Bedeutung von Activin für Kognition, Affekt und neuronales Überleben
-
Fang Zheng
Fang Zheng hat an der Nanjing Universität (China, 1986-1990) Physiologie studiert und 1999 an der National University Singapore in Neurophysiologie promoviert. Dazwischen hat sie im Rahmen eines Master-Programms am Shanghai Brain Research Institute (Chinesische Akademie für Wissenschaften) zusätzliche Kenntnisse auf dem Gebiet der Neuroanatomie erworben. Nach der Promotion hat sie als Postdoktorandin im Labor von Prof. Steve Johnson an der Oregon Health & Science University (USA) gearbeitet und sich dort elektrophysiologischen Untersuchungen an Hirnschnittpräparaten zugewandt. Seit 2003 leitet sie die Arbeitsgruppe für Hirnschnitt-Elektrophysiologie bei Prof. Christian Alzheimer, zunächst an der Universität zu Kiel und anschließend an der Friedrich-Alexander-Universität Erlangen-Nürnberg.
Andrea Link hat in Erlangen Molekulare Medizin studiert. Anschließend hat sie am Institut für Physiologie und Pathophysiologie unter der Leitung von Prof. Alzheimer die Effekte von Activin im ZNS während ihrer Promotion untersucht. Nach einer kurzen Phase als Postdoktorandin am selbigen Institut ist sie nun wissenschaftliche Koordinatorin des DFG-geförderten Graduiertenkollegs GRK2162 mit dem Schwerpunkt „
Neurodevelopment and Vulnerability of the Central Nervous System “.Christian Alzheimer hat in München Humanmedizin studiert und am Max-Planck-Institut für Psychiatrie promoviert. Nach der ärztlichen Approbation 1985 hat er als Wissenschaftler am Physiologischen Institut der Ludwig-Maximilians-Universität in München und am
Department of Physiology and Biophysics derUniversity of Washington in Seattle (USA) gearbeitet. Nach der Habilitation im Fach Physiologie 1995 und einem Heisenberg-Stipendium der Deutschen Forschungsgemeinschaft war er zunächst Professor für Physiologie und Direktor am Physiologischen Institut der Universität Kiel, bevor er 2008 auf einen Lehrstuhl für Physiologie der Friedrich-Alexander-Universität Erlangen-Nürnberg berufen wurde und dort seither das Institut für Physiologie und Pathophysiologie leitet. Derzeit fungiert er als Sprecher des Interdisziplinären Zentrums für Neurowissenschaften (IZN) der Universität. Sein Forschungsgebiet sind die zellulären und synaptischen Grundlagen höherer Hirnfunktionen im gesunden und erkrankten Gehirn.

Zusammenfassung
Activin ist ein multifunktionales regulatorisches Protein und gehört zur Transforming Growth Factor-β-Familie. Im adulten Gehirn fördert Activin neuronales Überleben bei akuter und chronischer Schädigung. Der Faktor reguliert neuronale Schaltkreise aber auch unter physiologischen Bedingungen. Activin soll hier als ein Master-Molekül vorgestellt werden, das erregende und hemmende Synapsen im ZNS in einer Weise beeinflusst, die kognitive Leistungen fördert und die Gemütslage stabilisiert. Activin unterstützt Lernen und Gedächtnis durch Verstärkung der synaptischen Plastizität, wobei NMDA-Rezeptoren und die Dendriten-Morphologie wichtige Angriffspunkte darstellen. Eine gentechnische Blockade des Activin-Signalwegs senkt über eine Veränderung der GABA-Wirkung das Angstverhalten. Außerdem mehren sich Hinweise, dass Activin als endogenes Antidepressivum und Mediator antidepressiver Therapien fungieren könnte.
Einführung
Der deutsche Hirnforscher Wolf Singer hat das Gehirn als „Orchester ohne Dirigent“ bezeichnet, als ein hochkomplexes, nichtlinear arbeitendes System mit der Fähigkeit zur dynamischen Selbstorganisation, das auch ohne jede übergeordnete Instanz in der Lage ist, Neurone aus all den Hirnregionen, die sich gerade mit einer Wahrnehmung oder Aufgabe beschäftigen, kurzfristig zu einem ad-hoc Ensemble zusammenzubringen (Singer, 2005). Als Mechanismus des Binding wird ein räumlich und zeitlich hochsynchrones bioelektrisches Erregungsmuster der beteiligten Nervenzellen angenommen. Die Frage stellt sich, wie es bei zeitlich ausgedehnteren Vorgängen aussieht, wenn es etwa bei der allmählichen Erkundung einer neuen, die Sinne vielfältig anregenden Umgebung zu plastischen Veränderungen in den beteiligten Hirnstrukturen kommt, die Lernen und Gedächtnis fördern und einer depressiven Verstimmung entgegenwirken. Noch längerfristig gedacht steht zu fragen, welche Prozesse dafür verantwortlich sind, dass sich die neuronalen Netzwerke, die für kognitive Leistungen oder emotionales Gleichgewicht sorgen, auch nach einer Auslenkung wieder im Grundzustand einpendeln. Auch hier scheint kein einzelner Dirigent am Werk zu sein, aber es gibt übergreifende Steuerungsmechanismen, die eine Vielzahl von Einzelprozessen miteinander koordinieren, um die erwünschten Effekte zu erzielen. Wir wollen in dieser Übersichtsarbeit einen derart zentral wirksamen, regulatorischen Faktor, nämlich Activin, vorstellen. Wir werden dabei herausarbeiten, dass Activin als eine Art Master-Molekül fungiert, das einzelne Elemente in neuronalen Schaltkreisen so aufeinander abstimmt, dass deren Performance in Hinblick auf unser Denken und Fühlen optimiert wird. Es ist bemerkenswert, dass Activin darüber hinaus sowohl im akut als auch chronisch erkrankten Gehirn ein beachtliches neuroprotektives Potenzial entfalten kann.
Activin, Activin-Rezeptoren und Signaltransduktion
Activin ist ein Mitglied der großen Transforming Growth Factor β (TGF-β) Familie (s. Kasten 1). Ursprünglich wurde Activin als endokrin wirksamer Faktor ovariellen Ursprungs beschrieben, der in der Hypophyse die Freisetzung des Follikel-stimulierenden Hormons (FSH) fördert und so als Gegenspieler des schon früher entdeckten Inhibins wirkt (daher der Name). Inzwischen wissen wir, dass Activin als multifunktionales regulatorisches Protein ein breites Spektrum von biologischen Wirkungen sowohl in der Embryonalentwicklung als auch in vielen ausdifferenzierten Geweben und Organen entfaltet, die insbesondere Proliferation, Differenzierung, Apoptose, Entzündungsprozesse, Immunantwort und Reparaturvorgänge betreffen.
