Lithiated oxazolinyloxiranes and oxazolinylaziridines: key players in organic synthesis
-
Saverio Florio
 ,
Leonardo Degennaro
,
Leonardo Degennaro

Abstract
This paper focuses on α-lithiated oxazolinyloxiranes and oxazolinylaziridines, their generation, reactions, and synthetic applications. The ability of the oxazoline ring in providing stabilization to such α-heterosubstituted carbanions either through electronic effects and coordinative action has been stressed as well as the contribution to the configurational stability or instability of such species. IR spectroscopic data, multinuclear NMR investigations, and ab initio calculations planned to get insights on chemical properties and structural features have been carried out. A number of new reactions including alkylations, addition reactions, hydroxyalkylations, cyclopropanations, lactonizations, and rearrangements have been discovered, giving access to a variety of substances including: aziridinolactones, epoxylactones, aryl alkanones, polysubstituted cyclopropanes, cyclopropanefused lactones, dihydro-oxazoloisoquinolines, diversely functionalized oxazolines, and products that can be derived from them by synthetic elaboration.
Introduction
Relatively stable lithiated intermediates in which the stability is provided by small-ring heterocycles including oxiranes [1] and aziridines [2], oxetanes [3] and azetidines [4], and tetrahydrofurans [5] have become important synthons useful for the construction of a variety of substances of interest in several fields over the last 20 or 15 years.
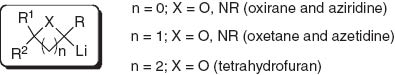
Lithiated small-ring heterocycles.
In this paper we will report on the generation, reactions and synthetic applications of α-lithiated oxazolinyloxiranes and oxazolinylaziridines.
One of the key features of the oxazoline system, which well deserves to be among the best functional groups in synthetic organic chemistry, is the stabilization it can provide to carbanions generated in the α position to it. Due to such a strong stabilizing power, α-lithiated oxazolinyl oxiranes and aziridines can be easily generated and used as key synthons for the synthesis of highly substituted epoxides and aziridines and products that can be derived from them. This manuscript reports numerous synthetic applications of those α-lithiated small ring heterocycles.
α-Lithiated oxazolinyloxiranes
Within the large family of α-lithiated oxiranes, for long time considered only fleeting intermediates in reactions of epoxides with strong bases and successively estimated useful synthons in synthetic organic chemistry because of their chamaleon-like character which gives them the ability to react either with electrophiles and with nucleophiles [6], α-lithiated oxazolinyloxiranes occupy a special place because of the their viable synthetic elaboration.
Subjected to lithiation (s-BuLi/TMEDA, THF, –100 °C) diphenyloxirane 1 generates lithiated species 1-Li which is stable at low temperature and could be successfully trapped with electrophiles to give functionalized oxazolinyloxiranes 2, elaborable to formyl epoxides 3 by using a known protocol [7].
Configurationally stable lithiated oxazolinyloxirane 4-Li, generated from the trans precursor 4, reacted regio- and stereospecifically with electrophiles, with complete retention of configuration to give 5 (Scheme 1) [8], while the lithiation-trapping sequence of the corresponding cis oxazolinyloxirane occurred with poor diastereoselectivity for steric reasons.

Synthesis of functionalized oxazolinyloxiranes 2 and formyl epoxides 3.
The α-lithiated oxazolinyloxiranes 6-Li, generated from the optically active oxazolinyloxiranes 6 proved to be configurationally unstable; indeed, they tend to epimerize and the addition of electrophiles ends up with the formation of diastereomeric mixtures of 7 (Scheme 2) [9].

