The function of lysosomes and their role in Parkinson’s disease
-
Friederike Zunke
Friederike Zunke studied Biochemistry (B.Sc.) and Biomedical Research (M.Res.) at the University of Kiel and St. George’s University of London, respectively. She obtained her PhD in Biochemistry in Prof. Paul Saftig’s group at the University of Kiel in 2015, and moved to Chicago to perform postdoctoral studies in Prof. Joseph Mazzulli’s laboratory. Since the end of 2016 she is independent junior group leader at the University of Kiel working on the molecular role of lysosomal function and protein aggregation pathways in Parkinson’s disease.



Abstract
Lysosomes are cellular organelles that are important for the degradation and recycling of various biomolecules. Specialized lysosomal membrane proteins, as well as soluble enzymes, are important for the efficient turn-over of lysosomal substrates. A deficiency in the degradative capacity of lysosomes leads to severe pathologies referred to as lysosomal storage disorders. There is increasing evidence for the importance of lysosomal function in neurodegenerative disorders, including Parkinson’s disease. One reason for this might be the vulnerability of neuronal cells. Since neurons do not undergo further cell division, non-degraded substrates accumulate in aging cells, causing a buildup of toxicity. Recent genomic screenings identified a number of lysosome-associated genes as potential risk factors for Parkinson’s disease, which are discussed in this review. Moreover, it is outlined how targeting lysosomal function might help in developing novel therapeutic strategies.
Zusammenfassung
Lysosomen sind membranumschlossene Zellorganelle, in denen lösliche Enzyme für den Abbau sowie das Recycling intrazellulärer als auch extrazellulärer Biomoleküle sorgen. Kommt es dagegen zu einer unvollständigen Degradation hat das schwerwiegende pathologische Konsequenzen und führt zu sogenannten lysosomalen Speichererkrankungen. Forschungsergebnisse der letzten Jahre deuten auf einen Zusammenhang zwischen lysosomaler Dysfunktion und dem Krankheitsverlauf neurodegenerativen Erkrankungen hin – so wie zum Beispiel beim Morbus Parkinson. Eine mögliche Erklärung hierfür ist, dass neuronale Zellen keine Zellteilung mehr durchlaufen und sich so über die Zeit lysosomale Substrate anhäufen. Interessanterweise zeigen genetische Untersuchungen von Parkinson Patienten eine Anhäufung lysosomaler Gene, welche als Risikofaktoren für die Erkrankung beschrieben und in diesem Review behandelt werden. Des Weiteren wird diskutiert, welche Rolle Lysosomen bei der Entwicklung neuartiger Therapien zur Behandlung der Parkinson Erkrankung spielen können.
Introduction
Recent studies have shown that lysosomal dysfunction is not only linked to rare lysosomal storage disorders, but also to age-dependent neurodegenerative diseases, such as Parkinson’s disease (Fraldi et al., 2016). Whereas most cases of Parkinson’s disease are idiopathic (~90 %), meaning they do not have a clear heritable genetic link, the remaining ~10 % of Parkinson’s disease cases are familial and show Mendelian inheritance of affected genetic variants (Kalia and Lang, 2015). In order to understand the underlying molecular disease pathways, single genetic variants are of particular value for scientific studies. Recent genetic meta-analyses of Parkinson’s patients (both familial and idiopathic) reveal enrichment in genetic risk loci related to lysosomal homeostasis (Chang et al., 2017; Robak et al., 2017). Hence, the majority of Parkinson’s disease-associated genes are linked to lysosomal protein trafficking or lysosomal function (Klein and Mazzulli, 2018). The following review will give an overview of the composition and function of lysosomes in general, their role in neurodegeneration with focus on Parkinson’s disease, and discuss potential new treatment strategies targeting lysosomal proteins or pathways.
The composition and function of lysosomes
Lysosomes were first described in the 1950 s by Christian DeDuve, and are membrane-surrounded cellular organelles ubiquitously found within the cytosol of mammalian cells (DeDuve et al., 1955). Lysosomes contain specialized membrane proteins, as well as luminal hydrolytic enzymes, that can break down many kinds of biomolecules, including intracellular and extracellular substrates. The importance of lysosomal function is highlighted by a number of diseases called lysosomal storage disorders (LSDs), that result from defects in soluble lysosomal proteins and proteins of the lysosomal membrane (Eskelinen et al., 2003).
Importantly, it has been realized that lysosomes are not only degradative organelles, but also play a pivotal role in cell metabolism. Hence, they are involved in processes of cell signaling, repair of the plasma membrane, defense against pathogens, cholesterol metabolism, cell death, and energy metabolism (Schulze and Sandhoff, 2011; Settembre et al., 2013). Lysosomes emerge from the fusion of endosomes, which are specialized vesicles with an acidic pH. The pH within the lysosomes reaches as low as pH 4.5–5.0, and is mainly mediated by the ATP-dependent vacuolar(v)-type H+ ATPase (Ohkuma et al., 1982) (Figure 1).
Lysosomal enzymes reach lysosomes via the secretory pathway, which means that they are synthesized in the endoplasmic reticulum (ER) as membrane-integrated or soluble glycoproteins and pass through the Golgi apparatus. Since the majority of lysosomal proteins are transported to lysosomes by the mannose-6-phosphate (M6P)-dependent pathway, proteins are tagged with an M6P recognition marker. This terminal residue is then recognized by M6P-receptors, which mediate protein delivery to lysosomes via clathrin coated vesicles (Kornfeld, 1992). It must be noted that M6P-independent lysosomal transport mechanisms also exist. Moreover, some lysosomal proteins can escape the lysosomal targeting pathways. They can be transported to the plasma membrane or outside the cell, where they are taken up and transported back to lysosomes by a process called endocytosis (Janvier and Bonifacino, 2005).
To date, 60 different lysosomal enzymes have been found to be responsible for the degradation of various substrates (Settembre et al., 2013). Depending on their degradation products, lysosomal enzymes are classified as proteases, nucleases, glucosidases, phosphatases, polysaccharide hydrolyzing enzymes or lipid degrading enzymes (Bainton, 1981). All require an acidic environment for optimal enzymatic function (De Duve and Beaufay, 1959).
The lysosomal membrane contains highly glycosylated membrane proteins, which contribute to the stability of the membrane, as well as the luminal glycocalyx (Schwake et al., 2013). Most abundant in the lysosomal membrane, and thus important for its integrity, are the lysosomal associated membrane proteins type-1 and -2 (LAMP-1, LAMP-2), and the lysosomal integral membrane proteins type-1 and -2 (LIMP-1, LIMP-2/SCARB2). Interestingly, the LIMP-2 protein has not only been shown to be important for the integrity of the lysosomal membrane, but also for the M6P-independent lysosomal transport of the lysosomal enzyme β-glucocerebrosidase (GCase) (Blanz et al., 2015; Reczek et al., 2007; Zunke et al., 2016).