Die TGF-β Familie
Die menschliche TGF-β-Familie besteht aus über 30 Mitgliedern. Dazu zählen die TGF-βs selbst (TGF-β1-3), die Activine und Inhibine, die Bone Morphogenetic Proteins (BMPs), die Growth and Differentiation Factors (GDFs), Nodal, Lefty und die Müllerian Inhibiting Substance (MIS). Es handelt sich um kleine sezernierte Signalmoleküle mit dimerer Struktur, welche die Entwicklung vieler Organe und Gewebe steuern. Im adulten Körper regulieren und koordinieren sie eine kaum überschaubare Vielfalt von Prozessen in diversen Geweben und Organsystemen, die insbesondere mit Gewebshomöostase und -reparatur, mit Entzündungsvorgängen und mit der Krebsentstehung zu tun haben.
Wenn wir von Activin im Singular reden, ist das eigentlich ungenau, denn der Faktor kommt in verschiedenen Varianten vor. Die am besten charakterisierten Activine bestehen aus zwei über Disulfid-Brücken verbundenen βA- oder βB-Untereinheiten und können als die Homodimere Activin A (βA/βA) bzw. Activin B (βB/βB) oder als das Heterodimer Activin AB (βA/βB) in Erscheinung treten. Die bisherigen Befunde sprechen dafür, dass im Gehirn Activin A die entscheidende Rolle spielt. Wenn wir im Folgenden der Einfachheit halber Activin schreiben, ist damit Activin A gemeint. Das in Neuronen oder auch Gliazellen gebildete und wahrscheinlich konstitutiv freigesetzte Activin entfaltet seine Wirkung über einen tetrameren Komplex aus Typ II Activin-Rezeptoren (ActRIIA, ActRIIB) und Typ I Activin-Rezeptoren (vor allem ActRIB, aber auch ActRIC), die jeweils intrazelluläre Serin-Threonin-Kinase-Domänen besitzen. Das Schema in Abb. 1 illustriert den Activin-Signalweg. Nach Bindung von Activin an Typ II-Rezeptoren werden Typ I-Rezeptoren rekrutiert, transphosphoryliert und dadurch in die Lage versetzt, selbst wiederum die intrazellulären Signalmoleküle SMAD2 und SMAD3 zu phosphorylieren. Nach deren Assoziation mit SMAD4 wird der ganze Komplex in den Kern transloziert und kann dort die Expression von bislang im Gehirn noch weitgehend unbekannten Zielgenen regulieren.
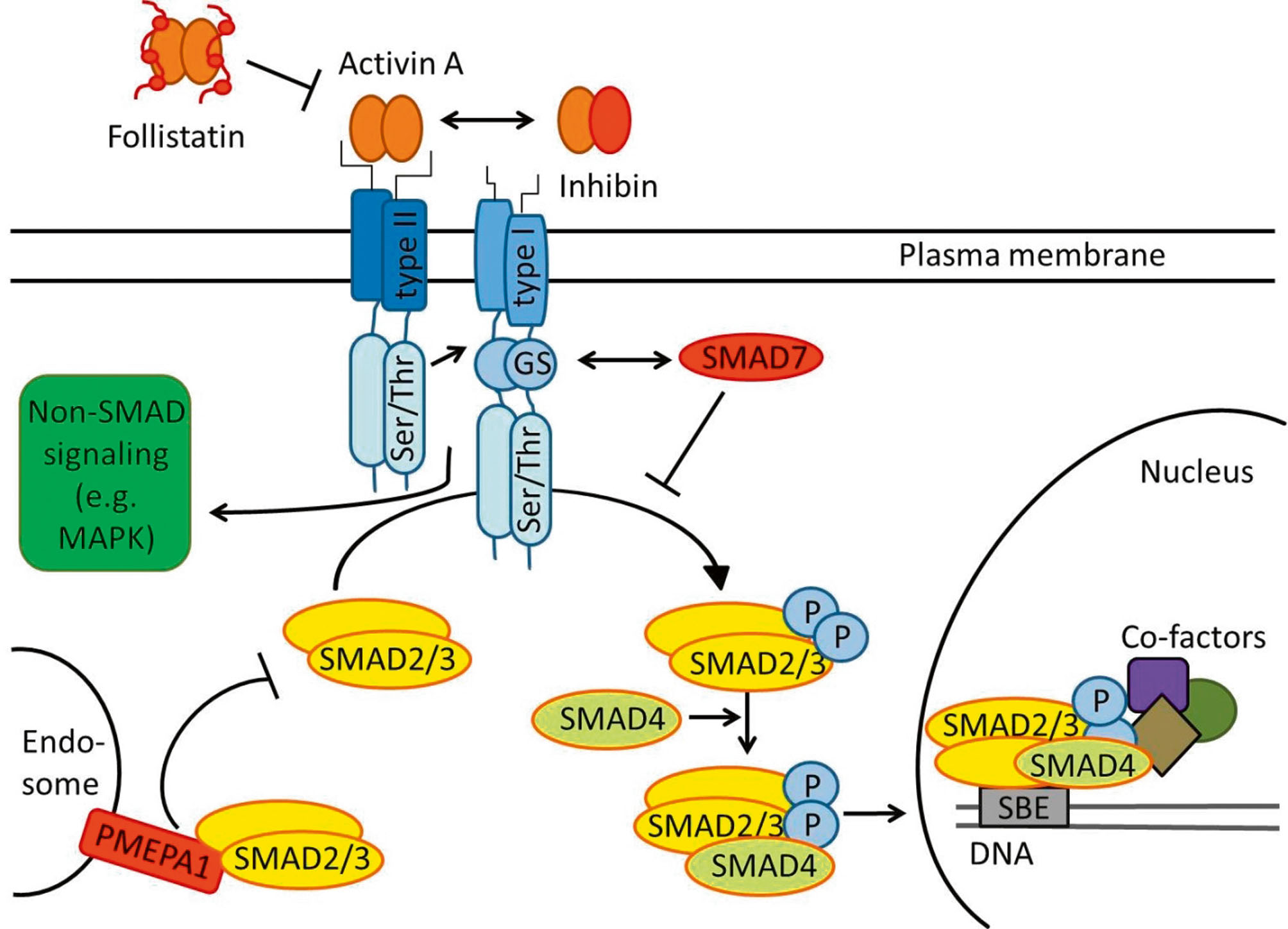
Schema der Activin-Signalwege. Ausmaß und Dauer der Wirkung von Activin werden auf mehreren Ebenen strikt kontrolliert (jeweils in Rot dargestellt). Dazu zählen auf der extrazellulären Seite Follistatin und Inhibin, im Zytosol SMAD7 und PMEPA1. Neben dem kanonischen Signalweg über SMAD-Proteine kann Activin auch auf andere Signalwege wie die MAP-Kinasen wirken. Weitere Erklärungen im Text. Abkürzungen: MAPK, mitogen activated protein kinase; PMEPA1, prostate transmembrane protein, androgen induced 1; SBE, SMAD-binding element.