Configurational instability of α-lithiated oxazolinyloxiranes 6-Li.
In the absence of epoxide-ring hydrogens, as in the case of the diphenyl epoxide 8, a remote lithiation occurs at the aryl group cis to the oxazolinyl group to give the arylalkanones 9 (Scheme 3) [10].

ortho-Lithiation of aryl oxazolinyloxiranes 8: synthesis of arylalkanones 9.
Interestingly, ortho-lithiated aryloxirane 10-Li, generated by lithium-bromine exchange performed on the corresponding ortho-bromo aryloxirane 10, gives the isoquinolines 11 (Scheme 4) [11].

ortho-Lithiated aryloxiranes 10-Li: preparation of isoquinolines 11.
An interesting aspect of the reactivity of α-lithiated oxiranes resides in the stereochemical outcome of their reactions with electrophiles which in principle can take place with retention or inversion of configuration or racemization.
A detailed multinuclear magnetic resonance investigation, jointly with an in situ IR study [12], demonstrated that α-lithiated oxazolinyloxiranes are thermally stable at low temperature but generally configurationally unstable. An indirect dynamic interconversion between two lithiated η3-aza-allyl enantiomeric monomers, namely (R)-η3-12-Li and (S)-η3-12-Li, mediated by a complex mixture of diastereomeric oxazoline-bridged dimeric species variously intra-aggregated, has been proposed as a possible mechanism responsible for the racemization 12-Li undergoes (Scheme 5).

Racemization of α-lithiated oxazolinyloxirane 12-Li.
Lithiation of racemic 2-oxazolinyl-3,3-diphenyloxirane 13 afforded interchangeable enantiomeric anions 13-Li and ent-13-Li [13], which reacted with (S)-2-p-tolylsulfinylbenzaldehyde 15 to give a 50:44:6 mixture of diastereoisomeric sulfinyl hydroxybenzyl oxazolinylepoxides 14a–c (65 % combined yield) (Scheme 6).

Reaction of α-lithiated 2-oxazolinyl-3,3-diphenyloxirane 13-Li with (S)-2-p-tolylsulfinylbenzaldehyde 15.
The absolute configuration of (1S,2S,SS)-14b, (1R,2S,SS)-14c and (1S,2R,SS)-14a was unequivocally determined by X-ray diffraction studies jointly with chemical correlations [13]. On the basis of these studies it was concluded that the sulfinyl group plays a significant role in the control of the configuration at the carbinolic carbon, determining a completely stereoselective evolution of 13-Li, yielding only 14a, and a highly stereoselective transformation of ent-13-Li to give an 88:12 mixture of 14b and 14c. It was also established that the influence of an additional stereogenic center in the nucleophile on the stereoselectivity of the process is very low.
As expected on the basis of the configurational stability of α-lithiated aryloxiranes, which proved to be dependent upon the substitution on the aryl group (Scheme 7) [1]c-d.

Configurational stability of α-lithiated aryloxiranes.
cis and trans Lithiated aryl oxazolinyloxiranes 16-Li and 19-Li are configurationally stable and, once generated, can be trapped with a series of electrophiles with complete retention of configuration [14, 15]. In particular, the trapping reaction of 16-Li with carbonyl compounds has been exploited for the preparation of α,β-epoxy-γ-butyrolactones 18 (Scheme 8).

Preparation of α,β-epoxy-γ-butyrolactones 18.
The capture of 19-Li by electrophiles furnished epoxides of the kind of 20 and 21 (Scheme 9).

Reaction of oxazolinyloxiranes 19-Li with electrophiles.
Almost optically pure α,β-epoxy-γ-butyrolactones 24 and 28 could be obtained starting from the chiral nonracemic oxazolinyloxirane 22 (dr trans/cis 90:10, er trans > 99:1) [16] (Scheme 10). The reaction of oxirane 23-Li with PhCHO takes place with a poor diastereoselective giving the easily separable epoxylactones 27 and 28, almost optically pure.

Synthesis of optically pure epoxylactones 27.
Synthesis of α,β-epoxy-γ-amino acids and α,β-epoxy-γ-butyrolactams
When lithiated (1R*,2S*)-1-methyl-1-oxazolinyloxirane 29-Li was reacted with (Z)-N-t-butyl- and N-cumylnitrones, diazaspiro[4.5]decanes diast-30 formed in good yields (42–71 %) and diastereoselectively (dr > 98/2) (Scheme 11). In CDCl3 as well as in THF-d8 spirocyclic compounds 30 equilibrate with the hydroxylamino forms 32 and diast-31. Trifluoroacetic acid (TFA)-catalyzed hydrolysis, carried out in dioxane/H2 O, afforded in good yields (40–94 %) and diastereoselectivity (dr > 98/2) 4,5-epoxy-1,2-oxazin-6-ones 33 (R1 = t-Bu) (Scheme 11) [16].

