A novel immune-associated prognostic signature based on the immune cell infiltration analysis for hepatocellular carcinoma
-
Xinrong Lin


Abstract
Objectives
Immune-related genes (IRGs) in hepatocellular carcinoma (HCC) are significantly associated with both tumor-infiltrating immune cells (TICs) and disease prognosis. Therefore, exploring the correlation between IRGs with HCC and its related mechanism will provide new evidence for the diagnosis and treatment of HCC.
Methods
The current paper analyzed the TICs in 374 HCC samples retrieved from the TCGA-LIHC dataset using ssGSEA and divided them according to the level of immune cell. A total of 177 differentially expressed genes (DEGs) were analyzed by protein-protein interaction (PPI) networks and univariate and multivariate Cox regression analyses.
Results
Four IRGs (C7, CTSV, MMP1, and VCAN) were found to be indicators of the immune prognosis for HCC according to the PPI network and Cox regression analyses of 177 DEGs, which was independently validated using an external dataset. A prognosis risk model was constructed for factors dependent on the four IRGs. Prognostic risk was associated with the subtype of infiltrating immune cells.
Conclusions
Four effective IRGs were identified as novel independent prognostic factors that were correlated with tumor immune infiltration in HCC. This signature may guide the choice of immunotherapy for HCC.
Introduction
Hepatocellular carcinoma (HCC) has a high incidence and high mortality [1]. The specific mechanism underlying HCC occurrence and development remains unclear due to the cancer’s high level of heterogeneity [2]. According to the recommended clinical guidelines, systemic therapy in the first-line treatment, including immunotherapy which has been found to have significant benefits [3]. However, its five-year survival rate is very low, primarily due to late-stage diagnosis [4]. Consequently, identifying reliable immune-related markers and prognostic indicators can enhance the HCC survival rate.
Increasing evidence has shown that the survival time of patients with HCC is closely related to the immune system [5]. Immune cells affect the proliferation, metastasis, and prognosis of several cancers [6, 7], including HCC [8]. Furthermore, immune cells are a crucial component of the tumor microenvironment with a substantial influence on the occurrence, metastasis, and prognosis of the tumor [9, 10]. Additionally, our findings depict that a high level of infiltration of immune cells (ICI) is a significant prognostic HCC factor.
Several studies have confirmed that intratumoral infiltration of dendritic cells (DCs) as well as inflammatory cells such as regulatory T cells and IL-17-producing cells are indicative of poor HCC patient prognosis [11]. Macrophages are also associated with both the prognosis and the numbers of tumor-infiltrating cells (TICs) in HCC patients [12].
Endothelin interacts with CD68, leading to macrophage recruitment [13]. This interaction also regulates Ga6 expression in cancer-associated fibroblasts (CAF) and mediates the M2 polarization of macrophages. Moreover, a higher neutrophil/lymphocyte ratio often indicates a better prognosis in HCC patients [14]. Moreover, tumor-associated CD4/CD8 double-positive T (DPT) cells have been identified with a synergistic PD-1 expression and HLA-DR and demonstrate more elevated levels of IFN-, TNF-, and PD-1 upon stimulation [15]. CD8T cells are markedly increased in patients with advanced HCC and are negatively associated with overall survival [16]. These cells influence HCC tumorigenesis and development. The high-density tumor-infiltrating B cell infiltration in tumors decreases significantly during HCC progression [17]. Numerous studies have shown the close relationship between immune-related genes (IRGs), ICI, cell types, and HCC prognosis [10, 18, 19]. Currently, there is no prognostic signature that can efficiently predict survival in HCC patients. Therefore, discovering a reliable immune biomarker for HCC prognosis based on ICI would be extremely useful.
We used single-sample gene set enrichment analysis (ssGSEA) and associated mechanisms to construct a prognostic signature for HCC, using the Cancer Genome Atlas (TCGA) database. Four IRG signatures were discovered to have a close correlation with overall survival of HCC patients. An independent International Cancer Genome Consortium (ICGC) dataset was used to confirm the significance of this prognostic signature. The results demonstrated that these four IRGs are reliable predictors of targeted and precise prognostic characteristics.
Methods
Data collection and grouping
Transcriptional group RNA-sequencing information and clinical messages were obtained from TCGA. The dataset comprised data on 371 HCC and 50 non-tumor tissues. The LIHC transcriptome data was grouped from TCGA based on ICI using the ssGSEA. This analysis was used to assess the infiltration levels of various immune cells, immune-associated signaling pathways, and functional activities. According to the median of ssGSEA results, the HCC samples were split into high-ICI (IMM_H) and low-ICI (IMM_L) groups. To determine the classification, the ESTIMATE algorithm was used to calculate various scores associated with the tumor microenvironment. These scores included Tumor Purity, Immune, ESTIMATE, and Stromal Scores. The box chart examination indicated that IMM_H was positively associated with these scores. No ethical approval was needed in this study since the data were retrieved from a public database.
Effectiveness of immune grouping
The ESTIMATE algorithm was used to identify differentially expressed genes (DEGs) in the HCC data. The algorithm determines the interstitial, estimated, immune, and tumor purity scores. The results were visualized by generating clustering heatmaps and statistical heatmaps (Supplementary Figures 1 and 2). We utilized human leukocyte antigen (HLA) genes to investigate the differences between the two types of immune cells. Additionally, the CIBERSort was used to measure the immune cell types and their association with the DEGs. These analyses comprehensively described the differences between the two groups in terms of ICI and gene expression levels.
Differentially expressed genes
After dividing the HCC samples into two groups, R software was used to identify DEGs related to immunity. A two-way Venn analysis was used to identify overlapping DEGs between the two groups. Functional enrichment of the DEGs was performed using Gene Ontology (GO) [20] and Kyoto Encyclopedia of Genes and Genomes (KEGG) [21] using the clusterProfiler package [22] in R with p<0.05 indicating statistical significance, and thus suggesting a potential biological value of the enriched terms in the DEGs.