Furthermore, various channel proteins and small molecule transporters, like the transmembrane protein 175 (TMEM175), a potassium channel, or the sialic acid transporter, Sialin, are major integral components of the lysosomal membrane (Eskelinen et al., 2003). Additionally, transporter proteins, like the lysosomal cholesterol transporter Niemann-Pick Type C1 (NPC1), are found in the lysosomal membrane and play an important role in cellular cholesterol homeostasis (Infante et al., 2008) (Figure 1).
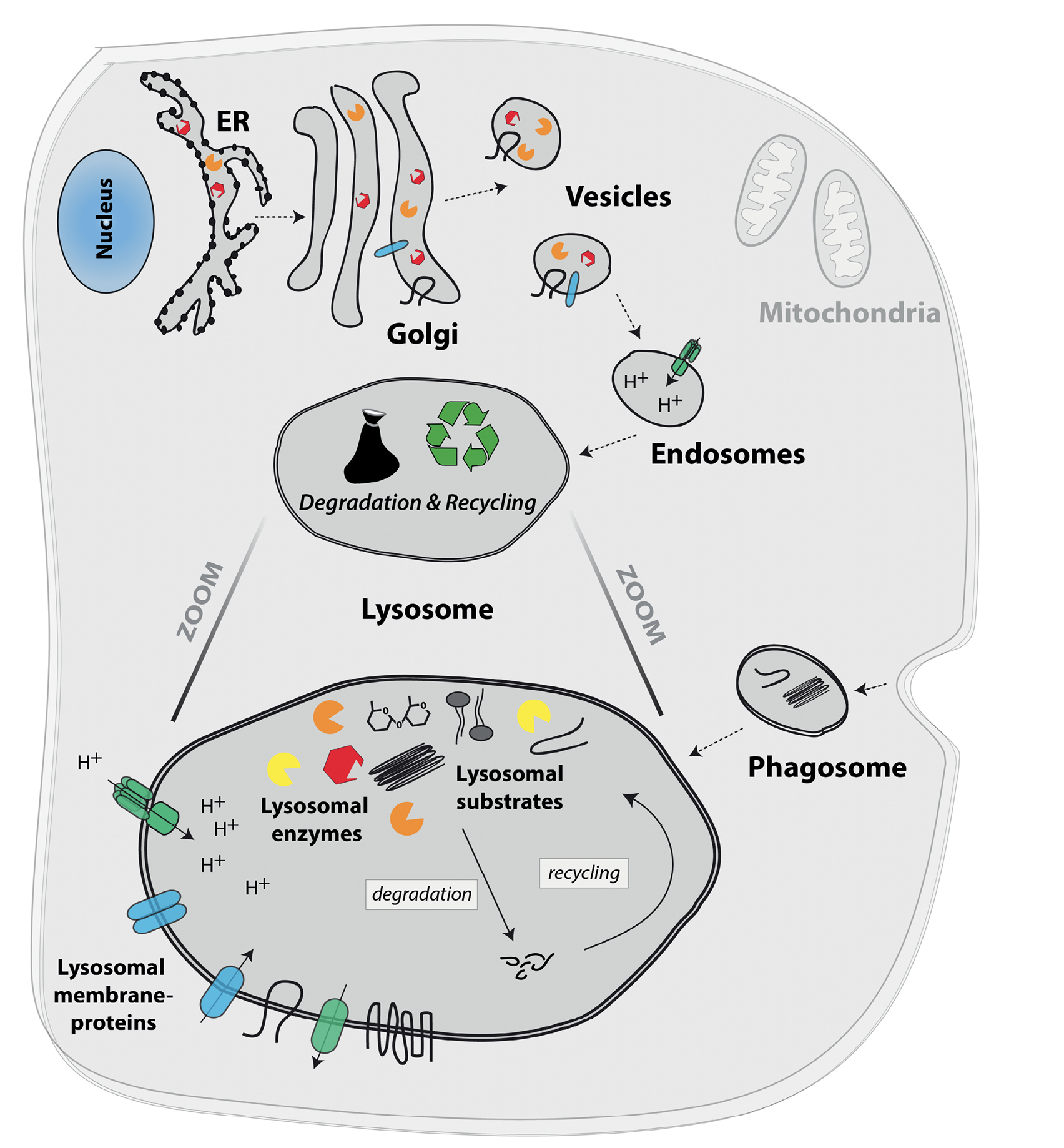
The eukaryotic cell and its organelles, highlighting the lysosome and its components.
The nucleus contains the majority of the cell’s genetic material and is the location of the DNA transcription process (DNA → RNA). Further protein translation (RNA → protein) is mediated at free ribosomes within the cytosol (not shown) or at ribosomes located at membranes of the endoplasmic reticulum (ER; indicated as black dots on ER membrane). The Golgi apparatus (Golgi) is important for further protein modification and sorting. Vesicles originating from the Golgi compartment can fuse with intracellular organelles, like the lysosome, or with the cell membrane. Mitochondria are the ‘powerhouse’ of the cell, generating energy by performing cellular respiration and producing ATP. The lysosome is the cell’s degradative organelle and arises from the fusion of Golgi-derived vesicles with endosomes, as indicated by dotted arrows. Lysosomes are not only responsible for degradation and recycling, but are also involved in pathogen defense, cell death, and energy metabolism. Their acidic pH (pH 4.5–5.0) is mainly maintained by the vacuolar-type H+ ATPase, which pumps H+ ions via the lysosomal membrane. A total of around 50 specialized lysosomal membrane proteins (blue and green molecules) are not only important for the maintenance and stability of the organelle, but also for the formation of the lysosomal glycocalyx at the inside of lysosomes, lysosomal protein transport, and the transport and exchange of biomolecules through the lysosomal membrane (mediated by a variety of channel and transporter proteins). Lysosomal substrates comprise a variety of intracellular as well as extracellular biomolecules including proteins, peptides, lipids, nucleic acids and carbohydrates. Substrates enter the lysosome via macro-, micro- and chaperone-mediated autophagy, as well as by fusion of lysosomes with phagosomes, which comprise extracellular material (indicated by dotted arrows). Around 60 different lysosomal enzymes (yellow, orange and red symbols) are responsible for the breakdown and recycling of specific substrates (shown in black and gray within the lysosome). These enzymes require an acidic pH and include proteases, nucleases, glucosidases, phosphatases, polysaccharide hydrolyzing enzymes, and lipid degrading enzymes.