Die Stärke und Dauer der Activin-Wirkung wird auf mehreren Ebenen strikt kontrolliert. Das schon erwähnte Inhibin kompetiert mit Activin um die Rezeptorbindung, während Follistatin im Extrazellulärraum Activin abfängt. SMAD7 verhindert die Phosphorylierung von SMAD2/3 durch die Typ I-Rezeptoren. Einen vergleichbaren Effekt übt das Protein PMEPA1 aus. Interessanterweise konnten wir PMEPA1 kürzlich als ein erstes Zielgen von Activin im Gehirn identifizieren (Link et al., 2016a). Hier ist offenbar eine Feedback-Hemmung des Activin-Signalwegs implementiert. Neben der kanonischen, SMAD-vermittelten Signaltransduktion kann Activin über seine Rezeptoren auch auf andere, etwa die von Mitogen-activated protein (MAP)-Kinase- und Phosphoinositid-3-Kinase (PI3K)-vermittelten Signalkaskaden Einfluss nehmen.
Activin und die molekularen Mechanismen von Lernen und Gedächtnis
Langdauernde Steigerungen in der Übertragungsfunktion von exzitatorischen Nervenzell-Synapsen, die Glutamat als Neurotransmitter benutzen, gelten als ein wesentlicher neurobiologischer Mechanismus von Lernen und Gedächtnisbildung. Die langfristige Erhöhung der synaptischen Effizienz wird als Langzeit-Potenzierung (long-term potentiation, LTP) bezeichnet. Sie stellt ein neuronales Korrelat der neugebildeten Gedächtnisspur dar. Damit LTP überhaupt initiiert werden kann, muss die Synapse mit einem besonderen Reizmuster aktiviert werden, das zur Öffnung eines Subtyps der Glutamat-Rezeptoren, nämlich der NMDA-Rezeptor-Kanäle führt. Das einströmende Ca2+ setzt eine Signalkaskade in Gang, die zur Etablierung von LTP führt. Die pharmakologische oder gentechnische Ausschaltung des NMDA-Rezeptors verhindert nicht nur LTP, sondern beeinträchtigt auch das Lernverhalten. Die kausale Verknüpfung von LTP und Gedächtnisbildung ist am intensivsten im Hippocampus untersucht worden, der essenzielle Bedeutung für das räumliche und vor allem das deklarative, also in Worten fassbare Gedächtnis besitzt. Mehrere experimentelle Befunde zeigen, dass Activin in Mäusen LTP und Lernen fördert (Krieglstein et al., 2011). Die Hemmung des Activin-Signalwegs durch eine gezielte postnatale Expression von dominant-negativen ActRIB-Rezeptoren (dnActRIB) rief eine signifikante Reduktion der hippocampalen LTP hervor (Muller et al., 2006). Ebenso verhinderte die Überexpression des extrazellulären Activin-Fängers Follistatin hippocampale LTP und führte zu Defiziten in einem kontextabhängigen Lerntest (Ageta et al., 2010). Activin-Rezeptoren finden sich in dendritischen Dornfortsätzen (Spines) und sind dort offenbar in der Lage, NMDA-Rezeptoren zu phosphorylieren, wahrscheinlich über einen nicht-SMAD-abhängigen Signalweg. Diese Befunde legen nahe, dass die Stärkung der LTP (und der Gedächtnisbildung) über eine Steigerung der NMDA-Rezeptor-Antwort auf freigesetztes Glutamat zustande kommt (Abb. 2). Dazu kommt, dass Activin auch die Zahl und Form der Spines in einer für die langdauernde Potenzierung günstigen Weise verändert, also auch das morphologische Substrat der Gedächtnisspur positiv beeinflussen kann.
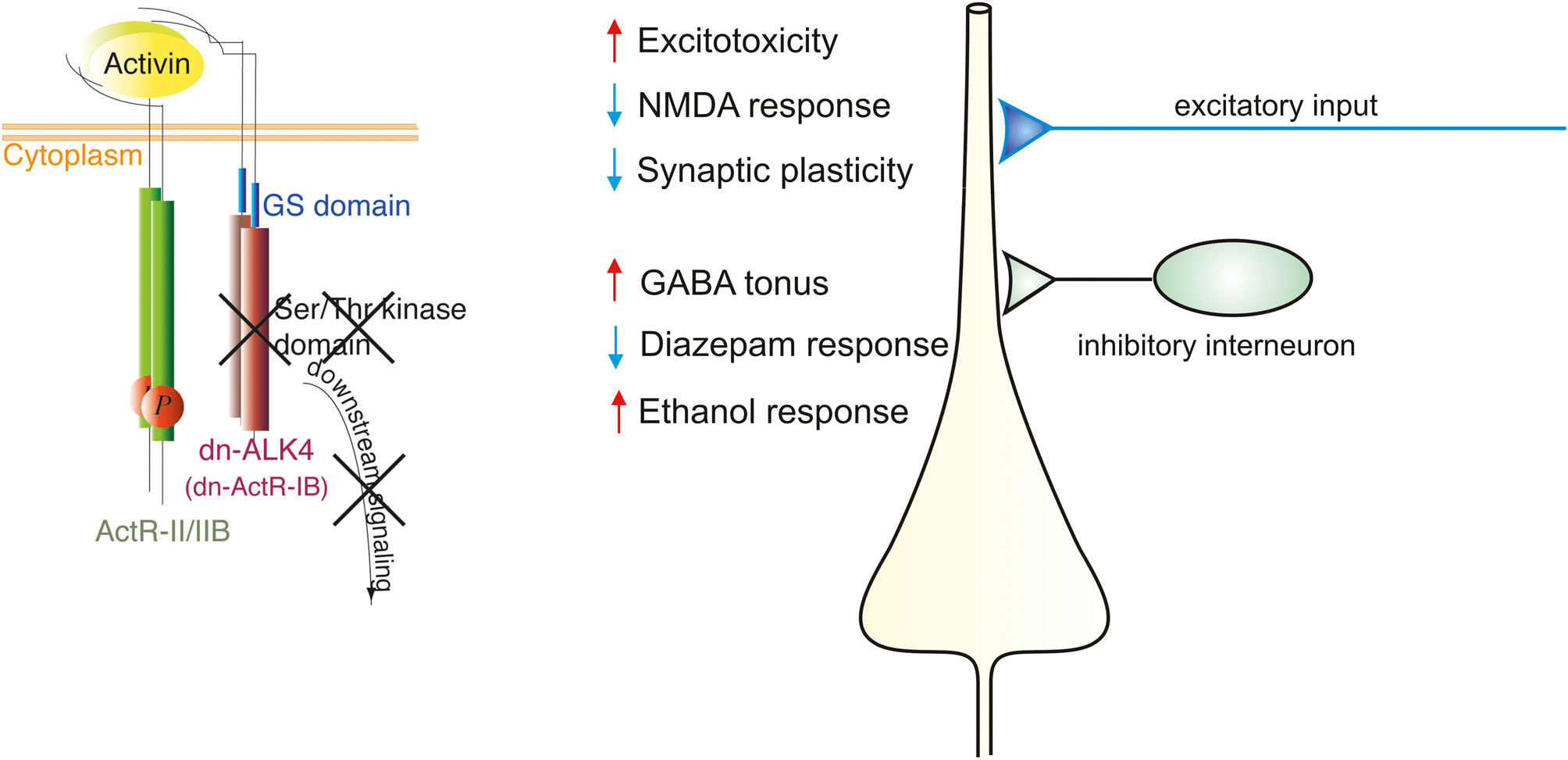
Einfluss von Activin auf die Physiologie und Pharmakologie von ZNS-Synapsen. Linkerhand ist der Activin-Rezeptorkomplex illustriert, der die dominant-negative Mutante des Activin-Rezeptors IB (dn-ActR-IB, auch als dn-ALK4 bezeichnet) enthält, bei dem die komplette Kinase-Domäne fehlt. Die Überexpression des mutanten Rezeptors blockiert den Activin-Signalweg. Die wichtigsten Folgen für die erregende und hemmende synaptische Übertragung sind rechterhand wiedergegeben.
