Nanoimmunotherapy – cloaked defenders to breach the cancer fortress
-
Gayathri Kandasamy
Gayathri Kandasamy received her M.Tech. degree in Medical Nanotechnology SASTRA Deemed University, Thanjavur, India. She joined as JRF in SASTRA Deemed University in June 2016. Currently, she is working in the Centre for Nanotechnology and Advanced Biomaterials (CeNTAB), SASTRA University, Thanjavur, India. Her research focuses on the development of novel core-shell polymeric polyplexes with multi-modal action against lung cancer.
Vadim Annenkov is the Principal Researcher at the Limnological Institute, Russia. He has more than 20 years of experience in synthesis of water-soluble functionalized polymers. During the last decade, his scientific interests have enlarged to encompass composite organo-inorganic systems inspired from the process of biosilicification. Prof. Annenkov’s research group have designed a stepwise method for mimic silaffins. He is currently studying these systems for the delivery of oligonucleotides for biomedical applications.
Uma Maheswari Krishnan is an Associate Dean and Professor at the Centre for Nanotechnology and Advanced Biomaterials (CeNTAB) and School of Chemical and Biotechnology, SASTRA Deemed University, India. She received her PhD degree in Applied Chemistry from Bharathiar University, India, in 2000 and worked as a research associate at University of Texas, USA, until 2002 and had training on thin film processing and clean room processes at the University of Arkansas, USA. Since 2003, she is in SASTRA Deemed University. Her lab works on different facets of smart drug delivery systems, nanobiosensors, electrophysiology, Alzheimer’s disease, and nanomaterials.


Abstract
Cancer continues to be ranked among the top causes of mortality in the world despite the advances made in science and technology. The sub-par performance of cancer therapeutic strategies is due to the transformation of the cancer from a proliferating mass of cells into an impregnable fortress that manipulates and controls the microenvironment to prevent access to any potential cytotoxic factor as well as circumvent the innate immune surveillance processes. Recruitment of the native immune cells to selectively recognize and kill cancer cells can serve to augment the cytotoxic effects of conventional cancer therapeutic approaches. In addition to annihilation of the cancer cells, the induction of memory in the immune cells prevents the possibility of cancer recurrence. However, despite the apparent benefits of cancer immunotherapy, there are several pitfalls that need to be addressed in order to extend these benefits to the clinic. In this context, engineered nanostructured carrier systems can be effectively employed for an activation and priming of the host immune system selectively against the target cancer cells. This has led to the emergence of “nanoimmunotherapy” as an important therapeutic approach against cancer. The use of multi-functional nanomaterials in combination with immunotherapy offers possible solutions to overcome the current limitations in cancer therapy and represents the next generation of “smart therapeutics,” which forms the prime focus of discussion in this review.
1 Introduction
Cytotoxic T-lymphocyte antigen (CTLA-4) and programmed cell death protein (PD-1) were discovered in 1987 as key targets that suppress the action of cytotoxic T lymphocytes in the tumor. Subsequently in 1993, it was successfully demonstrated in mouse models that removing the immunosuppressive environment through blocking these targets unleashes the immune system against cancer cells. These events represent major milestones that paved the way for the emergence of cancer immunotherapy as an important anti-cancer strategy [1], [2]. Encouraging results from clinical trials in 2010 have catapulted this therapeutic approach as a promising solution to conquer cancer. Anti-CTLA-4 antibody was approved for treatment of skin cancer in 2011, while in 2014, anti-PD-1 received approval for the treatment of melanoma [3]. Successful immunotherapy requires stimulating and activating the immune components in the first stage as well as imparting recognition and migration toward the target site for selective destruction of the cancer cells. The cancer microenvironment abounds with immunosuppressive factors that prevent the maturation and cytotoxic action of the immune cells. Further, it should reverse the immunosuppressive environment for selective elimination of the cancer cells [4]. Different approaches to accomplish these aspects have been attempted. Introduction of cytokines such as interleukin-2 (IL-2), IL-6, or interferon-α (immune stimulation); recruitment of dendritic cells or cytotoxic T lymphocytes programmed to attack the cancer cells through presentation of tumor-derived antigenic fragments (cancer vaccine) [5], [6]; or culturing the cells in the presence of cytokines or tumor antigens (adoptive therapy), overcome the immunosuppressive environment present in the tumor microenvironment by inhibiting the tumor-associated macrophages (TAMs) or the immune checkpoint targets in the cancer cells, namely, CTLA-4 and PD-1, or use of genetically engineered T-cell lymphocytes expressing chimeric anti-tumor antigen receptors for selective binding to cancer cells has been explored for destruction of cancer [7], [8]. Nanoparticle-based immunotherapy including antigen/adjuvant delivery vehicles to direct tumor antigen-specific T lymphocyte-targeting agents and their combinations has also been explored for cancer therapy [9], [10]. Currently, several targeted deliveries co-encapsulating therapeutic and imaging agents into a single platform have been reported [11], [12]. Cancer vaccination has shown better preclinical results and also has been investigated toward directed tumor destruction through strengthening the immune system [11], [13]. Metabolic reprogramming of the tumor-associated myeloid cells through inhibition of hypoxia-inducible factor 1α (HIF-1α), indoleamine-2,3-dioxygenase (IDO), fatty acid oxidase (FAO), and cyclooxygenase (COX-2) to alter their immunosuppressive function is also being explored [14]. Targeting cancer stem cells through the use of inhibitors to overcome the immunosuppressive environment is being investigated to aid immunotherapy [15]. In addition, immunotoxins that are formed by fusion of plant or bacterial toxins with antibodies derived against specific cancer cell markers have also been employed [16]. Viruses have also been used to deliver genes encoding for cancer-specific molecules into antigen-presenting cells to enable persistent immune response. Cell-based immunotherapy has been denoted as “living immunotherapy” and offers the possibility of personalized cancer therapy, as it employs cells derived from the patient. Sipuleucel-T, approved in 2010 for clinical use, was the first cell-based immunotherapy agent where dendritic cells, natural killer cells, B lymphocytes, and T lymphocytes isolated from the cancer patient were cultured in a medium conditioned with a fusion protein comprising domains from prostate-specific membrane antigen (PSMA), prostatic acid phosphates, and granulocyte-macrophage colony-stimulating factor (GM-CSF). These conditioned cells, when reintroduced into the patient, displayed directed tropism toward prostate cancer cells and caused reduction in the tumor volume [16].
2 The significance of the anti-angiogenic window in immunotherapy
The tumor microenvironment is very complex, and several types of immune cells have been detected in the tumor. These are collectively referred to as the TAMs and the myeloid-derived suppressor cells (MDSCs). TAMs have been found to infiltrate the tumor and transform from the pro-apoptotic M1 phenotype to the anti-apoptotic and pro-proliferative M2 phenotype [17]. Moreover, factors like vascular endothelial growth factor (VEGF) and chemotactic factors like C-C motif chemokine ligand 1 (CCL1), CCL22, CCL28, C-X-C motif chemokine ligand 5 (CXCL5), and CXCL12 that are enhanced in the hypoxic tumor environment lead to the recruitment of the T regulatory lymphocytes (Treg) and MDSCs that promote an immunosuppressive environment in the tumor, leading to cancer stemness and tumor progression [18], [19]. Several epigenetic modifications and the quiescent or active state of the tumor tissue determine the expression of these pro-proliferative factors. Hence, in order to potentiate the benefits of immunotherapy, inhibition of VEGF and the immunosuppressive factors has been attempted through co-delivery of anti-VEGF and chemotaxis inhibitors in the free form or using nanoparticles. The development of cross-species reactive nanobodies (a class of antibody) for immunotherapy of lung cancer has also been studied [20].
The dose of anti-VEGF factors when introduced along with immunotherapy components is a critical factor for the success of immunotherapy. Tumor vasculature is leaky and tortuous with abnormal branching unlike normal blood vessels [21], [22]. Therefore, it presents a challenge in the diffusion of therapeutic moieties. The main reason for the abnormal vasculature seen in tumors is the VEGF-A. Hence, anti-VEGF therapies are directed against this molecule or its receptors [23]. However, high doses of the anti-VEGF therapy completely disrupt the vascular network in the tumor tissue, thereby aggravating the hypoxic environment in the tumor, which prevents the conversion of the TAMs from the M2 to the M1 phenotype. In addition, the diffusion of the cytotoxic cells becomes further impaired due to lack of vasculature. Hence, it is essential to normalize the tumor vasculature rather than to totally destroy the same. Therefore, moderate doses of the anti-VEGF factor are critical to enable M2-to-M1 transformation of TAMs and penetrate natural killer and cytotoxic T cells to promote cancer cell death (Figure 1).

Importance of adequate dosing of anti-VEGF factors for reversal of immunosuppressive tumor environment for better cancer therapy.
Experimental evidence for the effectiveness of immunotherapy after tumor vascular normalization was demonstrated recently when the epidermal growth factor receptor inhibitor erlotinib was administered to different mouse models of cancer [18]. This treatment resulted in normalization of the tumor vasculature and transformation of the tumor microenvironment into an immune-supportive milieu as evidenced by the change in the phenotype of TAMs as well as improved vascular permeability. This resulted in the better retention of the anti-PD-L1 (programmed death protein ligand) in the tumor, leading to rapid and effective regression of tumors through the activation of cellular immune response. Several studies have demonstrated that vascular normalizing anti-angiogenic treatment aids in reprogramming the immunosuppressive tumor microenvironment and improves the efficacy of immunotherapy [24], [25]. Few studies have also shown that targeting the vasculature with sub-normalizing doses is superior to the high-dose anti-angiogenic therapy in polarizing TAMs from an immunosuppressive M2 phenotype toward an immunostimulatory M1 phenotype and enhances CD4+ and CD8+ T-cell infiltration into the tumor [24]. Several anti-VEGF therapeutic agents are available for clinical use, and several others are being evaluated in various clinical trials for cancer therapy. These include avastin, aflibercept, sunitinib, cediranib, sorafenib, nintedanib, cetuximab, etc. [26]. However, these strategies have to be used carefully, as excessive use of these agents will lead to adverse effects such as bleeding complications and thrombosis [27]. Nevertheless, clinical data reveal that patients undergoing anti-angiogenic therapy exhibit enhanced tumor perfusion and improved survival [28], [29]. In physiological conditions, the immune cells effectively detect and destroy foreign agents and abnormal cells. However, the hypoxic conditions and acidic milieu in the tumor tissue alter the nature of macrophages [28]. Therefore, modulation of the hypoxic environment is essential to enhance the performance of immunotherapeutic agents. In an interesting strategy, iron-oxo clusters and tetrabenzoatoporphyrin ligands were assembled to form an Fe-TBP complex which, when administered in cancer cells, reversed hypoxia by producing oxygen, thereby priming the cells for immunotherapy and photodynamic therapy [30].
The hypoxic tumor microenvironment presents several additional challenges. It is now recognized that cell response and function undergo a drastic transformation under hypoxic conditions. These changes are mediated by HIFs. In normoxic conditions, the α subunit of HIF undergoes proteasomal degradation followed by hydroxylation catalyzed by the prolyl hydroxylase domain (PHD) proteins and ubiquination reactions by the von Hippel-Lindau tumor suppressor. Hypoxic conditions suppress the PHD enzyme activity, thereby retarding the proteasomal degradation of HIF-α and subsequent chain of events, thus activating the HIF-mediated expression of VEGF-A [31]. Hypoxia also promotes the infiltration of TAMs, Treg, and MDSCs apart from over-expression of PD-L1 on tumor cells, all of which contribute to inhibition of T cell-mediated cytotoxicity [32]. Further, anti-angiogenic agents administered without regulation of doses have been found to augment the hypoxic environment and activate cancer stem cells in tumors and promote metastasis. The acidic pH in the hypoxic solid tumors reduces the therapeutic potential of anti-cancer strategies [32]. Therefore, a multi-pronged approach that simultaneously regulates angiogenesis, activates immune components, reverses hypoxia, and regulates cancer cell pH may offer greater therapeutic benefits.
Several drug candidates that target the pH regulator CIAX (extracellular carbonic anhydrase) are under active investigation to reverse acidic pH-mediated complications in cancer therapy. Some of the drugs such as SLC-0111, girentuximab, DTP-348, ADGMCAIX, and AZD3965 are currently under phase I/II/III clinical trials [32]. Bioreductive drugs or hypoxia-activated prodrugs (HAPs) represent a promising clinical strategy to overcome the pitfalls in cancer immunotherapy arising because of the hypoxic tumor microenvironment. HAPs under various stages of clinical investigation include tirapazamine, apaziquone, evofosfamide, tarloxitinib, banoxantrone, and porfiromycin [33]. Table 1 lists some additional strategies that are being employed to address hypoxia-associated hurdles in cancer therapy [33].
Strategies to address hypoxia-mediated therapeutic challenges in cancer.
| Therapeutic strategy | Therapeutic agents |
|---|---|
| Radiosensitization of hypoxic cells | Nimorazole, misonidazole |
| Enhanced oxygen delivery | Nicotinamide, efaproxiral, carbogen |
| Reduction of oxygen consumption | Atovaquone, metformin |
| HAPs | Evofosfamide, tarloxotinib bromide, tirapazamine |
| HIF inhibitors | Aryl sulfonamides |
| Other hypoxia-related mechanisms | Nitroglycerin, BKM120 |
3 Merits and demerits of immunotherapy
Immunotherapy has several advantages: it can be personalized for each patient, it can be used to treat both primary as well as secondary sites of tumors, and most importantly, it can prevent tumor recurrence owing to induction of memory cells. However, there are several demerits that have been associated with immunotherapy. A new category of disorders called immune-mediated adverse reactions that include endocrinopathies, colitis, nephritis, and hepatitis, have been identified with the use of immune checkpoint inhibitors [34], [35]. Systemic toxicity and inflammation have been associated with the administration of cytokines. Moreover, most immune therapies elicited only a weak response, which may be due to the combined effects of poor diffusivity into the tumor tissue as well as the inhibitory environment in the tumor to immune surveillance [3]. In an effort to overcome some of these limitations, the use of nanoparticles engineered to display the antigenic fragments or encapsulate the immunostimulants has been employed to reduce issues pertaining to off-targeting, poor penetration, and target specificity [36]. While drug and gene delivery systems require elaborate modifications to evade the immune system, nanoimmunoadjuvants are deliberately made visible to the immune system to activate their response [12]. The chemical nature of the nanoparticle as well as the surface modifications play an important role in the magnitude of recognition and response of the immune cells.
4 Nano-immunoadjuvants
Polymers of both natural and synthetic origin have long been explored for their adjuvant action. These include polysaccharides like dextran, inulin, mannan, zymosan, chitosan, etc., and synthetic polymers such as polyphosphazenes, polyanhydrides, polyacrylates, polylactides, polycaprolactones, etc. [37]. Converting them into nanoparticles encapsulated with or decorated with antigen-recognizing fragments or antigens can potentiate the components of the native immune system and confer tumor cell-specific tropism to enable destruction of the tumor by the innate immune cells. The engineered nanoparticle is endocytosed into the cells, leading to the presentation of the antigen fragments by the major histocompatibility complexes (MHCs). These in turn result in the production of pro-inflammatory cytokines and promote the maturation of dendritic cells that further triggers the natural killer cells and T lymphocytes, which are now directed toward the cancer cells and trigger their apoptosis [38]. Figure 2 depicts the immunogenic events mediated by an engineered nanocarrier.