Lysosomal dysfunction and neurodegenerative diseases
Genetic variants within genes encoding for lysosomal proteins resulting in less or non-functional protein (loss-of-function mutation) lead to lysosomal dysfunction and buildup of lysosomal substrates. This disease family is called lysosomal storage disorders (LSDs) and comprises over 70 distinct diseases, which in total have an incidence of ~1:7,000 live births (Gieselmann, 1995). The majority of LSDs are autosomal recessive and are caused by severe loss-of-function mutations within lysosomal trafficking components, lysosomal enzymes or membrane proteins (Platt et al., 2018). For instance, the most common LSD – Gaucher disease – is caused by mutations within the lysosomal enzyme GCase (gene name: GBA1). The resulting enzymatic dysfunction leads to the lysosomal accumulation of the non-degraded substrate glucosylceramide (a glycolipid), causing an enlargement of organs (organomegaly), anemia and bone malformation as well as, in some cases, neurological dysfunction (Pastores, 1997).
In a group of LSDs called neuronal ceroid lipofuscinosis (NCL), a lipopigment (lipofuscin) aggregates within lysosomal structures, resulting in a progressive loss of motor and cognitive abilities. NCLs are classified into ten different types caused by mutations within lysosomal enzymes – such as cathepsin D – but also membrane proteins (Bennett and Rakheja, 2013).
Interestingly, neurodegeneration is observed in nearly all LSDs, which emphasizes the importance of lysosomal degradation within neuronal cell homeostasis (Fraldi et al., 2016). There is increasing evidence for the contribution of lysosomal dysfunction in age-related neurodegenerative diseases, such as Huntington’s, Alzheimer’s and Parkinson’s diseases (Klein and Mazzulli, 2018; Pitcairn et al., 2019). The efficient degradation of lysosomal substrates (proteins, lipids, carbohydrates, etc.) is essential for neuronal survival, since neurons are particularly sensitive to alterations in the lysosomal degradation pathway. This is probably due to their post-mitotic state, since non-degraded lysosomal substrates will not be diluted by cell division, but accumulate over time until they reach critical concentrations. Studies have shown that abnormal protein accumulation can cause neurotoxicity and induce neuronal cell death (Hara et al., 2006; Komatsu et al., 2006). Since our brain cannot regenerate this cell type, the progressive loss of neurons results in neuropathology. Interestingly, a decrease in lysosomal activity could also be measured in postmortem brain samples derived from Parkinson’s disease patients (Dehay et al., 2010).
Parkinson’s disease is a progressive neurodegenerative disorder that affects about 1–2 % of the population above the age of 65. Parkinson’s disease symptoms comprise movement abnormalities, including tremor, bradykinesia (slowness of movement), and muscle rigidity (Kalia and Lang, 2015). During the course of the disease, non-motor symptoms may also arise, consisting of dementia, behavioral and digestive problems, as well as depression (Chaudhuri et al., 2006). These clinical manifestations result from the specific loss of neurons that produce the neurotransmitter dopamine. These so-called dopaminergic neurons are located within the substantia nigra, an area of the midbrain (Davie, 2008). Moreover, a histological hallmark of Parkinson’s disease is the intracellular aggregation of the cytosolic protein α-synuclein, which is a major component of Lewy body inclusions found in post-mortem brains of Parkinson’s disease patients (Baba et al., 1998; Braak and Del Tredici, 2017; Xia et al., 2008). It has been shown that intracellular α-synuclein aggregation leads to pathological manifestations and conveys cell toxicity (Lashuel and Lansbury, 2006; Lashuel et al., 2002). Importantly, accumulation of α-synuclein is a common feature observed in many LSDs (Shachar et al., 2011), further emphasizing the strong interplay between lysosomal function and α-synuclein aggregation.
The effect of lysosomal dysfunction on α-synuclein aggregation
α-Synuclein is a small cytosolic protein consisting of 140 amino acids encoded by the SNCA gene, and is (under physiological conditions) mainly found in the pre-synaptic terminals of neurons, where it is involved in synaptic neurotransmitter release and synaptic plasticity (George, 2002; Sulzer and Edwards, 2019). Recent studies further illuminate the relationship between lysosomal dysfunction and α-synuclein aggregation. Physiological α-synuclein can be degraded within lysosomes, where lysosomal cathepsins (CTSD and CTSB) are most likely involved in its degradation (Cuervo et al., 2004; Cullen et al., 2009; McGlinchey and Lee, 2015). Since α-synuclein is a very abundant and aggregation-prone protein, even subtle elevations in its concentration within the cell may drive the protein into pathological aggregates (Giasson et al., 1999). Hence, impairments in the lysosomal system have been shown to affect α-synuclein levels and aggregation. Moreover, multiple studies have demonstrated that metabolites like glycolipids that accumulate under lysosomal impairment can specifically interact with α-synuclein and further induce its aggregation (Mazzulli et al., 2011; Suzuki et al., 2015; Taguchi et al., 2017) (Figure 2). This process is thought to be mediated through a toxic structural conversion of α-synuclein oligomers resulting in amyloid fibril formation (Zunke et al., 2018) (Figure 2). Likewise, intra-lysosomal cholesterol may be an important modulator in Parkinson’s disease, as it has been shown to directly induce α-synuclein fibrilization (Bosco et al., 2006). Taken together, molecular data indicate that lysosomal storage of certain metabolites influences α-synuclein structure and aggregation capacity. This gives rise to the selective relationship between lysosomal dysfunction and α-synuclein aggregation, similar to that observed in Parkinson’s disease.