Synthesis of epoxy-1,2-oxazin-6-ones 33.
Lithiation of the optically active oxazolinyloxirane 34 (er = 98/2) [14], followed by the coupling of the corresponding lithiated intermediate 34-Li with N-tert-butyl- and N-cumylnitrones, gave spirocyclic compounds (-)-35 with complete diastereoselectivity. Hydrolysis (TFA) of (-)-35 furnished highly enantioenriched N-t-butyl- and N-cumyl-epoxy-1,2-oxazinones (-)-36, which could be converted to the corresponding α,β-epoxy-γ-amino acids (-)-37 (Scheme 12).

Synthesis of optically active α,β-epoxy-γ-amino acids (-)- 37.
Synthesis of polysubstituted oxazolinylcyclopropanes
The addition of lithiated aryl oxazolinyloxiranes 38-Li and 39-Li to tungsten Fischer carbene complexes 40 produced cyclopropanes 41 and 42, respectively (Scheme 13), which are likely the result of a Michael initiated ring closure (MIRC reaction) [17]. Oxidation of 41 furnished the ester 43.

Synthesis of polysubstituted oxazolinylcyclopropanes.
Lithiation of terminal oxazolinyloxirane: synthesis of trisubstituted oxazolinyloxiranes and (E)-dioxazolinyl-diphenylbut-2-ene-1,4-diol
A simple method of synthesis of trisubstituted oxazolinyloxiranes based on the stereospecific lithiation-trapping of a terminal α-substituted oxazolinyloxirane, good precursor to α-hydroxy-β-aminoalkanamides 50 of potential antifungal activity, was developed in our lab [18]. The required starting oxirane 46 was synthesized from 2-benzoyl-2-oxazoline 44 (Scheme 14). The trapping of the lithiated intermediate worked reasonably well with several electrophiles to give 47 and 49. It is interesting that in the absence of an external electrophile 44-Li undergoes an “eliminative dimerization” (the typical carbene-like reactivity often associate to α-lithiated ethers) [19] with a very high E diastereoselectivity to give the 1,4-dioxazolinylbutenediol 51.

Stereospecific lithiation-trapping of terminal α-substituted oxazolinyloxirane 46.
Conclusions
As you may have seen above, peculiar reactions such as deprotonation, stereoselective rearrangements, asymmetry induction and reductive alkylation make α-lithiated oxazolinyl oxiranes very useful building blocks for the preparation of a variety of substances. The oxazolinyl moiety is an added value as it is amenable to synthetic elaborations. Spectroscopic investigation as well as computational studies have significantly contributed to understand the very rich chemistry of this sort of lithiated epoxides.
α-Lithiated oxazolinylaziridines
Aziridines are valuable and widely used intermediates in synthetic chemistry [20, 21]. Numerous methods of synthesis of aziridines have been developed. A conceptually appealing strategy is the transformation of an already constructed aziridine scaffold through the generation of aziridinyl anions, followed by the reaction with electrophiles [22–25]. When treated with strong bases such as lithium amides or organolithiums, aziridines may undergo deprotonation at the aziridine ring carbons ending up with the formation of α-lithiated derivatives which can be captured with an electrophile to give a more substituted aziridine. Such a useful lithiation-electrophile trapping sequence, named aziridinyl anion methodology parallels the known oxiranyl anion methodology. The α-lithiated heterocycle-substituted aziridines, on their side, have an added value as the heterocyclic moiety can be synthetically elaborated.
α-Lithiated N-alkyl (aryl) oxazolinylaziridines: synthesis of aziridinolactones
Treatment of readily available N-phenyl oxazolinylaziridines 52 with n-BuLi at low temperature generates aziridinyllithiums 52-Li that react with electrophiles giving functionalized aziridines 53 and 54 of potential utility as ligands for asymmetric syntheses (Scheme 15) [26].

