Recent developments in the use of nanoparticles for treatment of biofilms
-


Abstract
Chronic infections have posed a tremendous burden on health care systems worldwide. Approximately 60% of chronic infections are estimated to be related to biofilms, in large part due to the extraordinary antibiotic resistance of biofilm bacteria. Nanoparticle (NP)-based therapies are viable approaches to treat biofilm-associated infections due to NPs’ unique chemical and physical properties, granted by their high surface area to volume ratio. The mechanism underlying the anti-biofilm activity of various types of NPs is actively under investigation. Simply comparing biofilm disruption or reduction rates is not adequate to describe the effectiveness of NPs; many other factors need to be taken into account, such as the NP type, bacterial strain, concentration of NPs, quantification methods, and the biofilm culture environment. This review focuses on recent research on the creation, characterization, and evaluation of NPs for the prevention or treatment of biofilm infections.
1 Introduction
Over 60% of infections are estimated to be related to biofilms. Biofilms are commonly defined as communities of surface associated bacteria encased in a self-produced, hydrated polymeric matrix attached to host tissue or an abiotic surface [1], [2]. The formation of biofilms proceeds once free-floating planktonic bacteria adhere onto surfaces. Medical devices commonly infected by biofilms include intravenous catheters, vascular prosthesis, prosthetic heart valves, urinary catheters, joint prostheses, cardiac pacemakers, and contact lenses. Biofilms also occur in dental caries, periodontitis, cystic fibrosis (CF), native valve endocarditis, chronic otitis media, and chronic wound infections [3], [4], [5], [6], [7], [8], [9], [10], [11]. Traditional approaches to combating biofilms usually involve a combination of conventional antibiotics with varied killing mechanisms. Conventional antibiotics fall into two categories, bacteriostatic or bactericidal. The former one prevents bacteria from dividing, and the latter kills bacteria. However, the actual therapeutic effect of antibiotics is highly related to their concentration. Antibiotics need to penetrate the bacterial cell membrane and accumulate until a sufficient therapeutic concentration is reached. This process can be dependent on bacterial structure, and bacterial cells are categorized as Gram-negative or Gram-positive based on their structure. Gram-negative cell walls have a thin peptidoglycan layer between the outer membrane and cytoplasmic membrane, while Gram-positive cell walls are composed of a thick layer of peptidoglycan and cytoplasmic membrane [12].
Under most circumstances, conventional antibiotics fail to remove biofilms completely because of the unique structure and properties of biofilms. Biofilm bacteria can be up to 1000-fold more tolerant to antibiotics compared to their free-floating (planktonic) counterparts, which can be removed by gentle rinsing [4]. This difference in antibiotic tolerance explains why patients with biofilm infections tend to suffer from chronic complications [1], [2], [13]. If a foreign body biomedical device is present at the site of infection, removal and replacement of that device through surgery are necessary, causing undesired pain in patients and adding considerable cost to the treatment. The process becomes more medically challenging when biofilms are present on complex implants such as artificial heart valves, catheters, and artificial prosthetics [14].
In the United States, there were roughly $42 billion in sales of antibiotics in 2009, which comprises 15–30% of drug expenditures among all therapeutic groups of drugs [15], [16], and the global consumption of antibiotics has increased by 36% from 2000 to 2010 [17]. The systemic administration of high-dose antibiotics often cannot achieve an adequate drug concentration at the biofilm-infected sites; however, increasing the dosage leads to adverse effects and potentially contributes to drug resistance of the bacteria. If sub-minimum inhibitory doses of antibiotics are administered, even more resistance is observed [18].
The failure of existing strategies to treat biofilm-associated infections necessitates the development of an improved drug delivery system and alternative strategies to overcome the limitations of conventional antibiotics including short half-life, low bioavailability, and systemic toxicity. Among all the drug delivery systems of interest, nanoparticles (NPs) have garnered significant attention from researchers. Since the success of the first liposomal pharmaceutical product, Doxil®, approved by the Food and Drug Administration (FDA) in 1995 [19], NPs have been studied extensively as carriers for drug delivery, imaging contrast agents for diagnosis, and pharmaceutical agents for therapy. The effectiveness of the NP formulation is determined by several parameters, such as physical particle properties, drug loading, drug release kinetics in vivo, circulation longevity, and the intrinsic toxicity of the carrier itself. Based on the intrinsic properties of the encapsulated drug, its desired action, and the method of administration, different particles designs rely on materials such as synthetic or natural polymers, metals, and lipids.
NPs allow for drug delivery with pre-determined kinetics [20], an often critical parameter in maintaining optimal dosing within the therapeutic window at the site of interest. For example, due to the concentration-dependent efficacy of aminoglycosides such as gentamicin, a single-burst release of the drug from the NPs is effective in killing all bacteria in a short period of time. Conversely, sustained release of beta-lactam is necessary because, in addition to concentration, the antibacterial efficacy of beta-lactam is dependent on prolonged exposure to the drug at a constant dose [21]. Depending on the intended kinetics and the chemistry of the design, therapeutic agents can either be encapsulated into or be attached onto NP surfaces.
The advantages of NPs are also dependent on the route of administration, with the potential for improved bioavailability and/or targetability of the drug compared to conventional therapeutics [22]. Potential routes of administration for NPs include local administration (e.g. aerosol, local injection, and wound dressing) and systemic administration (oral, intravenous, intraperitoneal, etc.). For example, in CF patients, NPs have been utilized to transport encapsulated antibiotics through the mucus barrier in the lungs to deposit therapeutic agents, which is a significant improvement over free antibiotics that are exhaled out with limited efficacy [23]. Due to the wealth of knowledge in the use of NPs for treatment of cancer, many biofilm-directed NP therapies have been transferred over from that field. Indeed, considering the hallmarks shared by cancer and infection-induced inflammation, such as increased vascular permeability [24], [25], it is convenient and practical to test the feasibility of the NP carriers designed for long circulation time and passive targeting in cancer treatments for efficacy in biofilm-associated infections. However, one cannot ignore the fact that biofilm infections and cancers are fundamentally different and the unique characteristics of biofilms need to be taken into consideration during the course of NP design, such as the decreased pH at the infection site [26]. With this in mind, here, we review the various NP strategies to treating biofilm infections within the preceding decade (Table 1).
Summary of the in vitro biofilm treatment results of the papers discussed.
| Biofilm type | Nanoparticles type | Active agent | Size (nm) | Zeta potential (mV) | Biofilm inhibition | Biofilm disruption | Biofilm bacteria viability | Reference |
|---|---|---|---|---|---|---|---|---|
| Streptococcus mutans | Chitosan | Erythrosine | 80.9±7.43 | 66.5±4.4 | – | – | 0% | [27] |
| Chitosan (low MW) | ~20 | – | – | – | 5% | [28] | ||
| Chitosan | DspB | 100–500 | – | >90% | – | – | [29] | |
| Pseudomonas aeruginosa | Chitosan | Erythrosine | 80.9±7.43 | 66.5±4.4 | – | – | 78% | [27] |
| Citrate-capped silver | Aztreonam | 10 | – | – | 98% | ~0% | [18] | |
| Chitosan coated silver | 55–278 | 51.1 | 65% | 0% | – | [30] | ||
| Silver | 20–30 | – | 67% | – | – | [31] | ||
| Silver made from Allophylus cobbe | Ampicillin | 5±4 | – | 69% | – | – | [32] | |
| Silver made from Allophylus cobbe | Vancomycin | 5±4 | – | 54% | – | – | [32] | |
| Silica | NO | 90±10 | – | – | – | 0.001% | [33] | |
| Liposome | 128.6±2.3 | 58.6±1.3 | 75% | – | – | [34] | ||
| PLGA nanoparticles coated with PL and DNase I | Ciprofloxacin | 251.9 | 28.9±1.43 | 100% | 95.00% | – | [35] | |
| PLGA | Ciprofloxacin and magnetic nanoparticles | 220.9±7.4 | – | – | – | 67% | [36] | |
| PLGA | Gentamicin | 241.3±12.4 | 0.7±0.2 | – | – | 3% | [37] | |
| PLGA, chitosan | Colistin (cationic antimicrobial peptides) | 300, become 6.4–8.3 μm after lactose coating | 12.4 | – | 50% | – | [23] | |
| PLGA, PVA | colistin (cationic antimicrobial peptides) | 300, become 6.4–8.3 μm after lactose coating | −7.14 | – | 50% | – | [23] | |
| PLGA, phosphatidylcholine | Levofloxacin | 240±50 | −26 | – | – | 5–10% | [38] | |
| Candida albicans | Chitosan | Erythrosine | 80.9±7.43 | 66.5±4.4 | – | – | 46% | [27] |
| Gold | Methylene blue | 26 | – | 82.20% | – | 4.60% | [39] | |
| Silica | NO | 90±10 | – | – | 0.10% | [33] | ||
| Actinobacillus actinomycet emcomitans | Chitosan | DspB | 100–500 | – | >90% | – | – | [29] |
| Staphylococcus epidermidis | Chitosan | DspB | 100–500 | – | >90% | – | – | [29] |
| Silica | NO | 90±10 | – | – | – | 1% | [33] | |
| CES-SPOION, with magnet | 13.8±2.1 | −15.4±0.5 | – | – | 62% | [40] | ||
| CES-SPOION, without magnet | 13.8±2.1 | −15.4±0.5 | – | – | 92% | [40] | ||
| PLL | 17 nm silver nanoparticles | 225.5 | −1.65 | 98% | – | – | [41] | |
| Enterococcus faecalis | Chitosan | Rose bengal | 60±20 | – | – | – | Decreased | [42] |
| Staphylococcus aureus | FeCl3-conjugated SPION | 23.32±1.17 | −34 | ~50% | – | – | [43] | |
| AgNO3-conjugated SPION | 193.72±6.15 | −40.1 | ~50% | – | – | [43] | ||
| ZnCl2-conjugated SPION | 19.67±0.72 | −43.2 | ~80% | – | – | [43] | ||
| FeCl3-conjugated SPION | Fructose | 27.69±1.20 | −34.0±3.40 | – | – | 0.00001% | [44] | |
| ZnCl2-conjugated SPION | Fructose | 19.67±0.72 | 40.1±0.40 | – | – | 0.00001% | [44] | |
| Chitosan coated silver | 55–278 | 51.1 | 22% or 65% at higher concentration | 0% | – | [30] | ||
| Silver made from Allophylus cobbe | Ampicillin | 5±4 | – | 49% | – | – | [32] | |
| Silver made from Allophylus cobbe | Vancomycin | 5±4 | – | 73% | – | – | [32] | |
| Silver made from Plumbago zeylanica | 63 | −31 | 99% or 100% | 78% | – | [45] | ||
| Silver-gold made from Plumbago zeylanica | 93 | −21 | 99% | 77% | – | [45] | ||
| Gold made from Plumbago zeylanica | Aggregated into gold triangles | – | 97% | 95% | – | [45] | ||
| Silica | NO | 90±10 | – | – | – | 1% | [33] | |
| Liposome | 128.6±2.3 | 58.6±1.3 | 43% | – | – | [34] | ||
| Streptococcus pneumoniae | Silver made from Allophylus cobbe | Ampicillin | 5±4 | – | 53% | – | – | [32] |
| Silver made from Allophylus cobbe | Vancomycin | 5±4 | – | 70% | – | – | [32] | |
| Shigella flexneri | Silver made from Allophylus cobbe | Ampicillin | 5±4 | – | 64% | – | – | [32] |
| Silver made from Allophylus cobbe | Vancomycin | 5±4 | – | 50% | – | – | [32] | |
| Acinetobacter baumannii | Silver made from Plumbago zeylanica | 63 | 98% | 88% | – | [45] | ||
| Silver-gold made from Plumbago zeylanica | 93 | 94% | 65% | – | [45] | |||
| Gold made from Plumbago zeylanica | Aggregated into gold triangles | 93% | 40% | – | [45] | |||
| Escherichia coli | Silver made from Plumbago zeylanica | 63 | 99% | 67% | – | [45] | ||
| Silver-gold made from Plumbago zeylanica | 93 | 98% | 61% | – | [45] | |||
| Gold made from Plumbago zeylanica | Aggregated into gold triangles | 93% | 60% | – | [45] | |||
| Silver-gold | 209±0.7 | −16±0.3 | 100% | – | – | [46] | ||
| Gold | 57±3 | −22±0.4 | ~80% | – | – | [46] | ||
| Silica | NO | 90±10 | – | – | – | 0.001% | [33] | |
| PLGA | Levofloxacin | 190±50 | – | – | – | 0.01%, recovered in 6 days | [38] | |
| Poly(caprolactone) | Levofloxacin | 110±40 | – | – | – | 0.01%, recovered in 6 days | [38] | |
| Helicobacter pylori | Pectin sulfate, RHL and PL | Amoxicillin | 181±18 | −32.89±4.17 | – | – | 18.60% | [47] |
2 Biofilm formation and resistance
2.1 Biofilm developmental cycle
The formation of the three-dimensional complex structure of the biofilm starts with the attachment of free-floating bacteria cells onto a living or non-living surface in a moist environment, followed by the secretion of extracellular polymeric substance (EPS) and the growth of the three-dimensional complex structures of biofilms [48]. EPS is the main component of all biofilms accounting for almost 90% of the entire biomass, and it is composed mainly of polysaccharides, proteins, nucleic acids, and lipids [49]. The EPS matrix physically holds the biofilm bacteria together, facilitating cell-cell communication and gene transfer and protecting bacteria from antibiotics and the host’s immune system. Moreover, the EPS matrix provides nutrients to the bacteria such as carbon-, nitrogen-, and phosphorus-containing compounds [49]. After the biofilm is mature, biofilm bacteria disperse from the biofilm community to form new colonies and gain better access to nutrients [48].
Different treatment strategies have been proposed for each stage of biofilm development. Anti-adhesion agents, such as mannosides, pilicides, and curlicides as well as anti-biofilm polysaccharides, can be used to prevent bacteria from adhering onto the surface [50], [51], [52], [53]. During microcolony formation and biofilm maturation, silver NPs, lytic phages, and EPS-degrading enzymes may be useful [32], [54], [55]. By manipulating dispersing signals, biofilm bacteria can be dispersed into surrounding environments before the natural dispersal occurs [56].
2.2 Biofilm antibiotic resistance
Within just 2 years of the introduction of penicillin, resistance to the antibiotic was noted [57]. The rise of antibiotic resistance has posed a tremendous burden on healthcare system [58], and biofilms play a potentially large role in contributing to this health crisis.
Treatment of biofilm-related diseases requires an underlying understanding of the mechanisms of biofilm antibiotic resistance. In general, antibiotics attack bacteria by disrupting bacterial cell wall biosynthesis, protein synthesis, as well as DNA replication and repair. It has been recognized that bacterial cells withstand these attacks through efflux pumps, deactivating enzymes, and reprogramming antibiotic targets [59]. Efflux pumps on bacterial cell membranes pump out the antibiotics to keep the antibiotic concentration low inside bacterial cells. Enzymes deactivate antibiotics by modifying their active components. Antibiotics targeted structures are modified to avoid recognition [57].