Activin, GABA-Rezeptoren und Angstverhalten
Bei den schon erwähnten Mäusen, deren Neurone im Vorderhirn dominant-negative Rezeptoren vom Typ ActRIB exprimieren (dnActRIB-Maus), fiel in Verhaltenstests eine markante Reduktion des spontanen Angstverhaltens auf (Zheng et al., 2009). Dieser überraschende Befund ist in Abb. 3 illustriert. Der Versuch ist so aufgebaut, dass die Maus die Wahl hat, in einer geschützten Umgebung (der Box) zu verbleiben, oder den erhöhten und hellbeleuchteten Balken, der von der Box ausgeht, zu erkunden. Es handelt sich um ein typisches Konfliktparadigma zwischen Sicherheitsbedürfnis einerseits und Neugier andererseits. Sind die normalen Mäuse recht vorsichtig und verlassen die sichere Box innerhalb einer fünfminütigen Testzeit im Schnitt nur zwei- bis dreimal und dann auch nur für eine kurze Weile, erwiesen sich die dnActRIB-Mäuse als deutlich risikofreudiger. Die Ängstlichkeit der normalen Mäuse ließ sich erheblich durch die Gabe von Diazepam senken, einem klinisch weithin eingesetzten Anxiolytikum, das zur Gruppe der Benzodiazepine gehört. Das sowieso schon geminderte Angstverhalten der dnActRIB-Mäuse ließ sich dagegen durch Diazepam nicht weiter reduzieren (Abb. 3).
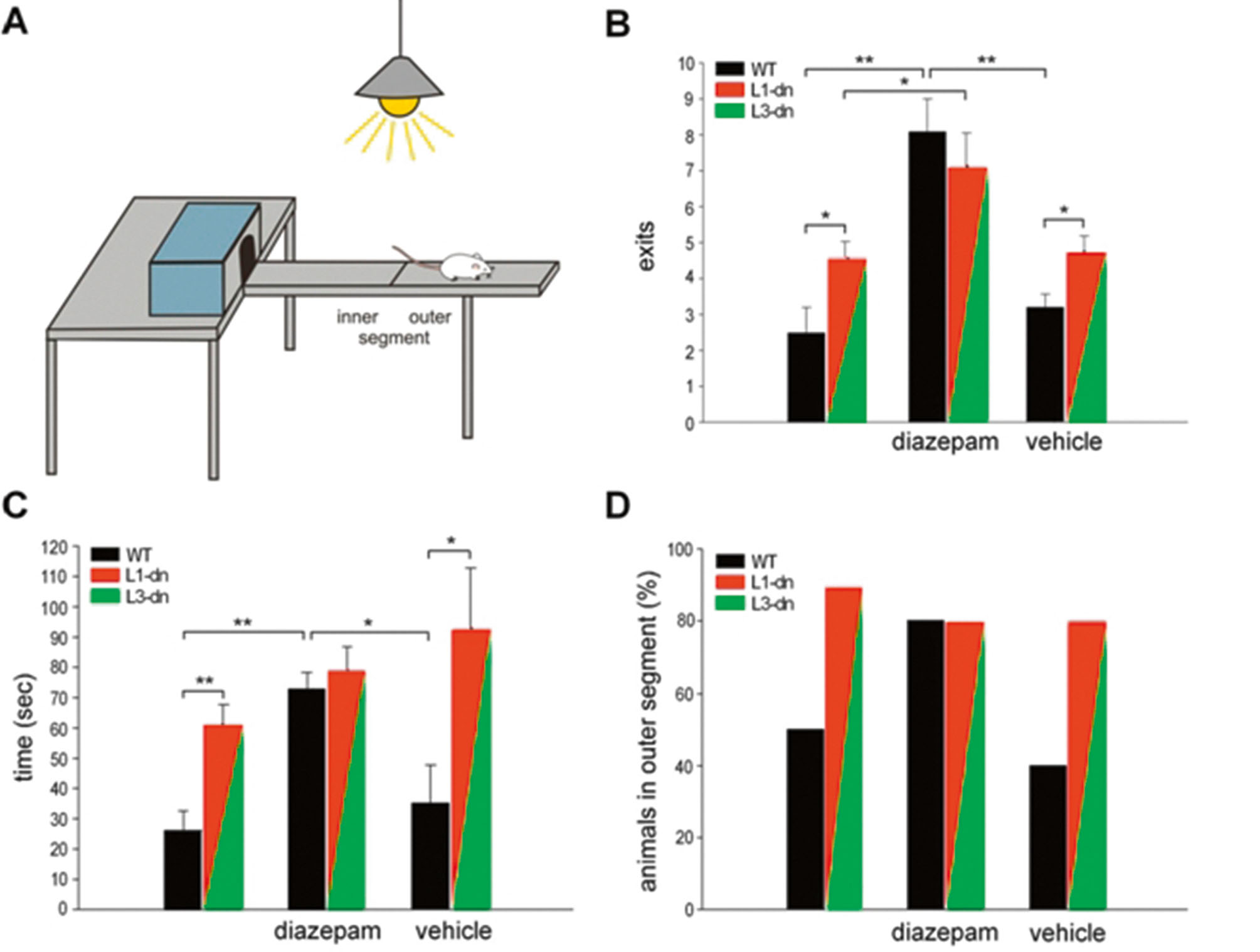
Blockade des Activin-Signalwegs vermindert spontanes Angstverhalten. A, Aufbau des Light-Dark-Exploration-Tests zur Untersuchung des Angstverhaltens von Mäusen. B-D, die Histogramme zeigen die Zahl der Exits der Mäuse aus der Box (B), die gesamten Exit-Zeit (C) und die Prozentzahl der Tiere, die das äußere Segment des Balkens betreten haben (D), jeweils über einen Zeitraum von 5 Minuten, für Kontrolltiere (WT, schwarze Säulen) und zwei dnActRIB-Mauslinien (L1-dn, L3-dn, rot-grüne Säulen), ohne Behandlung (linke Säulenpaare), mit Diazepam (mittlere Säulenpaare) bzw. reiner Trägersubstanz als Kontrolle (vehicle, rechte Säulenpaare). Die Befunde der beiden mutanten Mauslinien wurden zusammengefasst, da keine Unterschiede bestanden. * P < 0.05, ** P < 0.01 (nachgedruckt aus Zheng et al., 2009, mit freundlicher Genehmigung des Verlags).