Synthesis of functionalized N-phenyl oxazolinylaziridines 53 and 54.
Considering the stereochemistry, lithiation of 55 by using s-BuLi/TMEDA at –98 °C resulted in the formation of aziridinyllithiums 55-Li: deuteration occurred with retention of configuration to give aziridine 56 and the reaction with aldehydes provided spirocyclic compounds 57 and then aziridinolactones 58 [27]. It is worthy pointing out that the oxazolinyl ring plays a role in the deprotonation process exalting the acidity of the proton to be removed and providing stabilization to the resulting lithiated intermediate by chelation (Scheme 16) [14].

Regioselective lithiation of aziridine 55.
Lithiation of N-alkyl terminal aziridines
The role of the oxazolinyl ring as lithiation promoter was highlighted also with the chiral terminal N-trityl aziridine (S)-59 [28]. Subjected to deprotonation (s-BuLi/TMEDA at –70 °C), aziridine (S)-59 underwent surprisingly regioselective lithiation at the cis-β-position (Scheme 16) instead of at the more acidic α-position. The highly sterically demanding N-trityl group which, on the basis of NOESY experiments carried out on (S)-59, sets in a trans relationship with respect to the oxazoline ring, likely prevents lithiation at the α-position for steric reasons. The configurationally stable aziridinyllithium (S,S)-59-Li has been successfully used in the preparation of highly enantioenriched cis functionalized aziridines 60 and 61, which could be stereoselectively converted into the corresponding aziridino-γ-butyrolactones 62 (Scheme 17).

The sterically demanding of N-trityl group: asymmetric synthesis of aziridines 60.
In contrast, lithiation of the less hindered N-benzyl oxazolinylaziridine 63 occurred at the more acidic α-position producing almost exclusively α-functionalized aziridines 64, upon electrophilic trapping (Scheme 17). Not unespectedly, N-cumyl substituted oxazolinylaziridine 65 underwent lithiation with s-BuLi at both the α and β positions to give 66 and 67 in a 1:2 ratio (Scheme 18).

The effect of the N-substituent on the lithiation of aziridines 63 and 65.
Enantioenriched diastereomeric aziridines (R,R)-68 and (R,S)-70 have been regioselectively lithiated with n-BuLi in THF to give the α-lithiated aziridines (R,R)-68-Li and (R,S)-70-Li, which proved to be configurationally stable as testified by trapping them with a deuterium source (Schemes 19 and 20) [29]. This is in sharp contrast with the reported configurational instability of the corresponding α-lithiated oxazolinyloxiranes [12]. Such a peculiar behavior of the nitrogen bearing heterocycle has been rationalized on the basis of the investigated aziridine nitrogen dynamics and DFT calculations. A dynamically controlled lithiation has been proposed as a model supported by the estimation of the nitrogen inversion barrier by dynamic NMR experiments [29].

Configurational stability of (R,R)-68-Li.

Configurational stability of (R,S)-70-Li.
Lithiation of heterosubstituted oxazolinylaziridines
The ability of the oxazolinyl group as a promoter of α-lithiation of aziridines is stronger than that of other heterocycles [30]. Indeed, diastereomeric aziridines (R*,R*)-72 and (R*,S*)-74 are regioselectively lithiated only at the oxazoline-bearing carbon atom giving aziridinyllithiums (R*,R*)-72-Li and (R*,S*)-74-Li which can be trapped with a variable degree of retention of configuration to give 73 and 75 (Scheme 21) [30].

Regioselective lithiation of heterosubstituted oxazolinylaziridines (R*,R*)-72 and (R*,R*)-74.
Lithiation occurs at the other heterocycle-substituted carbon atom only in the absence of protons at the oxazoline-bearing carbon atom. In fact, aziridines (R*,S*)-76 were lithiated with n-BuLi in THF at –78 °C, producing aziridinyllithiums (R*,S*)-76-Li which could be trapped with a deuterium source again with a variable degree of stereoselectivity leading to the oxazolinyl aziridines 77 (Scheme 22).