PPI and immune-related gene
The protein-protein interaction (PPI) network was constructed with the STRING database [23] and visualized with Cytoscape software (Version 3.7.0, National Resource for Network Biology, USA). Only those nodes with interaction confidence intervals that were more significant than 0.7 were included [24]. Thus, the interactions between IRGs and potential functional relationships could be determined. Next, the survival package in R was used to evaluate IRGs in the training dataset using univariate Cox regression analysis, to determine the association between IRG expression and patient survival. We further screened the IRGs using a log-rank test, identifying genes significantly impacting patient prognosis. Then, a multi-factor Cox proportional hazards regression (PHR) model was developed to determine the risk score for patient survival in terms of the IRGs.
Immune-related gene prognostic signature
The clinical HCC data from TCGA were included in the univariate Cox PHR evaluation. The significance was set at p<0.05 to identify IRGs significantly connected with patient survival. Next, multivariate Cox regression was used to identify prognostic markers (age, sex, tumor grade, tumor status, tumor stage, and risk score) using the identified IRGs. The risk measures were determined for each sample depending on the outcomes of the above analysis. A Kaplan–Meier (KM) survival analysis was performed to equate the distinctions between the high and low-risk categories based on survivability to predict the risk scores and prognosis. Then, the risk score was examined to determine gene expression levels using the formula:
External validation
The International Cancer Society database was used as the validation dataset to confirm the predictive value of the immune-related gene risk score signature. According to the median risk measurement, the HCC patients were categorized into high- and low-risk categories in the validation cohort. A KM survival analysis was conducted in the validation cohort. Additionally, receiver operating characteristic (ROC) curves facilitated the assessment of risk measurement presentation signature in predicting patient outcomes.
Correlation analysis of TICs
The infiltration data from various cell types in the immune system was obtained from the Tumor Immune Assessment Resource (TIMER) databank. Pearson correlation analysis was also used.
Statistical analysis
R software (Version 3.6.1, The R Foundation, initially written by “R & R” of the Statistics Department of the University of Auckland.) was used for all analyses, with p-value <0.05 or |log2FC|>1 for assessment of significance.
Results
Analysis process
The analysis had the following steps (Figure 1):
Sample collection: Data on a total of 371 HCC and 50 adjacent tissues were retrieved from TCGA.
ICI analysis: The HCC tissues were grouped based on ICI levels: IMM_H (n=154) and IMM_L (n=217).
Identifying differentially expressed IRGs: A total of 177 IRGs were identified based on the screening criteria using R.
Functional enrichment analysis: GO and KEGG evaluations determined the probable characteristic mechanisms and signals associated with identified IRGs.
Protein-protein interaction (PPI) network construction: Overlapping DEGs were used to build a PPI network.
Cox regression analysis and prognosis construction of risk signature: This was used to verify the genes linked with prognosis using the PPI network DEGs. This led to the construction of a prognosis risk signature.
Assessment of the prognostic signature: This was evaluated using survival analysis, ROC analysis, principal component analysis (PCA), GSEA, and TIC correlation analysis.
Validation of the signature: The prognostic signature was validated with an external database.
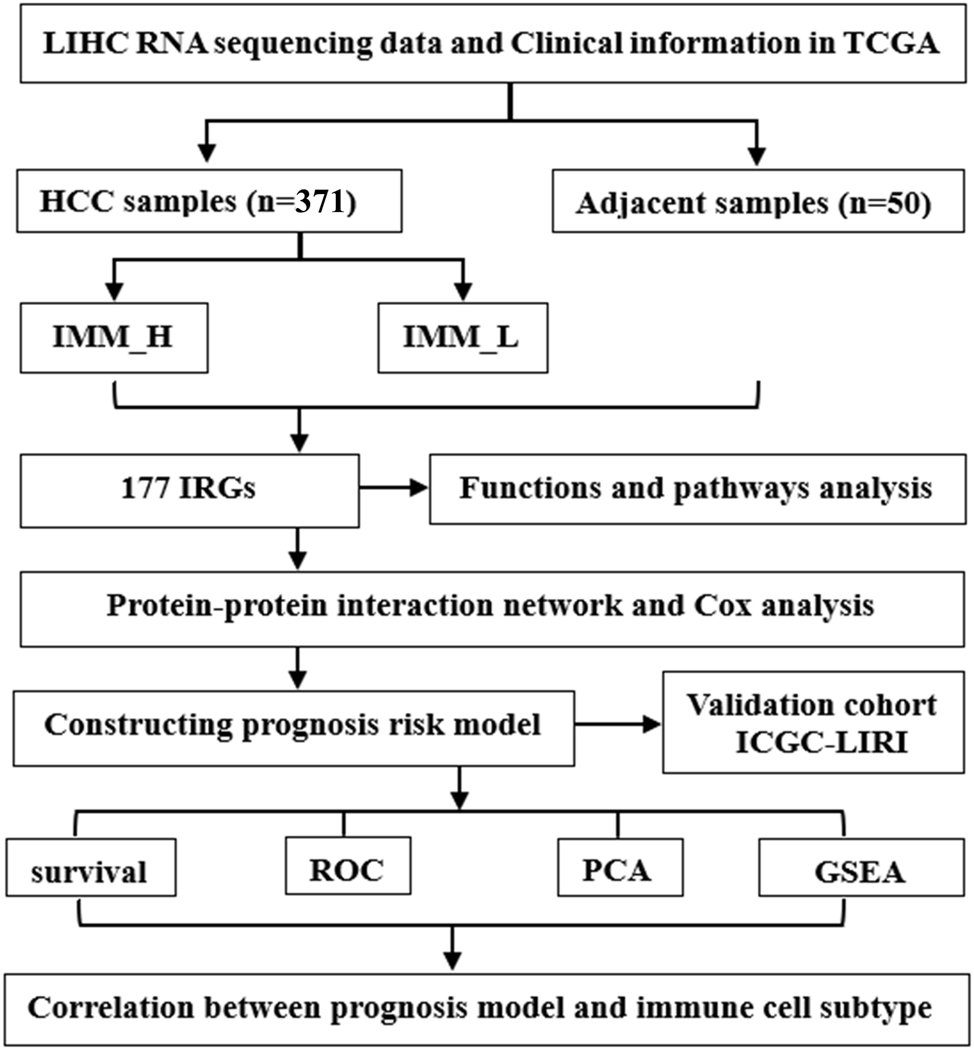
Workflow of the study.