Da Benzodiazepine anxiolytisch wirken, indem sie die Wirkung des hemmenden Neurotransmitters GABA am GABAA-Rezeptor verstärken, lag es nahe, in Hippocampusschnitten von normalen und dnActRIB-Mäusen die Funktion der GABAergen Synapsen vergleichend zu untersuchen (Zheng et al., 2009). Tatsächlich fehlte die typischerweise robuste Verstärkung des GABAA-Rezeptor-abhängigen Chloridstroms durch Diazepam in den Hippocampusneuronen, die keinen intakten Activin-Signalweg mehr besaßen. Daneben fand sich in diesen Neuronen ein deutlich stärkerer, durch extrasynaptisch lokalisierte GABAA-Rezeptoren hervorgerufener inhibitorischer Tonus (Abb. 2). Interessanterweise wird dieser GABA-Tonus mit der Regulation des Angstverhaltens in Zusammenhang gebracht. Schließlich stellte sich heraus, dass bei Fehlen des Activin-Signalwegs die GABAB-Rezeptor-vermittelte Aktivierung von Kaliumströmen durch G protein-activated, inwardly rectifying K+ (GIRK)-Kanäle verstärkt ist. Das passt ebenfalls gut ins Bild, weiß man doch, dass Substanzen, die den GABAB-Rezeptor positiv modulieren, das Angstverhalten mindern. Damit lassen sich für die beiden markanten Verhaltensauffälligkeiten der dnActRIB-Mäuse, also deren niedrige Ängstlichkeit und die fehlende anxiolytische Diazepam-Wirkung, die zugrunde liegenden Mechanismen auf Ebene der GABAA- und GABAB-Rezeptoren finden. Offensichtlich reguliert Activin die Funktion der beiden GABA-Rezeptoren in einer Weise, die Risikoscheu und Neugier in eine für das eigene Überleben vorteilhafte Balance bringen. Wir würden dementsprechend erwarten, dass das risikofreudige Draufgängertum der Mäuse, deren Activin-Signalweg unterbrochen ist, sie in der freien Wildbahn zur leichten Beute machen würde.
Activin, Alkohol und Suchtverhalten
Da Alkohol ein positiver allosterischer Modulator des GABAA-Rezeptors ist und über diesen Mechanismus einen Teil seiner zentralnervösen Wirkungen entfaltet, zu denen auch die Anxiolyse gehört, stellte sich angesichts der Vorbefunde zur Diazepam-Wirkung die Frage, ob Activin auch regulierend auf die Steigerung der GABAergen Hemmung durch Alkohol wirkt (Zheng et al., 2016). Während Activin, wie wir gerade gesehen haben, den Effekt von Diazepam am GABAA-Rezeptor fördert, ist es mit Alkohol genau anders herum. Im Vergleich zum Kontrollpräparat fand sich in Hippocampusneuronen von dnActRIB-Mäusen eine signifikante Linksverschiebung der Alkoholwirkung auf GABAA-Rezeptor-vermittelte synaptische Ströme (Abb. 2). Dadurch verstärkt Alkohol die GABAergen Synapsen bereits bei Konzentrationen ≤30 mM (17 mM entspricht 0.8‰), die normalerweise noch ohne potenzierende Wirkung sind. Auf der molekularen Ebene scheint Activin die Alkoholempfindlichkeit der GABAergen Synapsen über einen nicht-SMAD-abhängigen Signalweg zu dämpfen, der über die Proteinkinase C epsilon (PKCε) läuft. Auf der Verhaltensebene rief Alkohol in dnActRIB-Mäusen eine deutlich stärkere Sedierung hervor als in Kontrolltieren, das Suchtpotenzial von Alkohol war dagegen in den dnActRIB-Mäusen nicht verändert.
Eine kürzlich veröffentlichte Studie hat den kanonischen (SMAD-vermittelten) Activin-Signalweg für die charakteristischen Symptome nach Kokain-Entzug verantwortlich gemacht, also das immer stärker werdende Verlangen nach der Droge (Craving) und die damit verbundene Rückfallgefahr (Gancarz et al., 2015). Sieben Tage nach Kokain-Entzug fand sich im Nucleus accumbens, einem wichtigen Teil des mesolimbischen Belohnungssystems, eine deutliche Steigerung der SMAD3-Phosphorylierung. Nach Injektion von rekombinantem Activin in den Nucleus accumbens verabreichten sich die Mäuse deutlich mehr Kokain, während die Expression einer dominant-negativen Variante von SMAD3 (dnSMAD3) mittels eines viralen Vektors den gegenteiligen Effekt hervorrief. Interessanterweise unterdrückte dnSMAD3 auch die Zunahme von dendritischen Spines in Neuronen des Nucleus accumbens, die typischerweise mit der Ausbildung von Kokain-Craving assoziiert ist. Angesichts dieser Befunde scheint es denkbar, dass Kokain die Induktion plastischer Veränderungen durch Activin, die, wie wir oben gesehen haben, im Dienste von Lernen und Gedächtnis stehen, hier gleichsam für eigene Zwecke kapert.
Activin als endogenes Antidepressivum?
Angesichts der ausgeprägten Komorbidität von Angststörungen und Depression verwundert es nicht, dass Activin auch bei depressiven Erkrankungen eine Rolle spielt. Antidepressiva stimulieren den Activin-Signalweg ebenso wie die Elektrokrampftherapie (EKT), die bei schwerer, pharmakoresistenter Depression eingesetzt wird (Link et al., 2016b). Dass dies zu den therapeutischen Effekten der beiden Behandlungsformen beiträgt, wird durch Verhaltenstests unterstrichen, in denen die Injektion von rekombinantem Activin in den Gyrus dentatus des Hippocampus das depressionsartige Verhalten von Ratten signifikant reduzierte (Dow et al., 2005). Da es bei der EKT zu einer enormen, aber im Wesentlichen auf den Gyrus dentatus beschränkten Hochregulation von Activin kommt (Abb. 4c), stellt sich die Frage, ob der Faktor auf die in dieser Region stattfindende adulte Neurogenese Einfluss nehmen kann. In der Tat führt die Injektion von Activin zu einer deutlich gesteigerten Proliferation der neuronalen Stamm- und Vorläuferzellen im Gyrus dentatus. Wenn man der Hypothese folgt, wonach eine adäquate adulte Neurogenese im Gyrus dentatus für ein stabiles und ausgeglichenes Gemüt essenziell ist, könnte in der Steigerung der Neurogenese ein wichtiger antidepressiver Mechanismus liegen, der durch das massive Anschalten des Activin-Signalwegs angetrieben wird.