Synthesis of heterosubstituted oxazolinylaziridines 77.
Lithiation of N-sulfonyl- and phosphinylaziridines
The N-substitution may control the lithiation-trapping sequence of aziridines. Indeed, the lithiation of diastereomeric N-sulfonyl oxazolinylaziridines (R*,R*)-78 and (R*,S*)-83 takes place at the benzylic position and the resulting aziridinyllithiums (R*,R*)-78-Li and (R*,S*)-83-Li are both configurationally stable although differing in their reactivity. Aziridinyllithium (R*,R*)-78-Li could be trapped with electrophiles giving aziridines 79 only with low yield because of side reactions such as dearomatization, affording tricyclic aziridine 80, and ortho-lithiation furnishing aziridine 81 upon quenching with electrophiles (Scheme 23) [27]. Variable amounts of azirine 82 could also be observed.

Lithiation of N-sulfonylaziridine (R*,R*)-76.
In its turn, diastereomeric aziridinyllithium (R*,S*)-83-Li did not undergo dearomatization reaction likely because of the trans arrangement of the sulfinyl group and the C-Li bond, and could be trapped with ketones to furnish spirocyclic derivatives 84 which could be elaborated to aziridino-γ-lactones 85 (Scheme 24).

Lithiation of N-sulfonylaziridine (R*,S*)-83.
Moreover, the trans arrangement between the sulfonyl group and the C-Li bond in lithiated aziridine (R*,S*)-83-Li causes a fast trans β-elimination of lithium phenylsulfinate with formation of azirine 86 almost quantitatively.
The N-phosphinyl group cooperates with the oxazolinyl group in promoting deprotonation of diastereomeric aziridines (R*,S*)-87 and (R*,R*)-90 [27]. Diastereoisomer (R*,S*)-87 could be smoothly lithiated providing configurationally stable (R*,S*)-87-Li, which, upon trapping with acetone, gave first spirocyclic derivative 88 and aziridino-γ-lactone 89 after acidic hydrolysis. The lithiation-deuteration of diastereomer (R*,R*)-90 showed a different reactivity depending on the base used for the deprotonation: the use of s-BuLi gave a certain degree of epimerization furnishing, upon quenching with D2 O, a mixture of the epimers (R*R*)-91 and (R*S*)-92; the use of LDA gave a more pronounced epimerization and favored the N- to C-migration of the phosphinyl group leading to aziridine 93 (Scheme 25).