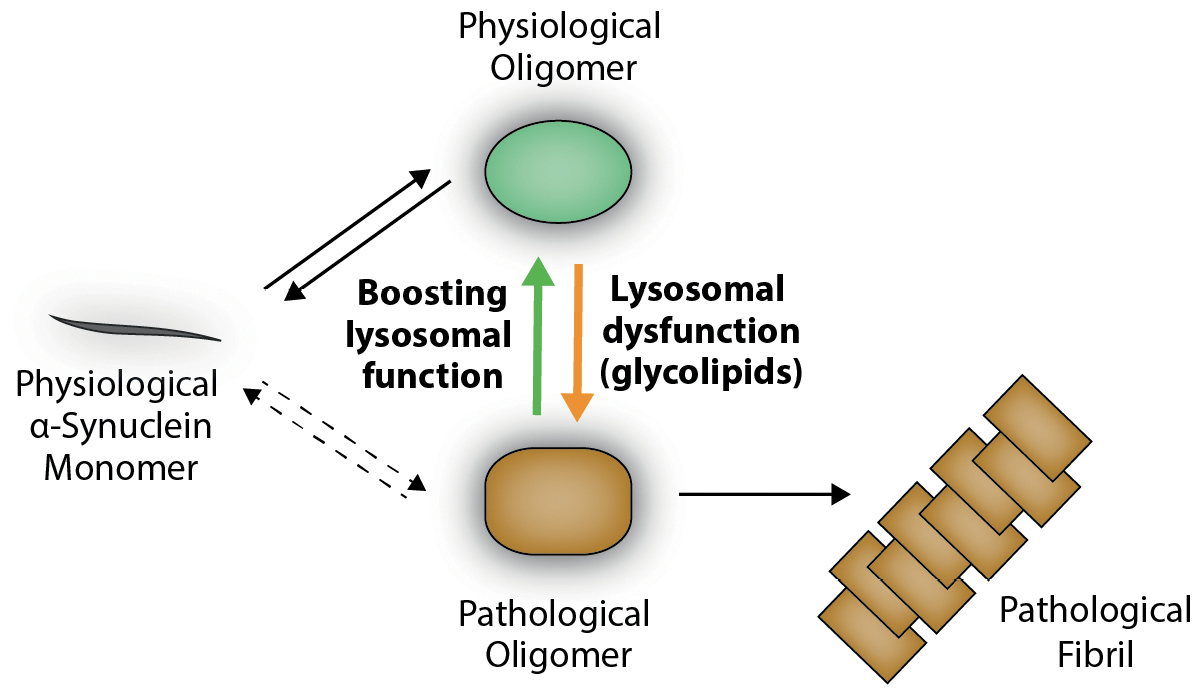
Schematic overview of α-synuclein aggregation.
Under physiological conditions, α-synuclein mainly exists as a monomer (gray), but can also form physiological oligomers (green) within the cytosol. Lysosomal dysfunction and aggregation of substrates like the GCase-substrate glucosylceramide (a glycolipid) have been shown to interfere with α-synuclein within lysosomes. This interaction of the protein with lysosomal glycolipids results in a structural change in the protein oligomer (orange). These pathological oligomers (orange) have been shown to be neurotoxic and induce pathological fibril formation (orange fibril), which is enhanced by the acidic pH within lysosomes. Interestingly, boosting lysosomal function or decreasing glycolipid levels has been shown to reverse pathological oligomers (orange) back to their physiological state (green). This molecular mechanism is utilized in substrate reduction therapy or by application of small compounds activating substrate-degrading enzymes (e. g. GCase) (modified from Riederer et al., 2019; Zunke et al., 2018).
Lysosomal risk factors in Parkinson’s disease
Over the years, numerous molecular, clinical and genetic studies have emphasized a central role for lysosomal pathways and proteins in the pathogenesis of Parkinson’s disease (Kalia and Lang, 2015; Klein and Mazzulli, 2018; Mazzulli et al., 2011; Pitcairn et al., 2019). Importantly, genetic meta-analyses of Parkinson’s disease patients reveal enrichment in genetic risk loci related to lysosomal function (Chang et al., 2017; Robak et al., 2017; Sidransky et al., 2009). Among these genetic risk factors are lysosomal hydrolases, such as GCase (GBA1), galactosylceramidase (GALC), sphingomyelin phosphodiesterase 1 (SMPD1), N-acylsphingosine Amidohydrolase 1 (ASAH1), and N-acetyl-glucosaminidase (NAGLU), as well as the two cathepsins B and D (CTSB, CTSD), which have been shown to degrade α-synuclein (Table 1). All enzymes degrade different lysosomal substrates comprised of various lipids (glycolipids, sphingomyelin, sphingosine) and proteins. Additionally, lysosomal membrane proteins have been added to the list of Parkinson’s disease risk genes, including the lysosomal integral membrane protein type-2 (LIMP-2), which is the lysosomal transport receptor of GCase, the transmembrane protein 175 (TMEM175) and the lysosome-associated membrane protein 3 (LAMP3). Furthermore, lysosomal transporter and channel proteins have been identified as risk genes and comprise cation-transporting ATPase 13A2 (ATP13A2), the proton pump ATPase H+ transporting V0 subunit A1 (ATP6V0A1), which is important for the regulation of lysosomal pH, the sialic acid exporter Sialin (SLC17A5), and the cholesterol transporter Niemann-Pick C protein (NPC1) (Table 1). Moreover, it is no surprise that two vacuolar protein sorting-associated proteins (VPS35/13C), that play an important role in vesicle-mediated protein trafficking, as well as two ras-related proteins (Rab7L1/39) crucial for vesicle fusion, are found on the list of lysosome-associated risk factors for Parkinson’s disease.
Of the lysosomal risk genes, GBA1 (GCase) has the highest prevalence and thus has been most intensively studied to date. The general mechanistic understanding is that slowed lysosomal digestion of GCase-substrates, including the glycolipid glucosylceramide, interferes with α-synuclein and accelerates its accumulation (Mazzulli et al., 2011; Zunke et al., 2018) (Figure 2). Data from genome studies indicate that mutations in GBA1 are linked to a ~20-fold increase in the risk of developing Parkinson’s disease (Beavan and Schapira, 2013). Additionally, genetic studies show that 5–10 % of Parkinson’s disease patients carry mutations in GBA1 (Lesage et al., 2011; McNeill et al., 2012; Sidransky et al., 2009).
For most other identified genetic risk factors shown in Table 1 there is still a lack of knowledge about their contribution to molecular disease mechanisms and disease progression. Future studies, for instance in induced-pluripotent stem cell (iPSC) derived patient neurons (Zunke and Mazzulli, 2019), will shed light on the impact of some of these pathology-associated genes. This will help further our understanding of Parkinson’s disease pathology and give rise to novel therapeutic approaches.
