference papers
- The energy landscape concept and its implications for synthesis planning
- Near-IR absorbing Bodipy functionalized SPIONs: a potential magnetic nanoplatform for diagnosis and therapy
- Glycerol acetals with antioxidant properties
- Lithiated oxazolinyloxiranes and oxazolinylaziridines: key players in organic synthesis
- Access to pyrrole-based heterocyclic compounds via addition of pyrrole to C=C and C=N bonds
- Hydrogenation of carboxylic acid derivatives with bifunctional ruthenium catalysts
- Generation of singlet oxygen (1O2) from hydrogen peroxide decomposition by in situ generated hypervalent iodoarene reagents
- Organometallic macrocycles and cages based on bis(amidinate) ligands
- Anisotropic core-shell Fe3 O4 @Au magnetic nanoparticles and the effect of the immunomagnetic separation volume on the capture efficiency
- Recent investigations of bioactive natural products from endophytic, marine-derived, insect pathogenic fungi and Thai medicinal plants
- Chemoecological studies on marine natural products: terpene chemistry from marine mollusks
- IUPAC Recommendations
- Abbreviations of polymer names and guidelines for abbreviating polymer names (IUPAC Recommendations 2014)
- IUPAC Technical Report
- Toward a comprehensive definition of oxidation state (IUPAC Technical Report)