Lithiation of N-phosphinylaziridines (R*,S*)-87 and (R*,R*)-90.
Concluding remarks
In conclusion, the chemistry and synthetic utility of α-lithiated oxazolinyl epoxides and aziridines have been highlighted in this paper. It has been shown that such lithiated species can be easily generated and captured with electrophiles leading to more functionalized derivatives. Spectroscopic and computational studies have proved their utility not only for the comprehension of the chemistry of aziridine and epoxides in general but also for the elucidation of the mechanisms of their lithiation and the stereochemistry of their reactions. Combining bench experiments with spectroscopic investigations and DFT calculations has contributed to enormously increase the synthetic potential of the lithiated oxazolines described in this manuscript.
Article note: A collection of invited papers based on presentations on the Chemical Synthesis theme at the 44th IUPAC Congress, Istanbul, Turkey, 11–16 August 2013.
Acknowledgments
We are grateful to the Interuniversities Consortium C.I.N.M.P.I.S., the University of Bari and the Italian MIUR, which funded most of the results described in this paper and to colleagues, researchers, and students for their valuable contribution to these investigations.
References
[1] (a) V. Capriati, S. Florio, R. Luisi. Chem. Rev.108, 1918–1942 (2008); (b) V. Capriati, S. Florio and A. Salomone, In Topics in Stereochemistry, Vol. 26: Stereochemical Aspects of 40 Organolithium Compounds; R. E. Gawley and J. S. Siegel, Eds.,Wiley-VCH, Weinheim, 2010, Chapter 4, p. 135; (c) V. Capriati, S. Florio, F. M. Perna, A. Salomone. Chem-A Eur.16, 9778–9788 (2010); (d) F. M. Perna, A. Salomone, M. Dammacco, S. Florio, V. Capriati. Chem.–Eur. J.17, 8216–8225 (2011); (e) A. Salomone, F. M. Perna, A. Falcicchio, S. O. Nilsson Lill, A. Moliterni, R. Michel, S. Florio, D. Stalke, V. Capriati. Chem. Sci.5, 528–538 (2014); (f) V. Capriati, S. Florio, F. M. Perna, A. Salomone, A. Abbotto, M. Amedjkouh, S. O. Nilson Lill. Chem.- Eur. J.15, 7958–7979 (2009); (g) R. Mansueto, F. M. Perna, A. Salomone, S. Florio, V. Capriati. Chem. Commun.49, 4911–4913 (2013).Suche in Google Scholar
[2] (a) S. Florio. Synthesis, 44, 2872–2884 (2012); (b) S. Florio, R. Luisi. Chem. Rev. 110, 5128 (2010); (c) M. C. De Ceglie, B. Musio, F. Affortunato, A. Moliterno, A. Altomare, S. Florio, R. Luisi. Chem.– Eur. J.17, 286–296 (2011).Suche in Google Scholar
[3] (a) D. I. Coppi, A. Salomone, F. M. Perna, V. Capriati. Chem. Commun.47, 9918–9920 (2011); (b) D. I. Coppi, A. Salomone, F. M. Perna, V. Capriati. Angew. Chem. Int. Ed.51, 7532–7536 (2012).Suche in Google Scholar
[4] D. M. Hodgson, J. Kloesges. Angew. Chem. Int. Ed. 49, 2900–2903 (2010).Suche in Google Scholar
[5] R. Mansueto, V. Mallardo, F. M. Perna, A. Salomone, V. Capriati. Chem. Commun. 49, 10160–10162 (2013).Suche in Google Scholar
[6] (a) V. Capriati, S. Florio. Chem.– Eur. J.16, 4152–4162 (2010); (b) Ref. 1b.Suche in Google Scholar
[7] S. Florio, V. Capriati, R. Luisi. Tetrahedron Lett. 37, 4781–4784 (1996).10.1016/0040-4039(96)00934-3Suche in Google Scholar
[8] A. Abbotto, V. Capriati, L. Degennaro, S. Florio, R. Luisi, A. Salomone. J. Org. Chem.66, 3049 (2001).Suche in Google Scholar
[9] R. Luisi, V. Capriati, C. Carlucci, L Degennaro, S. Florio. Tetrahedron59, 9707 (2003).10.1016/j.tet.2003.09.018Suche in Google Scholar
[10] F. M. Perna, V. Capriati, S. Florio, R. Luisi. Tetrahedron Lett.43, 7739 (2002).Suche in Google Scholar
[11] V. Capriati, S. Florio, R. Luisi, F. M. Perna. J. Org. Chem.72, 6316 (2007).Suche in Google Scholar