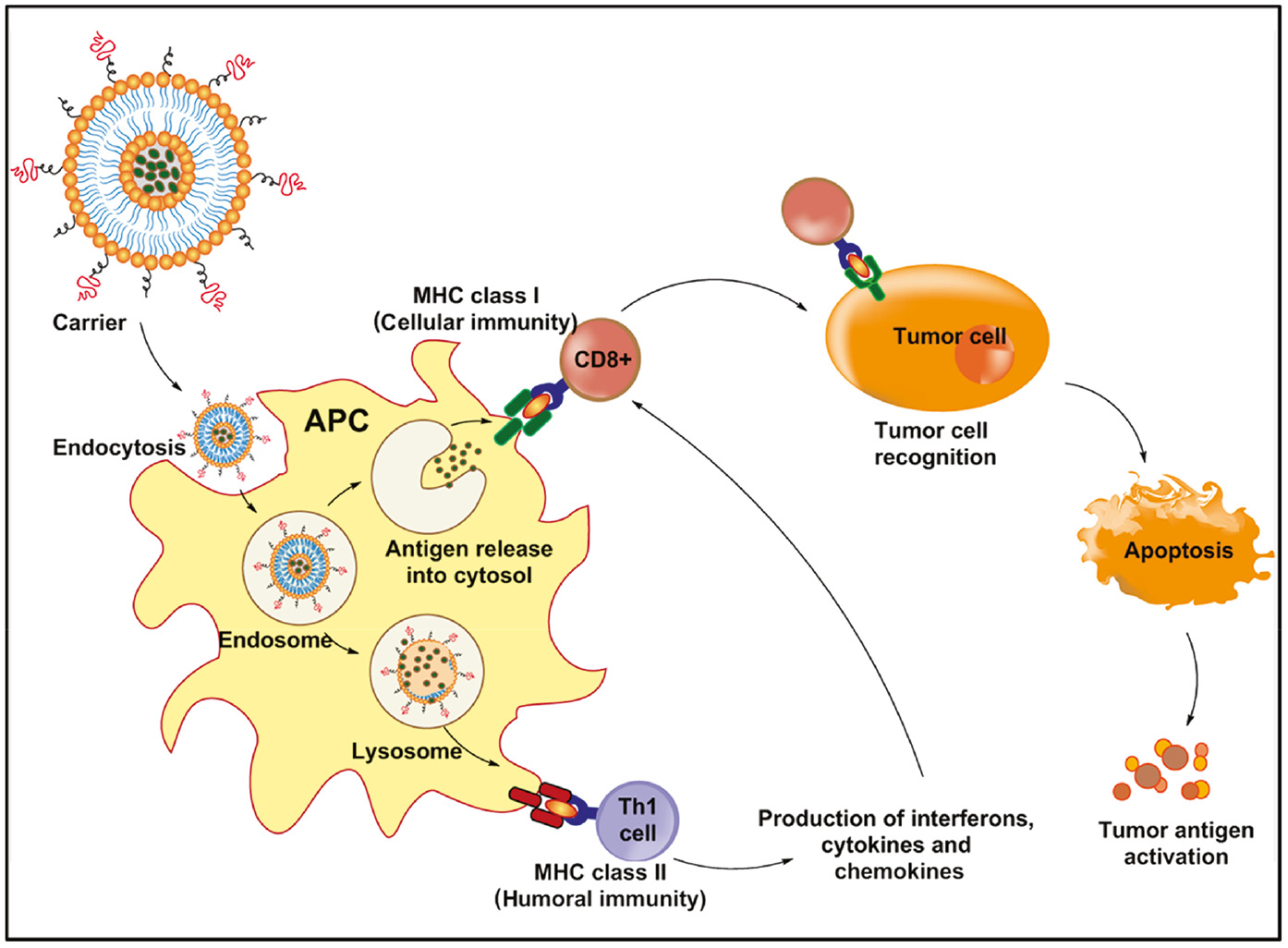
Schematic representation of immunotherapy mediated by engineered nanoparticles.
Nanoimmunotherapy enables site-specific delivery of the cargo. The nanoparticles can serve as immunoadjuvants themselves, thereby enhancing the therapeutic efficacy. They can be used to co-administer different immunoadjuvants or a combination of therapeutic molecules with an immunostimulant, thereby increasing efficacy [38]. Nanoparticles can be tailored to perform multiple functions – deliver immunostimulants, act as an immunoadjuvant, display tumor-specific antigen, as well as reverse the immunosuppressive tumor microenvironment [39], [40]. The size of the nanoparticle is a key determinant for its efficiency. Particles smaller than 5 nm do not reach the lymph nodes and are cleared in the blood. This does not augur well for immunotherapy, as the nanoparticle needs to reach the lymph nodes to interact with the immune cells. Nanoparticles bigger than 200 nm are phagocytosed by the antigen-presenting cells and eventually reach the lymph nodes after 18 h. Nanoparticles smaller than 200 nm directly drain in to the lymph nodes within 3 h [41]. Hence, nanoparticles in the range of 5–200 nm are generally employed for immunotherapy. Hydrophobic particles are more immunogenic when compared to their hydrophilic counterparts, but owing to their aggregation tendency, they require further modifications. Though nanoparticle-mediated immunotherapy is an attractive strategy, the stability of the nanoparticles in vivo remains a limiting factor. Several attempts to circumvent this shortcoming through intelligent surface modification strategies such as PEGylation are underway.
5 Cell-based immunotherapy
One of the major strategies in immunotherapy is to use immune cells and reprogram them toward tumor cells. Dendritic cells derived from the bone marrow form the mainstay of immunotherapy. Their maturation triggers the secretion of cytokines that activate the cytotoxic T lymphocytes and natural killer cells. In addition, the dendritic cells also present antigens for directing the cytotoxic cells to the specific target [42]. The use of nanoparticles to trigger the maturation of dendritic cells has been the most commonly employed strategy in immunotherapy. For instance, silver-coated gold nanorods were found to selectively internalize into dendritic cells, where they resided in the endosomes and caused maturation and activation of the dendritic cells [43]. In another approach, dendritic cells derived from the patient when incubated ex vivo in a medium containing tumor-derived antigens and reintroduced into the patient trigger an immune response against the tumor. Dendritic cells comprise a heterogeneous population of cells that respond to different cytokines and exhibit different pathogen-associated molecular patterns. The new generation of dendritic cell-based immunotherapy strives to use specific subsets of dendritic cells for better efficacy [44]. Many cancer patients do not express adequate levels of CD80 or CD86 in dendritic cells, and hence, these cells are not conducive for effective immunotherapy. The suppression of the Jak/STAT pathway by the protein tyrosine phosphatases TC-PTB and PTP1B has recently been discovered as a key factor that prevents dendritic cell maturation [45]. In a proof-of-concept study employing pancreatic cancer and lymphoma models, it was demonstrated that inhibition of these two phosphatases promoted maturation of dendritic cells by activation of the Jak/STAT signaling pathways and increased cytokine and cytotoxic T lymphocytes. This strategy can potentiate cell-based immunotherapy approaches for cancer therapy.
Instead of employing dendritic cell-mediated T-cell activation and proliferation that may be time-consuming and expensive, adoptive T-cell transfer (ACT) techniques involving K562 cells engineered to express co-stimulatory molecules have been employed for T-cell expansion ex vivo[46]. Nanostructured paramagnetic beads containing CD3 and CD28 that act as artificial antigen-presenting cells have also been employed for T-cell proliferation ex vivo. Such strategies have driven the rapid expansion of cytotoxic T lymphocytes that can be effective in destroying the tumor when reintroduced into the biological system. However, introduction of tumor-specific tropism of these ex vivo-expanded T lymphocytes is yet to be reproducibly realized. One of the dangers of using T cells conditioned with a common antigen (public antigen) is the adverse effects that may arise due to off-targeting. Therefore, the current paradigm in T cell-mediated immunotherapy is to identify the mutated antigen that is the signature of the cancer being addressed and condition the T cells with this neo-antigen [47]. This approach will mitigate several adverse effects associated with T cell-mediated immunotherapy and improve the anti-cancer efficacy of this therapeutic stratagem. In an interesting attempt, cytotoxic T lymphocytes were conjugated through disulfide bonds to cationic dextran-small interfering RNA (si-RNA) complexed redox-sensitive liposomes [48]. The inherent ability of the cytotoxic T cells to migrate to the tumor microenvironment and their accumulation in the tumor were found to elicit native immune response, which was successfully demonstrated in mice models of thymoma. Further, the high levels of reduced glutathione (GSH) in the tumor microenvironment lyse the disulfide bond, thereby releasing the liposome with its si-RNA cargo into the cells. Though this proof-of-concept study employed fluorescent si-RNA, this strategy can be successfully employed to develop synergistic combinations for multi-modal cancer therapeutics. Here, the cytotoxic T lymphocytes assumed a new role as carrier to deliver the nanoparticle to the tumor site. Cytotoxic T lymphocytes can lead to potential side effects due to a mismatch on the T-cell receptors expressed on the surface. Therefore, efforts to knock down unwanted T-cell receptors using gene editing techniques like CRISPR-CAS are also being explored [49]. Metabolic reprogramming of cytotoxic T lymphocytes to increase their anti-cancer potency through treatment with drugs like 2-deoxyglucose, metformin, rapamycin, and fenofibrate is also being interrogated and in the future may be integrated with nanoparticle-mediated delivery [50].
Cytokine-induced killer (CIK) cells are a sub-type of cytotoxic T lymphocytes produced by the peripheral blood lymphocytes in response to IL-2, IL-12, and IFN-γ [51]. CIK cells mediate tumor cell death through Fas-dependent apoptosis. The natural killer cells are the effector cells of CIK cells. The adjuvant action of several nanoparticles has been found to activate dendritic cells that trigger the CIK cells. This combination has been found to exhibit a more potent anti-cancer effect when compared with the action of either type of cell independently. Clinical trials involving adenovirus-activated dendritic cells in combination with CIK are currently underway for the treatment of renal cell carcinoma and high-risk soft tissue sarcoma. Further studies in realizing the potential of CIK cells for cancer immunotherapy regulated by nanoparticles are under investigation.
Natural killer cells are also a class of cytotoxic cells found in the peripheral blood. They do not require sensitization unlike the cytotoxic T lymphocytes. They also do not require antigen presentation by the MHCs [49]. As they do not attack the lung, liver, muscle, or kidney, natural killer cells have lesser adverse effects than cytotoxic T cells. Chimeric antigen receptor (CAR) modified natural killer cells, therefore, offer an effective and safer strategy to treat cancer. Attempts to optimize their expansion ex vivo and impart tumor specificity are currently underway.
Neutrophils are components of cellular immunity that have seldom been explored for immunotherapy. However, recently, denatured albumin nanoparticles decorated with an antibody against gp75 antigen and loaded with a photosensitizer were demonstrated to recruit neutrophils specifically to the tumor site [52]. The antibody-modified nanoparticles were selectively internalized by the neutrophils, which then caused tumor annihilation on irradiation due to a combination of immunotherapy and photodynamic therapy in mouse models of melanoma.
In a seminal work, the ability of macrolide drugs such as azithromycin, clarithromycin, tricyclic ketolide, etc. to selectively localize in macrophages was exploited to develop a novel strategy for the treatment of breast cancer [53]. Photo-responsive gold nanorods were conjugated using a poly(ethyleneglycol) linker to macrolide drugs. These particles were found to selectively internalize into TAMs. In a co-culture of TAMs and breast cancer cells, a greater percentage of cancer cell death was observed after irradiation with near infrared (NIR) radiation. The percentage of cell death was found to be significantly greater than those observed with cells treated with gold rods alone and irradiated with NIR or only macrolides. This indicates that the cytotoxic mechanism involves the activation of TAMs and is not solely due to gold nanorod NIR-mediated hyperthermia or cytotoxicity of macrolides. The heat generated in TAMs on NIR irradiation due to the photo-thermal effect of the gold nanorod-macrolide system activates the macrophages probably through the heat shock protein-mediated pathway. The activated macrophages release pro-inflammatory cytokines IL-6 and TNF-α, which induce the innate immune cell-mediated annihilation of the cancer cells in the vicinity (Figure 3). This report represents a milestone in the recruitment of TAMs for killing the cancer cells.

Macrolide modified gold nanorods for activation of immune response through the photothermal effect.
It is evident that the nanoparticle-mediated activation of the immune components can be synergistically used along with chemotherapeutic agents or other anti-cancer therapies for superior efficacy.
6 Virus-mediated immunotherapy
Viruses are nanodimensional structures that exhibit good specificity and excellent cell internalization. These aspects have been exploited in the development of engineered viruses for the treatment of high-grade glioblastoma [54]. Poliovirus chimera that selectively infects cells derived from the central nervous system was found to elicit an immune response against glioblastoma cells. Similarly, an adenoviral vector expressing IL-12 was employed for immunotherapy of brain cancer [54]. Papaya mosaic virus was employed for immunotherapy by direct administration into the tumor in mice induced with melanoma [55]. An increase in the cytotoxic T lymphocytes and a decrease in the suppressor cells were reported in the tumor site. A concomitant increase in the MHC expression was recorded because of the increase in IFN-γ levels post-treatment. The treatment resulted in a reduction in the tumor volume and also retarded lung metastasis. This model represents an interesting option for cancer immunotherapy, but further studies on the safety and risk factors involved in using viruses for therapy need to be carried out before translational attempts. To avoid risk factors associated with viral nanoparticles but to retain the positive characteristics associated with viruses, virus-like nanoparticles (VLPs) have been employed. Surface-modified VLPs derived from the Rous sarcoma virus have been employed for immunotherapy against colon cancer. The nanoparticles were modified with a recombinant single chain fragment variable (rscFv) attached to the vesicular membrane through glycol-inositol phosphate and recombinant human IL-12 fused with the hemagglutinin transmembrane domain [56]. The rscFv selectively bound to the receptors expressed on colon cancer cells, and IL-12 initiated the cascade of events mediating immune response.
Virosomes are another class of vesicular structures containing molecules derived from the viral capsid coat. These have been demonstrated to exhibit better cell internalization and highly efficient delivery of the therapeutic cargo, but unlike viruses are not infective. Influenza and Sendai viral capsid-derived virosomes have been most widely explored as carriers [57]. Because of their viral origin, virosomes can elicit an immune response and hence act as adjuvants. Plasmid DNAs encoding for tumor-associated antigens in prostate cancer have been successfully delivered using influenza virus-derived virosomes and have shown regression in tumor through activation of cytotoxic T lymphocytes. Sendai virus-derived virosomes have been found to internalize into the target cells through sialic acid moieties and stimulate dendritic cells to recruit natural killer cells for cancer cytotoxicity. Several oncolytic viruses are currently in clinical trials, though long-term effects of this therapy need to be monitored for future implementation.
7 Exosome-based immunotherapy
Exosomes are vesicular secretions by cells that have been used by the native system to transport signaling molecules as well as oligonucleotides to other cells. Their membrane composition, transport, and target specificity are determined by their cell of origin. Though their exact mechanism of transport is yet to be deciphered, these vesicular systems have garnered much attention as drug and gene delivery systems. Dendritic cell-derived exosomes have also been reported in the literature. Therefore, it was hypothesized that if dendritic cells were exposed to tumor-derived antigens, then the exosomal secretions from these primed dendritic cells may display these antigen fragments to activate T lymphocytes and natural killer cells. Such type of dendritic cell-derived exosomes, Dex, were found to contribute to tumor eradication mediated by activated cytotoxic T lymphocytes [58]. On similar lines, primed T cell-derived exosomes, Tex, were also found to be effective agents mediating immunotherapy. To improve the tumor-specific tropism of these carriers, surface-engineered exosomes containing tumor recognition motifs could be developed. A recent study has highlighted the importance of the type of MHC (type I or type II) on the exosomes for beneficial immunotherapy [59]. Mismatch or absence of MHC leads to contradictory effects, with increased tumor survival instead of regression. Though complete understanding of the formation and function of exosomes remains incomplete, these exosomes, independently or in combination, can be effective systems for immunotherapy. Engineered exosomes encapsulating specific immune stimulants and surface modified exosomes bearing antigen fragments are also being investigated for efficient immunotherapy against cancer and may represent the next generation of smart therapeutics [59].