However, these mechanisms are understood in planktonic bacteria, and there exists drastic difference between antibiotic resistance in the biofilm bacterial mode of growth and that of planktonic cells, indicating that the mechanisms behind them are dissimilar. It is unwise to confer the understanding about planktonic bacteria antibiotic resistance to biofilm bacteria without discretion. Although the mechanisms mentioned above may be present in biofilms, they likely work in synergy with multiple mechanisms that are distinct to the biofilm mode of growth. Although the exact mechanisms are still under investigation, several mechanisms have been hypothesized to explain the antibiotic resistance of biofilms. A few of the most prominent mechanisms are mentioned here, but a more thorough review written by Stewart [60] is available. It was originally suggested that the matrix structure of the biofilm may serve as a physical barrier to prevent antibiotics from diffusing inside; however, more recent studies have proved that the diffusion of antibiotics is not hindered by the biofilm matrix [61], [62]. An additional hypothesis is that after binding to polysaccharides, proteins, and DNA present in the biofilm EPS, the antibiotics reaching the target site may no longer be bioactive or may not achieve an adequate concentration to effectively kill bacteria [63]. Genetic components from lysed bacterial cells, such as plasmids, are retained inside the matrix, increasing the frequency of gene transfer between bacterial cells [53]. These plasmids may contain the genes beneficial to bacteria, such as genes for antibiotic resistance [64]. Umadevi et al. [65] have reported that most β-lactam antibiotics can be hydrolyzed by plasmid-encoded carbapenemases, furthering the evidence for gene transfer-related resistance mechanisms. Furthermore, at the interior of the biofilm, the bacteria are anaerobic and have very limited access to oxygen and nutrients, making antibiotics whose action relies on disrupting metabolic processes much less effective in bacterial killing [66]. For instance, penicillin interrupts bacterial cell wall synthesis, but if bacterial cells in the interior of the biofilm are not actively growing or synthesizing cell wall, then penicillin has no effect. In addition, bacterial cells inside biofilms may be at different growth phases; therefore, a subset of the bacterial population can survive from cell division-directed antibiotic attacks. According to persister theory, a small portion of bacterial cells become dormant cells that are tolerant to antibiotics, which then contribute to incomplete bacterial killing by interventions [2], [67], [68]. Quorum-sensing, a mechanism that bacteria use to control gene expression by sensing the local cell density [69], plays a role in biofilm formation [70], [71], [72], [73]. It is still unclear to what degree quorum-sensing is involved in biofilm antibiotic resistance, but the involvement of quorum-sensing in efflux pump genes has been reported by Maseda et al. [74]. A more recent review summarized the evidence that may indicate the relationship between quorum-sensing and antibiotic resistance [75]. Although only a few of the numerous hypotheses are mentioned here, it is readily apparent that biofilm antibiotic resistance is a complex, formidable challenge.
2.3 Nanoparticles in the treatment of biofilms
To combat bacterial infections, NPs made of polymers, silica, and lipids can be used as drug delivery carriers to potentiate current antibiotics, or metallic NPs can act as antimicrobial agents. When used as drug delivery carriers, NPs are expected to protect antimicrobial agents from deactivating enzymes such as β-lactamases, prevent electrostatic binding to EPS components such as DNA or polysaccharides, and release antimicrobial agents locally in a sustained and controlled manner to lower the incidence of systemic side effects as well as enhance antimicrobial efficacy. Consequently, NPs can improve the bioavailability and targeted delivery of antibiotics [76]. The treatment strategies of various NP formulations for biofilm-associated infections are summarized in Figure 1 [23], [33], [37], [42], [43], [44], [77], [78], [79], [80], [81], [82], [83].
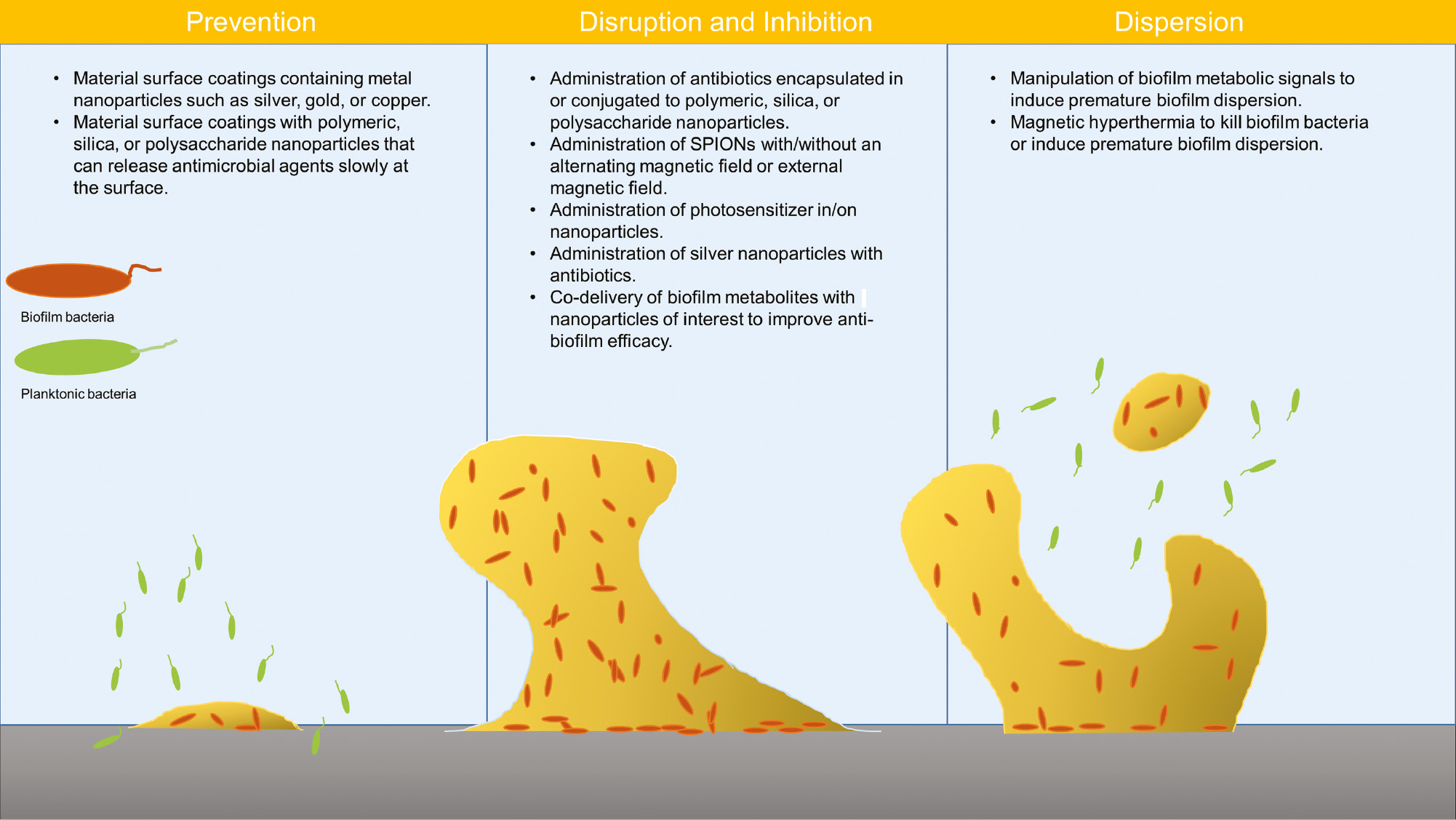
Applications of various nanoparticle treatments to either prevent, disrupt and inhibit, or disperse bacteria from biofilm infections.
2.4 In vitro biofilm quantification
In order to study the anti-biofilm efficacy of the NPs designed, it is necessary to understand the methods that can be used to quantify biofilms. Common methods used to assess the anti-biofilm effect of NPs are summarized in this section. The simplest method is the use of the bacterial growth rate, which can be measured by reading the optical density (OD) at 590 nm based on the fact that higher bacterial cell density causes more lightscattering.
Live/dead staining is a well-established method to evaluate bacterial cell viability. Bacterial cells with intact or damaged cytoplasmic membranes are stained with SYTO 9 (fluorescent green) and propidium iodide (fluorescent red) [84]. This method is time consuming, and in general, only a small portion of biofilm cultures can be examined using confocal laser microscopy. In order to investigate the overall biofilm bacterial cell viability after treatments of NPs with or without an alternating magnetic field, metabolic assays, such as the XTT assay and Alamar Blue assay, can be used to estimate bacterial viability as an indirect measurement [85].
The effect of NPs on biofilm formation can be assessed by staining biofilms with crystal violet and measuring the absorbance. Crystal violet stains the entire biomass, including extracellular matrixes as well as live and dead biofilm bacterial cells [86]. As EPS accounts for almost 90% of the biomass, the bacterial cell viability cannot be measured using crystal violet. In this case, a combination of live/dead staining and crystal violet staining is used to more thoroughly characterize in vitro biofilms. Additionally, because crystal violet sticks to tissue culture plastic, a blank control is critical. An alternate method for measurement of biomass quantifies the thickness of fluorescently-labeled biofilms using confocal microscopy.
3 Nanoparticle-based approaches for biofilm prevention or treatment
3.1 Silver nanoparticles
The disinfectant effect of silver compounds has been recognized since antiquity [87]. However, with the invention of penicillin, the era of antibiotics began and silver was largely forgotten. Now, with increasing concern about antibiotic resistance of bacterial infections and biofilms, silver’s anti-microbial capacity has again become a topic of interest to researchers, primarily in the form of silver NPs. Moreover, silver NPs are a broad spectrum antiseptic that are effective against Gram-negative and Gram-positive bacteria as well as fungi and viruses [88]. Silver NPs typically range from 1 to 100 nm in diameter, and they are composed of 20 to 15,000 silver atoms [89].
Silver NPs can be prepared through a number of methods. The most common chemical method reduces Ag+ (e.g. AgNO3) to Ag0 using a reducing agent such as sodium borohydride in the presence of a stabilizer like poly(N-vinyl-2pyrrolidone) to keep silver NPs from aggregating. Reducing agents and stabilizers must be selected carefully to limit reactivity and environmental impact while increasing particle stability [32]. More environmentally friendly ways to synthesize silver NPs include the use of living organisms like bacteria to produce the particles. For example, AgNO3 solution can be added to wet Bacillus licheniformis in a flask, and the mixture can be left on a shaker at 37°C for 24 h to produce particles. Cells are removed, and the silver NPs are purified [32]. Another method to prepare silver NPs with low environmental impact makes use of a biopolymer such as a starch as the stabilizer [79]. A solution of silver NPs in corn starch maintained colloidal stability (no particle aggregation) up to 90 days, as analyzed by absorption. Eighty-eight percent and 85% of Pseudomonas aeruginosa biofilm reduction were observed after 48 and 24 h of treatment, respectively, indicating an effective impedance of biofilm formation with these starch-stabilized particles [79].
Habash and colleagues [18] assessed the response of P. aeruginosa biofilms to treatments with citrate-capped silver NPs of various sizes (10 nm–100 nm), the antibiotic aztreonam, and the combination of silver NPs and antibiotics. Particle size was determined to be critical after treating preformed biofilms with silver NPs with various sizes at different concentrations. Habash et al. reported that silver NPs with small sizes (10 nm and 20 nm) were more effective than 100 nm particles with regard to biofilm biomass and biofilm bacterial cell viability reduction. However, no morphological change in biofilm structure or cellular morphology was observed [18]. Radzig and colleagues [87] reported that when 8.3 nm silver NPs were administered to Escherichia coli biofilms at an inhibitory concentration (for planktonic bacteria), the NPs failed to have an effect; however, when administered at a high concentration (100–150 μg/ml), the silver NPs killed most E. coli biofilm bacteria. Separately, Jena and colleagues [30] found that chitosan-coated silver NPs (sizes from 55 nm to 275 nm) did not disrupt 24-h-old P. aeruginosa and Staphylococcus aureus biofilms, but they did inhibit P. aeruginosa biofilm formation by 65%. A similar inhibitory effect on P. aeruginosa biofilm was also reported by Palanisamy et al. [31] using chemically synthesized silver NPs (20 nm–30 nm).
Several researchers have attempted combinatorial therapeutic strategies with both antibiotics and silver NPs. Interestingly, in one study, the antibiotic aztreonam administered alone did not eradicate biofilms and even stimulated biofilm growth, up to 250% of untreated biomass, after treatment was stopped [18]. Cationic antimicrobial peptides (CAMPs) are very promising in the treatment of multidrug-resistant P. aeruginosa lung infections experienced by CF patients. Nevertheless, there is an urgent need of inhalable formulations able to deliver the intact CAMP in conductive airways and to shield its interactions with airway mucus/bacterial biofilm, thus enhancing CAMP/bacteria interactions. Along these lines, the aim of this work was the design and development of nano-embedded microparticles (NEMs) for sustained delivery of CAMPs in the lung [23]. To this purpose, NPs made of poly(lactide-co-glycolide) (PLGA) containing a model CAMP, colistin (Col), were produced by emulsion/solvent diffusion technique. Engineering NPs with chitosan (CS) and poly(vinyl alcohol) (PVA) allowed to modulate surface properties and, in so doing, to improve NP transport through artificial CF mucus. In order to achieve a long-term stable dosage form useful for NP inhalation, NPs were spray-dried in different carriers (lactose or mannitol), thus producing NEM. The most promising NEM formulations were selected on the basis of bulk and flow properties, distribution of NPs in the carrier, and aerosolization performance upon delivery through a breath-actuated dry powder inhaler. Of note, selected Col-loaded NEMs were found to kill P. aeruginosa biofilm and to display a prolonged efficacy in biofilm eradication compared to the free Col. This effect was likely ascribable to the ability of NPs to penetrate into bacterial biofilm, as demonstrated by confocal analysis, and to sustain Col release inside it. Taken altogether, our results indicate that adequate engineering of PLGA NPs represents an enticing technological approach to harness novel antimicrobials for P. aeruginosa lung infection, such as CAMPs, especially in CF [23]. However, the combination of small silver NPs and aztreonam demonstrated rather promising results. The highest biomass reduction (~98%) and thickness reduction (~50%) as well as bacterial cell wall damage was observed after exposure to a combination of 10 nm silver NPs and aztreonam [18]. Furthermore, significantly higher reactive oxygen species (ROS) production was observed when silver NPs were combined with an antibiotic in comparison to the sole implementation of either agent alone [90].
Although the mechanism behind silver NPs’ antimicrobial ability is still unclear, it is generally believed that damage to the bacterial cells in the presence of silver NPs, as with other metal NPs, to a high degree, is induced by ROS such as hydroxyl radicals, superoxide anions, and hydrogen peroxide, which may cause damage to protein and DNA in bacteria [91]. Silver ions released from the silver NPs tend to react with thiol groups in protein and deactivate key respiratory enzymes to result in toxic ROS levels that damage and kill the cells [92]. It is also reported that the ROS generated on the surface of silver NPs can interact with cell membrane lipids, leading to the disruption of cell membrane functions [91]. Silver NPs are also known to be capable of binding and penetrating the bacterial cell wall, thus increasing cell wall permeability and cell death [93], [94]. Thus, the combination of silver NPs and conventional antibiotics appears promising for biofilm treatment.