[12] V. Capriati, S. Florio, R. Luisi, F. M. Perna, A. Spina. J. Org. Chem. 73, 9552–9564 (2008).Suche in Google Scholar
[13] J. L. Garcia Ruano, A. M. Martin-Castro, F. Tato, E. Torrente, M. G. Tocco, S. Florio, V. Capriati. Tetrahedron66, 1581–1585 (2010).10.1016/j.tet.2009.12.006Suche in Google Scholar
[14] V. Capriati, L. Degennaro, R. Favia, S. Florio, R. Luisi. Org. Lett.4, 1551 (2002).Suche in Google Scholar
[15] V. Capriati, R. Favia, S. Florio, R. Luisi. ARKIVOCxiv, 77 (2003).10.3998/ark.5550190.0004.e08Suche in Google Scholar
[16] V. Capriati, L. Degennaro, S. Florio, R. Luisi, P. Punzi. Org. Lett.8, 4803 (2006).Suche in Google Scholar
[17] S. Florio, F. M. Perna, R. Luisi, J. Barluenga, F. J. Fananas, F. Rodriguez. J. Org. Chem. 69, 9204–9207 (2004).Suche in Google Scholar
[18] L. Degennaro, V. Capriati, C. Carlucci, S. Florio, R. Luisi, I. Nuzzo, C. Cuocci. Tetrahedron65, 8745–8755 (2009).10.1016/j.tet.2009.08.040Suche in Google Scholar
[19] (a) D. M. Hodgson, C. D. Bray, N. D. Kindon. Org. Lett. 7, 2305–2308 (2005); (b) D. M. Hodgson, P. G. Humphreys, S. P. Hughes. Pure Appl. Chem.79, 269–279 (2007); (c) see ref 1f.Suche in Google Scholar
[20] (a) A. Padwa. in Comp. Heter. Chem III, A. R. Katrizky, C. A. Ramsden, E. F. V. Scriven, R. J. Taylor (Eds.), Vol. 1, p. 2, Elsevier (2008); (b) J. B. Sweeney. in Science of Synthesis, E. Schaumann, D. Enders, (Eds.), Vol. 40a, p. 643, Georg Thieme Verlag, Stuttgart, New York (2008); (c) A. Padwa, S. S. Murphee. Prog. Heterocycl. Chem.15, 75 (2003); (d) J. B. Sweeney. Chem. Soc. Rev. 31, 247 (2002); (e) B. Zwanenburg, P. ten Holte. Top. Curr. Chem.216, 93 (2001); (f) U. M. Lindstrom, P. Somfai. Synthesis 109–117, (1998); (g) P. Somfai, J. Ahman. Targets Heterocycl. Syst.3, 341 (1999); (h) J. B. Sweeney. in Science of Synthesis, E. Schaumann, D. Enders (Eds.), Vol. 40a, p. 643, Georg Thieme Verlag, Stuttgart, New York (2008); (i) W. McCoull, F. A. Davis. Synthesis 1347 (2000); (j) R. Pal, SH. Ghosh, K. Chandra, A. Basak. Chem. Rev. 107, 2080 (2007); (k) D. Tanner. Angew. Chem. Int. Ed. Engl. 33, 599 (1994); (l) X. E. Hu. Tetrahedron60, 2701 (2004).Suche in Google Scholar
[21] (a) X. E. Hu. Tetrahedron60, 2701 (2004); (b) M. D’hooghe, V. Van Speybroeck, M. Waroquier, N. De Kimpe. Chem. Commun. 1554 (2006).Suche in Google Scholar
[22] T. Satoh. Chem. Rev.96, 3303 (1996).10.1021/cr950081vSuche in Google Scholar PubMed
[23] For recent reviews of lithiated epoxide and aziridine chemistry, see: (a) Oxiranyl and aziridinyl anions as reactive intermediates in synthetic organic chemistry. S. Florio, Ed. Tetrahedron59, 9683 (2003); (b) D. M. Hodgson, C. D. Bray. in Aziridines and Epoxides in Organic Synthesis, A. K. Yudin (Ed.), p. 145, Wiley-VCH, Weinheim, Germany (2006); (c) D. M. Hodgson, C. D. Bray, P. G. Humphreys. Synlett 1–22 (2006); (d) D. M. Hodgson, P. G. Humphreys, S. P. Hughes. Pure. Appl. Chem.79, 269 (2007); (e) S. Florio, R. Luisi. Chem. Rev. 110, 5128–5157 (2010); (f) see ref 1b.Suche in Google Scholar
[24] (a) V. Capriati, S. Florio, R. Luisi. Chem. Rev.108, 1918 (2008); (b) F. Chemla, E. Vrancken. in The Chemistry of Organolithium Reagents, Z. Rappoport, I. Marek (Eds.), pp. 1165, Wiley and Sons, New York, 2004 (2004); (c) S. Florio. Synthesis44, 2872 (2012).Suche in Google Scholar
[25] For reviews on the chemistry of carbenoids see: (a) G. Boche, J. C. W. Lohrenz. Chem. Rev.101, 697 (2001); (b) V. Capriati, S. Florio. Chem. Eur. J.16, 4152–4162 (2010) and reference therein.Suche in Google Scholar