Lysosomal genes associated with Parkinson’s disease.
|
Gene name |
Protein name |
Function |
Reference |
|
Lysosomal enzymes |
|||
|
GBA1 |
β-Glucocerebrosidase (GCase) |
Degradation of lysosomal sphingolipids, mainly glucosylceramide |
(Chang et al., 2017; Nalls et al., 2014; Robak et al., 2017; Sidransky et al., 2009) |
|
GALC |
Galactocerebrosidase |
Degradation of lysosomal sphingolipids, mainly galactosylceramide |
(Chang et al., 2017) |
|
SMPD1 |
Sphingomyelin phosphodiesterase 1 |
Degradation of sphingomyelin |
(Gan-Or et al., 2015; Gan-Or et al., 2013; Robak et al., 2017) |
|
ASAH1 |
N-Acylsphingosine Amidohydrolase 1 |
Degradation of ceramide into sphingosine and free fatty acid |
(Robak et al., 2017) |
|
CTSB |
Cathepsin B |
Cysteine protease, involved in degradation of various substrates including α-synuclein |
(Chang et al., 2017) |
|
CTSD |
Cathepsin D |
Aspartyl protease, involved in degradation of various substrates including α-synuclein |
(Robak et al., 2017) |
|
NAGLU |
N-Acetylglucosaminidase |
Hydrolysis of terminal non-reducing N-acetyl-D-glucosamine |
(Winder-Rhodes et al., 2012) |
|
Lysosomal membrane proteins |
|||
|
TMEM175 |
Transmembrane protein 175 |
Potassium channel |
(Chang et al., 2017; Nalls et al., 2014) |
|
ATP13A2 |
Cation-transporting ATPase 13A2 |
Cation transporter |
(Ramirez et al., 2006) |
|
ATP6V0A1 |
ATPase H+ transporting V0 subunit A1 |
Component of the V-ATPase, acidification of lysosomes |
(Chang et al., 2017) |
|
LAMP3 |
Lysosome-associated membrane protein 3 |
Type I transmembrane glycoprotein, lysosomal glycocalyx |
(Pihlstrom et al., 2013; Simon-Sanchez et al., 2009) |
|
SCARB2 |
Lysosomal integral membrane protein type-2 (LIMP2) |
Type III transmembrane glycoprotein, lysosomal glycocalyx, lysosomal transport of GCase |
(Hopfner et al., 2011) |
|
SLC17A5 |
Sialin |
H+-coupled sialic acid exporter |
(Robak et al., 2017) |
|
NPC1 |
Niemann-Pick C1 protein |
Lysosomal cholesterol transporter |
(Kluenemann et al., 2013) |
|
Proteins involved in lysosomal trafficking/autophagy |
|||
|
VPS35 |
Vacuolar protein sorting-associated protein 35 |
Trafficking machinery, retromer complex component |
(Vilarino-Guell et al., 2011; Zimprich et al., 2011) |
|
VPS13C |
Vacuolar protein sorting-associated protein 13C |
Trafficking machinery, mitophagy, mitochondrial depolarization |
(Lesage et al., 2015) |
|
Rab7L1 |
Ras-related protein Rab-7L1 |
Retrograde trafficking pathway for recycling proteins |
(MacLeod et al., 2013; Nalls et al., 2014) |
|
Rab39B |
Ras-related protein Rab-39B |
Trafficking machinery, autophagy |
(Lesage et al., 2015; Mata et al., 2015) |
|
GAK |
Cyclin G-associated kinase |
Involved in the uncoating of clathrin-coated vesicles |
(Dumitriu et al., 2011) |
|
KAT8 |
Lysine acetyltransferase 8 |
Modulator of autophagic flux |
(Chang et al., 2017) |
Therapeutic approaches targeting lysosomes
As highlighted above, dysfunction in lysosomal pathways not only causes LSDs, but also plays a central role in neurodegenerative disorders, like Parkinson’s disease. A number of treatment strategies aiming to boost lysosomal function have been identified or are under development for LSDs, and may also provide novel opportunities for treating Parkinson’s disease. Considering the importance of cellular degradation in Parkinson’s disease etiology, strategies to enhance lysosomal function involve: (a) enzyme replacement to substitute dysfunctional enzymes; (b) chaperones to stabilize unstable or misfolded enzymes in order to increase their lysosomal activity; (c) small molecules that act as direct allosteric activators on lysosomal enzymes; (d) substrate reduction therapies in order to decrease lysosomal load; (e) cell therapies to replace injured cells; (f) gene therapy (Klein and Futerman, 2013).
The most common treatment strategy for certain LSDs is the administration of functional recombinant enzymes by repetitive infusions (enzyme replacement therapy) (Beck, 2018). Although this treatment has been shown to be effective in patients, enzyme replacement therapy has several disadvantages. First of all, the production of enzymes is very expensive and, secondly, recombinant enzymes are usually large proteins and thus difficult to transport across the blood-brain barrier. Hence, most enzyme replacement therapies are able to improve non-neuronopathic, but not neurological pathologies. Compared to enzyme replacement therapy, small activators of lysosomal enzymes have the potential to cross the blood-brain barrier and thus are able to treat neurological symptoms. Various small activators of lysosomal enzymes, including chaperones for GCase, are under development or already approved as drugs for the treatment of LSDs (Beck, 2018).
Interestingly, some of the compounds applied for LSDs are now tested for their effectiveness in neurodegenerative disorders, including Parkinson’s disease. Recent studies utilizing small activators/chaperones of the lysosomal enzyme GCase have been shown to improve Parkinson’s disease pathology by reducing α-synuclein aggregation in iPSC-derived neurons of Parkinson’s patients (Mazzulli et al., 2016), as well as in a transgenic Parkinson’s disease mouse model (Migdalska-Richards et al., 2016). The effect of GCase activators is due to increased enzymatic turn-over and a reduction in glucosylceramide, which has been shown to interfere with α-synuclein, resulting in neurotoxic oligomeric conformers (Zunke et al., 2018) (Figure 2: conversion from physiological (green) to pathological (orange) oligomer). Hence, a reduction in the glycolipid decreases the amount of toxic α-synuclein forms and thus α-synuclein-induced pathology (Figure 2: reversal of toxic form (orange) to physiological form (green)).
A similar reduction in α-synuclein aggregation could be observed after substrate reduction therapy. A small blood-brain-barrier-permeable molecule reducing the synthesis of the GCase-substrate glucosylceramide has been shown to reduce α-synuclein levels in Parkinson’s iPSC neurons (Zunke et al., 2018), as well as to improve cognitive symptoms in Parkinson’s disease mice (Sardi et al., 2017). Although substrate reduction is already in clinical use for some LSDs (e. g. Gaucher disease), this treatment option is still under investigation in clinical phases for Parkinson’s disease patients.
Likewise, therapies aiming to lower levels of other lysosomal molecules that might interfere with protein aggregation have shown to be beneficial for neurodegenerative disorders. Accordingly, reducing cholesterol levels in a mouse model of Parkinson’s disease also revealed a decrease in α-synuclein aggregation (Bar-On et al., 2006).