8 Microfluidic devices for immunotherapy
One of the major issues limiting many anti-cancer strategies is the poor in vitro-in vivo correlations of the results. Promising results demonstrated in vitro have not been reproduced in vivo in most cases. This has been now understood to be due to the lack of an appropriate model that mimics the complex tumor microenvironment. The receptor status of cells cultured in conventional two-dimensional (2D) methods and 3D models has been found to be significantly different. Apart from this, diffusion, internalization, and oxygen gradient also vary between the two systems, thereby reducing the 2D method irrelevant to investigate efficacy of anti-cancer strategies. Several extensive reviews on this topic have highlighted a wide range of diverse approaches to mimic the tumor microenvironment. This area is still evolving, and more exciting and exact mimics of tumor are on the anvil. Immunotherapy can also benefit immensely from the use of appropriate 3D models of cancer. Cell-based immunotherapy has been greatly limited by the retardation of T-lymphocyte entry into the cancer tissue through the stroma, thereby rendering the therapy ineffective. Hence, there is considerable interest in developing appropriate tumor 3D mimetics. One of the promising approaches involves the co-culture of different types of cells in 3D along with conditions that mimic fluid flow that is experienced by native tissues, which has been effectively integrated on a microfluidic platform [60]. The main advantage of this system is its portability, a versatile system that enables optical visualization as well as monitoring of the mechanical aspects of the immunotherapy such as cell adhesion, migration characteristics, as well as their interactions with the neighboring molecules and cells. The chemical environment in the microfluidic channels can be suitably altered to mimic various conditions that regulate the tumor microenvironment, thereby providing a flexible platform for evaluating different cell-based immunotherapeutic strategies on a real-time basis [61]. In a recent study, the movement of IFN-α-conditioned dendritic cells has been investigated using a microfluidic platform fabricated using polydimethylsiloxane [62]. Both cancer and immune cells were mixed with collagen and cultured in independent compartments fabricated in the microfluidic chip. The tendency of the dendritic cells to move between these compartments in response to the environmental cues was then monitored to assess its maturation and activation to present cancer antigens to the naive immune cells. This strategy is still in its fledgling stage with respect to immunotherapy, but it shows immense promise as a testing platform for evaluating immunotherapy strategies independently or as an adjuvant strategy and may also replace expensive and tedious in vivo models.
9 Polysaccharide nanoparticles for immunotherapy
Several polysaccharide-based systems have been explored for immunotherapy due to their inherent quality to be recognized by the immune system. Chief among them are alginate and chitosan. Alginate, an anionic polysaccharide that has been extensively explored for drug delivery applications, was used to conjugate a model antigen ovalbumin by a Schiff’s bond [63].
In order to specifically target dendritic cells, mannose-conjugated alginate nanoparticles were cross-linked with the ovalbumin-cross-linked alginate using calcium chloride. The system displayed pH-responsive release of ovalbumin with rapid release in the acidic pH due to the lysis of the Schiff’s bond. The mannose-alginate-ovalbumin-alginate nanoparticles preferentially internalized into dendritic cells due to the mannose modification. The dendritic cells treated with the mannose-alginate-ovalbumin-alginate nanoparticles were found to exhibit significantly high levels of CD40, CD80, and CD86, which are hallmarks of dendritic cell maturation. Enhanced expression of these markers has been demonstrated to activate the cytotoxic and helper T cells [63]. The cells treated with the mannose-alginate-ovalbumin-alginate nanoparticles were also found to secrete higher levels of the cytokines IL-12p70 and IL-4, which promote T-lymphocyte proliferation, as well as the tumor-suppressive cytokines TNF-α and IFN-γ. The higher cell uptake of the mannose-conjugated alginate nanoparticles was also cited to favor better antigen presentation by MHC-I. In vivo studies revealed a selective and longer residence time in the lymph nodes by the mannose-alginate-ovalbumin-alginate nanoparticles. Similarly, a higher reduction in tumor growth in the animals treated with the mannose-alginate-ovalbumin-alginate nanoparticles was recorded when compared with the moderate suppression observed with the free ovalbumin particles. However, because of the specific nature of the bond employed to link the antigen, this system, though promising, can be employed only for the introduction of amine-containing antigens. In another report, mannose-modified chitosan nanoparticles were employed to prime dendritic cells by delivering whole-cell lysates from melanoma [64]. The target-specific delivery ensured maturation of the dendritic cells and expression of IL-4 and IFN-γ. In vivo studies revealed superior suppression of the melanoma and resistance to recurrence of the tumor when again challenged with the melanoma cells.
The immunogenic nature of chitosan has been well exploited in the context of immunotherapy. Chitosan nanoparticles were employed for delivering the model antigen ovalbumin and the sodium salt of poly(inosinic)-poly(cytidylic acid) (poly I:C) that can target toll-like receptors (TLR3) in the endosomes of dendritic cells [65]. The cationic chitosan was found to effectively bind to the anionic dendritic cells through electrostatic interactions. The nanoparticle-treated dendritic cells expressed elevated levels of IL-1β, IL-6, IL-12p70, and TNF-α, indicating their maturation and activation. In in vivo studies performed in EG.7 lymphoma-induced mice, the chitosan nanoparticles containing ovalbumin and poly I:C reduced the tumor to the maximum extent when compared to the other groups. The poly I:C moiety serves as the ligand for TLRs, thereby promoting the immune activation. These superior anti-cancer effects of the chitosan system were attributed to the higher number of cytotoxic T lymphocytes found at the cancer site. The use of nanoparticles for activating the dendritic cells eliminates the need for external manipulation of these cells for inducing T-cell production and activation against specific targets.
Chitosan nanoparticles have also been employed to present MUC1 peptide sequence as the immune-stimulatory component [66]. The MUC1sequence is derived from the transmembrane protein mucin 1 found to be over-expressed in most cancers of epithelial origin. The MUC1 protein in normal cells is hyperglycosylated and confined to the basolateral side. However, in cancer cells, MUC1 is distributed throughout the cell surface and is hypoglycosylated. This modification in the glycosylation status and distribution alters its processing and signaling pathways activated by the MUC1 fragments that promote survival and metastasis of the cancer cells [67]. As MUC1 is aberrantly expressed in cancer cells, this could serve as a recognition paratope for the immune cells. In order to further potentiate the immune-stimulatory nature of MUC1, MUC1 was modified by glycosylating a threonine residue in the MUC1 sequence and introducing a tetanus toxoid-derived T-cell epitope. Chitosan nanoparticles formed by the addition of γ-glutamic acid and mixed with the modified MUC1 peptide elicited good antibody response, confirming its potential for cancer immunotherapy.
In the search for novel and effective immunostimulatory molecules, a novel fusion gene was developed composed of the NKG2D gene that selectively recognizes the MHC protein and IL-2, an immunoadjuvant [68]. A plasmid DNA constructed using this novel fusion gene, when delivered using chitosan nanoparticles in colon tumor-bearing mice, was found to effectively curtail tumor growth through activation of the natural killer and cytotoxic T lymphocytes. The results of this work open up new possibilities for the generation of designer genes for cancer immunotherapy.
10 Liposomes in cancer immunotherapy
Liposomes represent a versatile self-assembled vesicular system that has been extensively investigated for drug and gene delivery applications. These systems have also been effectively employed for immunotherapy. Cationic liposomes prepared from DOTAP (N-1-(2,3-dioleoyloxy) propyl N,N,N-trimethylammonium methyl sulfate) and cholesterol were used to complex the anionic cGAMP (2′,3′-cyclic guanosine monophosphate-adenosine monophosphate), a well-known agonist of the stimulator of interferon genes (STING) [69]. Free cGAMP is not effective in activating immune cells due to its poor internalization into the cells that also possess a negatively charged surface. Liposomal cGAMP was retained in the tumor tissue in orthotropic mouse models of melanoma and attracted the accumulation of antigen-presenting cells at the tumor site, resulting in better tumor inhibition. The added advantage of this strategy was that the liposomal delivery of cGAMP produced a memory effect that again suppressed the recurrence of tumor in mouse models. In another approach, the pH-responsive liposomal system prepared using egg phosphatidyl choline and dioleoylphosphatidylethanolamine (DOPE) was surface modified with the pH-responsive moiety 3-methylglutarylated poly(glycidol) either in the linear or hyperbranched form and also incorporated monophosphorylated lipid A (MPLA), an immunoadjuvant [70]. The antigenic ovalbumin was encapsulated in the liposomes. The substituted poly(glycidol) moieties were recognized by the dendritic cells, leading to the internalization of the liposomes through endocytosis. The hyperbranched modification masked the fusogenic DOPE, thereby retarding cell internalization when compared to the liposomes modified with the linear poly(glycidol) derivative. The acidic pH of the endosome promoted the destabilization of the poly(glycidol) derivative and phase transformation of DOPE, resulting in the perturbation of the endosomal membrane and subsequent release of ovalbumin in the cytosol. The MPLA moiety promoted dendritic cell maturation and subsequent events leading to the expression of MHC-I and -II molecules. The pH-responsive system showed good anti-tumor efficacy in in vivo models of lymphoma, indicating the potential of nanoparticle-mediated immunotherapy for cancer therapy.
In a recent report, macrophage membrane components were linked to the surface of ensantine-loaded pH-responsive liposomes [71]. The macrophage membrane component facilitated specific interactions with metastazing nodules in tumor tissue through α4-VCAM interactions mediated by the integrins. Though the activation of immune components was not investigated in this study, the liposomal system was demonstrated to significantly curtail the number of metastatic nodules in the lung metastatic model of breast cancer when compared to conventional drug-loaded liposomes and free drug.
Another approach in immunotherapy is to employ oligonucleotide sequences to bring about persistent expression of MHC genes in antigen-presenting cells. However, their translation is limited due to their poor in vivo stability as well as restricted cellular uptake. Hence, a liposomal system was employed for the delivery of mRNA to different antigen-presenting cells [72]. The system effectively transfected the mRNA for ovalbumin, which was manifested through the activation of cytotoxic T cells and natural killer cells when administered in the mouse models of melanoma. The study also demonstrated that introduction of lipopolysaccharides with the liposomal formulation further potentiated its immunotherapeutic effects. An mRNA for tumor antigen was used for producing persistent T-cell activation. The mRNA was protected using nucleotide derivatives to improve stability, but this modification resulted in the inhibition of IFN-γ, thereby rendering the immunotherapy ineffective. Hence, the mRNA was co-delivered with monophosphoryl lipid A, a TLR4 agonist that self-assembled to form liposomes [73]. The co-delivery showed excellent immune response in a tumor-bearing mouse model. A phospholipid monolayer containing a NIR probe and surface decorated with a fusion peptide comprising antigen peptide and domains from the natural high-density lipoprotein (HDL) receptor (SR-LB1) [74]. The SR-LB1 facilitates dendritic cell uptake while the peptide activates the immune response. Further potentiation of the immune response can be achieved by co-encapsulation of the cytosine-phosphate-guanosine (CpG) oligonucleotide. The system exhibited good anti-cancer efficacy in mouse models of melanoma. On similar lines, a synthetic high-density lipoprotein (sHDL) was prepared in the form of a disc. Cholesterol-linked CpG was incorporated into the sHDL disc along with a tumor-specific antigen peptide [75]. This system displayed efficient delivery of the immune stimulants to draining lymph nodes, thereby eliciting a strong T-lymphocyte response. The co-administration of anti-PD-L1 and anti-CTLA-4 that serve to overcome the immunosuppressive environment in the tumor tissue ensured regression of melanoma developed in mice models, and such combinatorial stratagems may offer greater benefits in annihilating cancer.
11 Polymeric carriers for immunotherapy
Poly(lactic acid-co-glycolic acid) (PLGA) is a biodegradable and biocompatible polymer with hydrolysable ester links that have been extensively employed for drug delivery applications. PLGA comprising 50:50 ratio of the constituent monomers was employed to encapsulate the model antigen ovalbumin and the TLR ligand poly I:C [65]. The PLGA nanoparticles were found to be effective in promoting the maturation of dendritic cells in mice models of lymphoma, leading to the activation of cytotoxic T lymphocytes through up-regulation of cytokines IL-1β, IL-6, IL-12p70, and TNF-α. This carrier can be explored for the delivery of other immunoadjuvants in the future. The use of novel strategies to stimulate the immune cells against cancer has resulted in the development of new immunomodulators. An anti-angiogenic agent nano-diamino-tetrac conjugated to PLGA, when administered to cancer cells, was found to specifically bind to the thyroid hormone binding site in αvβ3 integrin receptors that are over-expressed in cancer cells [76]. This binding inhibits the PD-L1 gene that has been implicated in the development of the immunosuppressive environment in cancer. The PD-L1 inhibitory action of PLGA-linked nano-diamino-tetrac was successfully demonstrated in many cancer cell lines stimulated by thyroxine. This strategy can be utilized for selective toxicity to cancer cells, as the αvβ3 integrin is over-expressed in cancer cells alone and not in normal cells. Further, in vivo validation of this strategy remains to be established. Recently, PLGA nanoparticles decorated with recognition motifs for tumor-derived peptide antigens were found to effectively capture these antigens, thus serving as a mimic of antigen-presenting cells [58]. These nanoparticles were subsequently captured by dendritic cells that moved to the lymphatic nodes and triggered the expansion and activation of cytotoxic T lymphocytes. This resulted in an abscopal effect where local delivery of the nanoparticles to the tumor enabled regression of the tumor at the primary site of administration as well as in metastatic sites as demonstrated in a mouse model of melanoma. Tumor-associated antigens ovalbumin and gp100 were encapsulated in PLGA nanoparticles and were found to increase therapeutic efficiency [77].
Another polyester that has been employed for immunoadjuvant delivery is poly(ethylene oxide)-co-poly(α-carboxylate-ε-caprolactone). The polymer was conjugated through an ester link to cucurbitacil-I, a STAT3 inhibitor [78]. The inhibition of STAT3 promotes a decrease in immunosuppressive factors including VEGF and IL-10. Consequently, the dendritic cells are activated, resulting in the increased expression of CD40 and CD86, indicative of dendritic cell maturation and elevation in IL-2 and IFN-γ, which suggests stimulation of cytotoxic T-lymphocyte response. The polymer conjugate displayed superior tumor cytotoxicity due to enhanced cellular uptake and lysis of the ester bond by intracellular esterase. This strategy holds promise for cancer therapy by making the tumor susceptible to the cytotoxic immune cells [79].
12 Inorganic nanoparticles for immunotherapy
Inorganic carriers like mesoporous silica have been extensively explored for drug delivery applications but are limited by several challenges when employed for immunotherapy. Chief among them is their poor ability to escape the endosome. In a recent seminal work, the synthesis of mesoporous silica was tailored to overcome the existing demerits in the carrier for immunotherapy [80]. A comparison was made of three different hollow mesoporous silica – double shelled mesoporous silica, a single shelled mesoporous organosilica, and a double shelled mesoporous organosilica. All three carriers were loaded with the model antigen ovalbumin. The double shelled mesoporous structures exhibited greater entrapment efficiency. It was found that both organosilica carriers exhibited superior endosomal escape capabilities due to their hydrophobic organic modifications. When administered in melanoma-bearing mice models, the double shelled mesoporous organosilica displayed the best tumor regression properties through activation of the innate immune response. This may be attributed to the higher loads and sustained release of the ovalbumin in cancer cells as well as the endosomal escape properties that enabled presentation of the antigen through the MHC to stimulate the cytotoxic immune cells. Thus, it becomes clear that the carrier properties play a major role in deciding the efficiency of immunotherapy. On similar lines, gold nanoparticles coated with proteins from tumor cell lysates were found to induce better maturation of dendritic cells and elicited better immune cell responses when compared with silica nanoparticles coated with similar cell lysates. These results suggest that the type of inorganic nanoparticle has a profound influence on its ability to activate the immune response. In another work employing inorganic particles, organically modified dendritic mesoporous silica nanoparticles with a tetrasulfide framework were used to deliver ovalbumin and a TLR9 agonist to antigen-presenting cells. The dendritic tetrasulfide-modified silica particles were capable of depleting the intracellular GSH levels that resulted in an increase in reactive oxygen species (ROS), which subsequently leads to the activation of cytotoxic T lymphocytes [81]. Extra-large pore-sized mesoporous silica particles have also been explored as adjuvants in immunotherapy for co-delivery of ovalbumin and a TLR9 agonist, but further pre-clinical trials are required to confirm the clinical potential of these systems [82].