Construction and verification of HCC groupings
An unsupervised hierarchical clustering algorithm was used to divide the 371 HCC cases retrieved from TCGA into IMM_H (n=154) and IMM_L (n=217) according to the median. ssGSEA was used to evaluate 24 types of infiltrating immune cells (Figure 2A). Conversely, IMM_L showed a positive correlation with tumor purity (Figure 2B). Thus, the IMM_H group had higher immunity features and lower tumor purity than the IMM_L group (p<0.05). Furthermore, the HLA family expression was higher in the IMM_H group than in the IMM_L group (Figure 2C). The CIBERSort algorithm was used to verify ICI in the two groups. The IMM_H group showed a greater variety of immunogenic cells (Figure 2D). The IMM_H group had higher ICI, lower tumor purity, and enhanced IRG expression, suggesting a more favorable immune microenvironment for novel immunotherapy interventions.

Development and verification of HCC grouping. (A) Grouping information of immune cells. (B) Between the groups, the box plot indicated a substantial distinction in Tumor Purity, ESTIMATE, Immune, and Stromal scores (p<0.01). (C) The levels of HLA homeostatic genes in IMM_H (red) were significantly greater than in IMM_L (blue) (p<0.01). (D) CIBERsort analysis showing the percentage distinction between the IMM_H (red) and the IMM_L (blue) for each immune cell.
Immune-related genes
We conducted a differential expression analysis between samples acquired from TCGA. We identified 1,833 DEGs, including 1,218 upregulated and 615 downregulated genes (Figure 3A) using the above criteria. Furthermore, a similar analysis was conducted between the two groups in the HCC samples. A total of 484 IRGs were identified with the same standards, including 398 upregulated and 86 downregulated genes (Figure 3B). A two-way Venn analysis found 177 common genes between the tumor and normal groups and the IMM_H and IMM_L groups (Figure 3C). These 177 p genes demonstrated the DEG intersection in the tumor and ICI analyses. Enrichment analyses were used to assess the molecular processes and pathways associated with the IRGs. GO analysis indicated that the IRGs were primarily enriched in the biological process category. In terms of cellular components, the IRGs were enriched in the extracellular matrix, collagen-containing extracellular matrices, and collagen trimer categories. In the molecular function category, enrichment was seen in the extracellular matrix structural constituents, receptor–ligand interactions, and regulatory activities (Figure 3D). Additionally, KEGG analysis showed that the IRGs were primarily associated with receptor interaction of cytokines, digestion, and absorption of proteins, and chemokine signaling (Figure 3E). A set of IRGs were found to be differentially expressed in HCC tissues. This information enhances the understanding of the immunological characteristics of HCC, generating novel insights into precision drug targets and prognostic markers.
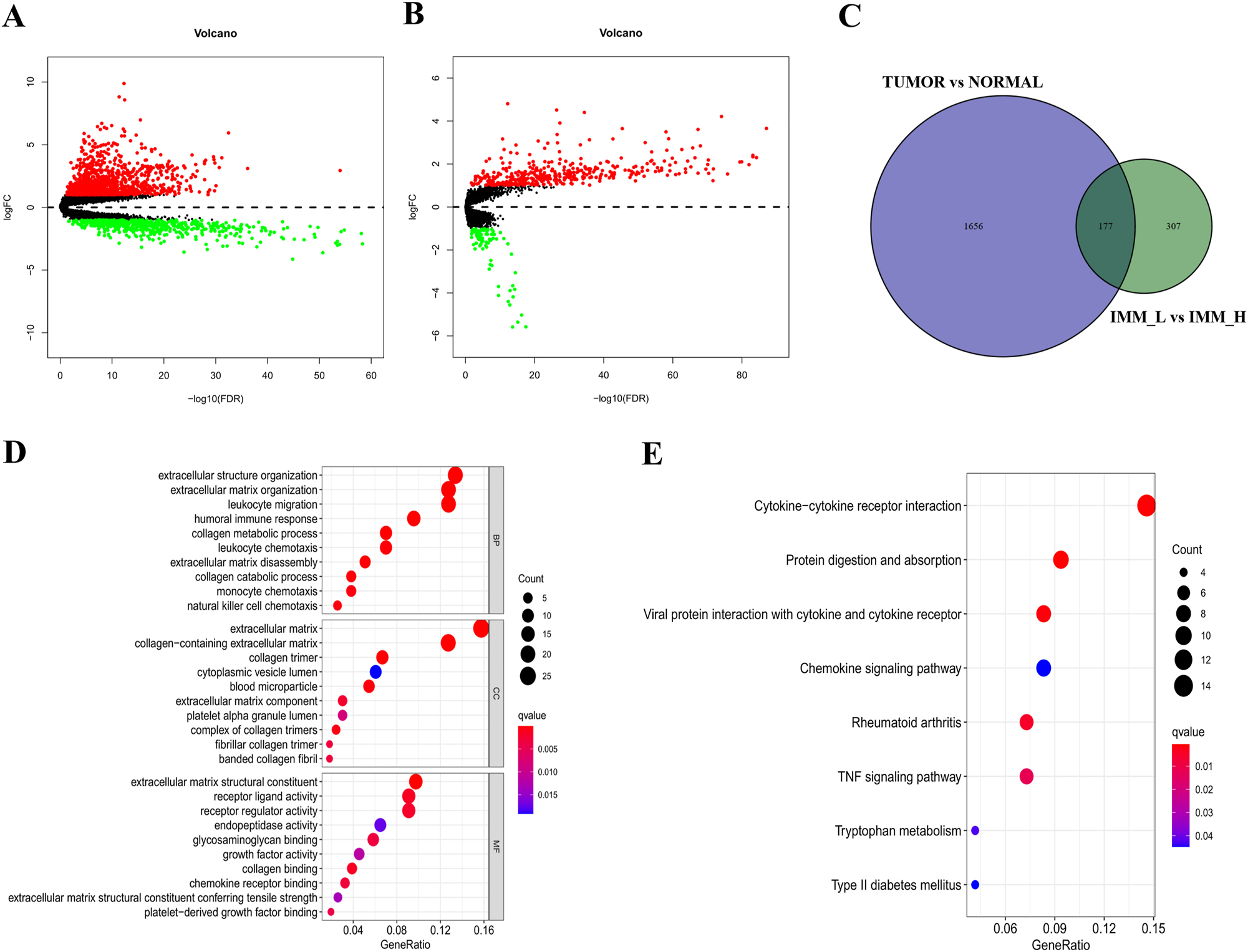
Analysis of IRGs. (A) Volcano plot indicating 1,218 upregulated and 615 downregulated DEGs between HCC and normal tissues. (B) Identification of 398 upregulated and 86 downregulated genes between IMM_H and IMM_L of HCC; red dots indicate upregulation and green dots downregulation (fold change>1, p=0.05). (C) 177 IRGs were identified by R software. (D and E) GO and KEGG enrichment of 177 IRGs, with p<0.05 used to show significant enrichment.