In summary, targeting lysosomal pathways in Parkinson’s disease pathology provides new possibilities to treat this progressive neurological disorder, for which no curative treatment strategy currently exists. The molecular and genetic evidence reviewed here will help us to further understand cellular disease mechanisms and direct future drug development.
About the author

Friederike Zunke studied Biochemistry (B.Sc.) and Biomedical Research (M.Res.) at the University of Kiel and St. George’s University of London, respectively. She obtained her PhD in Biochemistry in Prof. Paul Saftig’s group at the University of Kiel in 2015, and moved to Chicago to perform postdoctoral studies in Prof. Joseph Mazzulli’s laboratory. Since the end of 2016 she is independent junior group leader at the University of Kiel working on the molecular role of lysosomal function and protein aggregation pathways in Parkinson’s disease.
Funding
Funder Name: Deutsche Forschungsgemeinschaft, Funder Id: http://dx.doi.org/10.13039/501100001659, Grant Number: 125440785—SFB 877, project B11
References
Baba, M., Nakajo, S., Tu, P.H., Tomita, T., Nakaya, K., Lee, V.M., Trojanowski, J.Q., and Iwatsubo, T. (1998). Aggregation of α-synuclein in Lewy bodies of sporadic Parkinson’s disease and dementia with Lewy bodies. Am. J. Pathol. 152, 879–884.Search in Google Scholar
Bainton, D.F. (1981). The discovery of lysosomes. J. Cell Biol. 91, 66s–76 s.10.1083/jcb.91.3.66sSearch in Google Scholar
Bar-On, P., Rockenstein, E., Adame, A., Ho, G., Hashimoto, M., and Masliah, E. (2006). Effects of the cholesterol-lowering compound methyl-β-cyclodextrin in models of α-synucleinopathy. J. Neurochem. 98, 1032–1045.10.1111/j.1471-4159.2006.04017.xSearch in Google Scholar
Beavan, M.S., and Schapira, A.H. (2013). Glucocerebrosidase mutations and the pathogenesis of Parkinson disease. Ann. Med. 45, 511–521.10.3109/07853890.2013.849003Search in Google Scholar
Beck, M. (2018). Treatment strategies for lysosomal storage disorders. Dev. Med. Child Neurol. 60, 13–18.10.1111/dmcn.13600Search in Google Scholar
Bennett, M.J., and Rakheja, D. (2013). The neuronal ceroid-lipofuscinoses. Dev. Disabil. Res. Rev. 17, 254–259.10.1002/ddrr.1118Search in Google Scholar
Blanz, J., Zunke, F., Markmann, S., Damme, M., Braulke, T., Saftig, P., and Schwake, M. (2015). Mannose 6-phosphate-independent Lysosomal Sorting of LIMP-2. Traffic 16, 1127–1136.10.1111/tra.12313Search in Google Scholar
Bosco, D.A., Fowler, D.M., Zhang, Q., Nieva, J., Powers, E.T., Wentworth, P., Jr., Lerner, R.A., and Kelly, J.W. (2006). Elevated levels of oxidized cholesterol metabolites in Lewy body disease brains accelerate α-synuclein fibrilization. Nat. Chem. Biol. 2, 249–253.10.1038/nchembio782Search in Google Scholar
Braak, H., and Del Tredici, K. (2017). Neuropathological staging of brain pathology in sporadic Parkinson’s disease: separating the wheat from the chaff. J. Parkinsons Dis. 7, S71-S85.10.3233/JPD-179001Search in Google Scholar
Chang, D., Nalls, M.A., Hallgrimsdottir, I.B., Hunkapiller, J., van der Brug, M., Cai, F., International Parkinson’s Disease Genomics Consortium, 23andMe Research Team, et al. (2017). A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat. Genet. 49, 1511–1516.10.1038/ng.3955Search in Google Scholar
Chaudhuri, K.R., Healy, D.G., Schapira, A.H., and National Institute for Clinical Excellence (2006). Non-motor symptoms of Parkinson’s disease: diagnosis and management. Lancet Neurol. 5, 235–245.10.1016/S1474-4422(06)70373-8Search in Google Scholar
Cuervo, A.M., Stefanis, L., Fredenburg, R., Lansbury, P.T., and Sulzer, D. (2004). Impaired degradation of mutant α-synuclein by chaperone-mediated autophagy. Science 305, 1292–1295.10.1126/science.1101738Search in Google Scholar
Cullen, V., Lindfors, M., Ng, J., Paetau, A., Swinton, E., Kolodziej, P., Boston, H., Saftig, P., et al. (2009). Cathepsin D expression level affects α-synuclein processing, aggregation, and toxicity in vivo. Mol. Brain 2, 5.10.1186/1756-6606-2-5Search in Google Scholar
Davie, C.A. (2008). A review of Parkinson’s disease. Br. Med. Bull. 86, 109–127.10.1093/bmb/ldn013Search in Google Scholar
De Duve, C., and Beaufay, H. (1959). Tissue fractionation studies. 10. Influence of ischaemia on the state of some bound enzymes in rat liver. Biochem. J. 73, 610–616.10.1042/bj0730610Search in Google Scholar
De Duve, C., Pressman, B.C., Gianetto, R., Wattiaux, R., and Appelmans, F. (1955). Tissue fractionation studies. 6. Intracellular distribution patterns of enzymes in rat-liver tissue. Biochem. J. 60, 604–617.10.1042/bj0600604Search in Google Scholar
Dehay, B., Bove, J., Rodriguez-Muela, N., Perier, C., Recasens, A., Boya, P., and Vila, M. (2010). Pathogenic lysosomal depletion in Parkinson’s disease. J. Neurosci. 30, 12535–12544.10.1523/JNEUROSCI.1920-10.2010Search in Google Scholar
Eskelinen, E.L., Tanaka, Y., and Saftig, P. (2003). At the acidic edge: emerging functions for lysosomal membrane proteins. Trends Cell Biol. 13, 137–145.10.1016/S0962-8924(03)00005-9Search in Google Scholar
Fraldi, A., Klein, A. D., Medina, D.L., and Settembre, C. (2016). Brain disorders due to lysosomal dysfunction. Annu. Rev. Neurosci. 39, 277–295.10.1146/annurev-neuro-070815-014031Search in Google Scholar
George, J.M. (2002). The synucleins. Genome Biol. 3, REVIEWS3002.Search in Google Scholar