Nano-dimensional fluorescent diamond nanoparticles (nanodiamond) with nitrogen vacancies have also been explored for its immune activation properties using macrophage cells [83]. Enhanced expression of TNF-α was recorded in macrophages treated with the nanodiamonds. Activation of natural killer cells in a co-culture treated with the nanodiamonds also indicates its immune-stimulatory action. The nitrogen vacancies in the nanodiamond lattice impart fluorescence characteristics with an emission centered in the red region of the spectrum. This enables simultaneous visualization of the nanodiamond when administered in mice models. Surface modifications of the nanodiamond to modify its specificity have also been reported [83]. Inorganic nanoparticles show different capabilities based on the chemical composition of the cores [84], [85]. Magnetic core-shell nanoparticles comprising of iron oxide and zinc oxide were employed for delivering carcinoembryonic antigen to dendritic cells. In vivo experiments on mice models revealed the nanoparticle-mediated delivery delayed the tumor induction and improved the survival of the animals. The magnetic contrast enhancement properties of the iron oxide enabled visualization of the tumor tissue, thereby conferring theranostic properties to the carrier [86]. Other metallic oxides such as cuprous oxide, aluminum oxide, titanium oxide, zinc oxide, and cobalt oxide have also been found to elicit immune responses against different cancer models and hence offer promise as adjuvants in cancer immunotherapy [87]. However, further studies on their site-specificity and residence time have to be optimized for clinical translation.
13 Novel nanoparticles for immunotherapy
Recently, carbon nanotubes have also been explored as a nanovaccine. Two immunoadjuvants, namely, CpG oligonucleotide and α-CD40, along with ovalbumin were incorporated within multiwalled carbon nanotubes (MWCNTs) [88]. CpG is a well-known agonist of TLR9, while α-CD40 is an antibody specific for the tumor necrosis factor receptor CD40 found in antigen-presenting cells including dendritic cells. The MWCNT conjugate elicited excellent immune response in vitro and also reduced the progression of melanoma in mice models. The survival of mice was significantly improved post-treatment with the MWCNT conjugate. The MWCNT conjugate was found to localize in the lymph nodes, enhance the CD86 levels for T-cell proliferation, and also enhance the localization of the cytotoxic T cells in the vicinity of the tumor tissue, thereby contributing to the improved tumor regression effect.
Self-assembled nucleotide nanocapsules based on a DNA-RNA hybrid were employed for delivering CpG-short hairpin RNA (shRNA) sequence that served as adjuvants [89]. The encapsulation of the anionic immunoadjuvant into the anionic carrier was accomplished by coating the CpG-shRNA with a cationic peptide sequence. This system elicited superior cytotoxic T-lymphocyte response against colorectal cancer as well as prevented recurrence of the tumor due to induction of memory T cells. Another interesting strategy involved the use of inorganic magnesium pyrophosphate to complex the anionic CpG oligonucleotides to enhance their serum stability [90]. The system displayed superior activation of the innate immunity and improved localization and prolonged residence time in the melanoma tumor tissue, as well as reduced side effects such as splenomegaly that is commonly associated with CpG oligonucleotides. Further efforts in this direction could lead to the emergence of inorganic-organic hybrid structures for immunotherapy.
Hyaluronic acid conjugated to peptide-modified poly(ethylene glycol) was designed to deliver ovalbumin to the tumor microenvironment [91]. The peptide sequence was specifically recognized by matrix metalloproteinase-9 that is abundantly expressed in the tumor microenvironment. The lysis of the peptide enabled better CD44-mediated internalization of the nanoparticle mediated by hyaluronic acid. The in vivo studies employing the murine cervical cancer model demonstrated the anti-cancer efficacy of this nanoparticulate antigen delivering system, which can be attributed to the robust CD8+ response mediated by the antigen.
A novel strategy involving a scaffold fabricated using poly(ethylene glycol) and heparin was used to culture mesenchymal stromal cells engineered to produce bispecific antibody against CD33 and CD3 [92]. The scaffold when implanted in vivo supported the adhesion and proliferation of the cryogel macroporous engineered mesenchymal cells that released the bispecific antibody. This in turn recruited cytotoxic T lymphocytes due to the recognition of CD3 found on the T cells. The activated T cells exerted its cytotoxicity against acute myeloid leukemia cells that express CD33. This strategy enables long-term immunity against acute myeloid leukemia.
Targeting multiple targets can enhance the specificity of the nanosystem [93]. This formed the underlying hypothesis of a recent work where the nanoparticle was conjugated with cancer cell-specific ligand Herceptin2 (Her2) and calreticulin, which interacted with low-density lipoprotein receptors and subsequently activated the antigen-presenting cells for stimulation of the native immune response against the cancer tissue [94]. Studies carried out with both in vitro as well as in vivo models of breast cancer confirmed the effectiveness and specificity of this dual target-specific system. The repression of Treg in the tumor microenvironment is a key to reverse the immunosuppressive milieu, enabling efficient immunotherapy. The specific marker that distinguishes the Treg cells is the intracellular transcription factor FoxP3. In order to provide access to a FoxP3-inhibiting peptide (P60) to interact with FoxP3, it was conjugated to an aptamer that targeted CD28-expressing cytotoxic T cells [95]. This aptamer conjugate was found to potentiate immunotherapy in mouse models by effectively suppressing Treg action. This strategy can be explored in the future in combination with other therapeutic modalities for cancer treatment.
14 Microneedle-based immunotherapy
Recent advances in drug delivery have resulted in the development of microneedle-based systems that facilitate localized delivery of therapeutic cargo overcoming the transdermal barriers. In an innovative approach, degradable microneedle patch fabricated from hyaluronic acid and dextran nanoparticles encapsulated the immunoadjuvant programmed death-1 antibody (anti-PD-1) and glucose oxidase [96]. The enzyme converts glucose in blood to gluconic acid that reduces the pH, which leads to dissolution of the microneedle and release of the antibody. The anti-PD-1 activates the T-lymphocyte response against cancer cells. The microneedle patch was found to effectively treat melanoma in mouse models in a single application. The anti-cancer efficacy of the patch was further potentiated by co-delivery of two immunoadjuvants, namely, anti-PD-1 and anti-CTLA-4. This study opens up new vistas in exploring such systems for multi-modal therapy in combination with immunotherapy. The same group had also employed a dissolvable microneedle array comprising of hyaluronic acid modified with the indoleaminedioxygenase inhibitor 1-methyl-DL-tryptophan and containing nanocapsules with the immunoadjuvant anti-PD-1 [97]. The microneedle delivery of these two agents resulted in a synergistic anti-cancer effect on melanoma-induced mice models. On similar lines, two immunoadjuvants were co-delivered to melanoma cancer cells using a low-molecular weight poly(ethylene imine)-conjugated β-cyclodextrin carrier. The β-cyclodextrin possesses a hydrophobic interior and, hence, effectively retained the hydrophobic SB-505124, an inhibitor of the transforming growth factor-β (TGF-β), which is a key player in the development of the immunosuppressive tumor microenvironment [98]. The cationic poly(ethylene imine) was used to complex the anionic surface of the adenoviral vector that was used for transfecting the IL-2 gene to the target cells. Though adenoviral vectors can be independently employed for gene delivery, they cannot transfect cells that do not express the coxsackie-adenoviral vector (CAR), the major endocytotic route for these particles. Hence, they were complexed with poly(ethylene imine) to enable their transfection into melanoma cells. In vivo studies using melanoma models in mice revealed the ability of the nanoparticulate system to retard the tumor growth, increase IFN-γ levels, and activate natural killer and T lymphocytes apart from improving the survival rate of the animals.
As the field of nanotechnology continues, new classes of nanoparticles and modifications continue to emerge. Table 2 summarizes the different nanocarriers reported for immunotherapy.
Nanocarriers employed for immunotherapy [99].
| Nanocarrier | Immunomodulatory cargo | Cancer type | Salient features |
|---|---|---|---|
| Chitosan | IL-12 | Bladder cancer | − Induced cytokine expression − Prevented tumor recurrence |
| IL-12 plasmid | WEHI cancer cells | − Tumor regression | |
| Ovalbumin, alginate | In vitro | − Increased IFN-γ production | |
| Whole tumor lysate, mannose | Melanoma | − Tumor growth inhibition | |
| Liposome | Cisplatin, CpG | Melanoma | − Down regulation of Treg − Tumor regression − Prevention of recurrence |
| Hsp70 peptide complex | Breast cancer | − Improved immune response | |
| MUC1, TLR4 | Phase I, Phase II, and Phase III trials (lung cancer) | − Phase I: Well tolerated with no adverse effects − Phase II: Improved survival rate − Phase III: Improved survival rate only in combination with chemo- and radiotherapies | |
| HLA-B7 and β2 microglobulin DNA | Phase II and III (melanoma) | − Phase II: Improved therapeutic outcome − Phase III: Failure | |
| NY-ESO-1, MAGE-A3, tyrosinase, and TPTE RNA | Phase I clinical trials | − Ongoing trial | |
| Peptide mixture | Phase I clinical trials | − Successfully elicited T-cell response | |
| SOCS1, A20 si-RNA | Melanoma | − Increased cytokine production − Superior tumor regression | |
| E7 HPV | Lung cancer | − Treg suppression − Cytotoxic T-lymphocyte response | |
| OVA, TLR3/9 ligands | Mouse model of cancer | − Enhanced cytotoxic T-lymphocyte response | |
| PLGA | Paclitaxel, LPS | Melanoma | − Increased IL levels − Improved tumor regression |
| Whole tumor lysate | In vitro | − Higher IFN-γ levels − Reduced IL-10 levels − Higher T-cell proliferation | |
| WTL, CpG, poly I:C | TRAMP model | − Reduction in tumor volume − Increased cytotoxic T-lymphocyte response | |
| Ovalbumin, mannose | Mouse model of cancer | − Increased T-cell response | |
| Ovalbumin TLR3/7 ligands; CD40, CD11c, or DEC-205 ab | Mouse model of cancer | − Improved immune response | |
| TRP2180–188; TLR-4 ligand | Melanoma | − Improved tumor specific cytotoxic T-lymphocyte response | |
| Hgp10025–33 TRP2180–188 | Mouse model of cancer | − Improved tumor specific cytotoxic T-lymphocyte response | |
| Hyaluronic acid-PLGA + PLGA | Paclitaxel, CpG, IL-12 | Melanoma | − Improved tumor regression − Increased cytokine expression |
| PLGA-PEI | CpG, IL-10, si-RNA | Lymphoma | − Improved cytokine expression − Increased survival rate |
| mPEG-PLGA | Oxaliplatin | Pancreatic cancer | − Increased IL and IFN levels |
| PEI | IL-12 plasmid | Melanoma | − Increased IL permeation into tumor − Reduced tumor volume − Improved survival rate |
| γ-Poly(glutamic acid) | Ovalbumin | Mouse model of cancer | − TLR4-dependent immunogenicity |
| γ-Poly(glutamic acid)-poly(L-lysine) | Ovalbumin | Mouse model of cancer | − Improved immune response |
| Cholesteryl pullulan | HER2 fragment; NYESO-1 protein | Phase I trials | − Well tolerated − Antigen-specific immune responses elicited |
| Nanogel | Ovalbumin | Mouse model | − Increased MHC presentation |
| BSA-pyridine | Ovalbumin | In vitro | − Intrinsic immune stimulation property |
| Pyridyl disulfide | Palcitaxel, CpG | Melanoma | − Increased T-cell numbers − Reduced tumor growth |
| Poly(lactic acid) | IL-12, IL-18, TNF-α independently or in different combinations | Breast cancer | − Maximum tumor regression observed for the combination of IL-12 and TNF-α |
| Nanodiamond | CpG | Melanoma and breast cancer | − Tumor volume reduction − Increased IL-12 production |
| Silica | GM-CSF | In vitro | − Increased macrophage proliferation |
| Zinc oxide | Poly I:C | Melanoma | − Tumor growth retarded |
| Polysaccharide | Poly I:C | Melanoma | − Increased IL-6 levels |
| Poly(propylene sulfide) | CpG | Lymphoma and melanoma | − Increased cytokine levels − Prevention of tumor recurrence |
| Hydroxyethyl starch | IL-2 | Mouse model of cancer | − T cell-specific internalization |
| Nanolipogel | IL-2, TGF-β inhibitor | Melanoma | − Increased survival − Increased permeation of cytotoxic T lymphocytes into the tumor |
15 Multi-modal cancer therapy
Employing a combination of therapeutic strategies can be more effective than the use of a single approach. In addition, the use of such a multi-modal therapy can overcome the possibility of resistance that presently limits cancer therapy. The ability to co-deliver multiple therapeutic cargos to the target cancer cell using nanoparticles has stimulated interest in the development of unique therapeutic combinations for cancer treatment. Chemoimmunotherapy involves the use of a chemotherapeutic agent along with immunotherapy for the treatment of cancer. The tumor-suppressive and cytotoxic effects of immunotherapy drastically reduce the concentration of the chemotherapeutic agent for complete cancer cell kill. This in turn reduces the adverse effects associated with monotherapy using a chemotherapeutic agent. For instance, a fourth-generation polyamidoamine (PAMAM) dendrimer was linked with an oligonucleotide sequence containing unmethylated CpG sequences that served a dual purpose. They could not only activate the immune response but also complex with the cationic doxorubicin drug. In order to confer target specificity to the tumor microenvironment, the dendrimeric system was conjugated with an RNA aptamer with specific affinity to PSMA, a membrane protein over-expressed in prostate cancer cells [100]. The schematic representation of the system is shown in Figure 4.
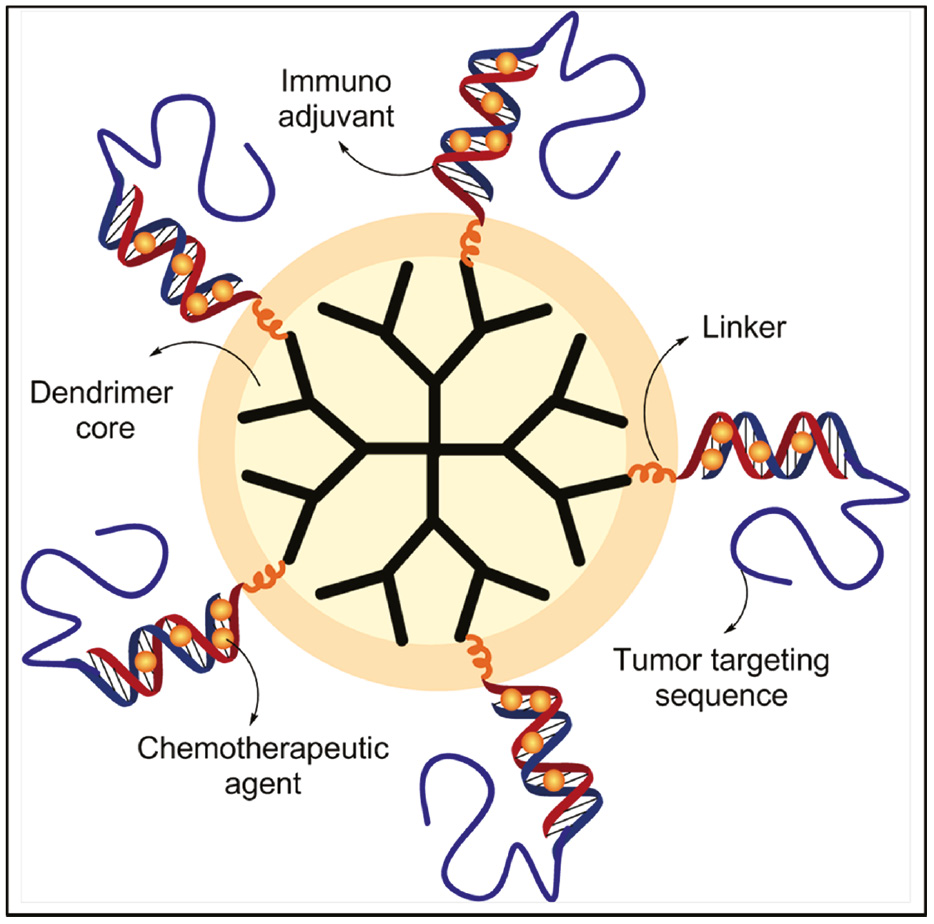
Schematic representation of a dendrimeric system for chemoimmunotherapy.