Immune-related gene prognostic signature
The STRING database and Cytoscape software were used to construct the PPI network to investigate the potential mechanism of the 177 DEGs. From the network, 82 key DEGs were identified with probable significant roles in HCC (Figure 4A). Univariate Cox regression analysis was conducted to identify 11 prognosis-linked IRGs (Figure 4B). Subsequently, multivariate Cox regression analysis of these 11 IRGs found that four IRGs, namely, C7, CTSV, MMP1, and VCAN, could be independent predictors of HCC patient outcomes (Figure 4C).

PPI network and Cox regression analyses. (A) The interaction network was constructed using nodes with an interaction confidence of >0.70. (B) The 82 IRGs underwent univariate Cox regression analysis, using p<0.05 as the level of significance. (C) Multivariate Cox regression analysis for determination of prognosis-associated genes.
We measured the risk score for every gene depending on their expression levels to determine the effects of the four IRGs on survival time and clinical prognosis in HCC patients, with the equation (Table 1):
Four prognosis-associated genes identified by multivariate Cox regression.
| Gene symbol | Coef | HR | HR.95L | HR.95H | p-Value |
|---|---|---|---|---|---|
| C7 | −0.18 | 0.84 | 0.71 | 0.99 | 0.034 |
| CTSV | 0.48 | 1.62 | 1.25 | 2.09 | 0.000 |
| MMP1 | 0.62 | 1.86 | 1.35 | 2.57 | 0.000 |
| VCAN | 0.35 | 1.42 | 1.08 | 1.86 | 0.013 |
In terms of the median measurement of risk, the HCC samples were divided into high- and low-risk categories. Then, we analyzed the impact of these risk groups on the overall survival proportion of HCC patients using KM curves. The high-risk group was found to have significantly worse OS (Figure 5A).

Risk score analysis in TCGA. (A) Survival curve for low- and high-risk subgroups. (B) ROC curves for determining overall survival according to the risk score. (C) Heatmap showing gene expression, risk score distribution, and survival.
We conducted a time-dependent ROC analysis to evaluate the prognostic capability of the four IRGs. The area under the ROC curve (AUC) for the gene risk score signature was 0.721 (Figure 5B), showing moderate diagnostic capabilities. This suggests that the gene signature has the potential to be a reliable predictive tool. A heatmap was constructed to assess the levels of the four IRGs in the high and low-risk categories. Furthermore, the results of these four immunogenic biomarkers were analyzed in the ICGC-LIRI cohort. The results revealed that patients with an elevated risk measurement in the ICGC-LIRI cohort also showed poorer survival outcomes compared with those with a low-risk determination (Figure 6A–C). This finding suggests that the use of the four immune genes as biomarkers improved the sensitivity and specificity of the prognostic signature.

Risk score assessment in ICGC. (A) Survival curve for low- and high-risk subgroups. (B) ROC curves for prediction of overall survival in terms of the risk score. (C) Heatmap showing gene expression, risk score distribution, and survival.
Construction of nomogram
According to the characteristics of the four immune-related genes, a nomogram was constructed to predict the survival outcomes of HCC patients (Figure 7). The estimated one-, three-, and five-year survival rates of HCC patients were determined using a vertical connection between the total point/each posterior axis. Furthermore, Cox regression analysis was used to evaluate the prognostic significance in relation to different clinical features. The tumor status, stage, T (tumor size), and risk score were found to be significantly linked with overall survival (p<0.01). Multivariate regression analysis showed that only the risk score was an independent predictor of survival outcome (p<0.01) (Table 2). This suggests that the risk score of the four IRGs provides valuable prognostic information, and acts as an independent predictor of patient outcomes. In summary, the nomogram incorporating the risk assessment with various clinical characteristics offers a comprehensive tool for forecasting the survival outcomes of HCC patients.

Nomogram to determine one-, three-, and five-year overall survival using data from the TCGA cohort.