3.2 Gold nanoparticles
Unlike silver, gold is not known to have intrinsic antimicrobial properties. However, the properties of nanoscale gold allow for robust particle functionalization, and researchers have investigated the possibility of using gold NPs in biofilm treatment. Like silver, gold NPs are most commonly synthesized by the reduction of gold salts via several well-established methods [43], [44], [76].
Using an antimicrobial plant extract from Plumbago zeylanica, silver NPs (~63 nm), gold NPs (aggregated into gold nanotriangles), and silver-gold bimetallic NPs (~93 nm) were synthesized from the bioreduction of silver nitrate (AgNO3) and chloroauric acid (HAuCl4) [45]. Gold NPs showed 97% S. aureus biofilm inhibition and disrupted 24-h-old S. aureus biofilms up to 95% and Acinetobacter baumannii biofilms up to 40%. Silver and bimetallic NPs outperformed gold NPs in all strains tested in terms of inhibition and disruption [45]. The anti-biofilm efficacy of gold-silver bimetallic NPs synthesized from γ-proteobacterium Shewanella oneidensis MR-1 was investigated by Ramasamy et al. [46]. E. coli, P. aeruginosa, Enterococcus faecalis, and S. aureus cultures were incubated with gold-silver bimetallic NPs (~209 nm) or gold NPs (~57 nm) for 24 h and further examined by crystal violet to determine biofilm inhibition. Gold-silver bimetallic NPs inhibited all strains effectively at 250 μM. E. coli biofilm formation was completely inhibited at particle concentrations as low as 10 μM [46].
The photosensitizer methylene blue (MB) was attached onto the surface of gold NPs (~26 nm after conjugation) to treat 24-h-old Candida albicans biofilms [39]. Photosensitizers can be activated under a light source and release highly ROS to damage bacteria. The conjugation was achieved through electrostatic interactions between negatively charged gold NPs and positively charged MB. After being treated with a 660 nm diode laser at 38.2 J/cm2 for 40 s every 6 h during a 24-h treatment, C. albicans biofilm growth was inhibited up to 63.2% by 20 μg/ml MB alone and 82.2% by MB-conjugated gold NPs containing an equivalent amount of MB, based on crystal violet staining results. The 2,3-Bis (2-methoxy-4-nitro-5-sulfophenyl)-5-([phenylamino] carbonyl)-2H-tetrazolium hydroxide (XTT) biofilm reduction assay revealed that 81.9% and 95.4% of the biofilm bacteria were dead under the same treatment conditions as above. Moreover, the structure of the MB-conjugated gold NP-treated biofilms was highly distorted compared to the untreated control biofilms when examined by SEM. Additionally, it was also found that MB increased bacterial cell membrane permeability [39]. Under the appropriate activating conditions, photosensitizers like MB can generate highly toxic ROS that cause bacteria cell death [95].
The use of gold NPs improved the anti-biofilm efficacy of the photosensitizer MB, and it is of continuing interest to investigate whether gold NPs can elicit similar results with photosensitizers such as rose bengal (RB) and erythrosine (ER). Interestingly, the chemically synthesized gold NPs failed to inhibit or disrupt A. baumannii and E. coli biofilms. Also, the chemically synthesized silver NPs and bimetallic NPs had poor performance in both biofilm inhibition and disruption [45]. The authors concluded that the anti-biofilm efficacy of gold NPs reported was due to the antimicrobial plant extract synthesis. While results with gold NPs are promising, further research into using gold NPs alone or as part of a combinatorial therapy is warranted.
3.3 Iron nanoparticles
Magnetic NPs have been studied extensively for use in the biomedical field due to their multifunctionality. They have the potential to facilitate magnetic resonance imaging (MRI), targeted drug delivery, and hyperthermia-based therapies [96]. In addition, magnetic NPs are considered biocompatible and are inexpensive.
Magnetic NPs can be synthesized through either co-precipitation or thermal decomposition. In the co-precipitation method, a base is added into an aqueous mixture of Fe3+ and Fe2+ salts at room temperature or elevated temperature in an oxygen-free environment, and a black magnetite precipitate is formed [97]. Co-precipitation is convenient to carry out and provides a considerable amount of magnetic NPs in a stable colloidal state [98]. However, synthesizing NPs with a uniform size remains a challenge using the co-precipitation method because the size distribution can be affected by a variety of parameters including temperature, pH, salts used, and concentrations [99].
Superparamagnetic iron oxide NPs (SPIONs) were used by Durmus et al. and Taylor et al. to treat methicillin-resistant S. aureus (MRSA) [43], [44]. The SPIONs were coated with dimercaptosuccinic acid (DMSA) before being chelated with Fe3+ from FeCl3 or Zn2+ from ZnCl2. DMSA is a chelator that is able to improve the solubility of SPION and conjugate with metal ions [100]. Twenty-four-hour-old S. aureus biofilms were treated with iron- or zinc-conjugated SPION or DMSA-coated SPION for 24 h in vitro. The OD as well as crystal violet staining revealed that zinc-conjugated SPION inhibited the biofilm growth effectively (~20% compared to control) at 0.01 mg/ml, the lowest concentration tested; all SPION inhibited biofilm growth at 1 mg/ml. On the contrary, the application of ZnCl2 alone actually stimulated biofilm growth. Combined with the result that SPIONs, as imaged by TEM, were found inside 48-h-old biofilm bacteria cells after 2 h of treatment, the authors concluded that SPION delivered the zinc and iron ions into biofilm bacteria cells where chemical balance was interrupted and eventually killed the biofilm bacteria [43].
Fructose was used along with the metal-conjugated SPION mentioned above by Durmus et al. to treat 24-h-old S. aureus biofilms for 24 h [44]. The anti-biofilm efficacy of SPION conjugated with or without metal ions was enhanced significantly when fructose was present. The use of fructose increased the amount of SPION deposited in the biofilms, leading to more ROS generation. Unconjugated SPION and 1 mM or 4 mM iron-conjugated SPION achieved a killing rate of up to 99.99999% when 1 mM fructose was present. In fact, all SPIONs, conjugated or not, performed better than did vancomycin. This result was confirmed with fluorescence microscopy images of cells with live/dead staining. Moreover, biomass was reduced by almost 50% after treatment with iron-conjugated SPION and fructose, similar to the reduction caused by vancomycin. Even 0.1 mg/ml SPION achieved a similar biofilm eradication effect as 1 mg/ml SPION alone when used with 100 mM fructose, indicating that the use of fructose can strengthen the anti-biofilm efficacy of SPION. In turn, this reduces the potential for systemic toxicity of SPION because a lower dose of SPION is required for a therapeutic effect. Promising results were also observed on Gram-negative biofilms such as P. aeruginosa and E. coli, suggesting a broad-spectrum anti-biofilm efficacy [44]. Salunke et al. [45] also reported that metal NPs reduced by fructose containing plant extract outperformed the chemically synthesized NPs.
Magnetic NPs, when directed using an external magnetic field, are capable of penetrating the full depth of biofilms, accumulating at the bottom, and killing bacterial cells deep inside biofilms. SPIONs were decorated with carboxyethylsilanetriol (CES) by Subbiahdoss et al. [40] to treat 24-h-old gentamicin-resistant Staphylococcus epidermidis biofilms. The graft of CES did not change the size of SPIONs (~13 nm) but lowered the zeta potential from +43 mV to −15 mV. With a magnet placed under the culture well where biofilms were growing, CES-SPIONs could be concentrated at the center of the wells. The cross-sections of live/dead stained biofilms further demonstrated that bacterial cells at the bottom of the biofilms accounted for most of the dead bacteria. The use of gentamicin did not change the anti-biofilm efficacy of SPIONs. Thirty-eight percent of mature biofilm bacteria cells were killed when treated with magnetically targeted CES-SPIONs, while only 9% were found dead when the magnetic field was not applied [40]. As there was no significant difference between the anti-biofilm efficacy of unmodified SPIONs and CES-SPIONs, it is possible that the ROS formation, caused by the extremely small size of NPs, attributes more to the anti-biofilm efficacy compared to the electrostatic interaction between SPIONs and biofilm bacterial cell walls [40].
Magnetic NP induced hyperthermia also facilitates anti-biofilm treatments. Up to 99.9% reduction was observed when planktonic S. aureus was heated in tryptic soy broth (TSB) for 20 min at 60°C [101]. When magnetic hyperthermia was implemented, a lower temperature (43°C) was needed to cause the same 99.9% reduction on S. aureus biofilms [14]. As for P. aeruginosa biofilms, when magnetic NP-mediated hyperthermia created a temperature rise of 5°C or more, the dispersion of preformed biofilms was induced. The hyperthermia did not show any toxic effects on the bacteria themselves but enhanced gentamicin treatments [102]. This conclusion was not unexpected, as planktonic bacteria are far more susceptible to conventional antibiotics than biofilm bacteria; therefore, the hyperthermia rendered the dispersed bacteria more susceptible to antibiotic treatment.
With their use as targeted delivery agents, magnetically targeted particles, or particles for inducing hyperthermia, iron oxide particles hold great promise. However, further investigation of the effect of size and chemical composition, as well as other formulation characteristics, is necessary. For example, in the case of Fe2O3 iron oxide NPs, 2 nm particles did not inhibit biofilm growth but actually induced its formation. It was hypothesized by Borcherding et al. that these very small Fe2O3 NPs may have been an iron source for biofilm bacteria growth in this study. In addition, Fe2O3 NPs were found to inhibit antimicrobial peptide function and deactivate ciprofloxacin [103], [104]. Additional studies into the effect of iron oxide NPs are necessary to understand their usefulness in biofilm treatment.
3.4 Copper nanoparticles
Copper NPs exhibit characteristics favorable to their utilization as therapeutic agents with antimicrobial/antifungal properties and have a lower cost compared to other metals such as silver and gold [105]. Copper NPs have found applications as coating agents on biomedical devices to inhibit the growth of pathogens [106], [107], [108]. Copper NPs can be used as industrial fungicides, in water purification, and as antifouling agents [107], [109]. A variety of research has been performed on the effect of copper NP use on the formation of biofilms based on their cell counts and ability to be cultured [110]. Given their ability to inhibit the formation of biofilms and limit the growth of pathogens, copper NPs demonstrate potential for their use in hospital and other healthcare facilities to prevent spreading of pathogens on equipment and nosocomial infections in patients.
A variety of methods for the synthesis of copper NPs exist in literature including the use of polyol synthesis, thermal plasma, inert-gas condensation, one-pot synthesis, and the solvothermal method [30], [32], [89], [91], [92]. Nonetheless, chemical reduction frequently appears as a prominent method for the synthesis of copper NPs. Generally, chemical reduction is a simple and economical method to produce metal NPs given the use of a reducing agent to reduce metal salts and a capping agent to curtail NP synthesis at a particular size. Due to copper’s sensitivity to air, copper NPs often form a surface oxide layer during synthesis [109], but some methods exist that utilize an environment void of oxygen in order to inhibit the formation of this layer. As with the other metallic NPs, the plethora of synthesis methods available provides flexibility in the ability of a facility to produce copper NPs and may allow for scaled-up manufacturing of copper NPs [105].
Many of the aforementioned studies utilized NPs in various experimental settings to assess the practical application of copper NPs as inhibitors against biofilm formation. The proliferation of pathogenic specimens quantifies the change in bacterial counts on agar plates after treatment with copper NPs [111]. This experiment involved the extraction of biofilm bacteria from the slurry of the tooth crown from a healthy, young male. The slurry was then suspended in phosphate-buffered saline (PBS) and dosed with 40 nm cupric oxide NPs at concentrations of 10, 50, or 100 μg/ml for 16 h at 37°C. These cultures were subsequently spread on agar plates, incubated for 3–5 days, and analyzed using crystal violet under light microscopy. The images showed that EPS biomass was inhibited by almost 61% when bacteria were treated with 50 μg/ml cupric NPs [111].
Shankar and Rhim [112] devised an environmentally friendly method to produce copper NPs through chemical reduction. They investigated the effect of copper salts and reducing agents on the size and shape of copper NPs and subsequently determined the effect of copper NPs on foodborne pathogens including the Gram-positive bacterium Listeria monocytogenes and the Gram-negative bacterium E. coli. Using two types of reducing agents, sodium hydroxide (NaOH) and ascorbic acid (AA), and three types of copper salts, they compared the effects of particles prepared with each reducing agent against the two types of bacteria through the use of an agar well diffusion method where zones of inhibition were measured. L. monocytogenes (10.5 mm–12.5 mm) were found to exhibit a greater susceptibility toward copper NPs compared to E. coli (11.7 mm–18.5 mm) due to a greater zone of growth inhibition. Additionally, NPs prepared using sodium hydroxide induced a more potent effect against both L. monocytogenes and E. coli than those synthesized using ascorbic acid [112].
LewisOscar et al. [107] assessed how antibiotic resistance of P. aeruginosa increases in more mature specimens and described how copper NPs help prevent the formation of biofilms at more nascent stages. The methodology employed utilized two control strains of P. aeruginosa as well as 17 other clinical isolates grown in Luria-Bertani (LB) agar at 37°C for 24 h. This study reported a maximum biofilm inhibition of 94% and exemplified the use of copper NPs as a cheaper alternative to silver NPs to impede the formation of biofilms [107]. Based on the current results, copper NPs appear to be best suited for biofilm prevention rather than biofilm eradication.
3.5 Liposomes
Biocompatible amphiphilic phospholipids form spherical vesicles readily, which are composed of one or more lipid bilayers. Liposomes can be categorized into multilamellar and unilamellar classes depending on the number of lipid bilayers, and they are capable of encapsulating hydrophilic or hydrophobic compounds [113]. High encapsulation efficiency synthesis methods of liposomal NPs include the Bangham, detergent-depletion, ether/ethanol injection, reverse phase evaporation, and emulsion methods [114]. The bilayer membrane structure grants liposomes exceptional permeability, which facilitates the cellular uptake of liposomes. Hydrophilic antimicrobial agents can be protected from deactivating factors in vivo by encapsulation into the large aqueous core of the liposomal NP with high drug loading.
The efficacy of the liposomal formulation of the antibiotic amikacin (Arikace®), a nebulized solution for CF treatment, was tested in vivo on P. aeruginosa biofilms by Meers et al. Uninfected rats were subject to inhalation of the same amount of free tobramycin and liposomal amikacin (~300 nm) for 60 or 80 min [115]. About 65% of the amikacin remained in liposomal form after deposition in the lung at an amount of 782 μg compared to 645 μg of free tobramycin, which is likely due to the fast clearance of the free aminoglycoside throughout the inhalation process. The free tobramycin only lasted fewer than 2 h until it was mostly removed from the lungs, while the liposomal amikacin exhibited a sustained drug release for up to 175 h after a burst release due to the transformation of liposomal amikacin into free form during inhalation. Nevertheless, the sustained release of amikacin lasted for only 48 h when incubated with diluted P. aeruginosa sputum from a patient with CF, which led to the speculation that the lytic components of biofilms may trigger the release of amikacin. Almost all amikacin was released over 24 h when liposomal amikacin was incubated with Di-rhamnolipid (RHL) and mono-rhamnolipid, indicating amikacin is more readily released at biofilm-infected areas. Rats infected with P. aeruginosa for 4 days were treated with equal doses of free amikacin and liposomal amikacin three times per week for 2 weeks. No obvious change of colony-forming units (CFUs) was seen in the free amikacin-treated group, while liposomal amikacin reduced CFUs by 40%. Over 1300 μg of amikacin was detected 3 days after treatment, indicating a sustained release of amikacin [115].