The targeted system exhibited superior cellular uptake that was manifested in higher cytotoxicity as well as higher expression levels of TNF-α, IL-1β, IL-6, and IL-12 both at the gene and protein levels. Interestingly, the aptamer and DNA oligonucleotide attached to the dendrimer displayed greater serum stability when compared with the free oligonucleotides. This was attributed to the change in conformation of the oligonucleotides on linking to the dendrimer that restricted the access of nucleases to the lytic bond. The in vivo efficacy of the targeted dendrimer hybrid was found to be superior to animals treated with monotherapy.
In another instance of chemoimmunotherapy, poly(L-histidine) was used to encapsulate Resiquimod (R848), an activator of TLR7 and TLR8 that promote the maturation of dendritic cell through activation of the myeloid differentiation factor-mediated pathway [101]. The chemotherapeutic agent doxorubicin was linked to hyaluronic acid with an acid-cleavable hydrazone bond and coated over the poly(L-histidine) nanoparticle. The poly(L-histidine) transforms into an ionized form at pH 6.8 encountered in the tumor microenvironment that releases R848, leading to the activation of the innate immune response characterized by increased expression of genes encoding for IFN-γ, IL-6, IL-12p40, and TNF-α. The hyaluronic acid mediated selective uptake in breast cancer cells through CD44 surface receptors. The acidic pH in the endosomes resulted in the lysis of the hydrazone bond, thereby releasing doxorubicin into the cells. In vivo studies confirmed the preferential accumulation of the core-shell nanoparticles in the tumor and the excellent tumor regression due to the combined effects of activated cytotoxic T lymphocytes and doxorubicin.
Gold nanorods conjugated with the TLR9 homing Y-shaped CpG oligonucleotides and the chemotherapeutic agent doxorubicin were employed for chemothermoimmunotherapy [102]. The gold nanorods were heated upon exposure to NIR radiation, which promoted the release of CpG and the intercalated doxorubicin. The immune activation was evidenced through elevated levels of TNF-α and IL-6. In vivo studies on mice induced with hepatocellular carcinoma revealed that the animals treated with CpG-Dox-gold nanorods and irradiated with NIR displayed the best anti-cancer effects due to the combined effects of hyperthermia, immune activation, and cytotoxicity induced by doxorubicin.
The photo-thermal effects of silica-coated gold nanorods were effectively synergized with immunotherapy to treat gastric cancer [103]. The system was used as a theranostic system where the tumors were simultaneously visualized through photoacoustic imaging. The photo-responsive nanoparticles were localized into CIK cells isolated from human volunteers. The killer cells homed into gastric cancer cells and destroyed them by generating a battery of cytokines such as IL-1, IL-2, IL-4, IL-12, IL-17, and IFN-γ, in combination with the photo-thermal effect on irradiation with NIR radiation. Photo-immunotherapy was accomplished through administration of Prussian blue nanoparticles that served as photosensitizers, which generated ROS on irradiation with NIR radiation in neuroblastoma-bearing mice. The intraperitoneal administration of anti-CTLA-4 antibodies in the animals primed the cytotoxic T cells toward the tumor tissues that were made more susceptible to cell death due to the photosensitizer-mediated oxidative stress. This study holds promise for the treatment of neuroblastoma, which is considered a challenge in treatment using conventional strategies.
A combination of the cytotoxic agent Nutlin 3a and the cytokine GM-CSF was encapsulated in pH-responsive sperimine-conjugated acetalated dextran nanoparticles [104]. The pH-responsive polymer enables endosomal escape leading to the release of the therapeutic cargo. Nutlin 3a interferes with p53-MDM2 interactions, thereby stabilizing it. This leads to activation of the apoptotic pathways and also reverses the immunosuppressive environment. Concurrently, the GM-CSF activates the innate immune system characterized by elevated levels of pro-inflammatory cytokines and accumulation of the cytotoxic T cells in the tumor tissue. This system displayed selective cytotoxicity toward breast cancer cells in a co-culture of immune cells and cancer cells. This strategy can be used only for p53 mutated cancer cells, but the approach can be extended to other cancer forms by employing a different cytotoxic drug. Another chemoimmunotherapy strategy involved the use of a pH tuned release of the cytotoxic drug paclitaxel and the cytokine IL-2 from chitosan-modified hydroxypropyl-β-cyclodextrin acrylate [105]. Apart from its chemotherapeutic effects, low doses of paclitaxel have also been found to activate the immune response and, hence, in combination with the pro-inflammatory IL-2 could lead to significant activation of the immune response against cancer. In order to realize co-delivery of these two entities in a single carrier without compromising their immunoadjuvant properties, the chitosan derivative was coated with erythrocyte membrane. Paclitaxel was encapsulated in the chitosan matrix, while IL-2 was loaded in the erythrocyte membrane layer. The erythrocyte membrane not only served to stabilize IL-2 but also promoted binding of the carrier to IL-2 receptors. The carrier displayed pH-responsive release of paclitaxel and IL-2, which reduced the immunosuppressive environment in B16-F10 tumor-induced mice. This was manifested through a reduction in Treg and an increase in the pro-inflammatory cytokines in the tumor. This resulted in the activation of cytotoxic T lymphocytes, which was confirmed with the increased expression of CD69. The reduction in tumor was attributed to the combined effects of immune activation as well as cytotoxicity mediated through paclitaxel.
A combination of photodynamic therapy and immunotherapy was realized through polymeric nanocarriers bearing a photosensitizer and an immunoadjuvant. Pheophorbide, a lipophilic photosensitizer, and ovalbumin were encapsulated in poly(ethylene imine) [63]. The use of the poly(ethylene imine) carrier improves cellular uptake and endosomal escape. The system, when administered in lymphoma-bearing mouse models, was found to display superior tumor regression properties when compared with the animals treated with free ovalbumin due to better internalization into the dendritic cells. Tumor regression was most prominent in animals treated with the polymeric carrier with the photosensitizer and immunoadjuvant on irradiation with electromagnetic radiation of wavelength 670 nm. This is due to the stimulation of the photosensitizer, leading to production of ROS in the cells that promotes endosomal escape, rapid release of the antigen from the polymeric matrix, maturation of the dendritic cell, and subsequent activation of the innate immune response against the tumor. Thus, this system may serve as an effective vaccine delivery system. Further studies on long-term toxicity and the ADME (absorption, distribution, metabolism, and excretion) characteristics of the polymeric carrier are warranted for the clinical translation of this approach. A similar strategy was employed using chlorinas as the photosensitizer moiety, which in combination with an IDO inhibitor was successfully demonstrated to reduce colorectal cancer [106]. The chlorin moiety was constructed to be part of a porous metal-organic framework. The inhibitor was entrapped in the pores. The system was highly effective in reducing tumor both at the primary and remote sites. The mechanism of cytotoxicity is initiated on irradiation of the metal-organic framework with electromagnetic radiation of 650 nm. This results in the generation of free radicals that mediate cell death. The debris of the dead cells are ingested by the immune cells and presented by MHC molecules, thereby activating the cytotoxic T lymphocytes. The release of the IDO inhibitor prevents the conversion of tryptophan to kyneurenin, thereby mitigating the immunosuppressive environment in the tumor tissue and making it susceptible to cytotoxic immune response.
Coordination complexes modified with phospholipids and cholesterol were loaded with the chemotherapeutic agent oxaliplatinin the core and the photosensitizer pyropheophorbide in the lipid shell were demonstrated to exhibit superior tumor cell kill by the synergistic action of the oxidative stress induced by the photosensitizer on irradiation and the cytotoxicity of the platinum-based chemotherapeutic agent [13]. Oxaliplatin was also found to elicit an immune response. This treatment induced an inflammatory environment in the tumor vicinity that activated the immune cells. Subsequent administration of anti-α-PD-1 potentiated the cytotoxic effects by also annihilating the secondary metastatic tumors in colorectal cancer models. This combination of photochemoimmunotherapy offers a better therapeutic option for cancer treatment.
Though several combination therapies involving immunotherapy have shown promise, in-depth studies on the possible adverse effects, fine-tuning of doses, and long-term effects on the immune performance need to be performed before the translation of this anti-cancer strategy [107]. The choice of the therapeutic agents being co-delivered needs to be made based on the effect on the molecular targets in cancer cells as well as in the immune cells. For instance, if an mammalian target of rapamycin (mTOR) inhibitor and a vaccine are co-delivered through a nanoparticle, the sequence of release becomes critical. If the vaccine is released first followed by the mTOR inhibitor, it favors cytotoxic T-cell activation and proliferation. However, if the sequence is reversed, then the therapy will lead to proliferation of Treg, which promotes immune suppression in the tumor microenvironment. Therefore, options of co-delivery of the therapeutic agents or the use of separate nano-carriers for mediating immunotherapy and other anti-cancer therapies need to be decided based on careful considerations of the pros and cons in each case. Nevertheless, nanoimmunotherapy independently or in combination represents a major advancement in combating cancer [108], [109].
16 Clinical aspects
Several attempts are being made to evaluate the therapeutic potential of immunotherapy against different forms of cancer. Monoclonal antibodies with specificity against molecules expressed on the surface of tumor cells comprise a major fraction of the immunotherapy interventions. Herceptin, also known as trastuzumab, is a Food and Drug Administration (FDA)-approved monoclonal antibody that targets the Her2 receptor in breast cancer. It up-regulates p21 and p27, both cell cycle inhibitors, and also activates immune response directed against Her2 over-expressing cancers [110], [111], [112]. Another widely used FDA-approved monoclonal antibody is Bevacizumab, commonly known as avastin, which inhibits angiogenesis by acting against VEGF-A. It is used to treat many types of epithelial cancers including lung and colorectal cancers [113], [114]. Rituximab marketed under the trade name Rituxan is an FDA-approved monoclonal antibody that has been used to treat B-cell malignancies [115], [116]. Table 3 lists the FDA-approved monoclonal antibodies that are available for the treatment of a wide range of malignancies. Bavituximab, a monoclonal antibody, is under clinical trials against different types of cancers such as lung cancer, glioblastoma, and liver cancer. This monoclonal antibody binds to phosphatidyl serine, which is aberrantly expressed in cancers and activates the immune response [118]. Immune modulators or checkpoint inhibitors have been used to regulate chemokine levels to trigger anti-cancer immune responses. These include CTLA-4 inhibitors like ipilimumab and tremelimumab, and PD-1 inhibitors such as atezolizumab, avelumab, and durvalumab [119], [120]. Several drug candidates targeting the protein targets such as CD137/4-1BB, CD27, GITR, and OX40 are under different phases of clinical trials [121], [122]. The adjuvant Bacillus Calmette-Guérin (BCG) has been approved for the treatment of bladder cancer, while another adjuvant, montanide, is currently in clinical trials for augmenting immune responses against cancer as part of adjuvant immunotherapy [123]. The cytokines IL-12 and IFN-α, which are key modulators of the immune response, are also being evaluated in clinical trials to assess their anti-cancer efficacy [124], [125]. A novel strategy employing oncolytic viruses that trigger the auto-destruction of cancer cells and activate the immune response against the tumor has also been included in the repertoire of immunotherapy arsenal. FDA has approved the use of Talimogene laherparepvec (T-VEC), marketed as Imlygic, for the treatment of metastatic melanoma [126]. Currently, several oncolytic viruses are under investigation for immunotherapy such as CG0070, Reolysin, CAVATAK, JX-594, and MV-NIS [127]. ACT therapy, wherein the patient’s T lymphocytes are removed, reprogrammed by treatment with chemicals to enhance their immune response against cancer cells, and reintroduced in the same patient, is also under active investigation. Tisagenlecleucel (Kymriah) and axicabtagene ciloleucel (Yescarta) have obtained regulatory approval for the treatment of lymphomas and leukemia and are currently in clinical trials to assess their therapeutic potential against other forms of solid tumors [128]. Cancer vaccines like Provenge and DCVax have obtained FDA approval for use against breast, brain, and pancreatic cancers, and several other potential candidates are in clinical trials [129]. Table 3 summarizes some of the major FDA-approved immunotherapy strategies and drug candidates that are under clinical trials.
| Treatment | FDA approved | Types of cancer | Currently in clinical trials |
|---|---|---|---|
| Monoclonal antibodies (mAbs) | Blincyto® Herceptin®, Perjeta®, Adcetris®, Rituxan®, Zevalin®, Avastin®, Erbitux®, Vectibix®, cyramza®, Gazyva®, Campath®, Rituxan®, Arzerra®, Kadcyla® | Breast, Hodgkin’s and non-Hodgkin’s lymphoma, lung, colorectal, chronic lymphocytic leukemia, gastric, kidney, brain, cervical, ovarian, acute lymphoblastic leukemia | Bavituximab, Ch14.18 (neuroblastoma), Rilotumumab |
| Checkpoint inhibitors | CTLA-4 inhibitors: tremelimumab, nivolumab, Ipilimumab PD-1/PD-L1 inhibitors: Atezolizumab, avelumab, durvalumab, nivolumab, pembrolizumab | Bladder, lung cancer, skin cancer, colorectal, kidney liver, melanoma, lymphoma | Drugs targeting CD137/4-1BB, CD27, GITR, and OX40 |
| Therapeutic cancer vaccines | Provenge®, NeuVax, DCVax, CDX-110, BioVaxID | Non-Hodgkin’s lymphoma, breast, brain, pancreatic, colorectal | – |
| Oncolytic virus immunotherapy | Talimogene laherparepvec, or T-VEC (Imlygic™) | Metastatic melanoma | CG0070, Reolysin, CAVATAK, JX-594, MV-NIS |
| ACT | Tisagenlecleucel (Kymriah®), axicabtagene ciloleucel (Yescarta®) | Non-Hodgkin lymphoma, B-cell acute lymphoblastic leukemia | Currently being tested in solid tumors and blood cancers |
| Cytokines | – | Melanoma, chronic myelogenous leukemia, Kaposi’s sarcoma, Follicular non-Hodgkin’s lymphoma | IL-2, IFN-α |
| Adjuvant immunotherapy | BCG | Bladder cancer | Montanide (currently in vaccine trials) |
17 Nanoparticle-assisted immunotherapies under clinical trials
Hafnium oxide containing nanoparticles referred to as NBTXR3 is currently being investigated in combination with the anti-PD-1 antibody nivolumab and radiotherapy for cancer therapy [130], [131]. Evaluation of the NBTXR3 administration on the cGAS-STING (cyclic GMP-AMP synthase-Stimulator of interferon genes) pathway, a key signaling cascade of the innate immune system activation, is underway. Table 4 lists clinical trials involving nanoparticles for immunotherapy.
Nanoparticle-mediated immunotherapies under clinical trials [132].
| Name | Nanoparticle | Antigen | Ref |
|---|---|---|---|
| Tecemotide (L-BLP25, stimuvax) | Liposome | MUC1 | [133], [134] |
| AS15 | Monophosphoryl lipid (also serves as an immunomodulator) | MAGE-A3 (melanoma antigen family A3), dHER2 | [135], [136] |
| Lipovaxin-MM | Liposome | Recombinant proteins from MM200 cells | [117], [132] |
| DepoVax (DPX) | Liposome | 7 HLA-A2 restricted TAAs or survivin peptides | [137] |
| RNA-LPX (RNA-lipoplexes) | Lipoplex | MAGEA3, tyrosinase, NY-ESO1, TPTE mRNA | [138] |
| OncoVAX – Id/IL-2 | Liposomes | Autologous idiotype protein | [132] |
| CHP (cholesteryl-bearing hydrophobized pullulan) | Cholesteryl pullulan ester | Recombinant MAGE-A4, truncated 146HER2, or NY-ESO-1 protein | [139] |
| ISCOMATRIX | Vesicle comprising saponin, cholesterol and dipalmitoyl phosphatidyl choline | Recombinant NY-ESO-1 protein, E6/E7 peptide | [140], [141] |
It is evident that the potential of nanoparticle-based immunotherapy is still to be extensively evaluated in clinical trials, but the encouraging pre-clinical trial results suggest that the number of nanoparticle-based immunotherapy clinical trials is bound to increase in the coming years.