The prognostic value of different clinical parameters.
| Variables | Univariate analysis | Multivariate analysis | ||
|---|---|---|---|---|
| HR (95 % CI) | p-Value | HR (95 % CI) | p-Value | |
| Age | 1.02 (0.99–1.05) | 0.107 | 1.02 (0.99–1.04) | 0.190 |
| Gender | 0.60 (0.34–1.06) | 0.078 | 0.97 (0.51–1.87) | 0.936 |
| Grade | 1.18 (0.80–1.75) | 0.400 | 1.10 (0.70–1.74) | 0.677 |
| Status | 2.65 (1.49–4.72) | 0.001 | 1.91 (1.02–3.55) | 0.042 |
| Stage | 1.72 (1.28–2.32) | 0.000 | 0.78 (0.15–3.96) | 0.764 |
| T | 1.65 (1.24–2.20) | 0.001 | 1.91 (0.42–8.67) | 0.399 |
| M | 4.15 (0.99–17.24) | 0.050 | 0.95 (0.17–5.18) | 0.954 |
| N | 3.42 (0.47–24.96) | 0.226 | 3.33 (0.08–143.94) | 0.531 |
| riskScore | 1.84 (1.42–2.40) | 0.000 | 1.71 (1.25–2.34) | 0.001 |
Validation of the predictive value
The KM-plotter tool was used for additional assessments. This tool determines the association between gene levels and OS. The log-rank test showed that the four IRGs were significantly associated with OS in HCC patients (p<0.01). This indicates that the levels of expression of these genes significantly influenced patient survival outcomes, supporting their prognostic potential as predictive biomarkers (Figure 8).

Validation of the predictive value of IRGs using the Kaplan Meier-plotter in LIHC.
Immune status and GSEA of prognostic risk signature
We conducted PCA to compare the distribution patterns of the high and low-risk categories based on IRG expression and genome-level signaling. The IRG expression levels did not differ significantly from the immunological measurements based on the genome-level signaling features (Figure 9A). However, a significant difference was observed between the two groups, with lower scores seen in the group with minimal risk (Figure 9B).

Immunological evaluation of the high- and low-risk groups using PCA and GSEA. (A) PCA of the high- and low-risk groups was performed using the whole protein-coding gene sets. (B) PCA of the high- and low-risk groups was performed using prognostic risk factors. (C and D) GO and KEGG enrichment analyses associated with risk levels.
Furthermore, GSEA was conducted on the high/low-risk signature datasets in TCGA-LIHC. GSEA indicated significant differences (NOM p<0.05) in the gene set enrichment from the MSigDB Collection. The most significantly enriched signaling bio-functions and network signals were identified following their normalized enrichment scores (NES). GSEA showed that genes were associated with modulation of the cell cycle transition between the G2 and M phases, spindle organization, complement activation, and humoral immune response (Figure 9C). The signaling pathways were primarily concentrated in base/nucleotide excision restoration, complement and coagulation cascades, and retinol metabolism (Figure 9D).
Correlation between the IRG prognostic signature and immune cell infiltration
Given the linkage between these IRGs and tumor immunity in HCC, data on immune cell infiltration were retrieved from the TIMER database. The analysis assessed the link between the IRG prognostic signature and immune cell infiltration in HCC. The results showed positive correlations between the risk measurement and infiltration of DCs, macrophages, neutrophils, and CD4+ T cells (Figure 10A–F). This suggests that the infiltration of these cell types is connected to HCC prognosis. The expression levels of the IRGs were associated with immune cell infiltration.

Associations of risk factors and infiltration of six types of immune cells. Associations were determined using PCA. (A) DCs. (B) Macrophages. (C) Neutrophils. (D) CD4+ T cells. (E) CD8+ T cells. (F) B cells.
Discussion
HCC is a common and deadly malignancy throughout the world, and is characterized by heterogeneity in both biological and clinical aspects [25, 26]. HCC tissues include many components, such as HCC cells, stromal cells, immune cells, and fibroblasts. The complex interactions between these cells lead to HCC evolution and complexity [27]. The heterogeneity of HCC manifests in multiple dimensions, including genetic, molecular, histological, and clinical aspects. HCC cells exhibit diverse phenotypes, genetic alterations, and signaling pathways, leading to tumor growth, invasion, metastasis, and variations in treatment response. Moreover, the tumor microenvironment, consisting of immune cells, stromal cells, and fibroblasts, contributes significantly to the complexity and heterogeneity of HCC. The connection between TICs and tumor cells is vital in HCC progression. TICs, including tumor-infiltrating lymphocytes, myeloid-derived suppressor cells, and tumor-associated macrophages, can modulate the immune response and influence tumor behavior. Tumor cells, in turn, can manipulate the immune microenvironment to evade immune surveillance and promote tumor proliferation. Studying the tumor heterogeneity of HCC and the interplay between TICs and HCC cells is crucial for advancing our understanding of underlying mechanisms and treatment of HCC. Thus, the heterogeneity of HCC and the intricate interactions between TICs and tumor cells are essential factors in HCC development and progression. Recently, TICs have emerged as key players in tumor immunotherapy.
This study explored the tumor heterogeneity of HCC and the interactions between TICs and HCC cells. By analyzing 374 samples from the TCGA database, we preliminarily identified 177 IRGs. Four prognostic IRGs were discovered through a series of analyses with the edgeR package. These IRGs have also been linked with the survival and long-term outcome of patients with breast cancer [28], lung cancer [29], and colon cancer [30]. As one of the well-known complement components, C7 plays an essential role in the immune response in vivo [31]. CTSV (also known as Cathepsin V, CTSL2) protein is a lysosomal enzyme abnormally expressed in various tumors. It is closely associated with the outcomes and disease progression of breast ductal cancer and HCC [32, 33]. MMP1 is a matrix metalloproteinase and is a zinc ion-dependent endoprotease and an oncogene. It is overexpressed expressed in various tumors and closely related to the tumor microenvironment [34]. MMP1 has been shown to regulate HCC progression and spread through the EMT and is also linked with HCC tumor immunity and prognosis [35]. VCAN is found mostly in the extracellular matrix and is closely related to tumor cell adhesion/migration and invasion. VCAN is significantly upregulated in HCC tissues and is linked with poor disease progression and outcome in HCC patients [36]. In conclusion, CTSV, MMP1, and VCAN are associated with tumor immune regulation and progression in HCC. This study identified C7 as an IRG and further supported the possibility of CTSV, MMP1, and VCAN as prognostic markers for HCC. We also found that this progression and outcome indicator was related to the infiltration of specific immune cell subtypes. This suggests that prognostic signature may predict increased ICI, indicating its important role in regulating the tumor immune microenvironment. For external validation, this study used TCGA data for screening as well as databases such as the GEO. The potential of C7, CTSV, MMP1, and VCAN as prognostic markers was systematically analyzed. However, this study also has limitations. Only database data were used for analysis and verification; in vivo and in vitro experiments were not undertaken. Therefore, together with in vitro verification of the above conclusions, we will continue to collect relevant clinical samples for multi-center confirmation to promote the clinical translation of the findings of this study.