As the biofilms in CF patients are surrounded by thick mucus, it is crucial to deliver a sufficient amount of antimicrobial agents through the mucus layer to the biofilm-infected sites, which is a size-dependent process. In a mixture with 1 μm polystyrene particles, 62% of fluorescent liposomes penetrated a layer of sputum in a period of 24 h as well as 60-h-old pure P. aeruginosa biofilms. In contrast, only 9% of the 1 μm polystyrene particles were recovered at the other end of the sputum layer, yet even a few 200 μm polystyrene particles were found to penetrate the sputum [115].
This size-dependent penetration was also mentioned in a separate study. Cationic, smaller sized (128 nm–141 nm), unilamellar particles penetrated both P. aeruginosa and S. aureus biofilms at a much higher rate with deeper penetration than larger, multilamellar particles [34]. The smaller, blank, cationic liposomes inhibited the growth of P. aeruginosa biofilms by 75% based on relative viability after 24-h exposure, and S. aureus biofilms were inhibited by 43%. It is hypothesized that the unilamellar cationic liposome particles may disrupt the electrostatic equilibrium of the colonies within the biofilm, allowing the anti-biofilm effects to be more potent [34].
Neutral and anionic liposomal tobramycin were prepared by dissolving a mixture of cholesterol and 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) and a mixture of 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) and 1,2-dipalmitoyl-sn-glycero-3-phosphoglycerol sodium salt (DPPG) in chloroform, respectively. However, neither liposomal tobramycin performed better than free tobramycin in this study [116].
Liposomal antibiotics have been studied as an alternative to traditional free antibiotics that only deliver low concentrations at the site of biofilm infections and are cleared quickly in the lungs. However, the clinical application of liposomal formulations is limited by a liposome’s weak physiochemical properties. Meer et al. reported that 35% of the amikacin did not arrive at the lungs in liposomal form in vivo. The side effects caused by the leakage of encapsulated antibiotics from liposomes are potentially unfavorable. Despite the promising results reported by Meer et al. there is still doubt as to whether liposomal antibiotics can penetrate the harsh conditions of the biofilm biomass in infected patients.
3.6 Chitosan nanoparticles
Chitosan is a natural polymer deacetylated from chitin. Its biocompatible, biodegradable, and inherent broad-spectrum antimicrobial properties make it suitable as a carrier for biofilm treatment [117]. Chitosan NPs have been effectively used against 24-h-old P. aeruginosa biofilms of six different strains derived from clinical cases [118]. Chitosan NPs carry a positive surface charge, while bacterial cell walls and biofilm EPS surfaces are negatively charged. Chitosan NPs are thus expected to have a higher affinity to infected areas than negatively charged NPs. One common method used to synthesize chitosan NPs is the ionic gelation method. Amine groups present in chitosan acetic acid solution react with polyanions like sodium tripolyphosphate (TTP) readily, and chitosan NPs are then formed through cross-linking [119], [120]. Preparing chitosan NPs for biofilms at neutral pH was proposed by Chávez de Paz et al. [28]. The anti-biofilm effect was examined through treatment of 24-h-old S. mutans biofilms with low molecular weight chitosan NPs. More than 95% of the biofilm bacterial cells were damaged after treatment, although no significant biofilm structure change was observed [28].
The photosensitizer ER was encapsulated into chitosan NPs to facilitate photodynamic therapy by Chen and coworkers [27]. ER-loaded chitosan NPs were used to treat S. mutans, P. aeruginosa, and C. albicans biofilms in vitro. Gram-positive S. mutans biofilms were eradicated completely based on viable cell count after incubation with ER-loaded chitosan NPs for 12 h at room temperature in the absence of light followed by 50 J cm−2dose. Under the same conditions, NP-treated Gram-negative P. aeruginosa biofilm cells were reduced only by around 28%, indicating a less effective treatment [27].
Another photosensitizer, RB, was conjugated onto chitosan NPs by carbodiimide chemistry to treat 24-h-old E. faecalis biofilms [42]. RB-loaded chitosan NPs increased the cell membrane permeability followed by cell content leakage after 15 min of incubation in a dark environment, which can be attributed to chitosan’s antimicrobial properties. Encapsulated RB showed a significantly higher cell uptake compared to free RB. It is worth noting that encapsulated RB eliminated biofilm completely when photodynamic therapy was divided. Two dosages of 20 J/cm2 were more effective than one 60 J/cm2 dosage. Although the time interval between the two dosages was not mentioned in this paper, it is likely that molecular oxygen was replenished during this time, enhancing the production of toxic highly reactive species around and inside the biofilm. Chitosan NPs also help slow down the release rate of singlet oxygen, elongating the bacterial killing effect [42].
In another study, linoleic acid modified chitosan NPs were used to immobilize the enzyme β-N-acetyl-glucosaminidase (DspB), which has been shown to be capable of inhibiting and detaching biofilms by degrading poly-β(1,6)-N-acetyl-glucosamine (PNAG), a major polysaccharide found in S. epidermidis, S. aureus, and Actinobacillus actinomycetemcomitans EPS [29]. At 37°C, free enzyme lost its activity completely after incubation for 7 h, yet encapsulated enzyme still possessed 33.4% activity. Immobilized enzyme was more stable upon temperature variation, which may result from stabilization of encapsulated enzyme into the chitosan NPs. Chitosan NPs showed a slightly anti-biofilm effect in biofilm treatment, and biofilm treated with immobilized DspB had the lowest absorbance reading at 590 nm to measure the amount of crystal violet after 1-h treatment. Longer treatment may result in biofilm eradication because immobilized enzyme remained active for longer than 7 h [29].
The biocompatibility, degradability, and antimicrobial nature of chitosan make it a strong candidate for biofilm antibiotic delivery. However, the hydrophobicity of chitosan prompts aggregation under biological condition and limits its use. Aggregation of chitosan NPs was observed after an hour in cell medium [121]. The addition of poly(ethylene glycol) (PEG) to the surface of chitosan NPs has been shown to be an effective way to overcome this drawback [122], which will likely have implications for the future of this drug delivery technology.
3.7 Poly(lactic-co-glycolic acid) nanoparticles
Poly(lactic-co-glycolic acid) (PLGA) is a biocompatible and degradable copolymer. The ester linkages in PLGA degrade through hydrolysis when water is present, and the polymer degradation products lactic acid and glycolic acid are natural byproducts of several metabolic pathways. Several drug delivery devices made of PLGA are FDA approved. A list of FDA-approved PLGA-based particles on the market is given elsewhere [123].
The use of PLGA NPs as a colloidal drug delivery system has been explored extensively. Numerous studies have shown that PLGA NPs possess the ability to deliver therapeutic agents in a controlled and sustained manner through polymer degradation and diffusion of therapeutic agents encapsulated while the degradation rate of PLGA NPs can be manipulated by changing the ratio of poly lactic acid (PLA) and poly glycolic acid (PGA), which controls the material’s hydrophobicity and crystallinity. Higher PGA ratios lead to higher crystallinity, while more PLA increases hydrophobicity [124]. Unmodified PLGA NPs, capped with COOH, possess a negative zeta potential because of the disassociation of the hydrogen of the carboxyl group in an aqueous environment. In the case of OH-capped PLGA NPs, the deprotonation of the hydroxyl group only happens under very basic conditions, which is not considered relevant for most biological applications. Generally, the size of PLGA NPs ranges from 100 nm to 400 nm. Longer sonication time during fabrication process can lead to a smaller size [125].
Ciprofloxacin (CPX) was encapsulated into PLGA NPs coated with poly(L-lysine) (PL) using an oil-in-water nano-precipitation method by Baelo et al. [35]. The PLGA core was functionalized with deoxyribonuclease I (DNase I) to facilitate the eradication of biofilm by degrading the extracellular DNA in EPS that is a key component in early stages of biofilm formation and abundant in EPS. Burst release and a slower sustained release after that were observed in an in vitro release study. PLGA-PL-CPX-DNase I, PLGA-CPX, PLGA-PL-CPX NPs, and free CPX were compared in terms of antimicrobial ability, prevention of biofilm formation, and eradication of preformed biofilm. The growth rate of biofilm was inhibited by PLGA-PL-CPX-DNase I NPs up to 80% at the minimum CPX concentration of 0.0078 μg/ml. The biomass of 2-day-old biofilm reduced by 95% after treatment with PLGA-PL-CPX-DNase I NPs. These results indicate that active DNase I on the surface of PLGA NPs helps increase the mobility of the particles and facilitate diffusion into the EPS [35].
In another study, gentamicin was loaded into PLGA NPs by Abdelghany et al. using a modified water-in-oil-in-water double emulsion to treat P. aeruginosa biofilms [37]. By adjusting the pH of the outer aqueous phase, drug-loading increased from 6.1 μg/ml to 22.4 μg/ml in comparison with a similar work done by Concepcion in 2006. Increased pH made gentamicin less hydrophilic, leading to higher entrapment in the hydrophobic PLGA core. After a burst release in the first 24 h, sustained release of gentamicin from PLGA NPs lasted for another 15 days at 37°C incubation. The existing biofilm was placed in the external chamber in a dialysis cell, and gentamicin-loaded PLGA NPs and free gentamicin were added into dialysis tubing, respectively. Half of the media was replaced with fresh media every hour to mimic the typical clearance rate of the lung. After 36 h of incubation, the CFU in the NP-treated biofilm was significantly lower than the free gentamicin-treated biofilm, suggesting that a sustained release of gentamicin from PLGA NPs is more effective in biofilm treatment than a single dosage of gentamicin. Gentamicin-loaded PLGA NPs and free gentamicin were delivered intraperitoneally to P. aeruginosa-infected mice. Peritoneal lavage was done in infected mice at certain time points, and CFUs were measured in each aspiration. After 48 h, a similar reduction was shown in CFU from both NP-encapsulated gentamicin and free gentamicin-treated mice. After 96 h, NP-encapsulated gentamicin managed to keep CFU levels low in the peritoneal lavage while the mice treated with free gentamicin returned to the same level as the positive control treated with saline buffer. The result of the in vivo test corresponds to the in vitro biofilm treatment results, indicating that these PLGA NPs improved the effectiveness of gentamicin by maintaining a working concentration of gentamicin at biofilm-infected areas [37].
Ciprofloxacin and magnetic NPs (CMX) were encapsulated into PLGA NPs by Xin and co-workers [36]. An external low-energy oscillating magnetic field was used to induce hypothermic conditions, which led to the disruption of the PLGA core. An external magnetic field was turned on and off alternatively for 6 h every other hour during the drug release experiment. The results showed that the CPX release rate surged when a magnetic field was on and plateaued when the magnetic field was off, suggesting a controlled release. CPX-CMX-loaded PLGA NPs released CPX for 14 days after being triggered by the magnetic field, and the total release amount was 95%. Mature biofilm was treated with CPX-CMX-loaded PLGA NPs, blank PLGA NPs, and free CPX to test the in vitro functionality of released CPX. XTT assay was conducted to measure the metabolic activity of biofilms after a 24-h treatment. The reduction of biological activity caused by triggered CPX-CMX-loaded NPs was 33.3%, slightly lower than free CPX due to the incomplete release of CPX [36].
Cationic antimicrobial peptides (CAMP) area promising treatment for P. aeruginosa lung infections. CAMP can cause disruption of Gram-negative cell walls by the interaction with anionic lipopolysaccharide on those cell walls. However, its cationic nature causes CAMP to react with anionic proteins and other polysaccharides of the lung extracellular environment, leading to poor bioavailability. Colistin, a model cationic antimicrobial peptide, was encapsulated into PLGA NPs coated with chitosan or PVA to achieve desired properties to overcome the mucus barrier and penetrate biofilms [23]. Colistin-loaded PLGA NPs were then embedded into water-soluble inert lactose microparticles using a co-spraying drying process to ensure enough NPs can be deposited onto lung-infected areas without being exhaled because of their small size. Both the size and zeta potential of CS-PLGA and PVA-PLGA NPs stayed consistent after the lactose coating was dissolved in water, suggesting that the lactose carrier did not significantly affect the properties of PLGA NPs. PVA-PLGA NPs showed a much slower burst release in the first 6 h compared to CS-PLGA NPs, yet both NPs released peptide for up to 15 days. Similar to other antimicrobial agents, free colistin had a robust anti-biofilm performance in the first 24 h, reducing 90% of biomass. Nevertheless, for high concentrations used in the biofilm treatment, biomass recovered completely after 72 h, with recovery being quicker at a lower concentration. Although not as strong as free colistin at the beginning, the anti-biofilm activity of PVA-PLGA particles and CS-PLGA particles was maintained after 72 h, and confocal microscopy imaging of rhodamine-encapsulated particles confirmed that both of them penetrate into biofilm. Interestingly, CS-PLGA NPs released twice as much colistin as PVA-PLGA NPs in the first 24 h in the release study. However, in biofilm treatment, CS-PLGA particles reduced approximately the same amount of biomass as PVA-PLGA particles after 24 h. The release kinetics of PLGA NPs may be changed after embedding into lactose [23].
Lipid-PLGA NPs were synthesized by Cheow and colleagues to encapsulate levofloxacin [38]. A lipid coat over the PLGA core was expected to help PLGA NPs penetrate sputum and target biofilm. Phosphatidylcholine (PC) was incorporated into the aqueous phase to generate the lipid coating. Levofloxacin-loaded lipid-PLGA NPs had an encapsulation efficiency of 21%. In the in vitro test, Lipid-PLGA NPs showed higher efficacy in eradicating P. aeruginosa biofilms compared to PLGA NPs. The author hypothesized that the incorporation of lipid may facilitate the diffusion of levofloxacin into the biofilm matrix. In another study, rhoamnolipid, a biosurfactant produced by P. aeruginosa, has shown the potential to trigger the release of low solubility-low permeability antibiotic such as Levofloxacin and calcein from lipid-PLGA NPs by disrupting the lipid coating. However, neither an in vivo nor in vitro biofilm susceptibility test was conducted in the study [81].
Instead of antibiotics, Iannitelli et al. [126] encapsulated carvacrol (Car.) into PLGA NPs using an oil-in-water single emulsion method. This was done to see its capability as an adjunctive therapy to promote the eradication of biofilm when an antibiotic was co-delivered. Car. is present in essential oil and able to inhibit the growth of several bacterial strains. Due to the drug’s hydrophobic nature, PLGA NPs were used to improve its bioavailability. Sixty percent of Car. was released after 3 h in an in vitro drug release study, and 95% was released in the first 24 h. Based on a significant reduction of elasticity and mechanical stability from rheological testing, Iannitelli and colleagues suggested the architecture of 2-day-old biofilm was altered, which made it more susceptible to antimicrobial agents [126].
PLGA NPs provide a robust drug delivery system. Most PLGA-encapsulated antibiotics show a biphasic sustained release profile in vitro. With the decoration of PEG on the surface, the biocompatibility of PLGA NPs can further be improved and the circulation time elongated, if the application necessitates this. The low encapsulation efficiency, especially of hydrophilic agents, is the main disadvantage of PLGA NPs.