18 Concluding remarks
Immunotherapy represents a remarkable advancement in cancer therapy that harnesses the immune system to treat cancer at the primary as well as metastatic sites apart from offering protection from tumor recurrence. However, this therapy is limited to treatment of only a few forms of cancer due to poor immune response. The use of nanoparticles for stimulating the tailored response of the immune components against cancer as well as for multi-modal therapy has opened up newer vistas of cancer therapy. The appropriate choice of the nanocarrier, size and lipophilicity, surface modifications, and therapeutic cargo are important for highly effective cancer therapy. A better understanding of the tumor microenvironment coupled with the rapid advances in nanotechnology promises an exciting future for conquering cancer through optimal use of the body’s defense system.
About the authors

Gayathri Kandasamy received her M.Tech. degree in Medical Nanotechnology SASTRA Deemed University, Thanjavur, India. She joined as JRF in SASTRA Deemed University in June 2016. Currently, she is working in the Centre for Nanotechnology and Advanced Biomaterials (CeNTAB), SASTRA University, Thanjavur, India. Her research focuses on the development of novel core-shell polymeric polyplexes with multi-modal action against lung cancer.

Vadim Annenkov is the Principal Researcher at the Limnological Institute, Russia. He has more than 20 years of experience in synthesis of water-soluble functionalized polymers. During the last decade, his scientific interests have enlarged to encompass composite organo-inorganic systems inspired from the process of biosilicification. Prof. Annenkov’s research group have designed a stepwise method for mimic silaffins. He is currently studying these systems for the delivery of oligonucleotides for biomedical applications.

Uma Maheswari Krishnan is an Associate Dean and Professor at the Centre for Nanotechnology and Advanced Biomaterials (CeNTAB) and School of Chemical and Biotechnology, SASTRA Deemed University, India. She received her PhD degree in Applied Chemistry from Bharathiar University, India, in 2000 and worked as a research associate at University of Texas, USA, until 2002 and had training on thin film processing and clean room processes at the University of Arkansas, USA. Since 2003, she is in SASTRA Deemed University. Her lab works on different facets of smart drug delivery systems, nanobiosensors, electrophysiology, Alzheimer’s disease, and nanomaterials.
Acknowledgments
The authors wish to thank the Department of Science and Technology (INT/RUS/RSF/P-10) and the Russian Science Federation (6-45-02001) for funding and the SASTRA Deemed University for infrastructural support.
References
[1] Couzin-Frankel J. The avenger. Sci. Mag. 2013, 90, 215–241.10.1126/science.353.6296.212Suche in Google Scholar PubMed
[2] Chan BA, Hughes BGM. Targeted therapy for non-small cell lung cancer: current standards and the promise of the future. Transl. Lung Cancer Res. 2015, 4, 36–54.Suche in Google Scholar
[3] Farkona S, Diamandis EP, Blasutig IM. Cancer immunotherapy: the beginning of the end of cancer. BMC Med. 2016, 14, 1–18.10.1186/s12916-016-0623-5Suche in Google Scholar PubMed PubMed Central
[4] Yuzhakova DV, Shirmanova MV, Sergeeva TF, Zagaynova EV, Lukyanov KA. Immunotherapy of cancer (Review). Sovrem. Tehnol. v Med. 2016, 8, 173–182.10.17691/stm2016.8.1.23Suche in Google Scholar
[5] Matsiko A. Cancer immunotherapy making headway. Nat. Mater. 2018, 17, 472.10.1038/s41563-018-0091-8Suche in Google Scholar PubMed
[6] Song W, Musetti SN, Huang L. Nanomaterials for cancer immunotherapy. Biomaterials 2017, 148, 16–30.10.1016/j.biomaterials.2017.09.017Suche in Google Scholar PubMed PubMed Central
[7] Morgan MM, Johnson BP, Livingston MK, Schuler LA, Alarid ET, Sung KE, Beebe DJ. Personalized in vitro cancer models to predict therapeutic response: challenges and a framework for improvement. Pharmacol. Ther. 2016, 165, 79–82.10.1016/j.pharmthera.2016.05.007Suche in Google Scholar PubMed PubMed Central
[8] Shelper TB, Lovitt CJ, Avery VM. Assessing drug efficacy in a miniaturized pancreatic cancer in vitro 3D cell culture model. Assay Drug Dev. Technol. 2016, 14, 367–380.10.1089/adt.2016.737Suche in Google Scholar PubMed
[9] Shao K, Singha S, Clemente-Casares X, Tsai S, Yang Y, Santamaria P. Nanoparticle-based immunotherapy for cancer. ACS Nano 2015, 9, 16–30.10.1021/nn5062029Suche in Google Scholar PubMed
[10] Irvine DJ, Hanson MC, Rakhra K, Tokatlian T. Synthetic Nanoparticles for vaccines and immunotherapy. Chem. Rev. 2015, 115, 11109–11146.10.1021/acs.chemrev.5b00109Suche in Google Scholar PubMed PubMed Central
[11] Conniot J, Silva JM, Fernandes JG, Silva LC, Gaspar R, Brocchini S, Florindo HF, Barata TS. Cancer immunotherapy: nanodelivery approaches for immune cell targeting and tracking. Front. Chem. 2014, 2, 1–27.10.3389/fchem.2014.00105Suche in Google Scholar PubMed PubMed Central
[12] Melero I, Gaudernack G, Gerritsen W, Huber C, Parmiani G, Scholl S, Thatcher N, Wagstaff J, Zielinski C, Faulkner I, Mellstedt H. Therapeutic vaccines for cancer: an overview of clinical trials. Nat. Rev. Clin. Oncol. 2014, 11, 509–524.10.1038/nrclinonc.2014.111Suche in Google Scholar PubMed
[13] He C, Duan X, Guo N, Chan C, Poon C, Weichselbaum RR, Lin W. Core-shell nanoscale coordination polymers combine chemotherapy and photodynamic therapy to potentiate checkpoint blockade cancer immunotherapy. Nat. Commun. 2016, 7, 12499.10.1038/ncomms12499Suche in Google Scholar PubMed PubMed Central
[14] Porta C, Sica A, Riboldi E. Tumor-associated myeloid cells: new understandings on their metabolic regulation and their influence in cancer immunotherapy. FEBS J. 2018, 285, 717–733.10.1111/febs.14288Suche in Google Scholar PubMed
[15] Fiori ME, Villanova L, De Maria R. Cancer stem cells: at the forefront of personalized medicine and immunotherapy. Curr. Opin. Pharmacol. 2017, 35, 1–11.10.1016/j.coph.2017.04.006Suche in Google Scholar PubMed
[16] Geresu MA, Sultan AF, Ahmed SK, Kassa GM. Immunotherapy against cancer: a comprehensive review. J. Cancer Res. Exp. Oncol. 2016, 8, 15–25.10.5897/JCREO2015.0124Suche in Google Scholar
[17] Zanganeh S, Spitler R, Hutter G, Ho JQ, Pauliah M, Mahmoudi M. Tumor-associated macrophages, nanomedicine and imaging: the axis of success in the future of cancer immunotherapy. Immunotherapy 2017, 9, 819–835.10.2217/imt-2017-0041Suche in Google Scholar PubMed
[18] Chen Q, Xu L, Chen J, Yang Z, Liang C, Yang Y, Liu Z. Tumor vasculature normalization by orally fed erlotinib to modulate the tumor microenvironment for enhanced cancer nanomedicine and immunotherapy. Biomaterials 2017, 148, 69–80.10.1016/j.biomaterials.2017.09.021Suche in Google Scholar PubMed
[19] Nagarsheth N, Wicha MS, Zou W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat. Rev. Immunol. 2017, 17, 559–572.10.1038/nri.2017.49Suche in Google Scholar PubMed PubMed Central
[20] Kazemi-Lomedasht F, Pooshang-Bagheri K, Habibi-Anbouhi M, Hajizadeh-Safar E, Shahbazzadeh D, Mirzahosseini H, Behdani M. In vivo immunotherapy of lung cancer using cross-species reactive vascular endothelial growth factor nanobodies. Iran. J. Basic Med. Sci. 2017, 20, 489–496.Suche in Google Scholar
[21] Azzi S, Hebda JK, Gavard J. Vascular permeability and drug delivery in cancers. Front. Oncol. 2013, 3, 1–14.10.3389/fonc.2013.00211Suche in Google Scholar PubMed PubMed Central
[22] Yang W-H, Xu J, Mu J-B, Xie J. Revision of the concept of anti-angiogenesis and its applications in tumor treatment. Chronic Dis. Transl. Med. 2017, 3, 33–40.10.1016/j.cdtm.2017.01.002Suche in Google Scholar PubMed PubMed Central
[23] Nagy JA, Chang SH, Dvorak AM, Dvorak HF. Why are tumour blood vessels abnormal and why is it important to know? Br. J. Cancer 2009, 100, 865–869.10.1038/sj.bjc.6604929Suche in Google Scholar
[24] Huang Y, Yuan J, Righi E, Kamoun WS, Ancukiewicz M, Nezivar J, Santosuosso M, Martin JD, Martin MR, Vianello F, Leblanc P, Munn LL, Huang P, Duda DG, Fukumura D, Jain RK, Poznansky MC. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc. Natl. Acad. Sci. 2012, 109, 17561–17566.10.1073/pnas.1215397109Suche in Google Scholar
[25] Park J, Kim I, Han S, Kim I. Normalization of tumor vessels by Tie2 activation and Ang2 inhibition enhances drug delivery and produces a favorable tumor microenvironment. Cancer Cell 2016, 30, 953–967.10.1016/j.ccell.2016.10.018Suche in Google Scholar
[26] Al-Abd AM, Alamoudi AJ, Abdel-Naim AB, Neamatallah TA, Ashour OM. Anti-angiogenic agents for the treatment of solid tumors: potential pathways, therapy and current strategies – a review. J. Adv. Res. 2017, 8, 591–605.10.1016/j.jare.2017.06.006Suche in Google Scholar
[27] Elice F, Rodeghiero F. Side effects of anti-angiogenic drugs. Thromb. Res. 2012, 129, S50–S53.10.1016/S0049-3848(12)70016-6Suche in Google Scholar
[28] Jain RK. Antiangiogenesis strategies revisited: from starving tumors to alleviating hypoxia. Cancer Cell 2014, 26, 605–622.10.1016/j.ccell.2014.10.006Suche in Google Scholar PubMed PubMed Central
[29] Campesato LF, Merghoub T. Antiangiogenic therapy and immune checkpoint blockade go hand in hand. Ann. Transl. Med. 2017, 5, 497.10.21037/atm.2017.10.12Suche in Google Scholar PubMed PubMed Central
[30] Lan G, Ni K, Xu Z, Veroneau SS, Song Y, Lin W. Nanoscale metal-organic framework overcomes hypoxia for photodynamic therapy primed cancer immunotherapy. J. Am. Chem. Soc. 2018, 140, 5670–5673.10.1021/jacs.8b01072Suche in Google Scholar PubMed PubMed Central
[31] McKenna DJ, Errington R, Pors K. Current challenges and opportunities in treating hypoxic prostate tumors. J. Cancer Metastasis Treat. 2018, 4, 1–12.10.20517/2394-4722.2017.54Suche in Google Scholar
[32] McDonald PC, Chafe SC, Dedhar S. Overcoming hypoxia-mediated tumor progression: combinatorial approaches targeting pH regulation, angiogenesis and immune dysfunction. Front. Cell Dev. Biol. 2016, 4, 1–16.10.3389/fcell.2016.00027Suche in Google Scholar PubMed PubMed Central
[33] Dubois LJ, Niemans R, Van Kuijk SJA, Panth KM, Parvathaneni NK, Peeters SGJA, Zegers CML, Rekers NH, Van Gisbergen MW, Biemans R, Lieuwes NG, Spiegelberg L, Yaromina A, Winum JY, Vooijs M, Lambin P. New ways to image and target tumour hypoxia and its molecular responses. Radiother. Oncol. 2015, 116, 352–357.10.1016/j.radonc.2015.08.022Suche in Google Scholar PubMed
[34] Li Y, Li F, Jiang F, Lv X, Zhang R, Lu A, Zhang G. A mini-review for cancer immunotherapy: molecular understanding of PD-1/PD-L1 pathway & translational blockade of immune checkpoints. Int. J. Mol. Sci. 2016, 17, Doi: 10.3390/ijms17071151.10.3390/ijms17071151Suche in Google Scholar PubMed PubMed Central
[35] Del Paggio JC. Immunotherapy: cancer immunotherapy and the value of cure. Nat. Rev. Clin. Oncol. 2018, 15, 268–270.10.1038/nrclinonc.2018.27Suche in Google Scholar PubMed
[36] Zhou Q, Zhang L, Wu H. Nanomaterials for cancer therapies. Nanotechnol. Rev. 2017, 6, 473–496.10.1515/ntrev-2016-0102Suche in Google Scholar
[37] Shakya AK, Nandakumar KS. Applications of polymeric adjuvants in studying autoimmune responses and vaccination against infectious diseases. J. R. Soc. Interface 2013, 10, 20120536.10.1098/rsif.2012.0536Suche in Google Scholar PubMed PubMed Central
[38] Fontana F, Liu D, Hirvonen J, Santos HA. Delivery of therapeutics with nanoparticles: what’s new in cancer immunotherapy? Wiley Interdiscip. Rev. Nanomedicine Nanobiotechnology 2017, 9, DOI: 10.1002/wnan.1421.10.1002/wnan.1421Suche in Google Scholar PubMed
[39] Jin W, Cui D. Cancer nano-immunoengineering: the marriage of immunoengineering and nanotechnology for cancer therapy. Nano Biomed. Eng. 2016, 8, 105–107.10.5101/nbe.v8i2.p105-107Suche in Google Scholar
[40] Sau S, Alsaab HO, Bhise K, Alzhrani R, Nabil G, Iyer AK. Multifunctional nanoparticles for cancer immunotherapy: a groundbreaking approach for reprogramming malfunctioned tumor environment. J. Control. Release 2018, 274, 24–34.10.1016/j.jconrel.2018.01.028Suche in Google Scholar PubMed PubMed Central
[41] Luo M, Samandi LZ, Wang Z, Chen ZJ, Gao J. Synthetic nanovaccines for immunotherapy. J. Control. Release 2017, 263, 200–210.10.1016/j.jconrel.2017.03.033Suche in Google Scholar PubMed PubMed Central
[42] Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat. Rev. Cancer 2012, 12, 265–277.10.1007/978-3-7091-1300-4_4Suche in Google Scholar
[43] Vang KB, Safina I, Darrigues E, Nedosekin D, Nima ZA, Majeed W, Watanabe F, Kannarpady G, Kore RA, Casciano D, Zharov VP, Griffin RJ, Dings RPM, Biris AS. Modifying Dendritic Cell Activation with Plasmonic Nano Vectors. Sci. Rep. 2017, 7, 5513.10.1038/s41598-017-04459-1Suche in Google Scholar PubMed PubMed Central
[44] Garg AD, Coulie PG, Van den Eynde BJ, Agostinis P. Integrating next-generation dendritic cell vaccines into the current cancer immunotherapy landscape. Trends Immunol. 2017, 38, 577–593.10.1016/j.it.2017.05.006Suche in Google Scholar PubMed
[45] Penafuerte C, Feldhammer M, Mills JR, Vinette V, Pike KA, Hall A, Migon E, Karsenty G, Pelletier J, Zogopoulos G, Tremblay ML. Downregulation of PTP1B and TC-PTP phosphatases potentiate dendritic cell-based immunotherapy through IL-12/IFNγ signaling. Oncoimmunology 2017, 6, 1–14.10.1080/2162402X.2017.1321185Suche in Google Scholar PubMed PubMed Central