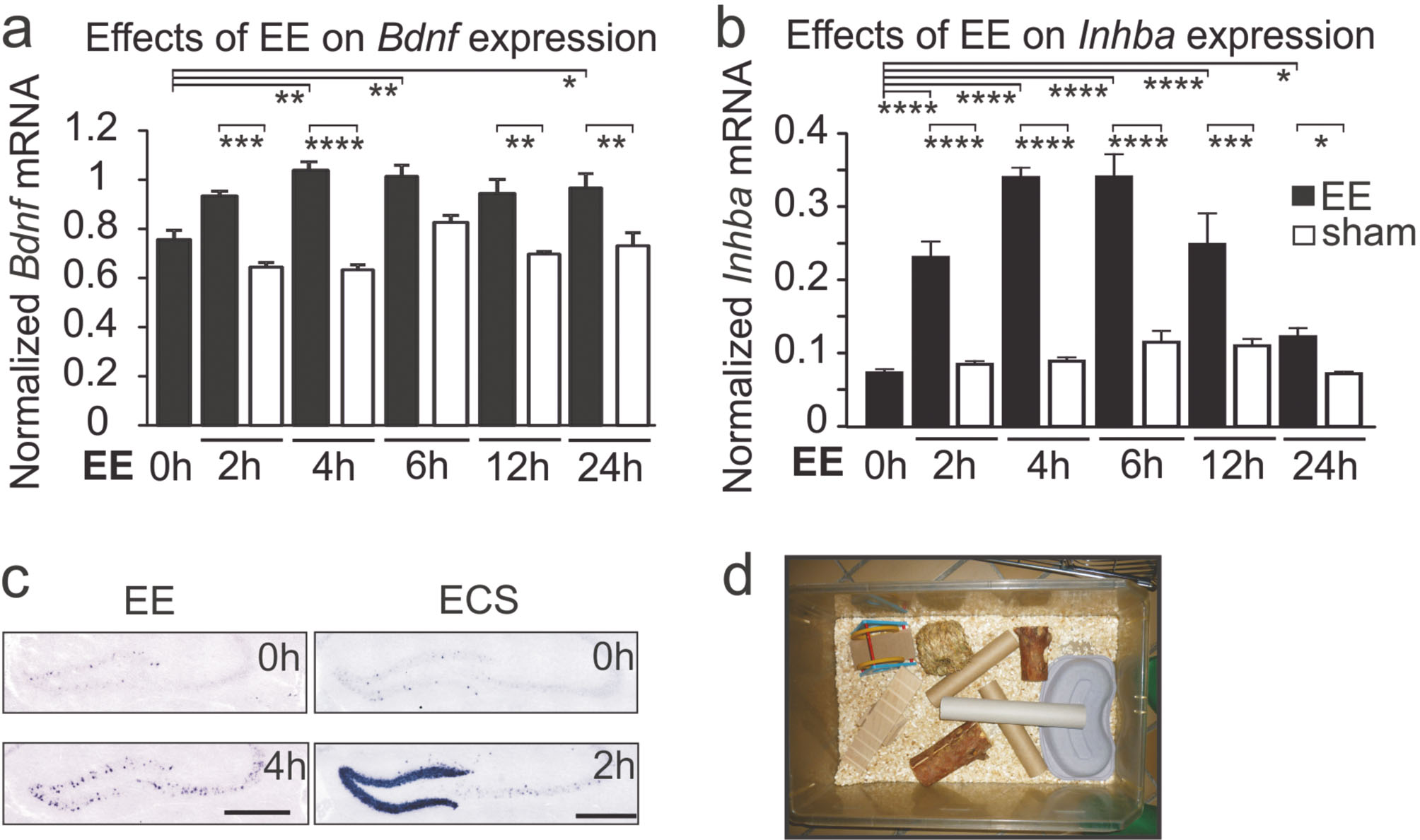
Exploration einer stimulierenden Umgebung (Enriched Environment, EE) erhöht die Expression von Bdnf-mRNA (a) und von Inhba-mRNA, die für die Activin βA-Untereinheit kodiert (b). Die mRNA-Spiegel wurden mittels RT-qPCR bestimmt und auf den Mittelwert von Tbp, Hprt und Rpl13a normalisiert. Die Mäuse wurden in Käfige gesetzt, die mit Spielzeug, Tunneln und Verstecken ausgestattet waren (d). Die Funktionalität der Activin-Hochregulation wurde anhand erhöhter SMAD2/3-Phosphorylierung nachgewiesen (nicht gezeigt). Die in situ-Hybridisierung der Activin βA-mRNA zeigt eine über den Gyrus dentatus und die CA3-Region punktförmig verstreute Hochregulation nach vierstündigem Aufenthalt im Spielkäfig (c, links). Dagegen beobachtete man nach Electroconvulsive Seizures (ECS), einem Nager-Modell für die Elektrokrampftherapie bei Patienten mit schwerer Depression, einen massiven, aber hauptsächlich auf den Gyrus dentatus beschränkten Anstieg der βA-mRNA (c, rechts). (Modifiziert nach Link et al., 2016a)
Interessanterweise kann das Activin-System auch ohne antidepressive Medikation oder EKT angeregt werden. Bringt man Mäuse aus ihrem normalen „Käfig-Alltag“ in eine stimulierende, mit Spielzeug, Röhren und Verstecken angereicherte Umgebung, ein sog. Enriched Environment (EE), so sieht man bereits nach 2 Stunden einen erheblichen Anstieg von Activin, der nach 4–6 Stunden sein Maximum erreicht und über 24 Stunden wieder abklingt (s. Abb. 4) (Link et al., 2016a). Da bekannt ist, dass durch EE allein schon antidepressive Wirkungen erzielt werden können, liegt es nahe, die hierbei auftretende Steigerung des Activin-Signalwegs als endogenen antidepressiven Mechanismus anzusehen. Neben Activin wird im gleichen experimentellen Setting auch die Expression des Brain-Derived Neurotrophic Factor (BDNF) erhöht, wenngleich der Anstieg schwächer ausfällt. Die Parallele ist insofern bemerkenswert, als BDNF seit Langem nicht nur eine prominente Rolle bei synaptischer Plastizität, Lernen und Gedächtnis spielt, sondern ebenso als wichtiger Mediator antidepressiver Effekte hervorgetreten ist. Die starke Überlappung der Wirkspektren lässt vermuten, dass die beiden Faktoren in der Lage sein sollten, synergistische Effekte sowohl auf kognitive Leistungen wie auf affektives Verhalten auszuüben.