3.8 Other polymeric particles
In addition to PLGA NPs, many investigators have had success in using other polymeric particles. Amoxicillin (AMX) was loaded into lipid polymer NPs containing pectin sulfate (PECS) by Cai et al. [47] to treat Helicobacter pylori biofilms and prevent biofilm adhesion. PECS was dissolved in water and then mixed with CaCl2 to form electrolyte complexes before it was emulsified with a mixture of rhamnolipid and phospholipids (phosphatidylcholine, PC) in methylene dichloride. The particles were then freeze dried and soaked in an AMX aqueous solution to load AMX into NPs. The potential of pectin to inhibit the adhesion of H. pylori onto host cells such as gastric epithelial AGS cells and red blood cells was studied by Song and Li [127]. Rhamnolipid is a bacterial surfactant produced by P. aeruginosa and has been found to be involved in several stages of biofilm development. It is hypothesized that rhamnolipid may have the ability to induce biofilm EPS breakdown as well as fuse with the EPS of biofilms so that the antibiotic can be released inside the EPS. Cheow and Hadinoto [81] incorporated rhamnolipid into PLGA NPs as mentioned before. In this research, AMX-loaded RHL-PC lipid polymer NPs had a mean size of 181 nm and a negative zeta potential. Due to the solubility of AMX in water, the majority of the AMX was released in the first 2 h and the rest was released at a relatively stable rate for another 22 h. Live/dead staining was used to visualize biofilm bacteria by confocal scanning laser microscopy and quantify viability by fluorescence intensity. A percentage of 82.4 of biofilm bacteria was not viable after treatment with AMX-loaded RHL-PC lipid polymer NPs. The minimum inhibitory concentration of AMX to mature H. pylori biofilms was reduced by nearly 7-fold after treatment with RHL-PC lipid polymer NPs for 24 h at 37°C. Fluorescein isothiocyanate was used to tag polysaccharides in EPS as an indicator of the change of the amount of EPS after mature biofilms were treated with RHL-PC lipid polymer NPs for 24 h. The fluorescence intensity of the treated group accounted for 15% of the control group, suggesting a significant EPS loss, which coincides with the previous two results that more susceptibility was observed after treatment [127].
In addition, broad spectrum antimicrobial activity against intraoral biofilm of dental resin composite containing 1% w/w quaternary ammonium polyethylenimine (QPEI) NPs in vivo was observed by Beyth et al. [128]. A specimen of resin composite containing QPEI NPs and resin composite control were recovered from 10 volunteers after letting biofilm buildup on the composite for 4 h. Confocal laser scanning microscopy revealed that the viability of biofilm bacteria decreased significantly compared to the control group, while the thickness of biofilms on QPEI incorporated resin composite increased. This contradicts established knowledge that an anti-biofilm effect of bactericidal-immobilized material is only observed when biofilm bacteria come into contact with that material directly. The author thus hypothesized that the stress caused by QPEI NPs in the resin composite interacting with biofilm bacteria cell wall may electrostatically trigger a programmed cell death of other biofilm bacteria that were not in contact with the resin composite [128].
Another polymeric NP system consisting of silver NPs attached to poly(L-lactic acid) (PLLA) particles was created by Taheri et al. [41] using an emulsion method with sodium dodecyl sulfate (SDS) and PVA as surfactants. Although SDS stabilized silver-loaded PLLA NPs exhibited a smaller size (~90 nm) compared to those stabilized with PVA (~225 nm), more silver NPs (~17 nm after the synthesis process) were found attached to the PVA group under both SEM and TEM. About 0.6% of silver was found in the hybrid NPs using XPS. Thus, only the PVA stabilized NPs were further immobilized onto a thin layer of allylamine plasma polymer (AApp) through electrostatic interaction between COO- groups on the surface of the particles and NH3+ groups on the AApp film to test their capability of inhibiting S. epidermidis biofilm formation, and Safranin-O staining showed that up to 98% of biofilm development was inhibited compared to control treated with blank PLLA NPs. The author attributed this result to the combination of PLLA and silver NPs. Lactic acid, which degrades from PLLA, increases the permeability of the bacterial cell wall, allowing more silver ions into the bacterial cells. Additionally, the degradation resulted in local pH drop that accelerated oxidation and dissolution of silver NPs [41].
A study by Kho et al. [129] focused on encapsulation of LEX into poly(caprolactone) (PCL) NPs at an encapsulation efficiency of 5–10%. LEX-loaded PCL NPs were then spray-dried with lactose and leucine to be delivered by dry-powder inhalers in lung infection treatments. The inclusion of lactose and leucine prevented the melting of PCL during the spray-drying process, increased re-dispersability of PCL NPs, and may facilitate the deposition of PCL NPs onto lung tissues. In vitro E. coli biofilm treatment results showed that the encapsulation of LEX into PCL NPs maintained the anti-biofilm efficacy of free LEX. A total of 99.9% biofilm bacterial cells were killed after 24 h of incubation, which was determined using the drop plate method [129]. The drop plate method enumerates viable bacterial cells but has not been widely standardized [130], [131], [132]. Biofilms need to be removed from attached surfaces and disaggregated before using this method to measure viability. More details about utilizing this method are described elsewhere by Zelver et al. [133].
3.9 Silica nanoparticles
Biodegradable materials are expected to provide a way to deliver antimicrobial agents to a biofilm in a controlled manner. However, as revealed by in vitro experiments, organic biodegradable NPs tend to start releasing the active agent (attached or encapsulated) molecules once in contact with water. In some cases, burst release is caused by antimicrobial agents attached onto the NP surface like PLGA NPs. Yet in other cases, like liposomes, relatively weak mechanical properties are responsible for premature leakage of encapsulated molecules. Thus, stronger and biocompatible but not biodegradable silica NPs become an option. In terms of anti-biofilm uses, silica NPs are commonly paired up with nitric oxide (NO) as a carrier to deliver NO donor molecules. NO is an endogenous free radical and is involved in several biological processes [134]. In terms of the antimicrobial effects, researchers expect NO and its byproducts to eradicate the biofilm without giving rise to resistance like antibiotics. Another advantage of NO is its broad-spectrum antimicrobial capability and low toxicity at working concentration [135].
NO releasing inorganic-organic hybrid silica NPs were synthesized in a novel one-pot method reported by Shin et al. [136]. Generally, silica NPs were prepared through cocondensation of tetramethoxysilane (TMOS) or tetraethoxysilane (TEOS), and various aminoalkoxysilanes such as N-(6-aminohexyl)aminopropyltrimethoxysilane (AHAP3), N-(2-aminoethyl)-3-aminopropyltrimethoxysilane (AEAP3), and (aminoethylamino-methyl)phenethyltrimethoxysilane (AEMP3) were used in this study with ethanol, water, and ammonia. Aminoalkoxysilanes provided functional amine groups that were further converted to N-diazeniumdiolate NO donors under high pressures of NO and TMOS or TEOS as the initial silicon alkoxide backbone. Particle sizes tended to pseudolinearly decrease with increasing AEAP3 concentration, and this characteristic made silica NP size tunable. When NP size was smaller, the NO release rate was faster possibly because water in smaller NPs requires a decreased distance to diffuse to NO donors. The highest total NO amount released from the AHAP3/TMOS combination was almost three times that of surface grafted control silica NPs. The longest half-life of NO release was up to 12 h, achieved by AEAP3/TEOS NPs. Higher NO amounts and longer release times were attributed to the one-pot method used to prepare silica NPs, as NO donors precursors were distributed evenly throughout silica NPs. On the other hand, the amount of NO donor precursors in surface grafted NPs was limited by surface area and exposure to water [136].
The method mentioned above was used to prepare silica NPs using methylaminopropyltrimethoxysilane (MAP3) or AHAP3 as a NO donor precursor. MAP3 silica NPs released much more NO than AHAP3 NPs in a shorter period. Both NPs showed more than 99% killing of P. aeruginosa biofilm bacterial cells. Yet, MAP3 NPs were 1000-fold more effective than AHAP3 NPs, indicating that a higher concentration of NO released rapidly at the biofilm infection site may be more effective than a lower concentration of NO released continuously due to NO diffusion into biofilms, which are facilitated by high concentrations. MAP3 NPs were then used to treat against biofilms of P. aeruginosa, E. coli, S. aureus, S. epidermidis, and C. albicans, and 99.999% reduction in Gram-negative biofilm, 99.9% in fungal biofilm, and 99% in Gram-positive biofilms were observed, demonstrating a broad-spectrum anti-biofilm capability. To be noted, more susceptible Gram-positive biofilms usually appeared more resistant to NO releasing NPs in this study. The authors hypothesized that electrostatic interaction between silica NPs and biofilms may play a role in these experiments [33].
Similar results were reported by Slomberg et al. [137] where Gram-negative biofilms were more susceptible to NO released from silica NPs than Gram-positive ones. Surface-grafting silica NPs with different sizes and shapes represented by the aspect ratio were used to treat P. aeruginosa and S. aureus biofilms in this study. The aspect ratio of the particles was tuned by varying reaction temperature and ammonia concentration. Furthermore, more effective biofilm bacterial killing was observed from smaller (14 nm) and more rod-like (AR4) NPs. Although higher fluorescent intensity from rhodamine B isothiocyanate labeled silica NPs was observed inside 150 nm NP-treated biofilm under confocal microscopy after 60 min, 14 nm NPs dispersed biofilm in 14 min and led to bacterial cell death. Fourteen-nanometer NPs also diffused into biofilm faster than larger 150 nm particles. As a result, this NP strategy showed a rapid release of NO in high concentration from silica NPs under 20 nm was promising in Gram-negative biofilm eradication. Regarding shape, a large portion of dead bacterial cells were found after 15 min of treatment with rod-like AR4 and AR8 NPs, and the biofilm was dispersed after 60 min. Spherical NPs were confined in a limited area in the biofilm, and the death of cells was not observed until 60 min later. As this group of NPs has a much bigger size, it is more likely that AR4 and AR8 released sufficient NO and dispersed the biofilm [137].
In addition to NO donors, chlorhexidine was encapsulated into mesoporous silica NPs by submerging silica NPs in chlorhexidine ethanolic solution [138]. Chlorhexidine released for 80 h and was effective against mono-species biofilms including S. mutans, Streptococcus sobrinus, Fusobacterium nucleatum, A. actinomycetemcomitans, and E. faecalis. However, chlorhexidine released from silica NPs was unable to eradicate 72-h-old multispecies biofilm [138]. Paired with NO, silica NPs are expected to remove the biofilm without giving rise to biofilm bacterial antibiotic resistance. Similar to conventional antibiotics, a sufficient amount of NO needs to be delivered to biofilms in order to eradicate them. The size, shape, and synthesis method all affect the anti-biofilm efficacy of silica NPs. Thus, it is paramount to find the optimum combination of these parameters in order to achieve biofilm annihilation.
3.10 Importance of size and surface charge
Regardless of material composition, NP size affects the uptake of NPs in biofilms and is therefore a critical parameter in the design of an effective treatment. Forier et al. studied the penetration cut-off size for polymer NPs on Burkholderia multivorans and P. aeruginosa biofilms using PEG-coated polystyrene NPs [115]. The size of fluorescent polystyrene NPs ranged from 40 nm to 555 nm, and the zeta potential ranged from −6 mV to −9 mV, similar to poly(vinyl acetate)-coated polymeric NPs. This PEG coating was meant to minimize the interaction between hydrophobic NPs and components of biofilm so that NP size was the only variable in this research. The NP concentration inside the P. aeruginosa biofilm decreased gradually as particle size became larger than 100 nm. Sixty-one percent of the 40 nm NPs were detected, suggesting the penetration rate of NPs is size dependent, and a smaller size (<~100 nm) is favorable in P. aeruginosa biofilm treatment. In another study conducted by Meer et al. 1 μm fluorescent polystyrene beads were unable to pass through P. aeruginosa biofilm, revealing that there is an effective size for NP penetration into P. aeruginosa biofilms [115]. A similar result was also found in B. multivorans biofilm except that a sharp decrease was observed when NPs were larger than 100 nm. Forier et al. [139] hypothesized that the size limit of channels inside B. multivorans biofilm may be more uniform than P. aeruginosa biofilm.
Polystyrene NPs with different surface charges but similar sizes (~ 200 nm) were used by Messiaen et al. [116] to study the mobility of these NPs inside Burkholderia cepacia complex (Bcc) biofilms. Positively charged NPs (~30 mV) were modified from fluorescent carboxylate-modified polystyrene FluoSpheres (~−48 mV) with N, N-dimethylethylenediamine (DMEDA). Single particle analysis revealed the sluggish movement of negatively charged NPs inside the Bcc biofilms. On the other hand, positively charged NPs were hindered by the negatively charged fibrous structures within the biofilm (hypothesized to be eDNA) [116].
4 Conclusion
Chronic infections are most typically a result of biofilm infections, which are extremely problematic to eradicate. Furthermore, antibiotic resistance is a pressing problem for the medical industry, and its impact is only worsened by biofilm bacterial infections. With the use of nanotechnology in antibiotic drug delivery, the impact of traditional antibiotics on biofilm infections can be improved due to targeted or localized high doses of the pharmaceuticals being delivered to the biofilm bacteria. Polymeric, liposomal, and silica NP drug delivery systems can provide a way to potentiate current antibiotics. Additionally, metal NPs may become a viable alternative to traditional antibiotics in treating infections or as delivery vehicles enabling photosensitizer-based or hyperthermia-based treatments. The integration of NPs into biomaterials to prevent bacterial adhesion and biofilm formation further extends the application of NPs [140]. NPs can be embedded into the polymer matrix or coated onto the surface [141], [142], [143]. A surface made of silver is suggested to be less effective than a coating incorporated with silver NPs, as the surface silver tends to be deactivated by protein anions [144]. Considering the extensive usage of polymer in the healthcare industry, the development of polymer/NP composite can relieve the pressure of hospital acquired infections by inhibiting the infection occurring on the surface made of plastics, such as catheters, which account for more than 80% of hospital-acquired urinary infections [145]. Future investigations of NPs in biofilm treatments may include mechanisms of anti-biofilm action, mechanisms of biofilm antibiotic resistance, passive/active targeting, stimulus-responsive NPs, NPs with novel killing mechanisms, aggregation under biological conditions, coating processes, and the scale-up of production processes. It is highly likely that NPs will play an indispensable role in combating biofilm infections, including their prevention, disruption, and removal in the medical industry.