In summary, the study identified a prognostic signature consisting of four IRGs for HCC. Investigating these aspects will contribute to advancing HCC research and developing innovative diagnostic and therapeutic approaches. Understanding these interactions is crucial for revealing the mechanisms underlying tumor development and progression and for developing new and more effective diagnostic and therapeutic procedures. Additionally, this research may facilitate the development of personalized treatment strategies and improve patient outcomes.
Funding source: National Natural Science Foundation of China
Award Identifier / Grant number: 81472995
Acknowledgments
We thank expert native English-speaking editors from MJ Language Editing Services, Shenzhen, China (www.mjeditor.com), for editing the English text of a draft of this manuscript.
-
Research ethics: Not applicable.
-
Informed consent: Informed consent was obtained from all individuals included in this study.
-
Author contributions: XRL wrote the paper; CT and FP assisted in the preparation of the manuscript and editing. RW coordinated all the research activities, from the design of the experiment to the final correction of the manuscript. All authors have accepted responsibility for the entire content of this manuscript and approved its submission.
-
Competing interests: Authors state no conflict of interest.
-
Research funding: The work was supported by grants from the National Natural Science Foundation of China (No. 81772995).
-
Data availability: The TCGA (https://portal.gdc.cancer.gov/), ICGC (https://dcc.icgc.org/), GEO (https://www.ncbi.nlm.nih.gov/geo/), GO (https://geneontology.org/), KEGG (https://www.kegg.jp/) and STRING (https://geneontology.org/) material is public available.
References
1. Ganesan, P, Kulik, LM. Hepatocellular carcinoma: new developments. Clin Liver Dis 2023;27:85–102. https://doi.org/10.1016/j.cld.2022.08.004.Suche in Google Scholar PubMed
2. Kawamura, Y, Kobayashi, M, Shindoh, J, Kobayashi, Y, Kasuya, K, Sano, T, et al.. Pretreatment heterogeneous enhancement pattern of hepatocellular carcinoma may Be a useful new predictor of early response to Lenvatinib and overall prognosis. Liver Cancer 2020;9:275–92. https://doi.org/10.1159/000505190.Suche in Google Scholar PubMed PubMed Central
3. Wen, N, Cai, Y, Li, F, Ye, H, Tang, W, Song, P, et al.. The clinical management of hepatocellular carcinoma worldwide: a concise review and comparison of current guidelines: 2022 update. Biosci Trends 2022;16:20–30. https://doi.org/10.5582/bst.2022.01061.Suche in Google Scholar PubMed
4. Nagaraju, GP, Dariya, B, Kasa, P, Peela, S, El-Rayes, BF. Epigenetics in hepatocellular carcinoma. Semin Cancer Biol 2022;86:622–32. https://doi.org/10.1016/j.semcancer.2021.07.017.Suche in Google Scholar PubMed
5. Chen, D, Liu, J, Zang, L, Xiao, T, Zhang, X, Li, Z, et al.. Integrated machine Learning and bioinformatic analyses constructed a novel stemness-related classifier to predict prognosis and immunotherapy responses for hepatocellular carcinoma patients. Int J Biol Sci 2022;18:360–73. https://doi.org/10.7150/ijbs.66913.Suche in Google Scholar PubMed PubMed Central
6. Cao, MD, Song, YC, Yang, ZM, Wang, DW, Lin, YM, Lu, HD. Identification of osteosarcoma metastasis-associated gene biomarkers and potentially targeted drugs based on bioinformatic and experimental analysis. Onco Targets Ther 2020;13:8095–107. https://doi.org/10.2147/ott.s256617.Suche in Google Scholar PubMed PubMed Central
7. Ji, S, Chen, H, Yang, K, Zhang, G, Mao, B, Hu, Y, et al.. Peripheral cytokine levels as predictive biomarkers of benefit from immune checkpoint inhibitors in cancer therapy. Biomed Pharmacother 2020;129:110457. https://doi.org/10.1016/j.biopha.2020.110457.Suche in Google Scholar PubMed
8. Llovet, JM, Castet, F, Heikenwalder, M, Maini, MK, Mazzaferro, V, Pinato, DJ, et al.. Immunotherapies for hepatocellular carcinoma. Nat Rev Clin Oncol 2022;19:151–72. https://doi.org/10.1038/s41571-021-00573-2.Suche in Google Scholar PubMed
9. Oura, K, Morishita, A, Tani, J, Masaki, T. Tumor immune microenvironment and immunosuppressive therapy in hepatocellular carcinoma: a review. Int J Mol Sci 2021;22:5801. https://doi.org/10.3390/ijms22115801.Suche in Google Scholar PubMed PubMed Central
10. Xiao, H, Wang, B, Xiong, HX, Guan, JF, Wang, J, Tan, T, et al.. A novel prognostic index of hepatocellular carcinoma based on immunogenomic landscape analysis. J Cell Physiol 2021;236:2572–91. https://doi.org/10.1002/jcp.30015.Suche in Google Scholar PubMed
11. Jiang, S, Zhang, J, Bian, J, Zhang, L, Xu, Y, Zhao, H, et al.. Novel nomograms based on immune and stromal scores for predicting the disease-free and overall survival of patients with hepatocellular carcinoma undergoing radical surgery. J Surg Oncol 2020;122:1569–79. https://doi.org/10.1002/jso.26197.Suche in Google Scholar PubMed
12. Zhou, ZJ, Xin, HY, Li, J, Hu, ZQ, Luo, CB, Zhou, SL. Intratumoral plasmacytoid dendritic cells as a poor prognostic factor for hepatocellular carcinoma following curative resection. Cancer Immunol Immunother 2019;68:1223–33. https://doi.org/10.1007/s00262-019-02355-3.Suche in Google Scholar PubMed