Activin als neuroprotektiver Faktor
Historisch gesehen wurde Activin im Gehirn zunächst als neurotropher und -protektiver Faktor charakterisiert. Dieser Befund hat sich inzwischen in einer Vielzahl von Modellen der akuten und chronischen Hirnschädigung bewahrheitet. Bei der akuten exzitotoxischen Hippocampusläsion der Maus kommt es zu einem raschen Anstieg von endogenem Activin, wodurch das Ausmaß der Läsion begrenzt wird (Muller et al., 2006). Wird rekombinantes Activin über eine osmotische Minipumpe in den Ventrikel prä-appliziert, verhindert dies den exzitotoxischen Zelltod im Hippocampus (Tretter et al., 2000). Von besonderem klinischem Interesse ist der Befund aus einem Schlaganfall-Modell der Maus, wonach die einmalige intracerebroventrikuläre Gabe von Activin selbst sechs Stunden nach dem schädigenden Ereignis noch ausreicht, um das Läsionsvolumen zu reduzieren (Mukerji et al., 2009). Auch in Parkinsonmodellen erwies sich Activin als neuroprotektiv, ein Hinweis, dass der Faktor auch bei chronisch neurodegenerativen Erkrankungen therapeutisches Potenzial besitzen könnte. In einer eleganten Studie haben Hilmar Bading und Mitarbeiter kürzlich einen sehr interessanten Mechanismus entdeckt, bei dem das sequenzielle Zusammenwirken von BDNF und Activin die Aktivierung von neurotoxisch wirkenden, extrasynaptischen NMDA-Rezeptoren reduziert (Lau et al., 2015). Die Signalkette ist in Abb. 5 schematisch illustriert. Nach diesen Befunden würde jede Beeinträchtigung des BDNF-Activin-Synergismus die heikle Balance zwischen den synaptischen NMDA-Rezeptoren, die das neuronale Überleben fördern, und den extrasynaptischen NMDA-Rezeptoren, die dem neuronalen Zelltod Vorschub leisten, zugunsten letzterer verschieben. Besteht eine derartige Dysbalance über längere Zeit, könnte sie einen wichtigen pathogenetischen Mechanismus für neurodegenerative Erkrankungen darstellen.
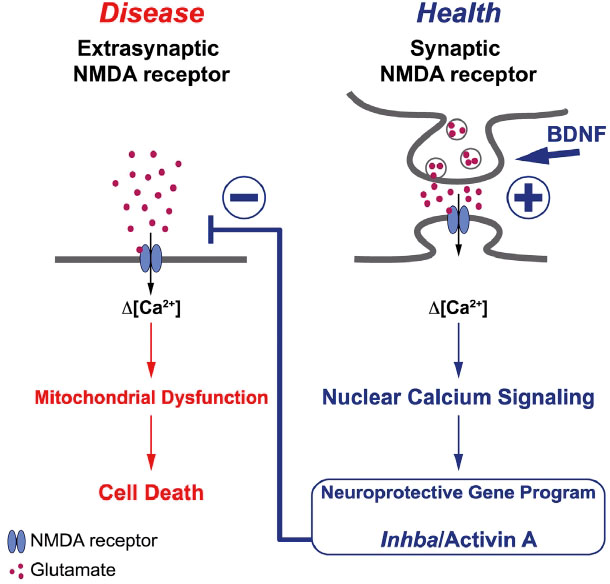
Schema der neuroprotektiven Interaktion zwischen BDNF und Activin. Durch den Synergismus der beiden Faktoren können die Neurone vor der Aktivierung der neurotoxisch wirkenden extrasynaptischen NMDA-Rezeptoren geschützt werden. (Nachgedruckt aus Lau et al., 2015, mit freundlicher Genehmigung des Verlags)
Ausblick
Die allmähliche Entschlüsselung der unerwartet vielfältigen Wirkungen von Activin im gesunden und erkrankten Gehirn des Erwachsenen zeigt eine Reihe von erstaunlichen Parallelen zu dem Wirkspektrum des etablierten und eingehend erforschten Proteins BDNF. Das reicht von synaptischer Plastizität, Lernen und Gedächtnis über Suchtverhalten, Gemütserkrankungen bis hin zu neurodegenerativen Krankheiten. Inwieweit die Effekte, welche die beiden Faktoren bei jeweils vergleichbarer Zielrichtung in den unterschiedlichen Bereichen ausüben, auf weitgehend unabhängigen oder eng miteinander abgestimmten Signalwegen beruhen, ist eine spannende Frage für die zukünftige Forschung.
Über die Autoren

Fang Zheng hat an der Nanjing Universität (China, 1986-1990) Physiologie studiert und 1999 an der National University Singapore in Neurophysiologie promoviert. Dazwischen hat sie im Rahmen eines Master-Programms am Shanghai Brain Research Institute (Chinesische Akademie für Wissenschaften) zusätzliche Kenntnisse auf dem Gebiet der Neuroanatomie erworben. Nach der Promotion hat sie als Postdoktorandin im Labor von Prof. Steve Johnson an der Oregon Health & Science University (USA) gearbeitet und sich dort elektrophysiologischen Untersuchungen an Hirnschnittpräparaten zugewandt. Seit 2003 leitet sie die Arbeitsgruppe für Hirnschnitt-Elektrophysiologie bei Prof. Christian Alzheimer, zunächst an der Universität zu Kiel und anschließend an der Friedrich-Alexander-Universität Erlangen-Nürnberg.

Andrea Link hat in Erlangen Molekulare Medizin studiert. Anschließend hat sie am Institut für Physiologie und Pathophysiologie unter der Leitung von Prof. Alzheimer die Effekte von Activin im ZNS während ihrer Promotion untersucht. Nach einer kurzen Phase als Postdoktorandin am selbigen Institut ist sie nun wissenschaftliche Koordinatorin des DFG-geförderten Graduiertenkollegs GRK2162 mit dem Schwerpunkt „Neurodevelopment and Vulnerability of the Central Nervous System“.

Christian Alzheimer hat in München Humanmedizin studiert und am Max-Planck-Institut für Psychiatrie promoviert. Nach der ärztlichen Approbation 1985 hat er als Wissenschaftler am Physiologischen Institut der Ludwig-Maximilians-Universität in München und am Department of Physiology and Biophysics der University of Washington in Seattle (USA) gearbeitet. Nach der Habilitation im Fach Physiologie 1995 und einem Heisenberg-Stipendium der Deutschen Forschungsgemeinschaft war er zunächst Professor für Physiologie und Direktor am Physiologischen Institut der Universität Kiel, bevor er 2008 auf einen Lehrstuhl für Physiologie der Friedrich-Alexander-Universität Erlangen-Nürnberg berufen wurde und dort seither das Institut für Physiologie und Pathophysiologie leitet. Derzeit fungiert er als Sprecher des Interdisziplinären Zentrums für Neurowissenschaften (IZN) der Universität. Sein Forschungsgebiet sind die zellulären und synaptischen Grundlagen höherer Hirnfunktionen im gesunden und erkrankten Gehirn.
Danksagung:
Die Autoren danken Frau Prof. Sabine Werner, Institute of Molecular Health Sciences, ETH Zürich, für hilfreiche Diskussionen und Kommentare zu dem Manuskript. Folgende Institutionen und Stiftungen haben unsere Forschung über die Jahre maßgeblich gefördert: Deutsche Forschungsgemeinschaft, DFG-GRK2162/1, Bundesministerium für Bildung und Forschung, Johannes und Frieda Marohn-Stiftung, Emerging Field Initiative der FAU, Dr. Ernst und Anita Bauer-Stiftung und Jürgen Manchot-Stiftung.