Giasson, B.I., Uryu, K., Trojanowski, J.Q., and Lee, V.M. (1999). Mutant and wild type human α-synucleins assemble into elongated filaments with distinct morphologies in vitro. J. Biol. Chem. 274, 7619–7622.10.1074/jbc.274.12.7619Search in Google Scholar
Gieselmann, V. (1995). Lysosomal storage diseases. Biochim. Biophys. Acta 1270, 103–136.10.1016/0925-4439(94)00075-2Search in Google Scholar
Hara, T., Nakamura, K., Matsui, M., Yamamoto, A., Nakahara, Y., Suzuki-Migishima, R., Yokoyama, M., Mishima, K., et al. (2006). Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441, 885–889.10.1038/nature04724Search in Google Scholar
Infante, R.E., Radhakrishnan, A., Abi-Mosleh, L., Kinch, L.N., Wang, M.L., Grishin, N.V., Goldstein, J.L., and Brown, M.S. (2008). Purified NPC1 protein: II. Localization of sterol binding to a 240-amino acid soluble luminal loop. J. Biol. Chem. 283, 1064–1075.10.1074/jbc.M707944200Search in Google Scholar
Janvier, K., and Bonifacino, J.S. (2005). Role of the endocytic machinery in the sorting of lysosome-associated membrane proteins. Mol. Biol. Cell 16, 4231–4242.10.1091/mbc.e05-03-0213Search in Google Scholar
Kalia, L.V., and Lang, A.E. (2015). Parkinson’s disease. Lancet 386, 896–912.10.1016/S0140-6736(14)61393-3Search in Google Scholar
Klein, A. D., and Futerman, A.H. (2013). Lysosomal storage disorders: old diseases, present and future challenges. Pediatr. Endocrinol. Rev. 11 (Suppl. 1), 59–63.Search in Google Scholar
Klein, A. D., and Mazzulli, J.R. (2018). Is Parkinson’s disease a lysosomal disorder? Brain 141, 2255–2262.10.1093/brain/awy147Search in Google Scholar
Komatsu, M., Waguri, S., Chiba, T., Murata, S., Iwata, J., Tanida, I., Ueno, T., Koike, M., et al. (2006). Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441, 880–884.10.1038/nature04723Search in Google Scholar
Kornfeld, S. (1992). Structure and function of the mannose 6-phosphate/insulinlike growth factor II receptors. Annu. Rev. Biochem. 61, 307–330.10.1146/annurev.bi.61.070192.001515Search in Google Scholar
Lashuel, H.A., and Lansbury, P.T., Jr. (2006). Are amyloid diseases caused by protein aggregates that mimic bacterial pore-forming toxins? Q. Rev. Biophys. 39, 167–201.10.1017/S0033583506004422Search in Google Scholar
Lashuel, H.A., Petre, B.M., Wall, J., Simon, M., Nowak, R.J., Walz, T., and Lansbury, P.T., Jr. (2002). Alpha-synuclein, especially the Parkinson’s disease-associated mutants, forms pore-like annular and tubular protofibrils. J. Mol. Biol. 322, 1089–1102.10.1016/S0022-2836(02)00735-0Search in Google Scholar
Lesage, S., Anheim, M., Condroyer, C., Pollak, P., Durif, F., Dupuits, C., Viallet, F., Lohmann, et al. (2011). Large-scale screening of the Gaucher’s disease-related glucocerebrosidase gene in Europeans with Parkinson’s disease. Hum. Mol. Genet. 20, 202–210.10.1093/hmg/ddq454Search in Google Scholar PubMed
Mazzulli, J.R., Xu, Y.H., Sun, Y., Knight, A.L., McLean, P.J., Caldwell, G.A., Sidransky, E., Grabowski, G.A., et al. (2011). Gaucher disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 146, 37–52.10.1016/j.cell.2011.06.001Search in Google Scholar PubMed PubMed Central
Mazzulli, J.R., Zunke, F., Tsunemi, T., Toker, N.J., Jeon, S., Burbulla, L.F., Patnaik, S., Sidransky, et al. (2016). Activation of β-Glucocerebrosidase Reduces Pathological α-Synuclein and Restores Lysosomal Function in Parkinson’s Patient Midbrain Neurons. J. Neurosci. 36, 7693–7706.10.1523/JNEUROSCI.0628-16.2016Search in Google Scholar PubMed PubMed Central
McGlinchey, R.P., and Lee, J.C. (2015). Cysteine cathepsins are essential in lysosomal degradation of α-synuclein. Proc. Natl. Acad. Sci. U.S. A. 112, 9322–9327.10.1073/pnas.1500937112Search in Google Scholar PubMed PubMed Central
McNeill, A., Duran, R., Hughes, D.A., Mehta, A., and Schapira, A.H. (2012). A clinical and family history study of Parkinson’s disease in heterozygous glucocerebrosidase mutation carriers. J. Neurol. Neurosurg. Psychiatry 83, 853–854.10.1136/jnnp-2012-302402Search in Google Scholar PubMed PubMed Central
Migdalska-Richards, A., Daly, L., Bezard, E., and Schapira, A.H. (2016). Ambroxol effects in glucocerebrosidase and α-synuclein transgenic mice. Ann. Neurol. 80, 766–775.10.1002/ana.24790Search in Google Scholar PubMed PubMed Central
Ohkuma, S., Moriyama, Y., and Takano, T. (1982). Identification and characterization of a proton pump on lysosomes by fluorescein-isothiocyanate-dextran fluorescence. Proc. Natl. Acad. Sci. USA 79, 2758–2762.10.1073/pnas.79.9.2758Search in Google Scholar PubMed PubMed Central
Pastores, G.M. (1997). Gaucher’s Disease. Pathological features. Baillieres Clin. Haematol. 10, 739–749.10.1016/B978-0-12-385157-4.00100-7Search in Google Scholar
Pitcairn, C., Wani, W.Y., and Mazzulli, J.R. (2019). Dysregulation of the autophagic-lysosomal pathway in Gaucher and Parkinson’s disease. Neurobiol. Dis. 122, 72–82.10.1016/j.nbd.2018.03.008Search in Google Scholar PubMed PubMed Central
Platt, F.M., d’Azzo, A., Davidson, B.L., Neufeld, E.F., and Tifft, C.J. (2018). Lysosomal storage diseases. Nat. Rev. Dis. Primers 4, 27.10.1038/s41572-018-0025-4Search in Google Scholar PubMed
Reczek, D., Schwake, M., Schroder, J., Hughes, H., Blanz, J., Jin, X., Brondyk, W., Van, P.S., et al. (2007). LIMP-2 is a receptor for lysosomal mannose-6-phosphate-independent targeting of beta-glucocerebrosidase. Cell 131, 770–783.10.1016/j.cell.2007.10.018Search in Google Scholar PubMed