[26] S. Florio, L. Troisi, V. Capriati, G. Ingrosso. Tetrahedron Lett.40, 6101 (1999).Suche in Google Scholar
[27] R. Luisi, V. Capriati, S. Florio, P. Di Cunto, B. Musio. Tetrahedron61, 3251–3260 (2005).10.1016/j.tet.2005.01.045Suche in Google Scholar
[28] R. Luisi, V. Capriati, P. Di Cunto, S. Florio, R. Mansueto. Org. Lett.9, 3295 (2007).Suche in Google Scholar
[29] L. Degennaro, R. Mansueto, E. Carenza, R. Rizzi, S. Florio, L. M. Pratt, R. Luisi. Chem. Eur. J. 12, 1761–1765 (2011).Suche in Google Scholar
[30] L. Troisi, C. Granito, C. Carlucci, F. Bona, S. Florio. Eur. J. Org. Chem. 775 (2006).10.1002/ejoc.200500570Suche in Google Scholar
©2014 IUPAC & De Gruyter Berlin/Boston
Artikel in diesem Heft
- Frontmatter
- Preface
- 44th IUPAC World Chemistry Congress: Clean Energy Through Chemistry
- Conference papers
- The energy landscape concept and its implications for synthesis planning
- Near-IR absorbing Bodipy functionalized SPIONs: a potential magnetic nanoplatform for diagnosis and therapy
- Glycerol acetals with antioxidant properties
- Lithiated oxazolinyloxiranes and oxazolinylaziridines: key players in organic synthesis
- Access to pyrrole-based heterocyclic compounds via addition of pyrrole to C=C and C=N bonds
- Hydrogenation of carboxylic acid derivatives with bifunctional ruthenium catalysts
- Generation of singlet oxygen (1O2) from hydrogen peroxide decomposition by in situ generated hypervalent iodoarene reagents
- Organometallic macrocycles and cages based on bis(amidinate) ligands
- Anisotropic core-shell Fe3 O4 @Au magnetic nanoparticles and the effect of the immunomagnetic separation volume on the capture efficiency
- Recent investigations of bioactive natural products from endophytic, marine-derived, insect pathogenic fungi and Thai medicinal plants
- Chemoecological studies on marine natural products: terpene chemistry from marine mollusks
- IUPAC Recommendations
- Abbreviations of polymer names and guidelines for abbreviating polymer names (IUPAC Recommendations 2014)
- IUPAC Technical Report
- Toward a comprehensive definition of oxidation state (IUPAC Technical Report)
Artikel in diesem Heft
- Frontmatter
- Preface
- 44th IUPAC World Chemistry Congress: Clean Energy Through Chemistry
- Conference papers
- The energy landscape concept and its implications for synthesis planning
- Near-IR absorbing Bodipy functionalized SPIONs: a potential magnetic nanoplatform for diagnosis and therapy
- Glycerol acetals with antioxidant properties
- Lithiated oxazolinyloxiranes and oxazolinylaziridines: key players in organic synthesis
- Access to pyrrole-based heterocyclic compounds via addition of pyrrole to C=C and C=N bonds
- Hydrogenation of carboxylic acid derivatives with bifunctional ruthenium catalysts
- Generation of singlet oxygen (1O2) from hydrogen peroxide decomposition by in situ generated hypervalent iodoarene reagents
- Organometallic macrocycles and cages based on bis(amidinate) ligands
- Anisotropic core-shell Fe3 O4 @Au magnetic nanoparticles and the effect of the immunomagnetic separation volume on the capture efficiency
- Recent investigations of bioactive natural products from endophytic, marine-derived, insect pathogenic fungi and Thai medicinal plants
- Chemoecological studies on marine natural products: terpene chemistry from marine mollusks
- IUPAC Recommendations
- Abbreviations of polymer names and guidelines for abbreviating polymer names (IUPAC Recommendations 2014)
- IUPAC Technical Report
- Toward a comprehensive definition of oxidation state (IUPAC Technical Report)