Literatur
Ageta H., Ikegami S., Miura M., Masuda M., Migishima R., Hino T., Takashima N., Murayama A., Sugino H., Setou M., Kida S., Yokoyama M., Hasegawa Y., Tsuchida K., Aosaki T., Inokuchi K. (2010). Activin plays a key role in the maintenance of long-term memory and late-LTP. Learn Mem 17: 176–185.10.1101/lm.16659010Search in Google Scholar PubMed
Dow A.L., Russell D.S., Duman R.S. (2005). Regulation of activin mRNA and Smad2 phosphorylation by antidepressant treatment in the rat brain: effects in behavioral models. J Neurosci. 25: 4908–4916.10.1523/JNEUROSCI.5155-04.2005Search in Google Scholar PubMed PubMed Central
Gancarz A.M., Wang Z.J., Schroeder G.L., Damez-Werno D., Braunscheidel K.M., Mueller L.E., Humby M.S., Caccamise A., Martin J.A., Dietz K.C., Neve R.L., Dietz D.M. (2015). Activin receptor signaling regulates cocaine-primed behavioral and morphological plasticity. Nat Neurosci. 18: 959–961.10.1038/nn.4036Search in Google Scholar PubMed PubMed Central
Krieglstein K., Zheng F., Unsicker K., Alzheimer C. (2011). More than being protective: functional roles for TGF-beta/activin signaling pathways at central synapses. Trends Neurosci. 34: 421–429.10.1016/j.tins.2011.06.002Search in Google Scholar PubMed
Lau D., Bengtson C.P., Buchthal B., Bading H. (2015). BDNF Reduces Toxic Extrasynaptic NMDA Receptor Signaling via Synaptic NMDA Receptors and Nuclear-Calcium-Induced Transcription of inhba/Activin A. Cell Rep 12: 1353–1366.10.1016/j.celrep.2015.07.038Search in Google Scholar PubMed
Link A.S., Kurinna S., Havlicek S., Lehnert S., Reichel M., Kornhuber J., Winner B., Huth T., Zheng F., Werner S., Alzheimer C. (2016a). Kdm6b and Pmepa1 as Targets of Bioelectrically and Behaviorally Induced Activin A Signaling. Mol Neurobiol 53: 4210–4225.10.1007/s12035-015-9363-3Search in Google Scholar PubMed
Link A.S., Zheng F., Alzheimer C. (2016b). Activin Signaling in the Pathogenesis and Therapy of Neuropsychiatric Diseases. Front Mol Neurosci. 9: 32.10.3389/fnmol.2016.00032Search in Google Scholar PubMed PubMed Central
Mukerji S.S., Rainey R.N., Rhodes J.L., Hall A.K. (2009). Delayed activin A administration attenuates tissue death after transient focal cerebral ischemia and is associated with decreased stress-responsive kinase activation. J Neurochem 111: 1138–1148.10.1111/j.1471-4159.2009.06406.xSearch in Google Scholar PubMed PubMed Central
Muller M.R., Zheng F., Werner S., Alzheimer C. (2006). Transgenic Mice Expressing Dominant-negative Activin Receptor IB in Forebrain Neurons Reveal Novel Functions of Activin at Glutamatergic Synapses. J Biol Chem 281: 29076–29084.10.1074/jbc.M604959200Search in Google Scholar PubMed
Singer W. (2005). Das Gehirn – ein Orchester ohne Dirigent. MaxPlanckFORSCHUNG 2/2005: 15–18.Search in Google Scholar
Tretter Y.P., Hertel M., Munz B., ten Bruggencate G., Werner S., Alzheimer C. (2000). Induction of activin A is essential for the neuroprotective action of basic fibroblast growth factor in vivo. Nat Med 6: 812–815.10.1038/77548Search in Google Scholar PubMed
Zheng F., Adelsberger H., Muller M.R., Fritschy J.M., Werner S., Alzheimer C. (2009). Activin tunes GABAergic neurotransmission and modulates anxiety-like behavior. Mol Psychiatry 14: 332–346.10.1038/sj.mp.4002131Search in Google Scholar PubMed
Zheng F., Puppel A., Huber S.E., Link A.S., Eulenburg V., van Brederode J.F., Muller C.P., Alzheimer C. (2016). Activin Controls Ethanol Potentiation of Inhibitory Synaptic Transmission Through GABAA Receptors and Concomitant Behavioral Sedation. Neuropsychopharmacology 41: 2024–2033.10.1038/npp.2015.372Search in Google Scholar PubMed PubMed Central
© 2017 by De Gruyter
Articles in the same Issue
- Frontmatter
- Übersichtsartikel
- Schlaf, Nahrungsaufnahme und Fortbewegung – Koordination von angeborenem Verhalten durch den lateralen Hypothalamus
- To eat? To sleep? To run? Coordination of innate behaviors by lateral hypothalamus
- Apollos Fluch und Segen: Musizieren als Neuroplastizitätsmotor
- Apollos Gift and Curse: Making Music as a model for Adaptive and Maladaptive Plasticity
- Lust an Gewalt: appetitive Aggression als Teil der menschlichen Natur
- Lust for violence: Appetitive aggression as a fundamental part of human nature
- Bedeutung von Activin für Kognition, Affekt und neuronales Überleben
- Role of activin in cognitive functions, affective behavior and neuronal survival
- Neuronale Vielfalt in der Netzhaut
- Neuronal Diversity In The Retina
- Forschungsförderung
- DFG-Sonderforschungsbereich SFB1193 „Neurobiologie der Resilienz gegenüber stressinduzierter psychischer Dysfunktion: Mechanismen verstehen und Prävention fördern“
- Rezension
- Norman Sieroka: Leibniz, Husserl, and the Brain
- Nachrichten
- Nachrichten
Articles in the same Issue
- Frontmatter
- Übersichtsartikel
- Schlaf, Nahrungsaufnahme und Fortbewegung – Koordination von angeborenem Verhalten durch den lateralen Hypothalamus
- To eat? To sleep? To run? Coordination of innate behaviors by lateral hypothalamus
- Apollos Fluch und Segen: Musizieren als Neuroplastizitätsmotor
- Apollos Gift and Curse: Making Music as a model for Adaptive and Maladaptive Plasticity
- Lust an Gewalt: appetitive Aggression als Teil der menschlichen Natur
- Lust for violence: Appetitive aggression as a fundamental part of human nature
- Bedeutung von Activin für Kognition, Affekt und neuronales Überleben
- Role of activin in cognitive functions, affective behavior and neuronal survival
- Neuronale Vielfalt in der Netzhaut
- Neuronal Diversity In The Retina
- Forschungsförderung
- DFG-Sonderforschungsbereich SFB1193 „Neurobiologie der Resilienz gegenüber stressinduzierter psychischer Dysfunktion: Mechanismen verstehen und Prävention fördern“
- Rezension
- Norman Sieroka: Leibniz, Husserl, and the Brain
- Nachrichten
- Nachrichten