Riederer, P., Berg, D., Casadei, N., Cheng, F., Classen, J., Dresel, C., Jost, W., Kruger, R., et al. (2019). α-Synuclein in Parkinson’s disease: causal or bystander? J. Neural Transm. (Vienna) 126, 815–840.10.1007/s00702-019-02025-9Search in Google Scholar PubMed
Robak, L.A., Jansen, I. E., van Rooij, J., Uitterlinden, A.G., Kraaij, R., Jankovic, J., International Parkinson’s Disease Genomics Consortium, Heutink, P., et al. (2017). Excessive burden of lysosomal storage disorder gene variants in Parkinson’s disease. Brain 140, 3191–3203.10.1093/brain/awx285Search in Google Scholar PubMed PubMed Central
Sardi, S.P., Viel, C., Clarke, J., Treleaven, C.M., Richards, A.M., Park, H., Olszewski, M.A., Dodge, J.C., et al. (2017). Glucosylceramide synthase inhibition alleviates aberrations in synucleinopathy models. Proc. Natl. Acad. Sci. U.S. A. 114, 2699–2704.10.1073/pnas.1616152114Search in Google Scholar PubMed PubMed Central
Schulze, H., and Sandhoff, K. (2011). Lysosomal lipid storage diseases. Cold Spring Harb. Perspect. Biol. 3.10.1101/cshperspect.a004804Search in Google Scholar PubMed PubMed Central
Schwake, M., Schroder, B., and Saftig, P. (2013). Lysosomal membrane proteins and their central role in physiology. Traffic 14, 739–748.10.1111/tra.12056Search in Google Scholar PubMed
Settembre, C., Fraldi, A., Medina, D.L., and Ballabio, A. (2013). Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat. Rev. Mol. Cell Biol. 14, 283–296.10.1038/nrm3565Search in Google Scholar PubMed PubMed Central
Shachar, T., Lo Bianco, C., Recchia, A., Wiessner, C., Raas-Rothschild, A., and Futerman, A.H. (2011). Lysosomal storage disorders and Parkinson’s disease: Gaucher disease and beyond. Mov. Disord. 26, 1593–1604.10.1002/mds.23774Search in Google Scholar PubMed
Sidransky, E., Nalls, M.A., Aasly, J.O., Aharon-Peretz, J., Annesi, G., Barbosa, E.R., Bar-Shira, A., Berg, D., et al. (2009). Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N. Engl. J. Med. 361, 1651–1661.10.1056/NEJMoa0901281Search in Google Scholar PubMed PubMed Central
Sulzer, D., and Edwards, R.H. (2019). The physiological role of α-synuclein and its relationship to Parkinson’s Disease. J. Neurochem. 150, 475–486.10.1111/jnc.14810Search in Google Scholar PubMed PubMed Central
Suzuki, M., Fujikake, N., Takeuchi, T., Kohyama-Koganeya, A., Nakajima, K., Hirabayashi, Y., Wada, K., and Nagai, Y. (2015). Glucocerebrosidase deficiency accelerates the accumulation of proteinase K-resistant α-synuclein and aggravates neurodegeneration in a Drosophila model of Parkinson’s disease. Hum. Mol. Genet. 24, 6675–6686.10.1093/hmg/ddv372Search in Google Scholar PubMed
Taguchi, Y.V., Liu, J., Ruan, J., Pacheco, J., Zhang, X., Abbasi, J., Keutzer, J., Mistry, P.K., et al. (2017). Glucosylsphingosine promotes α-synuclein pathology in mutant GBA-associated Parkinson’s disease. J. Neurosci. 37, 9617–9631.10.1523/JNEUROSCI.1525-17.2017Search in Google Scholar PubMed PubMed Central
Xia, Q., Liao, L., Cheng, D., Duong, D.M., Gearing, M., Lah, J.J., Levey, A.I., and Peng, J. (2008). Proteomic identification of novel proteins associated with Lewy bodies. Front. Biosci. 13, 3850–3856.10.2741/2973Search in Google Scholar PubMed PubMed Central
Zunke, F., Andresen, L., Wesseler, S., Groth, J., Arnold, P., Rothaug, M., Mazzulli, J.R., Krainc, D., et al. (2016). Characterization of the complex formed by beta-glucocerebrosidase and the lysosomal integral membrane protein type-2. Proc. Natl. Acad. Sci. USA 113, 3791–3796.10.1073/pnas.1514005113Search in Google Scholar PubMed PubMed Central
Zunke, F., and Mazzulli, J.R. (2019). Modeling neuronopathic storage diseases with patient-derived culture systems. Neurobiol. Dis. 127, 147–162.10.1016/j.nbd.2019.01.018Search in Google Scholar PubMed PubMed Central
Zunke, F., Moise, A.C., Belur, N.R., Gelyana, E., Stojkovska, I., Dzaferbegovic, H., Toker, N.J., Jeon, S., et al. (2018). Reversible conformational conversion of α-synuclein into toxic assemblies by glucosylceramide. Neuron 97, 92–107 e110.10.1016/j.neuron.2017.12.012Search in Google Scholar PubMed PubMed Central
© 2020 Walter de Gruyter GmbH, Berlin/Boston
Articles in the same Issue
- Titelseiten
- Review Article
- Food reward and gut-brain signalling
- Synapses: Multitasking Global Players in the Brain
- Apolipoprotein E: Cholesterol metabolism and Alzheimer’s pathology
- The enteric nervous system: “A little brain in the gut”
- The function of lysosomes and their role in Parkinson’s disease
- Rezension
- Konrad Lehmann: Das schöpferische Gehirn Auf der Suche nach der Kreativität – eine Fahndung in sieben Tagen
- Obituary
- Prof. Dr. med. Dr. med. h.c. Georg W. Kreutzberg
- Nachrichten der Gesellschaft
- EU-Projektförderung 2020 Neurowissenschaften
Articles in the same Issue
- Titelseiten
- Review Article
- Food reward and gut-brain signalling
- Synapses: Multitasking Global Players in the Brain
- Apolipoprotein E: Cholesterol metabolism and Alzheimer’s pathology
- The enteric nervous system: “A little brain in the gut”
- The function of lysosomes and their role in Parkinson’s disease
- Rezension
- Konrad Lehmann: Das schöpferische Gehirn Auf der Suche nach der Kreativität – eine Fahndung in sieben Tagen
- Obituary
- Prof. Dr. med. Dr. med. h.c. Georg W. Kreutzberg
- Nachrichten der Gesellschaft
- EU-Projektförderung 2020 Neurowissenschaften