References
[1] Costerton JW, Stewart PS, Greenberg EP. Bacterial biofilms: a common cause of persistent infections. Science. 1999, 284, 1318–1322.10.1126/science.284.5418.1318Search in Google Scholar PubMed
[2] Lewis K. Riddle of biofilm resistance. Antimicrob. Agents Chemother. 2001, 45, 999–1007.10.1128/AAC.45.4.999-1007.2001Search in Google Scholar PubMed PubMed Central
[3] Abidi SH, Sherwani SK, Siddiqui TR, Bashir A, Kazmi SU. Drug resistance profile and biofilm forming potential of Pseudomonas aeruginosa isolated from contact lenses in Karachi-Pakistan. BMC Ophthalmol. 2013, 13, 57.10.1186/1471-2415-13-57Search in Google Scholar PubMed PubMed Central
[4] Donlan RM. Biofilms and device-associated infections. Emerg. Infect. Dis. 2001, 7, 277–281.10.3201/eid0702.010226Search in Google Scholar PubMed PubMed Central
[5] Høiby N, Ciofu O, Bjarnsholt T. Pseudomonas aeruginosa biofilms in cystic fibrosis. Future Microbiol. 2010, 5, 1663–1674.10.2217/fmb.10.125Search in Google Scholar PubMed
[6] Santos APA, Watanabe E, de Andrade D. Biofilm on artificial pacemaker: fiction or reality? Arq. Bras. Cardiol. 2011, 97, e113–e120.10.1590/S0066-782X2011001400018Search in Google Scholar PubMed
[7] Song Z, Borgwardt L, Høiby N, Wu H, Sørensen TS, Borgwardt A. Prosthesis infections after orthopedic joint replacement: the possible role of bacterial biofilms. Orthop. Rev. 2013, 5, 65–71.10.4081/or.2013.e14Search in Google Scholar PubMed PubMed Central
[8] Tollefson DF, Bandyk DF, Kaebnick HW, Seabrook GR, Towne JB. Surface biofilm disruption. Enhanced recovery of microorganisms from vascular prostheses. Arch. Surg. Chic. Ill 1960. 1987, 122, 38–43.Search in Google Scholar
[9] Tran PL, Lowry N, Campbell T, Reid TW, Webster DR, Tobin E, Aslani A, Mosley T, Dertien J, Colmer-Hamood JA, Hamood AN. An organoselenium compound inhibits Staphylococcus aureus biofilms on hemodialysis catheters in vivo. Antimicrob. Agents Chemother. 2012, 56, 972–978.10.1128/AAC.05680-11Search in Google Scholar PubMed PubMed Central
[10] Wessman M, Bjarnsholt T, Eickhardt-Sørensen SR, Johansen HK, Homøe P. Mucosal biofilm detection in chronic otitis media: a study of middle ear biopsies from Greenlandic patients. Eur. Arch. Oto-Rhino-Laryngol. Off. J. Eur. Fed. Oto-Rhino-Laryngol. Soc. EUFOS Affil. Ger. Soc. Oto-Rhino-Laryngol. Head Neck Surg. 2015, 272, 1079–1085.10.1007/s00405-014-2886-9Search in Google Scholar PubMed
[11] Wu H, Moser C, Wang HZ, Høiby N, Song ZJ. Strategies for combating bacterial biofilm infections. Int. J. Oral Sci. 2015, 7, 1–7.10.1038/ijos.2014.65Search in Google Scholar PubMed PubMed Central
[12] Hajipour MJ, Fromm KM, Ashkarran AA, Jimenez de Aberasturi D, de Larramendi IR, Rojo T, Serpooshan V, Parak WJ, Mahmoudi M. Antibacterial properties of nanoparticles. Trends Biotechnol. 2012, 30, 499–511.10.1016/j.tibtech.2012.06.004Search in Google Scholar PubMed
[13] Spoering AL, Lewis K. Biofilms and planktonic cells of Pseudomonas aeruginosa have similar resistance to killing by antimicrobials. J. Bacteriol. 2001, 183, 6746–6751.10.1128/JB.183.23.6746-6751.2001Search in Google Scholar PubMed
[14] Kim MH, Yamayoshi I, Mathew S, Lin H, Nayfach J, Simon SI. Magnetic nanoparticle targeted hyperthermia of cutaneous Staphylococcus aureus infection. Ann. Biomed. Eng. 2013, 41, 598–609.10.1007/s10439-012-0698-xSearch in Google Scholar PubMed
[15] Andersson DI, Hughes D. Antibiotic resistance and its cost: is it possible to reverse resistance? Nat. Rev. Microbiol. 2010, 8, 260–271.10.1038/nrmicro2319Search in Google Scholar PubMed
[16] Ray PC, Khan SA, Singh AK, Senapati D, Fan Z. Nanomaterials for targeted detection and photothermal killing of bacteria. Chem. Soc. Rev. 2012, 41, 3193.10.1039/c2cs15340hSearch in Google Scholar PubMed
[17] Van Boeckel TP, Gandra S, Ashok A, Caudron Q, Grenfell BT, Levin SA, Laxminarayan R. Global antibiotic consumption 2000 to 2010: an analysis of national pharmaceutical sales data. Lancet Infect. Dis. 2014, 14, 742–750.10.1016/S1473-3099(14)70780-7Search in Google Scholar PubMed
[18] Habash MB, Park AJ, Vis EC, Harris RJ, Khursigara CM. Synergy of silver nanoparticles and aztreonam against Pseudomonas aeruginosa PAO1 biofilms. Antimicrob. Agents Chemother. 2014, 58, 5818–5830.10.1128/AAC.03170-14Search in Google Scholar PubMed
[19] Barenholz Y. Doxil®–the first FDA-approved nano-drug: lessons learned. J. Control. Release Off. J. Control. Release Soc. 2012, 160, 117–134.10.1016/j.jconrel.2012.03.020Search in Google Scholar
[20] Hans ML, Lowman AM. Biodegradable nanoparticles for drug delivery and targeting. Curr. Opin. Solid State Mater. Sci. 2002, 6, 319–327.10.1016/S1359-0286(02)00117-1Search in Google Scholar
[21] Lacy MK, Nicolau DP, Nightingale CH, Quintiliani R. The pharmacodynamics of aminoglycosides. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 1998, 27, 23–27.10.1086/514620Search in Google Scholar PubMed
[22] Gref R, Minamitake Y, Peracchia MT, Trubetskoy V, Torchilin V, Langer R. Biodegradable long-circulating polymeric nanospheres. Science 1994, 263, 1600–1603.10.1126/science.8128245Search in Google Scholar PubMed
[23] d’Angelo I, Casciaro B, Miro A, Quaglia F, Mangoni ML, Ungaro F. Overcoming barriers in Pseudomonas aeruginosa lung infections: engineered nanoparticles for local delivery of a cationic antimicrobial peptide. Colloids Surf. B Biointerfaces. 2015, 135, 717–725.10.1016/j.colsurfb.2015.08.027Search in Google Scholar PubMed
[24] Pelgrift RY, Friedman AJ. Nanotechnology as a therapeutic tool to combat microbial resistance. Adv. Drug Deliv. Rev. 2013, 65, 1803–1815.10.1016/j.addr.2013.07.011Search in Google Scholar PubMed
[25] Maeda H. Vascular permeability in cancer and infection as related to macromolecular drug delivery, with emphasis on the EPR effect for tumor-selective drug targeting. Proc. Jpn. Acad. Ser. B. 2012, 88, 53–71.10.2183/pjab.88.53Search in Google Scholar PubMed PubMed Central
[26] Radovic-Moreno A F, Lu TK, Puscasu VA, Yoon CJ, Langer R, Farokhzad OC. Surface charge-switching polymeric nanoparticles for bacterial cell wall-targeted delivery of antibiotics. ACS Nano. 2012, 6, 4279–4287.10.1021/nn3008383Search in Google Scholar PubMed PubMed Central
[27] Chen CP, Chen CT, Tsai T. Chitosan nanoparticles for antimicrobial photodynamic inactivation: characterization and in vitro investigation. Photochem. Photobiol. 2012, 88, 570–576.10.1111/j.1751-1097.2012.01101.xSearch in Google Scholar PubMed
[28] Chávez de Paz LE, Resin A, Howard KA, Sutherland DS, Wejse PL. Antimicrobial effect of chitosan nanoparticles on streptococcus mutans biofilms. Appl. Environ. Microbiol. 2011, 77, 3892–3895.10.1128/AEM.02941-10Search in Google Scholar PubMed PubMed Central
[29] Tan Y, Ma S, Liu C, Yu W, Han F. Enhancing the stability and antibiofilm activity of DspB by immobilization on carboxymethyl chitosan nanoparticles. Microbiol. Res. 2015, 178, 35–41.10.1016/j.micres.2015.06.001Search in Google Scholar PubMed
[30] Jena P, Mohanty S, Mallick R, Jacob B, Sonawane A. Toxicity and antibacterial assessment of chitosan-coated silver nanoparticles on human pathogens and macrophage cells. Int. J. Nanomedicine 2012, 7, 1805–1818.10.2147/IJN.S28077Search in Google Scholar PubMed PubMed Central
[31] Palanisamy NK, Ferina N, Amirulhusni AN, Mohd-Zain Z, Hussaini J, Ping LJ, Durairaj R. Antibiofilm properties of chemically synthesized silver nanoparticles found against Pseudomonas aeruginosa. J. Nanobiotechnology 2014, 12, 2.10.1186/1477-3155-12-2Search in Google Scholar PubMed PubMed Central
[32] Kalishwaralal K, BarathManiKanth S, Pandian SRK, Deepak V, Gurunathan S. Silver nanoparticles impede the biofilm formation by Pseudomonas aeruginosa and Staphylococcus epidermidis. Colloids Surf. B Biointerfaces 2010, 79, 340–344.10.1016/j.colsurfb.2010.04.014Search in Google Scholar PubMed
[33] Hetrick EM, Shin JH, Paul HS, Schoenfisch MH. Anti-biofilm efficacy of nitric oxide-releasing silica nanoparticles. Biomaterials 2009, 30, 2782–2789.10.1016/j.biomaterials.2009.01.052Search in Google Scholar PubMed PubMed Central
[34] Dong D, Thomas N, Thierry B, Vreugde S, Prestidge CA, Wormald PJ. Distribution and Inhibition of Liposomes on Staphylococcus aureus and Pseudomonas aeruginosa Biofilm. PLoS One 2015, 10, e0131806.10.1371/journal.pone.0131806Search in Google Scholar PubMed PubMed Central
[35] Baelo A, Levato R, Julián E, Crespo A, Astola J, Gavaldà J, Engel E, Mateos-Timoneda MA, Torrents E. Disassembling bacterial extracellular matrix with DNase-coated nanoparticles to enhance antibiotic delivery in biofilm infections. J. Controlled Release 2015, 209, 150–158.10.1016/j.jconrel.2015.04.028Search in Google Scholar PubMed
[36] Hua X, Tan S, Bandara HMHN, Fu Y, Liu S, Smyth HDC. Externally controlled triggered-release of drug from PLGA micro and nanoparticles. PLoS One 2014, 9, e114271.10.1371/journal.pone.0114271Search in Google Scholar PubMed PubMed Central
[37] Abdelghany SM, Quinn DJ, Ingram RJ, Gilmore BF, Donnelly RF, Taggart CC, Scott CJ. Gentamicin-loaded nanoparticles show improved antimicrobial effects towards Pseudomonas aeruginosa infection. Int. J. Nanomedicine 2012, 7, 4053–4063.10.2147/IJN.S34341Search in Google Scholar PubMed PubMed Central
[38] Cheow WS, Chang MW, Hadinoto K. The roles of lipid in anti-biofilm efficacy of lipid–polymer hybrid nanoparticles encapsulating antibiotics. Colloids Surf. Physicochem. Eng. Asp. 2011, 389, 158–165.10.1016/j.colsurfa.2011.08.035Search in Google Scholar
[39] Khan S, Alam F, Azam A, Khan AU. Gold nanoparticles enhance methylene blue-induced photodynamic therapy: a novel therapeutic approach to inhibit Candida albicans biofilm. Int. J. Nanomedicine 2012, 7, 3245–3257.10.2147/IJN.S31219Search in Google Scholar PubMed PubMed Central
[40] Subbiahdoss G, Sharifi S, Grijpma DW, Laurent S, van der Mei HC, Mahmoudi M, Busscher HJ. Magnetic targeting of surface-modified superparamagnetic iron oxide nanoparticles yields antibacterial efficacy against biofilms of gentamicin-resistant staphylococci. Acta Biomater. 2012, 8, 2047–2055.10.1016/j.actbio.2012.03.002Search in Google Scholar PubMed
[41] Taheri S, Baier G, Majewski P, Barton M, Förch R, Landfester K, Vasilev K. Synthesis and surface immobilization of antibacterial hybrid silver-poly(l-lactide) nanoparticles. Nanotechnology. 2014, 25, 305102.10.1088/0957-4484/25/30/305102Search in Google Scholar PubMed
[42] Shrestha A, Hamblin MR, Kishen A. Photoactivated rose bengal functionalized chitosan nanoparticles produce antibacterial/biofilm activity and stabilize dentin-collagen. Nanomedicine Nanotechnol. Biol. Med. 2014, 10, 491–501.10.1016/j.nano.2013.10.010Search in Google Scholar PubMed PubMed Central
[43] Taylor EN, Kummer KM, Durmus NG, Leuba K, Tarquinio KM, Webster TJ. Superparamagnetic iron oxide nanoparticles (SPION) for the treatment of antibiotic-resistant biofilms. Small 2012, 8, 3016–3027.10.1002/smll.201200575Search in Google Scholar PubMed
[44] Durmus NG, Taylor EN, Kummer KM, Webster TJ. Enhanced efficacy of superparamagnetic iron oxide nanoparticles against antibiotic-resistant biofilms in the presence of metabolites. Adv. Mater. 2013, 25, 5706–5713.10.1002/adma.201302627Search in Google Scholar PubMed
[45] Salunke GR, Ghosh S, Santosh Kumar RJ, Khade S, Vashisth P, Kale T, Chopade S, Pruthi V, Kundu G, Bellare JR, Chopade BA. Rapid efficient synthesis and characterization of silver, gold, and bimetallic nanoparticles from the medicinal plant Plumbago zeylanica and their application in biofilm control. Int. J. Nanomedicine 2014, 9, 2635–2653.10.2147/IJN.S59834Search in Google Scholar PubMed PubMed Central
[46] Ramasamy M, Lee JH, Lee J. Potent antimicrobial and antibiofilm activities of bacteriogenically synthesized gold-silver nanoparticles against pathogenic bacteria and their physiochemical characterizations. J. Biomater. Appl. 2016, 31, 366–378.10.1177/0885328216646910Search in Google Scholar PubMed
[47] Cai J, Huang H, Song W, Hu H, Chen J, Zhang L, Li P, Wu R, Wu C. Preparation and evaluation of lipid polymer nanoparticles for eradicating H. pylori biofilm and impairing antibacterial resistance in vitro. Int. J. Pharm. 2015, 495, 728–737.10.1016/j.ijpharm.2015.09.055Search in Google Scholar PubMed
[48] Stoodley P, Sauer K, Davies DG, Costerton JW. Biofilms as complex differentiated communities. Annu. Rev. Microbiol. 2002, 56, 187–209.10.1146/annurev.micro.56.012302.160705Search in Google Scholar PubMed
[49] Flemming HC, Wingender J. The biofilm matrix. Nat. Rev. Microbiol. 2010, 8, 623–633.10.1038/nrmicro2415Search in Google Scholar PubMed