[46] Neal LR, Bailey SR, Wyatt MM, Bowers JS, Majchrzak K, Nelson MH, Haupt C, Paulos CM, Varela JC. The basics of artificial antigen presenting cells in t cell-based cancer immunotherapies. J. Immunol. Res. Ther. 2017, 2, 68–79.Suche in Google Scholar
[47] Bethune MT, Joglekar AV. Personalized T cell-mediated cancer immunotherapy: progress and challenges. Curr. Opin. Biotechnol. 2017, 48, 142–152.10.1016/j.copbio.2017.03.024Suche in Google Scholar PubMed
[48] Wayteck L, Dewitte H, De Backer L, Breckpot K, Demeester J, De Smedt SC, Raemdonck K. Hitchhiking nanoparticles: reversible coupling of lipid-based nanoparticles to cytotoxic T lymphocytes. Biomaterials 2016, 77, 243–254.10.1016/j.biomaterials.2015.11.016Suche in Google Scholar PubMed
[49] Liu D, Tian S, Zhang K, Xiong W, Lubaki NM, Chen Z, Han W. Chimeric antigen receptor (CAR)-modified natural killer cell-based immunotherapy and immunological synapse formation in cancer and HIV. Protein Cell 2017, 8, 861–877.10.1007/s13238-017-0415-5Suche in Google Scholar PubMed PubMed Central
[50] Dugnani E, Pasquale V, Bordignon C, Canu A, Piemonti L, Monti P. Integrating T cell metabolism in cancer immunotherapy. Cancer Lett. 2017, 411, 12–18.10.1016/j.canlet.2017.09.039Suche in Google Scholar PubMed
[51] Gao X, Mi Y, Guo N, Xu H, Xu L, Gou X, Jin W. Cytokine-induced killer cells as pharmacological tools for cancer immunotherapy. Front. Immunol. 2017, 8, 774.10.3389/fimmu.2017.00774Suche in Google Scholar PubMed PubMed Central
[52] Chu D, Zhao Q, Yu J, Zhang F, Zhang H, Wang Z. Nanoparticle targeting of neutrophils for improved cancer immunotherapy. Adv. Healthc. Mater. 2016, 5, 1088–1093.10.1002/adhm.201500998Suche in Google Scholar PubMed PubMed Central
[53] Dreaden EC, Mwakwari SC, Austin LA, Kieffer MJ, Oyelere AK, El-Sayed MA. Small molecule-gold nanorod conjugates selectively target and induce macrophage cytotoxicity towards breast cancer cells. Small 2012, 8, 2819–2822.10.1002/smll.201200333Suche in Google Scholar PubMed PubMed Central
[54] Lyon JG, Mokarram N, Saxena T, Carroll SL, Bellamkonda RV. Engineering challenges for brain tumor immunotherapy. Adv. Drug Deliv. Rev. 2017, 114, 19–32.10.1016/j.addr.2017.06.006Suche in Google Scholar PubMed PubMed Central
[55] Lebel MÈ, Chartrand K, Tarrab E, Savard P, Leclerc D, Lamarre A. Potentiating cancer immunotherapy using papaya mosaic virus-derived nanoparticles. Nano Lett. 2016, 16, 1826–1832.10.1021/acs.nanolett.5b04877Suche in Google Scholar PubMed
[56] Deo VK, Kato T, Park EY. Virus-like particles displaying recombinant short-chain fragment region and interleukin 2 for targeting colon cancer tumors and attracting macrophages. J. Pharm. Sci. 2016, 105, 1614–1622.10.1016/j.xphs.2016.02.011Suche in Google Scholar PubMed
[57] Saga K, Kaneda Y. Virosome presents multimodel cancer therapy without viral replication. Biomed Res. Int. DOI: 10.1155/2013/764706.10.1155/2013/764706Suche in Google Scholar PubMed PubMed Central
[58] Syn NL, Wang L, Chow EKH, Lim CT, Goh BC. Exosomes in cancer nanomedicine and immunotherapy: prospects and challenges. Trends Biotechnol. 2017, 35, 665–676.10.1016/j.tibtech.2017.03.004Suche in Google Scholar PubMed
[59] Bell BM, Kirk ID, Hiltbrunner S, Gabrielsson S, Bultema JJ. Designer exosomes as next-generation cancer immunotherapy. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 163–169.10.1016/j.nano.2015.09.011Suche in Google Scholar PubMed
[60] Adriani G, Pavesi A, Tan AT, Bertoletti A, Thiery JP, Kamm RD. Microfluidic models for adoptive cell-mediated cancer immunotherapies. Drug Discov. Today 2016, 21, 1472–1478.10.1016/j.drudis.2016.05.006Suche in Google Scholar PubMed PubMed Central
[61] Boussommier-Calleja A, Li R, Chen MB, Wong SC, Kamm RD. Microfluidics: a new tool for modeling cancer-immune interactions. Trends in Cancer 2016, 2, 6–19.10.1016/j.trecan.2015.12.003Suche in Google Scholar PubMed PubMed Central
[62] Parlato S, De Ninno A, Molfetta R, Toschi E, Salerno D, Mencattini A, Romagnoli G, Fragale A, Roccazzello L, Buoncervello M, Canini I, Bentivegna E, Falchi M, Bertani FR, Gerardino A, Martinelli E, Natale C, Paolini R, Businaro L, Gabriele L. 3D microfluidic model for evaluating immunotherapy efficacy by tracking dendritic cell behaviour toward tumor cells. Sci. Rep. 2017, 7, 1–16.10.1038/s41598-017-01013-xSuche in Google Scholar PubMed PubMed Central
[63] Zhang C, Zhang J, Shi G, Song H, Shi S, Zhang X, Huang P, Wang Z, Wang W, Wang C, Kong D, Li C. A light responsive nanoparticle-based delivery system using pheophorbide a graft polyethylenimine for dendritic cell-based cancer immunotherapy. Mol. Pharm. 2017, 14, 1760–1770.10.1021/acs.molpharmaceut.7b00015Suche in Google Scholar PubMed
[64] Shi G-N, Zhang C-N, Xu R, Niu J-F, Song H-J, Zhang X-Y, Wang W-W, Wang Y-M, Li C, Wei X-Q, Kong D-L. Enhanced antitumor immunity by targeting dendritic cells with tumor cell lysate-loaded chitosan nanoparticles vaccine. Biomaterials 2017, 113, 191–202.10.1016/j.biomaterials.2016.10.047Suche in Google Scholar PubMed
[65] Han HD, Byeon Y, Jang JH, Jeon HN, Kim GH, Kim MG, Pack CG, Kang TH, Jung ID, Lim YT, Lee YJ, Lee JW, Shin BC, Ahn HJ, Sood AK, Park YM. In vivo stepwise immunomodulation using chitosan nanoparticles as a platform nanotechnology for cancer immunotherapy. Sci. Rep. 2016, 6, 38348.10.1038/srep38348Suche in Google Scholar PubMed PubMed Central
[66] Chen P-G, Huang Z-H, Sun Z-Y, Gao Y, Liu Y-F, Shi L, Chen Y-X, Zhao Y-F, Li Y-M. Chitosan nanoparticles based nanovaccines for cancer immunotherapy. Pure Appl. Chem. 2018, 89, 931–939.10.1515/pac-2016-0913Suche in Google Scholar
[67] Bafna S, Kaur S, Batra SK. Membrane-bound mucins: the mechanistic basis for alterations in the growth and survival of cancer cells. Oncogene 2010, 29, 2893–2904.10.1038/onc.2010.87Suche in Google Scholar PubMed PubMed Central
[68] Tan L, Han S, Ding S, Xiao W, Ding Y, Qian L, Wang C, Gong W. Chitosan nanoparticle-based delivery of fused NKG2D-IL-21 gene suppresses colon cancer growth in mice. Int. J. Nanomedicine 2017, 12, 3095–3107.10.2147/IJN.S128032Suche in Google Scholar PubMed PubMed Central
[69] Koshy ST, Cheung AS, Gu L, Graveline AR, Mooney DJ. Liposomal delivery enhances immune activation by STING agonists for cancer immunotherapy. Adv. Biosyst. 2017, 1, 1600013.10.1002/adbi.201600013Suche in Google Scholar PubMed PubMed Central
[70] Yuba E, Harada A, Sakanishi Y, Watarai S, Kono K. A liposome-based antigen delivery system using pH-sensitive fusogenic polymers for cancer immunotherapy. Biomaterials 2013, 34, 3042–3052.10.1016/j.biomaterials.2012.12.031Suche in Google Scholar PubMed
[71] Cao H, Dan Z, He X, Zhang Z, Yu H, Yin Q, Li Y. Liposomes coated with isolated macrophage membrane can target lung metastasis of breast cancer. ACS Nano 2016, 10, 7738–7748.10.1021/acsnano.6b03148Suche in Google Scholar PubMed
[72] Oberli MA, Reichmuth AM, Dorkin JR, Mitchell MJ, Fenton OS, Jaklenec A, Anderson DG, Langer R, Blankschtein D. Lipid nanoparticle assisted mRNA delivery for potent cancer immunotherapy. Nano Lett. 2017, 17, 1326–1335.10.1021/acs.nanolett.6b03329Suche in Google Scholar PubMed PubMed Central
[73] Verbeke R, Lentacker I, Wayteck L, Breckpot K, Van Bockstal M, Descamps B, Vanhove C, De Smedt SC, Dewitte H. Co-delivery of nucleoside-modified mRNA and TLR agonists for cancer immunotherapy: restoring the immunogenicity of immunosilent mRNA J. Control. Release 2017, 266, 287–300.10.1016/j.jconrel.2017.09.041Suche in Google Scholar PubMed
[74] Qian Y, Jin H, Qiao S, Dai Y, Huang C, Lu L, Luo Q, Zhang Z. Targeting dendritic cells in lymph node with an antigen peptide-based nanovaccine for cancer immunotherapy. Biomaterials 2016, 98, 171–183.10.1016/j.biomaterials.2016.05.008Suche in Google Scholar PubMed
[75] Kuai R, Ochyl LJ, Bahjat KS, Schwendeman A, Moon JJ. Designer vaccine nanodiscs for personalized cancer immunotherapy. Nat. Mater. 2017, 16, 489–496.10.1038/nmat4822Suche in Google Scholar PubMed PubMed Central
[76] Lin HY, Chin YT, Nana AW, Shih YJ, Lai HY, Tang HY, Leinung M, Mousa SA, Davis PJ. Actions of l-thyroxine and nano-diamino-tetrac (Nanotetrac) on PD-L1 in cancer cells. Steroids 2016, 114, 59–67.10.1016/j.steroids.2016.05.006Suche in Google Scholar PubMed
[77] Solbrig CM, Saucier-Sawyer JK, Cody V, Saltzman WM, Hanlon DJ. Polymer nanoparticles for immunotherapy from encapsulated tumor-associated antigens and whole tumor cells. Mol. Pharm. 2007, 4, 47–57.10.1021/mp060107eSuche in Google Scholar PubMed
[78] Garg SM, Vakili MR, Molavi O, Lavasanifar A. Self-associating poly(ethylene oxide)-block-poly(α-carboxyl-ε-caprolactone) drug conjugates for the delivery of STAT3 Inhibitor JSI-124: potential application in cancer immunotherapy. Mol. Pharm. 2017, 14, 2570–2584.10.1021/acs.molpharmaceut.6b01119Suche in Google Scholar PubMed
[79] Parveen S, Sahoo SK. Polymeric nanoparticles for cancer therapy. J. Drug Target. 2008, 16, 108–123.10.1080/10611860701794353Suche in Google Scholar PubMed
[80] Yang Y, Lu Y, Abbaraju PL, Zhang J, Zhang M, Xiang G, Yu C. Multi-shelled dendritic mesoporous organosilica hollow spheres: roles of composition and architecture in cancer immunotherapy. Angew. Chemie - Int. Ed. 2017, 56, 8446–8450.10.1002/anie.201701550Suche in Google Scholar PubMed
[81] Lu Y, Yang Y, Gu Z, Zhang J, Song H, Xiang G, Yu C. Glutathione-depletion mesoporous organosilica nanoparticles as a self-adjuvant and co-delivery platform for enhanced cancer immunotherapy. Biomaterials 2018, 175, 82–92.10.1016/j.biomaterials.2018.05.025Suche in Google Scholar PubMed
[82] Cha BG, Jeong JH, Kim J. Extra-large pore mesoporous silica nanoparticles enabling co-delivery of high amounts of protein antigen and toll-like receptor 9 agonist for enhanced cancer vaccine efficacy. ACS Cent. Sci. 2018, 4, 484–492.10.1021/acscentsci.8b00035Suche in Google Scholar PubMed PubMed Central
[83] Suarez-Kelly LP, Campbell AR, Rampersaud IV, Bumb A, Wang MS, Butchar JP, Tridandapani S, Yu L, Rampersaud AA, Carson WE. Fluorescent nanodiamonds engage innate immune effector cells: a potential vehicle for targeted anti-tumor immunotherapy. Nanomed. Nanotechnol. Biol. Med. 2017, 13, 909–920.10.1016/j.nano.2016.12.005Suche in Google Scholar PubMed PubMed Central
[84] Giner-Casares JJ, Henriksen-Lacey M, Coronado-Puchau M, Liz-Marzán LM. Inorganic nanoparticles for biomedicine: where materials scientists meet medical research. Mater. Today 2015, 19, 19–28.10.1016/j.mattod.2015.07.004Suche in Google Scholar
[85] Grigore ME. Organic and inorganic nano-systems used in cancer treatment. J. Med. Res. Heal. Educ. 2017, 1, 1–8.Suche in Google Scholar
[86] Cho NH, Cheong TC, Min JH, Wu JH, Lee SJ, Kim D, Yang JS, Kim S, Kim YK, Seong SY. A multifunctional core-shell nanoparticle for dendritic cell-based cancer immunotherapy. Nat. Nanotechnol. 2011, 6, 675–682.10.1038/nnano.2011.149Suche in Google Scholar PubMed
[87] Evans ER, Bugga P, Asthana V, Drezek R. Metallic nanoparticles for cancer immunotherapy. Mater. Today, DOI: 10.1016/j.mattod.2017.11.022.10.1016/j.mattod.2017.11.022Suche in Google Scholar PubMed PubMed Central
[88] Hassan HAFM, Smyth L, Wang JTW, Costa PM, Ratnasothy K, Diebold SS, Lombardi G, Al-Jamal KT. Dual stimulation of antigen presenting cells using carbon nanotube-based vaccine delivery system for cancer immunotherapy. Biomaterials 2016, 104, 310–322.10.1016/j.biomaterials.2016.07.005Suche in Google Scholar PubMed PubMed Central
[89] Zhu G, Mei L, Vishwasrao HD, Jacobson O, Wang Z, Liu Y, Yung BC, Fu X, Jin A, Niu G, Wang Q, Zhang F, Shroff H, Chen X. Intertwining DNA-RNA nanocapsules loaded with tumor neoantigens as synergistic nanovaccines for cancer immunotherapy. Nat. Commun. DOI: 10.1038/s41467-017-01386–7.10.1038/s41467-017-01386-7Suche in Google Scholar PubMed PubMed Central
[90] Settanni G, Zhou J, Suo T, Schöttler S, Landfester K, Schmid F, Mailänder V. DNA-inorganic hybrid nanovaccine for cancer immunotherapy. Nanoscale 2016, 8, 6684–6692.10.1039/C5NR08821FSuche in Google Scholar
[91] Shin JM, Oh SJ, Kwon S, Deepagan VG, Lee M, Song SH, Lee HJ, Kim S, Song KH, Kim TW, Park JH. A PEGylated hyaluronic acid conjugate for targeted cancer immunotherapy. J. Control. Release, 2017, 267, 181–190.10.1016/j.jconrel.2017.08.032Suche in Google Scholar PubMed
[92] Aliperta R, Welzel PB, Bergmann R, Freudenberg U, Berndt N, Feldmann A, Arndt C, Koristka S, Stanzione M, Cartellieri M, Ehninger A, Ehninger G, Werner C, Pietzsch J, Steinbach J, Bornhäuser M, Bachmann MP. Cryogel-supported stem cell factory for customized sustained release of bispecific antibodies for cancer immunotherapy. Sci. Rep. 2017, 7, 1–16.10.1038/srep42855Suche in Google Scholar PubMed PubMed Central
[93] Weiden J, Tel J, Figdor CG. Synthetic immune niches for cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 212–219.10.1038/nri.2017.89Suche in Google Scholar PubMed
[94] Yuan H, Jiang W, Von Roemeling CA, Qie Y, Liu X, Chen Y, Wang Y, Wharen RE, Yun K, Bu G, Knutson KL, Kim BYS. Multivalent bi-specific nanobioconjugate engager for targeted cancer immunotherapy. Nat. Nanotechnol. 2017, 12, 763–769.10.1038/nnano.2017.69Suche in Google Scholar PubMed