13. Song, D, Wang, Y, Zhu, K, Tian, L, Gao, Q, Zhou, J, et al.. DCK is a promising prognostic biomarker and correlated with immune infiltrates in hepatocellular carcinoma. World J Surg Oncol 2020;18:176. https://doi.org/10.1186/s12957-020-01953-1.Suche in Google Scholar PubMed PubMed Central
14. Yang, F, Wei, Y, Han, D, Li, Y, Shi, S, Jiao, D, et al.. Interaction with CD68 and regulation of GAS6 expression by endosialin in fibroblasts drives recruitment and polarization of macrophages in hepatocellular carcinoma. Cancer Res 2020;80:3892–905. https://doi.org/10.1158/0008-5472.can-19-2691.Suche in Google Scholar PubMed
15. Li, SC, Xu, Z, Deng, YL, Wang, YN, Jia, YM. Higher neutrophil-lymphocyte ratio is associated with better prognosis of hepatocellular carcinoma. Medicine 2020;99:e20919. https://doi.org/10.1097/md.0000000000020919.Suche in Google Scholar
16. Zheng, B, Wang, D, Qiu, X, Luo, G, Wu, T, Yang, S, et al.. Trajectory and functional analysis of PD-1 high CD4+ CD8+ T cells in hepatocellular carcinoma by single-cell cytometry and transcriptome sequencing. Adv Sci 2020;7:2000224. https://doi.org/10.1002/advs.202000224.Suche in Google Scholar PubMed PubMed Central
17. Liu, X, Li, M, Wang, X, Dang, Z, Jiang, Y, Wang, X, et al.. PD-1(+) TIGIT(+) CD8(+) T cells are associated with pathogenesis and progression of patients with hepatitis B virus-related hepatocellular carcinoma. Cancer Immunol Immunother 2019;68:2041–54. https://doi.org/10.1007/s00262-019-02426-5.Suche in Google Scholar PubMed
18. Mei, J, Wang, R, Xia, D, Yang, X, Zhou, W, Wang, H, et al.. BRCA1 is a novel prognostic indicator and associates with immune cell infiltration in hepatocellular carcinoma. DNA Cell Biol 2020;39:1838–49. https://doi.org/10.1089/dna.2020.5644.Suche in Google Scholar PubMed
19. Hu, Y, Sun, H, Zhang, H, Wang, X. An immunogram for an individualized assessment of the antitumor immune response in patients with hepatocellular carcinoma. Front Oncol 2020;10:1189. https://doi.org/10.3389/fonc.2020.01189.Suche in Google Scholar PubMed PubMed Central
20. Gene Ontology, C. The Gene Ontology (GO) project in 2006. Nucleic Acids Res 2006;34:D322–6. https://doi.org/10.1093/nar/gkj021.Suche in Google Scholar PubMed PubMed Central
21. Kanehisa, M, Furumichi, M, Tanabe, M, Sato, Y, Morishima, K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res 2017;45:D353–D61. https://doi.org/10.1093/nar/gkw1092.Suche in Google Scholar PubMed PubMed Central
22. Yu, G, Wang, LG, Han, Y, He, QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 2012;16:284–7. https://doi.org/10.1089/omi.2011.0118.Suche in Google Scholar PubMed PubMed Central
23. Szklarczyk, D, Gable, AL, Lyon, D, Junge, A, Wyder, S, Huerta-Cepas, J, et al.. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res 2019;47:D607–D13. https://doi.org/10.1093/nar/gky1131.Suche in Google Scholar PubMed PubMed Central
24. Leira, Y, Mascarenhas, P, Blanco, J, Sobrino, T, Mendes, JJ, Machado, V, et al.. Network protein interaction in the link between stroke and periodontitis interplay: a pilot bioinformatic analysis. Genes 2021;12:787. https://doi.org/10.3390/genes12050787.Suche in Google Scholar PubMed PubMed Central
25. Llovet, JM, Pinyol, R, Kelley, RK, El-Khoueiry, A, Reeves, HL, Wang, XW, et al.. Molecular pathogenesis and systemic therapies for hepatocellular carcinoma. Nat Cancer 2022;3:386–401. https://doi.org/10.1038/s43018-022-00357-2.Suche in Google Scholar PubMed PubMed Central
26. Molina-Sanchez, P, Ruiz de Galarreta, M, Yao, MA, Lindblad, KE, Bresnahan, E, Bitterman, E, et al.. Cooperation between distinct cancer driver genes underlies intertumor heterogeneity in hepatocellular carcinoma. Gastroenterology 2020;159:2203–20 e14. https://doi.org/10.1053/j.gastro.2020.08.015.Suche in Google Scholar PubMed PubMed Central
27. Liu, S, Wang, W, Zhao, Y, Liang, K, Huang, Y. Identification of potential key genes for pathogenesis and prognosis in prostate cancer by integrated analysis of gene expression profiles and the cancer genome Atlas. Front Oncol 2020;10:809. https://doi.org/10.3389/fonc.2020.00809.Suche in Google Scholar PubMed PubMed Central
28. Ahmed, W, Malik, MFA, Saeed, M, Haq, F. Copy number profiling of Oncotype DX genes reveals association with survival of breast cancer patients. Mol Biol Rep 2018;45:2185–92. https://doi.org/10.1007/s11033-018-4379-1.Suche in Google Scholar PubMed
29. Zhou, H, Xiang, Q, Hu, C, Zhang, J, Zhang, Q, Zhang, R. Identification of MMP1 as a potential gene conferring erlotinib resistance in non-small cell lung cancer based on bioinformatics analyses. Hereditas 2020;157:32. https://doi.org/10.1186/s41065-020-00145-x.Suche in Google Scholar PubMed PubMed Central
30. Kurebayashi, Y, Kubota, N, Sakamoto, M. Immune microenvironment of hepatocellular carcinoma, intrahepatic cholangiocarcinoma and liver metastasis of colorectal adenocarcinoma: relationship with histopathological and molecular classifications. Hepatol Res 2021;51:5–18. https://doi.org/10.1111/hepr.13539.Suche in Google Scholar PubMed