[50] Cegelski L, Pinkner JS, Hammer ND, Cusumano CK, Hung CS, Chorell E, Aberg V, Walker JN, Seed PC, Almqvist F, Chapman MR, Hultgren SJ. Small-molecule inhibitors target Escherichia coli amyloid biogenesis and biofilm formation. Nat. Chem. Biol. 2009, 5, 913–919.10.1038/nchembio.242Search in Google Scholar PubMed PubMed Central
[51] Chorell E, Bengtsson C, Sainte-Luce Banchelin T, Das P, Uvell H, Sinha AK, Pinkner JS, Hultgren SJ, Almqvist F. Synthesis and application of a bromomethyl substituted scaffold to be used for efficient optimization of anti-virulence activity. Eur. J. Med. Chem. 2011, 46, 1103–1116.10.1016/j.ejmech.2011.01.025Search in Google Scholar PubMed PubMed Central
[52] Han Z, Pinkner JS, Ford B, Obermann R, Nolan W, Wildman SA, Hobbs D, Ellenberger T, Cusumano CK, Hultgren SJ, Janetka JW. Structure-based drug design and optimization of mannoside bacterial FimH antagonists. J. Med. Chem. 2010, 53, 4779–4792.10.1021/jm100438sSearch in Google Scholar PubMed PubMed Central
[53] Qin Z, Yang L, Qu D, Molin S, Tolker-Nielsen T. Pseudomonas aeruginosa extracellular products inhibit staphylococcal growth, and disrupt established biofilms produced by Staphylococcus epidermidis. Microbiol. Read. Engl. 2009, 155(Pt 7), 2148–2156.10.1099/mic.0.028001-0Search in Google Scholar PubMed
[54] Boyd A, Chakrabarty AM. Role of alginate lyase in cell detachment of Pseudomonas aeruginosa. Appl. Environ. Microbiol. 1994, 60, 2355–2359.10.1128/aem.60.7.2355-2359.1994Search in Google Scholar PubMed PubMed Central
[55] Hughes KA, Sutherland IW, Jones MV. Biofilm susceptibility to bacteriophage attack: the role of phage-borne polysaccharide depolymerase. Microbiol. Read. Engl. 1998, 144(Pt 11), 3039–3047.10.1099/00221287-144-11-3039Search in Google Scholar PubMed
[56] Kostakioti M, Hadjifrangiskou M, Hultgren SJ. Bacterial biofilms: development, dispersal, and therapeutic strategies in the dawn of the postantibiotic era. Cold Spring Harb. Perspect. Med. 2013, 3, a010306.10.1101/cshperspect.a010306Search in Google Scholar PubMed PubMed Central
[57] Walsh C. Molecular mechanisms that confer antibacterial drug resistance. Nature. 2000, 406, 775–781.10.1038/35021219Search in Google Scholar PubMed
[58] Brooks BD, Brooks AE. Therapeutic strategies to combat antibiotic resistance. Adv. Drug Deliv. Rev. 2014, 78, 14–27.10.1016/j.addr.2014.10.027Search in Google Scholar PubMed
[59] Walsh C, and Wright G. Introduction: antibiotic resistance. Chem. Rev. 2005, 105, 391–394.10.1021/cr030100ySearch in Google Scholar PubMed
[60] Stewart PS. Mechanisms of antibiotic resistance in bacterial biofilms. Int. J. Med. Microbiol. 2002, 292, 107–113.10.1078/1438-4221-00196Search in Google Scholar PubMed
[61] Anderl JN, Franklin MJ, Stewart PS. Role of antibiotic penetration limitation in Klebsiella pneumoniae biofilm resistance to ampicillin and ciprofloxacin. Antimicrob. Agents Chemother. 2000, 44, 1818–1824.10.1128/AAC.44.7.1818-1824.2000Search in Google Scholar PubMed PubMed Central
[62] Walters MC, Roe F, Bugnicourt A, Franklin MJ, Stewart PS. Contributions of antibiotic penetration, oxygen limitation, and low metabolic activity to tolerance of Pseudomonas aeruginosa biofilms to ciprofloxacin and tobramycin. Antimicrob. Agents Chemother. 2003, 47, 317–323.10.1128/AAC.47.1.317-323.2003Search in Google Scholar PubMed PubMed Central
[63] Taylor PK, Yeung ATY, Hancock REW. Antibiotic resistance in Pseudomonas aeruginosa biofilms: towards the development of novel anti-biofilm therapies. J. Biotechnol. 2014, 191, 121–130.10.1016/j.jbiotec.2014.09.003Search in Google Scholar PubMed
[64] Singh R, Paul D, Jain RK. Biofilms: implications in bioremediation. Trends Microbiol. 2006, 14, 389–397.10.1016/j.tim.2006.07.001Search in Google Scholar PubMed
[65] Umadevi S, Joseph NM, Kumari K, Easow JM, Kumar S, Stephen S, Srirangaraj S, Raj S. Detection of extended spectrum beta lactamases, ampc beta lactamases and metallobetalactamases in clinical isolates of ceftazidime resistant Pseudomonas Aeruginosa. Braz. J. Microbiol. 2011, 42, 1284–1288.10.1590/S1517-83822011000400006Search in Google Scholar PubMed PubMed Central
[66] Drenkard E. Antimicrobial resistance of Pseudomonas aeruginosa biofilms. Microbes Infect. 2003, 5, 1213–1219.10.1016/j.micinf.2003.08.009Search in Google Scholar PubMed
[67] Lewis K. Persister cells and the riddle of biofilm survival. Biochem. Biochemistry (Moscow). 2005, 70, 267–274.10.1007/s10541-005-0111-6Search in Google Scholar PubMed
[68] Dawson CC, Intapa C, Jabra-Rizk MA. ‘Persisters’: survival at the cellular level. PLoS Pathog. 2011, 7, e1002121.10.1371/journal.ppat.1002121Search in Google Scholar PubMed PubMed Central
[69] Høiby N, Bjarnsholt T, Givskov M, Molin S, Ciofu O. Antibiotic resistance of bacterial biofilms. Int. J. Antimicrob. Agents. 2010, 35, 322–332.10.1016/j.ijantimicag.2009.12.011Search in Google Scholar PubMed
[70] Davies DG, Parsek MR, Pearson JP, Iglewski BH, Costerton JW, Greenberg EP. The involvement of cell-to-cell signals in the development of a bacterial biofilm. Science 1998, 280, 295–298.10.1126/science.280.5361.295Search in Google Scholar PubMed
[71] Parsek MR, Greenberg EP. Sociomicrobiology: the connections between quorum sensing and biofilms. Trends Microbiol. 2005, 13, 27–33.10.1016/j.tim.2004.11.007Search in Google Scholar PubMed
[72] Rasamiravaka T, El Jaziri M. Quorum-sensing mechanisms and bacterial response to antibiotics in P. aeruginosa. Curr. Microbiol. 2016, 73, 747–753.10.1007/s00284-016-1101-1Search in Google Scholar PubMed
[73] Singh PK, Schaefer AL, Parsek MR, Moninger TO, Welsh MJ, Greenberg EP. Quorum-sensing signals indicate that cystic fibrosis lungs are infected with bacterial biofilms. Nature 2000, 407, 762–764.10.1038/35037627Search in Google Scholar PubMed
[74] Maseda H, Sawada I, Saito K, Uchiyama H, Nakae T, Nomura N. Enhancement of the mexAB-oprM efflux pump expression by a quorum-sensing autoinducer and its cancellation by a regulator, MexT, of the mexEF-oprN efflux pump operon in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2004, 48, 1320–1328.10.1128/AAC.48.4.1320-1328.2004Search in Google Scholar PubMed PubMed Central
[75] Rasamiravaka T, Jaziri ME. Quorum-sensing mechanisms and bacterial response to antibiotics in P. aeruginosa. Curr. Microbiol. 2016, 73, 747–753.10.1007/s00284-016-1101-1Search in Google Scholar
[76] De Jong WH, Borm PJ. Drug delivery and nanoparticles: applications and hazards. Int. J. Nanomed. 2008, 3, 133–149.10.2147/IJN.S596Search in Google Scholar
[77] Gao L, Giglio KM, Nelson JL, Sondermann H, Travis AJ. Ferromagnetic nanoparticles with peroxidase-like activity enhance the cleavage of biological macromolecules for biofilm elimination. Nanoscale 2014, 6, 2588–2593.10.1039/C3NR05422ESearch in Google Scholar PubMed PubMed Central
[78] Thukkaram M, Sitaram S., Kannaiyan SK, Subbiahdoss G. Antibacterial efficacy of iron-oxide nanoparticles against biofilms on different biomaterial surfaces. Int. J. Biomater. 2014, 2014, 716080.10.1155/2014/716080Search in Google Scholar PubMed PubMed Central
[79] Mohanty S, Mishra S, Jena P, Jacob B, Sarkar B, Sonawane A. An investigation on the antibacterial, cytotoxic, and antibiofilm efficacy of starch-stabilized silver nanoparticles. Nanomed. Nanotechnol. Biol. Med. 2012, 8, 916–924.10.1016/j.nano.2011.11.007Search in Google Scholar PubMed
[80] Jaiswal S, Bhattacharya K, McHale P, Duffy B. Dual effects of β-cyclodextrin-stabilised silver nanoparticles: enhanced biofilm inhibition and reduced cytotoxicity. J. Mater. Sci. Mater. Med. 2015, 26, 1–10.10.1007/s10856-014-5367-1Search in Google Scholar PubMed
[81] Cheow WS, Hadinoto K. Lipid-polymer hybrid nanoparticles with rhamnolipid-triggered release capabilities as anti-biofilm drug delivery vehicles. Particuology, 2012, 10, 327–333.10.1016/j.partic.2011.08.007Search in Google Scholar
[82] Sun L, Zhang C, Li P. Characterization, antibiofilm, and mechanism of action of novel PEG-stabilized lipid nanoparticles loaded with terpinen-4-ol. J. Agric. Food Chem. 2012, 60, 6150–6156.10.1021/jf3010405Search in Google Scholar PubMed
[83] Huang YC, Li RY, Chen JY, Chen JK. Biphasic release of gentamicin from chitosan/fucoidan nanoparticles for pulmonary delivery. Carbohydr. Polym. 2016, 138, 114–122.10.1016/j.carbpol.2015.11.072Search in Google Scholar PubMed
[84] Berney M, Hammes F, Bosshard F, Weilenmann HU, Egli T. Assessment and interpretation of bacterial viability by using the LIVE/DEAD BacLight Kit in combination with flow cytometry. Appl. Environ. Microbiol. 2007, 73, 3283–3290.10.1128/AEM.02750-06Search in Google Scholar PubMed PubMed Central
[85] Welch K, Cai Y, Strømme M. A method for quantitative determination of biofilm viability. J. Funct. Biomater. 2012, 3, 418–431.10.3390/jfb3020418Search in Google Scholar PubMed PubMed Central
[86] Peeters E, Nelis HJ, Coenye T. Comparison of multiple methods for quantification of microbial biofilms grown in microtiter plates. J. Microbiol. Methods 2008, 72, 157–165.10.1016/j.mimet.2007.11.010Search in Google Scholar PubMed
[87] Radzig MA, Nadtochenko VA, Koksharova OA, Kiwi J, Lipasova VA, Khmel IA. Antibacterial effects of silver nanoparticles on gram-negative bacteria: influence on the growth and biofilms formation, mechanisms of action. Colloids Surf. B Biointerfaces 2013, 102, 300–306.10.1016/j.colsurfb.2012.07.039Search in Google Scholar PubMed
[88] Murphy M, Ting K, Zhang X, Soo C, Zheng Z. Current development of silver nanoparticle preparation, investigation, and application in the field of medicine, current development of silver nanoparticle preparation, investigation, and application in the field of medicine. J. Nanomater. J. Nanomater. 2015, 2015, e696918.Search in Google Scholar
[89] Williams DF. On the mechanisms of biocompatibility. Biomaterials 2008, 29, 2941–2953.10.1016/j.biomaterials.2008.04.023Search in Google Scholar PubMed
[90] Gurunathan S, Han JW, Kwon DN, Kim JH. Enhanced antibacterial and anti-biofilm activities of silver nanoparticles against Gram-negative and Gram-positive bacteria. Nanoscale Res. Lett. 2014, 9, 373.10.1186/1556-276X-9-373Search in Google Scholar PubMed PubMed Central
[91] Kim JS, Kuk E, Yu KN, Kim JH, Park SJ, Lee HJ, Kim SH, Park YK, Park YH, Hwang CY, Kim YK, Lee YS, Jeong DH, Cho MH. Antimicrobial effects of silver nanoparticles. Nanomedicine 2007, 3, 95–101.10.1016/j.nano.2006.12.001Search in Google Scholar PubMed
[92] Su HL, Chou CC, Hung DJ, Lin SH, Pao IC, Lin JH, Huang FL, Dong RX, Lin JJ. The disruption of bacterial membrane integrity through ROS generation induced by nanohybrids of silver and clay. Biomaterials 2009, 30, 5979–5987.10.1016/j.biomaterials.2009.07.030Search in Google Scholar PubMed
[93] Gogoi SK, Gopinath P, Paul A, Ramesh A, Ghosh SS, Chattopadhyay A. Green fluorescent protein-expressing Escherichia coli as a model system for investigating the antimicrobial activities of silver nanoparticles. Langmuir 2006, 22, 9322–9328.10.1021/la060661vSearch in Google Scholar PubMed
[94] Sondi I, Salopek-Sondi B. Silver nanoparticles as antimicrobial agent: a case study on E. coli as amodel for Gram-negative bacteria. J. Colloid Interface Sci. 2004, 275, 177–182.10.1016/j.jcis.2004.02.012Search in Google Scholar PubMed
[95] Wilson M, Dobson J, Harvey W. Sensitization of oral bacteria to killing by low-power laser radiation. Curr. Microbiol. 1992, 25, 77–81.10.1007/BF01570963Search in Google Scholar PubMed
[96] Gupta AK, Gupta M. Synthesis and surface engineering of iron oxide nanoparticles for biomedical applications. Biomaterials 2005, 26, 3995–4021.10.1016/j.biomaterials.2004.10.012Search in Google Scholar PubMed
[97] Massart R. Preparation of aqueous magnetic liquids in alkaline and acidic media. IEEE Trans. Magn. 1981, 17, 1247–1248.10.1109/TMAG.1981.1061188Search in Google Scholar
[98] Salunkhe AB, Khot VM, Pawar SH. Magnetic hyperthermia with magnetic nanoparticles: a status review. Curr. Top. Med. Chem. 2014, 14, 572–594.10.2174/1568026614666140118203550Search in Google Scholar PubMed
[99] Lu AH, Salabas EL, Schüth F. Magnetic nanoparticles: synthesis, protection, functionalization, and application. Angew. Chem. Int. Ed. 2007, 46, 1222–1244.10.1002/anie.200602866Search in Google Scholar PubMed
[100] Yantasee W, Warner CL, Sangvanich T, Addleman RS, Carter TG, Wiacek RJ, Fryxell GE, Timchalk C, Warner MG. Removal of heavy metals from aqueous systems with thiol functionalized superparamagnetic nanoparticles. Environ. Sci. Technol. 2007, 41, 5114–5119.10.1021/es0705238Search in Google Scholar PubMed
[101] Kennedy J, Blair IS, McDowell DA, Bolton DJ. An investigation of the thermal inactivation of Staphylococcus aureus and the potential for increased thermotolerance as a result of chilled storage. J. Appl. Microbiol. 2005, 99, 1229–1235.10.1111/j.1365-2672.2005.02697.xSearch in Google Scholar PubMed