[95] Lozano T, Soldevilla MM, Casares N, Villanueva H, Bendandi M, Lasarte JJ, Pastor F. Targeting inhibition of Foxp3 by a CD28 2′-fluro oligonucleotide aptamer conjugated to P60-peptide enhances active cancer immunotherapy. Biomaterials 2016, 91, 73–80.10.1016/j.biomaterials.2016.03.007Suche in Google Scholar PubMed
[96] Wang C, Ye Y, Hochu GM, Sadeghifar H, Gu Z. Enhanced cancer immunotherapy by microneedle patch-assisted delivery of anti-PD1 antibody. Nano Lett. 2016, 16, 2334–2340.10.1021/acs.nanolett.5b05030Suche in Google Scholar PubMed
[97] Ye Y, Wang J, Hu Q, Hochu GM, Xin H, Wang C, Gu Z. Synergistic transcutaneous immunotherapy enhances antitumor immune responses through delivery of checkpoint inhibitors. ACS Nano 2016, 10, 8956–8963.10.1021/acsnano.6b04989Suche in Google Scholar PubMed
[98] Jiang J, Zhang Y, Peng K, Wang Q, Hong X, Li H, Fan G, Zhang Z, Gong T, Sun X. Combined delivery of a TGF-β inhibitor and an adenoviral vector expressing interleukin-12 potentiates cancer immunotherapy. Acta Biomater. 2017, 61, 114–123.10.1016/j.actbio.2017.05.009Suche in Google Scholar PubMed
[99] Graciotti M, Berti C, Klok HA, Kandalaft L. The era of bioengineering: how will this affect the next generation of cancer immunotherapy? J. Transl. Med. 2017, 15, 1–16.10.1186/s12967-017-1244-2Suche in Google Scholar PubMed PubMed Central
[100] Lee IH, An S, Yu MK, Kwon HK, Im SH, Jon S. Targeted chemoimmunotherapy using drug-loaded aptamer-dendrimer bioconjugates. J. Control. Release 2011, 155, 435–441.10.1016/j.jconrel.2011.05.025Suche in Google Scholar PubMed
[101] Liu Y, Qiao L, Zhang S, Wan G, Chen B, Zhou P, Zhang N, Wang Y. Dual pH-responsive multifunctional nanoparticles for targeted treatment of breast cancer by combining immunotherapy and chemotherapy. Acta Biomater. 2018, 66, 310–324.10.1016/j.actbio.2017.11.010Suche in Google Scholar PubMed
[102] Tao Y, Ju E, Liu Z, Dong K, Ren J, Qu X. Engineered, self-assembled near-infrared photothermal agents for combined tumor immunotherapy and chemo-photothermal therapy. Biomaterials 2014, 35, 6646–6656.10.1016/j.biomaterials.2014.04.073Suche in Google Scholar PubMed
[103] Yang Y, Zhang J, Xia F, Zhang C, Qian Q, Zhi X, Yue C, Sun R, Cheng S, Fang S, Jin W, Yang Y, Cui D. Human CIK cells loaded with au nanorods as a theranostic platform for targeted photoacoustic imaging and enhanced immunotherapy and photothermal therapy. Nanoscale Res. Lett. 2016, 11, 285.10.1186/s11671-016-1468-8Suche in Google Scholar PubMed PubMed Central
[104] Bauleth-Ramos T, Shahbazi MA, Liu D, Fontana F, Correia A, Figueiredo P, Zhang H, Martins JP, Hirvonen JT, Granja P, Sarmento B, Santos HA. Nutlin-3a and cytokine co-loaded spermine-modified acetalated dextran nanoparticles for cancer chemo-immunotherapy. Adv. Funct. Mater. 2017, 27, 1703303.10.1002/adfm.201703303Suche in Google Scholar
[105] Song Q, Yin Y, Shang L, Wu T, Zhang D, Kong M, Zhao Y, He Y, Tan S, Guo Y, Zhang Z. Tumor microenvironment responsive nanogel for the combinatorial antitumor effect of chemotherapy and immunotherapy. Nano Lett. 2017, 17, 6366–6375.10.1021/acs.nanolett.7b03186Suche in Google Scholar PubMed
[106] Lu K, He C, Guo N, Chan C, Ni K, Weichselbaum RR, Lin W. Chlorin-based nanoscale metal-organic framework systemically rejects colorectal cancers via synergistic photodynamic therapy and checkpoint blockade immunotherapy. J. Am. Chem. Soc. 2016, 138, 12502–12510.10.1021/jacs.6b06663Suche in Google Scholar PubMed PubMed Central
[107] Gotwals P, Cameron S, Cipolletta D, Cremasco V, Crystal A, Hewes B, Mueller B, Quaratino S, Sabatos-Peyton C, Petruzzelli L, Engelman JA, Dranoff G. Prospects for combining targeted and conventional cancer therapy with immunotherapy. Nat. Rev. Cancer 2017, 17, 286–301.10.1038/nrc.2017.17Suche in Google Scholar PubMed
[108] Qian H, Liu B, Jiang X. Application of nanomaterials in cancer immunotherapy. Mater. Today Chem. 2018, 7, 53–64.10.1016/j.mtchem.2018.01.001Suche in Google Scholar
[109] Hu C-MJ, Aryal S, Zhang L. Nanoparticle-assisted combination therapies for effective cancer treatment. Ther. Deliv. 2010, 1, 323–334.10.4155/tde.10.13Suche in Google Scholar PubMed
[110] Vu T, Sliwkowski MX, Claret FX. Personalized drug combinations to overcome trastuzumab resistance in HER2-positive breast cancer. Biochim. Biophys. Acta - Rev. Cancer 2014, 1846, 353–365.10.1016/j.bbcan.2014.07.007Suche in Google Scholar PubMed PubMed Central
[111] Mayer IA. Treatment of HER2-positive metastatic breast cancer following initial progression. Clin. Breast Cancer 2012, 9, 1–16.10.3816/CBC.2009.s.005Suche in Google Scholar PubMed PubMed Central
[112] Damodaran S, Olson EM. Targeting the human epidermal growth factor receptor 2 pathway in breast cancer. Hosp. Pract. 2013, 40, 7–15.10.3810/hp.2012.10.997Suche in Google Scholar PubMed PubMed Central
[113] Morihira K, Hara R, Kawahara S, Nishimori T, Nakamura N, Kusama H, Kuwajima I. Enantioselective total synthesis of taxol. J. Am. Chem. Soc. 1998, 120, 12980–12981.10.1021/ja9824932Suche in Google Scholar
[114] Jackson MW, Rusthoven CG, Fisher CM, Schefter TE. Clinical potential of bevacizumab in the treatment of metastatic and locally advanced cervical cancer: current evidence. Onco. Targets. Ther. 2014, 7, 751–759.10.2147/OTT.S49429Suche in Google Scholar PubMed PubMed Central
[115] Dotan E, Aggarwal C, Smith MR. Impact of rituximab (Rituxan) on the treatment of B-cell non-Hodgkin’s lymphoma. P T 2010, 35, 148–157.Suche in Google Scholar
[116] Salles G, Barrett M, Foà R, Maurer J, O’Brien S, Valente N, Wenger M, Maloney DG. Rituximab in B-cell hematologic malignancies: a review of 20 years of clinical experience. Adv. Ther. 2017, 34, 2232–2273.10.1007/s12325-017-0612-xSuche in Google Scholar PubMed PubMed Central
[117] Shi J, Kantoff PW, Wooster R, Farokhzad OC. Cancer nanomedicine: progress, challenges and opportunities. Nat. Rev. Cancer 2017, 17, 20–37.10.1038/nrc.2016.108Suche in Google Scholar PubMed PubMed Central
[118] Chalasani P, Marron M, Roe D, Clarke K, Iannone M, Livingston RB, Shan JS, Stopeck AT. A phase I clinical trial of bavituximab and paclitaxel in patients with HER2 negative metastatic breast cancer. Cancer Med. 2015, 4, 1051–1059.10.1002/cam4.447Suche in Google Scholar PubMed PubMed Central
[119] Elsegood CL, Tirnitz-Parker JE, Olynyk JK, Yeoh GC. Immune checkpoint inhibition: prospects for prevention and therapy of hepatocellular carcinoma. Clin. Transl. Immunol. 2017, 6, e161.10.1038/cti.2017.47Suche in Google Scholar PubMed PubMed Central
[120] Davies M, Duffield EA. Safety of checkpoint inhibitors for cancer treatment: strategies for patient monitoring and management of immune-mediated adverse events. ImmunoTargets Ther. 2017, 6, 51–71.10.2147/ITT.S141577Suche in Google Scholar PubMed PubMed Central
[121] Sturgill ER, Redmond WL. TNFR agonists: a review of current biologics targeting OX40, 4-1BB, CD27, and GITR. Am. J. Hematol. Oncol. 2017, 11, 4–15.Suche in Google Scholar
[122] Mayes PA, Hance KW, Hoos A. The promise and challenges of immune agonist antibody development in cancer. Nat. Rev. Drug Discov. 2018, 17, 509–527.10.1038/nrd.2018.75Suche in Google Scholar PubMed
[123] Novotny V, Froehner M, Ollig J, Koch R, Zastrow S, Wirth MP. Impact of adjuvant intravesical bacillus Calmette-Guérin treatment on patients with high-grade T1 bladder cancer. Urol. Int. 2016, 96, 136–141.10.1159/000443705Suche in Google Scholar PubMed
[124] Floros T, Tarhini AA. Anticancer cytokines: biology and clinical effects of interferon-α2, interleukin (IL)-2, IL-15, IL-21, and IL-12. Semin. Oncol. 2015, 42, 539–548.10.1053/j.seminoncol.2015.05.015Suche in Google Scholar PubMed PubMed Central
[125] Chin KL, Anis FZ, Sarmiento ME, Norazmi MN, Acosta A. Role of interferons in the development of diagnostics, vaccines, and therapy for tuberculosis. J. Immunol. Res. DOI: 10.1155/2017/5212910.10.1155/2017/5212910Suche in Google Scholar PubMed PubMed Central
[126] Bommareddy PK, Patel A, Hossain S, Kaufman HL. Talimogene Laherparepvec (T-VEC) and other oncolytic viruses for the treatment of melanoma. Am. J. Clin. Dermatol. DOI: 10.1007/s40257-016-0238–9.10.1007/s40257-016-0238–9Suche in Google Scholar
[127] Pol J, Buqu A, Saut C, Galluzzi L. Trial Watch – Oncolytic viruses and cancer therapy. Oncoimmunology 2016, 5, 91–97.10.1080/2162402X.2015.1117740Suche in Google Scholar PubMed PubMed Central
[128] Zheng PP, Kros JM, Li J. Approved CAR T cell therapies: ice bucket challenges on glaring safety risks and long-term impacts. Drug Discov. Today 2018, 23, 1175–1182.10.1016/j.drudis.2018.02.012Suche in Google Scholar PubMed
[129] Kudrin A. Cancer vaccines: what do we need to measure in clinical trials? Hum. Vaccines Immunother. 2014, 10, 3236–3240.10.4161/hv.27586Suche in Google Scholar PubMed PubMed Central
[130] Cushman TR, Gomez D, Kumar R, Likacheva A, Chang JY, Cadena AP, Paris S, Welsh JW. Combining radiation plus immunotherapy to improve systemic immune response. J. Thorac. Dis. 2018, 10, S468–S479.10.21037/jtd.2018.01.130Suche in Google Scholar PubMed PubMed Central
[131] Bonvalot S, Le Pechoux C, De Baere T, Kantor G, Buy X, Stoeckle E, Terrier P, Sargos P, Coindre JM, Lassau N, Sarkouh RA, Dimitriu M, Borghi E, Levy L, Deutsch E, Soria JC. First-in-human study testing a new radioenhancer using nanoparticles (NBTXR3) activated by radiation therapy in patients with locally advanced soft tissue sarcomas. Clin. Cancer Res. 2017, 23, 908–917.10.1158/1078-0432.CCR-16-1297Suche in Google Scholar PubMed
[132] Grippin AJ, Sayour EJ, Mitchell DA. Translational nanoparticle engineering for cancer vaccines. Oncoimmunology 2017, 6, e1290036.10.1080/2162402X.2017.1290036Suche in Google Scholar PubMed PubMed Central
[133] Chesson CB, Zloza A. Nanoparticles: augmenting tumor antigen presentation for vaccine and immunotherapy treatments of cancer. Nanomedicine 2017, 12, 2693–2706.10.2217/nnm-2017-0254Suche in Google Scholar PubMed PubMed Central
[134] Wurz GT, Kao C, Wolf M, Degregorio MW. Tecemotide: an antigen-specific cancer immunotherapy. Hum. Vaccin. Immunother. 2014, 10, 3383–3393.10.4161/hv.29836Suche in Google Scholar PubMed PubMed Central
[135] Brichard VG, Godechal Q. MAGE-A3-specific anticancer immunotherapy in the clinical practice. Oncoimmunology 2013, 2, e25995.10.4161/onci.25995Suche in Google Scholar PubMed PubMed Central
[136] Gérard C, Baudson N, Ory T, Louahed J. Tumor mouse model confirms MAGE-A3 cancer immunotherapeutic as an efficient inducer of long-lasting anti-tumoral responses. PLoS One 2014, DOI: 10.1371/journal.pone.0094883.10.1371/journal.pone.0094883Suche in Google Scholar PubMed PubMed Central
[137] Karkada M, Quinton T, Blackman R, Mansour M. Tumor inhibition by DepoVax-based cancer vaccine is accompanied by reduced regulatory/suppressor cell proliferation and tumor infiltration. ISRN Oncol. 2013, 2013, 753427.10.1155/2013/753427Suche in Google Scholar PubMed PubMed Central
[138] Gilboa E. A quantum leap in cancer vaccines? J. Immunother. Cancer 2016, 4, 3–4.10.1186/s40425-016-0192-3Suche in Google Scholar PubMed PubMed Central
[139] Miyauchi K, Tsuchikawa T, Wada M, Abiko T, Kyogoku N, Shichinohe T, Miyahara Y, Kageyama S, Ikeda H, Shiku H, Hirano S. Clinical relevance of antigen spreading pattern induced by CHP-MAGE-A4 cancer vaccination. Immunotherapy 2016, 8, 527–540.10.2217/imt-2016-0007Suche in Google Scholar PubMed
[140] Morelli AB, Becher D, Koernig S, Silva A, Drane D, Maraskovsky E. ISCOMATRIX: a novel adjuvant for use in prophylactic and therapeutic vaccines against infectious diseases. J. Med. Microbiol. 2012, 61, 935–943.10.1099/jmm.0.040857-0Suche in Google Scholar PubMed
[141] Wilson NS, Yang B, Morelli AB, Koernig S, Yang A, Loeser S, Airey D, Provan L, Hass P, Braley H, Couto S, Drane D, Boyle J, Belz GT, Ashkenazi A, Maraskovsky E. ISCOMATRIX vaccines mediate CD8+ T-cell cross-priming by a MyD88-dependent signaling pathway. Immunol. Cell Biol. 2012, 90, 540–552.10.1038/icb.2011.71Suche in Google Scholar PubMed PubMed Central
©2018 Walter de Gruyter GmbH, Berlin/Boston
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Artikel in diesem Heft
- Frontmatter
- In this issue
- Regular articles
- Design of an integrated optics for transglutaminase conformational change
- Carboxymethyl cellulose-grafted graphene oxide for efficient antitumor drug delivery
- Reviews
- Copper and copper nanoparticles: role in management of insect-pests and pathogenic microbes
- Nanoimmunotherapy – cloaked defenders to breach the cancer fortress
- Nanotechnology for the oil and gas industry – an overview of recent progress
Artikel in diesem Heft
- Frontmatter
- In this issue
- Regular articles
- Design of an integrated optics for transglutaminase conformational change
- Carboxymethyl cellulose-grafted graphene oxide for efficient antitumor drug delivery
- Reviews
- Copper and copper nanoparticles: role in management of insect-pests and pathogenic microbes
- Nanoimmunotherapy – cloaked defenders to breach the cancer fortress
- Nanotechnology for the oil and gas industry – an overview of recent progress