31. Wurzner, R. Deficiencies of the complement MAC II gene cluster (C6, C7, C9): is subtotal C6 deficiency of particular evolutionary benefit? Clin Exp Immunol 2003;133:156–9. https://doi.org/10.1046/j.1365-2249.2003.02230.x.Suche in Google Scholar PubMed PubMed Central
32. Xia, Y, Ge, M, Xia, L, Shan, G, Qian, H. CTSV (cathepsin V) promotes bladder cancer progression by increasing NF-kappaB activity. Bioengineered 2022;13:10180–90. https://doi.org/10.1080/21655979.2022.2061278.Suche in Google Scholar PubMed PubMed Central
33. Jing, JWS, Ma, J, Yu, L, Zhou, H. Elevated CTSL2 expression is associated with an adverse prognosis in hepatocellular carcinoma. Int J Clin Exp Pathol 2018;11:4035–43.Suche in Google Scholar
34. Yu, J, Xu, Z, Guo, J, Yang, K, Zheng, J, Sun, X. Tumor-associated macrophages (TAMs) depend on MMP1 for their cancer-promoting role. Cell Death Discov 2021;7:343. https://doi.org/10.1038/s41420-021-00730-7.Suche in Google Scholar PubMed PubMed Central
35. Dai, L, Mugaanyi, J, Cai, X, Dong, M, Lu, C, Lu, C. Comprehensive bioinformatic analysis of MMP1 in hepatocellular carcinoma and establishment of relevant prognostic model. Sci Rep 2022;12:13639. https://doi.org/10.1038/s41598-022-17954-x.Suche in Google Scholar PubMed PubMed Central
36. Wang, MQ, Li, YP, Xu, M, Tian, Y, Wu, Y, Zhang, X, et al.. VCAN, expressed highly in hepatitis B virus-induced hepatocellular carcinoma, is a potential biomarker for immune checkpoint inhibitors. World J Gastrointest Oncol 2022;14:1933–48. https://doi.org/10.4251/wjgo.v14.i10.1933.Suche in Google Scholar PubMed PubMed Central
Supplementary Material
This article contains supplementary material (https://doi.org/10.1515/oncologie-2023-0360).
© 2023 the author(s), published by De Gruyter, Berlin/Boston
This work is licensed under the Creative Commons Attribution 4.0 International License.
{kind=link}
{kind=link}
Artikel in diesem Heft
- Frontmatter
- Review Articles
- Advances in ferroptosis of cancer therapy
- Immunotherapy in hepatocellular carcinoma: an overview of immune checkpoint inhibitors, drug resistance, and adverse effects
- The role of matrix metalloproteinase-2 in the metastatic cascade: a review
- The tumor microenvironment: a key player in multidrug resistance in cancer
- Robotic vs. laparoscopic approach in obese patients with endometrial cancer: which is the best? A mini-review
- SLC25 family with energy metabolism and immunity in malignant tumors
- Research Articles
- Catalase expression is an independent prognostic marker in liver hepatocellular carcinoma
- A novel immune-associated prognostic signature based on the immune cell infiltration analysis for hepatocellular carcinoma
- AKAP12 inhibits the proliferation of ovarian cancer by activating the Hippo pathway
- AQP1 as a novel biomarker to predict prognosis and tumor immunity in glioma patients
- Exosomal circular RNA NT5E driven by heterogeneous nuclear ribonucleoprotein A1 induces temozolomide resistance by targeting microRNA-153 in glioma cells
- miR‐30a‐3p inhibits the proliferation of laryngeal cancer cells by targeting DNMT3a through regulating DNA methylation of PTEN
- Disulfidptosis-related long non-coding RNAs predict prognosis and indicate therapeutic response in non-small cell lung carcinoma
- Case Report
- Primary retroperitoneal choriocarcinoma with lung and liver metastasis in a male patient: case report
- Short Commentary
- Clinical pharmacy services in cancer patients with hypertension
Artikel in diesem Heft
- Frontmatter
- Review Articles
- Advances in ferroptosis of cancer therapy
- Immunotherapy in hepatocellular carcinoma: an overview of immune checkpoint inhibitors, drug resistance, and adverse effects
- The role of matrix metalloproteinase-2 in the metastatic cascade: a review
- The tumor microenvironment: a key player in multidrug resistance in cancer
- Robotic vs. laparoscopic approach in obese patients with endometrial cancer: which is the best? A mini-review
- SLC25 family with energy metabolism and immunity in malignant tumors
- Research Articles
- Catalase expression is an independent prognostic marker in liver hepatocellular carcinoma
- A novel immune-associated prognostic signature based on the immune cell infiltration analysis for hepatocellular carcinoma
- AKAP12 inhibits the proliferation of ovarian cancer by activating the Hippo pathway
- AQP1 as a novel biomarker to predict prognosis and tumor immunity in glioma patients
- Exosomal circular RNA NT5E driven by heterogeneous nuclear ribonucleoprotein A1 induces temozolomide resistance by targeting microRNA-153 in glioma cells
- miR‐30a‐3p inhibits the proliferation of laryngeal cancer cells by targeting DNMT3a through regulating DNA methylation of PTEN
- Disulfidptosis-related long non-coding RNAs predict prognosis and indicate therapeutic response in non-small cell lung carcinoma
- Case Report
- Primary retroperitoneal choriocarcinoma with lung and liver metastasis in a male patient: case report
- Short Commentary
- Clinical pharmacy services in cancer patients with hypertension