[102] Nguyen TK, Duong HTT, Selvanayagam R, Boyer C, Barraud N. Iron oxide nanoparticle-mediated hyperthermia stimulates dispersal in bacterial biofilms and enhances antibiotic efficacy. Sci. Rep. 2015, 5, 18385.10.1038/srep18385Search in Google Scholar PubMed PubMed Central
[103] Borcherding J, Baltrusaitis J, Chen H, Stebounova L, Wu CM, Rubasinghege G, Mudunkotuwa IA, Caraballo JC, Zabner J, Grassian VH, Comellas AP. Iron oxide nanoparticles induce Pseudomonas aeruginosa growth, induce biofilm formation, and inhibit antimicrobial peptide function. Environ. Sci. Nano 2014, 1, 123–132.10.1039/c3en00029jSearch in Google Scholar PubMed PubMed Central
[104] Masadeh MM, Karasneh GA, Al-Akhras MA, Albiss BA, Aljarah KM, Al-Azzam SI, Alzoubi KH. Cerium oxide and iron oxide nanoparticles abolish the antibacterial activity of ciprofloxacin against gram positive and gram negative biofilm bacteria. Cytotechnology 2015, 67, 427–435.10.1007/s10616-014-9701-8Search in Google Scholar PubMed PubMed Central
[105] Ramyadevi J, Jeyasubramanian K, Marikani A, Rajakumar G, Rahuman AA. Synthesis and antimicrobial activity of copper nanoparticles. Mater. Lett. 2012, 71, 114–116.10.1016/j.matlet.2011.12.055Search in Google Scholar
[106] Ghasemian E, Naghoni A, Rahvar H, Kialha M, Tabaraie B. Evaluating the effect of copper nanoparticles in inhibiting Pseudomonas aeruginosa and Listeria monocytogenes biofilm formation. Jundishapur J. Microbiol. 2015, 8, e17430.10.5812/jjm.17430Search in Google Scholar PubMed
[107] LewisOscar F, MubarakAli D, Nithya C, Priyanka R, Gopinath V, Alharbi NS, Thajuddin N. One pot synthesis and anti-biofilm potential of copper nanoparticles (CuNPs) against clinical strains of Pseudomonas aeruginosa. Biofouling 2015, 31, 379–391.10.1080/08927014.2015.1048686Search in Google Scholar PubMed
[108] Ren G, Hu D, Cheng EWC, Vargas-Reus MA, Reip P, Allaker RP. Characterisation of copper oxide nanoparticles for antimicrobial applications. Int. J. Antimicrob. Agents 2009, 33, 587–590.10.1016/j.ijantimicag.2008.12.004Search in Google Scholar PubMed
[109] Kruk T, Szczepanowicz K, Stefańska J, Socha RP, Warszyński P. Synthesis and antimicrobial activity of monodisperse copper nanoparticles. Colloids Surf. B Biointerfaces 2015, 128, 17–22.10.1016/j.colsurfb.2015.02.009Search in Google Scholar PubMed
[110] Dwidjosiswojo Z, Richard J, Moritz MM, Dopp E, Flemming HC, Wingender J. Influence of copper ions on the viability and cytotoxicity of Pseudomonas aeruginosa under conditions relevant to drinking water environments. Int. J. Hyg. Environ. Health 2011, 214, 485–492.10.1016/j.ijheh.2011.06.004Search in Google Scholar PubMed
[111] Tabrez S, Khan, Ahamed M, Al-Khedhairy A, Musarrat J. Biocidal effect of copper and zinc oxide nanoparticles on human oral microbiome and biofilm formation. Mater. Lett. 2013, 97, 67–70.10.1016/j.matlet.2013.01.085Search in Google Scholar
[112] Shankar S, Rhim JW. Effect of copper salts and reducing agents on characteristics and antimicrobial activity of copper nanoparticles. Mater. Lett. 2014, 132, 307–311.10.1016/j.matlet.2014.06.014Search in Google Scholar
[113] Shek PN, Yung BY, Stanacev NZ. Comparison between multilamellar and unilamellar liposomes in enhancing antibody formation. Immunology 1983, 49, 37–44.Search in Google Scholar PubMed
[114] Chang HI, Yeh MK. Clinical development of liposome-based drugs: formulation, characterization, and therapeutic efficacy. Int. J. Nanomedicine 2012, 7, 49–60.10.2147/IJN.S26766Search in Google Scholar PubMed PubMed Central
[115] Meers P, Neville M, Malinin V, Scotto AW, Sardaryan G, Kurumunda R, Mackinson C, James G, Fisher S, Perkins WR. Biofilm penetration, triggered release and in vivo activity of inhaled liposomal amikacin in chronic Pseudomonas aeruginosa lung infections. J. Antimicrob. Chemother. 2008, 61, 859–868.10.1093/jac/dkn059Search in Google Scholar PubMed
[116] Messiaen AS, Forier K, Nelis H, Braeckmans K, Coenye T. Transport of nanoparticles and tobramycin-loaded liposomes in Burkholderia cepacia complex biofilms. PLoS One 2013, 8, e79220.10.1371/journal.pone.0079220Search in Google Scholar PubMed PubMed Central
[117] Rabea EI, Badawy MET, Stevens CV, Smagghe G, Steurbaut W. Chitosan as antimicrobial agent: applications and mode of action. Biomacromolecules 2003, 4, 1457–1465.10.1021/bm034130mSearch in Google Scholar PubMed
[118] Machul A, Mikołajczyk D, Regiel-Futyra A, Heczko PB, Strus M, Arruebo M, Stochel G, Kyzioł A. Study on inhibitory activity of chitosan-based materials against biofilm producing Pseudomonas aeruginosa strains. J. Biomater. Appl. 2015, 30, 269–278.10.1177/0885328215578781Search in Google Scholar PubMed
[119] Ali SW, Rajendran S, Joshi M. Synthesis and characterization of chitosan and silver loaded chitosan nanoparticles for bioactive polyester. Carbohydr. Polym. 2011, 83, 438–446.10.1016/j.carbpol.2010.08.004Search in Google Scholar
[120] Del Carpio-Perochena A, Bramante CM, Duarte MAH, de Moura MR, Aouada FA, Kishen A. Chelating and antibacterial properties of chitosan nanoparticles on dentin. Restor. Dent. Endod. 2015, 40, 195–201.10.5395/rde.2015.40.3.195Search in Google Scholar PubMed PubMed Central
[121] de Salamanca AE, Diebold Y, Calonge M, García-Vazquez C, Callejo S, Vila A, Alonso MJ. Chitosan nanoparticles as a potential drug delivery system for the ocular surface: toxicity, uptake mechanism and in vivo tolerance. Investig. Opthalmology Vis. Sci. 2006, 47, 1416.10.1167/iovs.05-0495Search in Google Scholar PubMed
[122] Hassani Najafabadi A, Abdouss M, Faghihi S. Synthesis and evaluation of PEG-O-chitosan nanoparticles for delivery of poor water soluble drugs: ibuprofen. Mater. Sci. Eng. C 2014, 41, 91–99.10.1016/j.msec.2014.04.035Search in Google Scholar PubMed
[123] Mundargi RC, Babu VR, Rangaswamy V, Patel P, Aminabhavi TM. Nano/micro technologies for delivering macromolecular therapeutics using poly(d,l-lactide-co-glycolide) and its derivatives. J. Controlled Release 2008, 125, 193–209.10.1016/j.jconrel.2007.09.013Search in Google Scholar PubMed
[124] Jain RA. The manufacturing techniques of various drug loaded biodegradable poly(lactide-co-glycolide) (PLGA) devices. Biomaterials 2000, 21, 2475–2490.10.1016/S0142-9612(00)00115-0Search in Google Scholar PubMed
[125] Dinarvand R, Sepehri N, Manoochehri S, Rouhani H, Atyabi F. Polylactide-co-glycolide nanoparticles for controlled delivery of anticancer agents. Int. J. Nanomedicine 2011, 6, 877–895.10.2147/IJN.S18905Search in Google Scholar PubMed
[126] Iannitelli A, Grande R, Di Stefano A, Di Giulio M, Sozio P, Bessa LJ, Laserra S, Paolini C, Protasi F, Cellini L. Potential antibacterial activity of carvacrol-loaded poly(DL-lactide-co-glycolide) (PLGA) nanoparticles against microbial biofilm. Int. J. Mol. Sci. 2011, 12, 5039–5051.10.3390/ijms12085039Search in Google Scholar PubMed
[127] Song HY, Li Y. Can eradication rate of gastric Helicobacter pylori be improved by killing oral Helicobacter pylori? World J. Gastroenterol. 2013, 19, 6645–6650.10.3748/wjg.v19.i39.6645Search in Google Scholar PubMed
[128] Beyth N, Yudovin-Farber I, Perez-Davidi M, Domb AJ, Weiss EI. Polyethyleneimine nanoparticles incorporated into resin composite cause cell death and trigger biofilm stress in vivo. Proc. Natl. Acad. Sci. USA. 2010, 107, 22038–22043.10.1073/pnas.1010341107Search in Google Scholar
[129] Kho K, Cheow WS, Lie RH, Hadinoto K. Aqueous re-dispersibility of spray-dried antibiotic-loaded polycaprolactone nanoparticle aggregates for inhaled anti-biofilm therapy. Powder Technol. 2010, 203, 432–439.10.1016/j.powtec.2010.06.003Search in Google Scholar
[130] Chen CY, Nace GW, Irwin PL. A 6 x 6 drop plate method for simultaneous colony counting and MPN enumeration of Campylobacter jejuni, Listeria monocytogenes, and Escherichia coli. J. Microbiol. Methods 2003, 55, 475–479.10.1016/S0167-7012(03)00194-5Search in Google Scholar PubMed
[131] Herigstad B, Hamilton M, Heersink J. How to optimize the drop plate method for enumerating bacteria. J. Microbiol. Methods 2001, 44, 121–129.10.1016/S0167-7012(00)00241-4Search in Google Scholar PubMed
[132] Naghili H, Tajik H, Mardani K, Razavi Rouhani SM, Ehsani A, Zare P. Validation of drop plate technique for bacterial enumeration by parametric and nonparametric tests. Vet. Res. Forum 2013, 4, 179–183.Search in Google Scholar PubMed
[133] Zelver N, Hamilton M, Pitts B, Goeres D, Walker D, Sturman P, Heersink J. Measuring antimicrobial effects on biofilm bacteria: from laboratory to field. Methods Enzymol. 1999, 310, 608–628.10.1016/S0076-6879(99)10047-8Search in Google Scholar PubMed
[134] Ignarro LJ, Buga GM, Wood KS, Byrns RE, Chaudhuri G. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc. Natl. Acad. Sci. USA. 1987, 84, 9265–9269.10.1073/pnas.84.24.9265Search in Google Scholar PubMed PubMed Central
[135] Carpenter AW, Slomberg DL, Rao KS, Schoenfisch MH. Influence of scaffold size on bactericidal activity of nitric oxide releasing silica nanoparticles. ACS Nano 2011, 5, 7235–7244.10.1021/nn202054fSearch in Google Scholar PubMed PubMed Central
[136] Shin JH, Metzger SK, Schoenfisch MH. Synthesis of nitric oxide-releasing silica nanoparticles. J. Am. Chem. Soc. 2007, 129, 4612–4619.10.1021/ja0674338Search in Google Scholar PubMed
[137] Slomberg DL, Lu Y, Broadnax AD, Hunter RA, Carpenter AW, Schoenfisch MH. Role of size and shape on biofilm eradication for nitric oxide-releasing silica nanoparticles. ACS Appl. Mater. Interfaces 2013, 5, 9322–9329.10.1021/am402618wSearch in Google Scholar PubMed
[138] Seneviratne CJ, Leung KC, Wong CH, Lee SF, Li X, Leung PC, Lau CB, Wat E, Jin L. Nanoparticle-encapsulated chlorhexidine against oral bacterial biofilms. PLoS One 2014, 9, e103234.10.1371/journal.pone.0103234Search in Google Scholar PubMed PubMed Central
[139] Forier K, Messiaen AS, Raemdonck K, Nelis H, De Smedt S, Demeester J, Coenye T, Braeckmans K. Probing the size limit for nanomedicine penetration into Burkholderia multivorans and Pseudomonas aeruginosa biofilms. J. Controlled Release 2014, 195, 21–28.10.1016/j.jconrel.2014.07.061Search in Google Scholar PubMed
[140] Paladini F, Pollini M, Sannino A, Ambrosio L. Metal-based antibacterial substrates for biomedical applications. Biomacromolecules 2015, 16, 1873–1885.10.1021/acs.biomac.5b00773Search in Google Scholar PubMed
[141] Jones DS, Djokic J, Gorman SP. The resistance of polyvinylpyrrolidone-iodine-poly(-caprolactone) blends to adherence of Escherichia coli. Biomaterials 2005, 26, 2013–2020.10.1016/j.biomaterials.2004.06.001Search in Google Scholar PubMed
[142] Yuan Y, Ai F, Zang X, Zhuang W, Shen J, Lin S. Polyurethane vascular catheter surface grafted with zwitterionic sulfobetaine monomer activated by ozone. Colloids Surf. B Biointerfaces 2004, 35, 1–5.10.1016/j.colsurfb.2004.01.005Search in Google Scholar PubMed
[143] Zhang W, Zhang YH, Ji JH, Zhao J, Yan Q, Chu PK. Antimicrobial properties of copper plasma-modified polyethylene. Polymer 2006, 47, 7441–7445.10.1016/j.polymer.2006.08.057Search in Google Scholar
[144] Monteiro DR, Gorup LF, Takamiya AS, Ruvollo-Filho AC, de Camargo ER, Barbosa DB. The growing importance of materials that prevent microbial adhesion: antimicrobial effect of medical devices containing silver. Int. J. Antimicrob. Agents 2009, 34, 103–110.10.1016/j.ijantimicag.2009.01.017Search in Google Scholar PubMed
[145] Curtis LT. Prevention of hospital-acquired infections: review of non-pharmacological interventions. J. Hosp. Infect. 2008, 69, 204–219.10.1016/j.jhin.2008.03.018Search in Google Scholar PubMed PubMed Central
©2017 Walter de Gruyter GmbH, Berlin/Boston
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Articles in the same Issue
- Frontmatter
- In this issue
- Reviews
- Recent developments in the use of nanoparticles for treatment of biofilms
- Biomaterial-based nanoreactors, an alternative for enzyme delivery
- The role of defects and dimensionality in influencing the charge, capacitance, and energy storage of graphene and 2D materials
- Hydrogel based 3D carriers in the application of stem cell therapy by direct injection
- Magnetic targeting with superparamagnetic iron oxide nanoparticles for in vivo glioma
- Nanomaterials for cancer therapies
Articles in the same Issue
- Frontmatter
- In this issue
- Reviews
- Recent developments in the use of nanoparticles for treatment of biofilms
- Biomaterial-based nanoreactors, an alternative for enzyme delivery
- The role of defects and dimensionality in influencing the charge, capacitance, and energy storage of graphene and 2D materials
- Hydrogel based 3D carriers in the application of stem cell therapy by direct injection
- Magnetic targeting with superparamagnetic iron oxide nanoparticles for in vivo glioma
- Nanomaterials for cancer therapies