Advances in the prerequisite and consequence of STING downstream signalosomes
-

 and
and

Abstract
The cyclic GMP-AMP synthase (cGAS)-stimulator of interferon genes (STING) pathway is an evolving DNA-sensing mechanism involved in innate immunity and pathogen defense that has been optimized while remaining conserved. Aside from recognizing pathogens through conserved motifs, these receptors also detect aberrant or misplaced self-molecules as possible signs of perturbed homeostasis. Upon binding external or self-derived DNA, a mobile secondary messenger 2′3′-cyclic GMP-AMP (cGAMP) is produced by cGAS and in turn activates its adapter STING in the endoplasmic reticulum (ER). Resting-state or activated STING protein is finely restricted by multiple degradation machineries. The post-translational changes of the STING protein, along with the regulatory machinery of the secret routes, limit the onset, strength and sustention of STING signal. STING experiences a conformational shift and relocates with TBK1 from the ER to perinuclear vesicles containing transcription factors, provoking the transcription activity of IRF3/IFN-I and NF-κB pathways, as well as to initiate a number of cellular processes that have been shown to alter the immune landscape in cancer, such as autophagy, NLRP3 inflammasome, ER stress, and cell death. STING signal thus serves as a potent activator for immune mobilization yet also triggers immune-mediated pathology in tissues. Recent advances have established the vital role of STING in immune surveillance as well as tumorigenic process. This review provides an overview of the disparate outcomes of cancer attributed to the actions of pleiotropic and coordinated STING downstream signalosomes, along with the underlying mechanisms of STING function in pathologies, providing therapeutic implications for new approaches in hunt for the next generation of cancer immunotherapy base on STING.
A chronic DNA leak that occurs as tumors grow due to a variety of causes, such as hyperproliferation, genomic instability, genetic alterations or damage to the mitochondria arising at an exponential rate. The front line of defense for finely probing the immediate environment and preserving host integrity are pattern recognition receptors (PRR). One of them, the evolutionarily conserved DNA sensor cyclic GMP-AMP synthase (cGAS) catalyzes the synthesis of an atypical cyclic di-nucleotide second messenger 2′3′-cyclic GMP-AMP (cGAMP) once binding on cytosolic DNA. cGAMP transits through the surroundings in the tumor microenvironment to initiate the activation of its endoplasmic reticulum (ER)-located receptor Stimulator of interferon genes (STING, also known as MITA [1], ERIS [2], MPYS [3]) in the tumor infiltrating cells (Figure 1). The complex nature of cGAS-STING signaling is emphasized by this distinct two-step PRR pathway, which offers extra layers of control. STING situates the membrane of the ER with its four-pass transmembrane domain (TMD), long cytoplasmic ligand-binding domain (LBD), and C-terminal tail (CTT) enabling heterotypic interactions with TANK-binding kinase 1 (TBK1) and Interferon regulatory factor 3 (IRF3) to produce downstream type I interferons (IFN-I) and other cytokines [4], [5], [6]. Featuring a bilayer form and head-to-head and side-to-side connections by LBD in its autoinhibitory state on the ER, Apo-STING limits TBK1 recruitment [7]. Once the LBD domain binds to cGAMP, the dimeric STING protein experiences a conformational shift that causes the LBD to seal around cGAMP and rotates the cytosolic domain 180° regarding to its TMD [8] (Figure 1). When the CTT is released from its autoinhibitory binding site, the oligomerization interface of STING is exposed. This enables STING to generate disulfide bridges that secure stabilized linear polymers via STING’s cysteine 148 at the ER, resulting in irreversible STING activation [9].
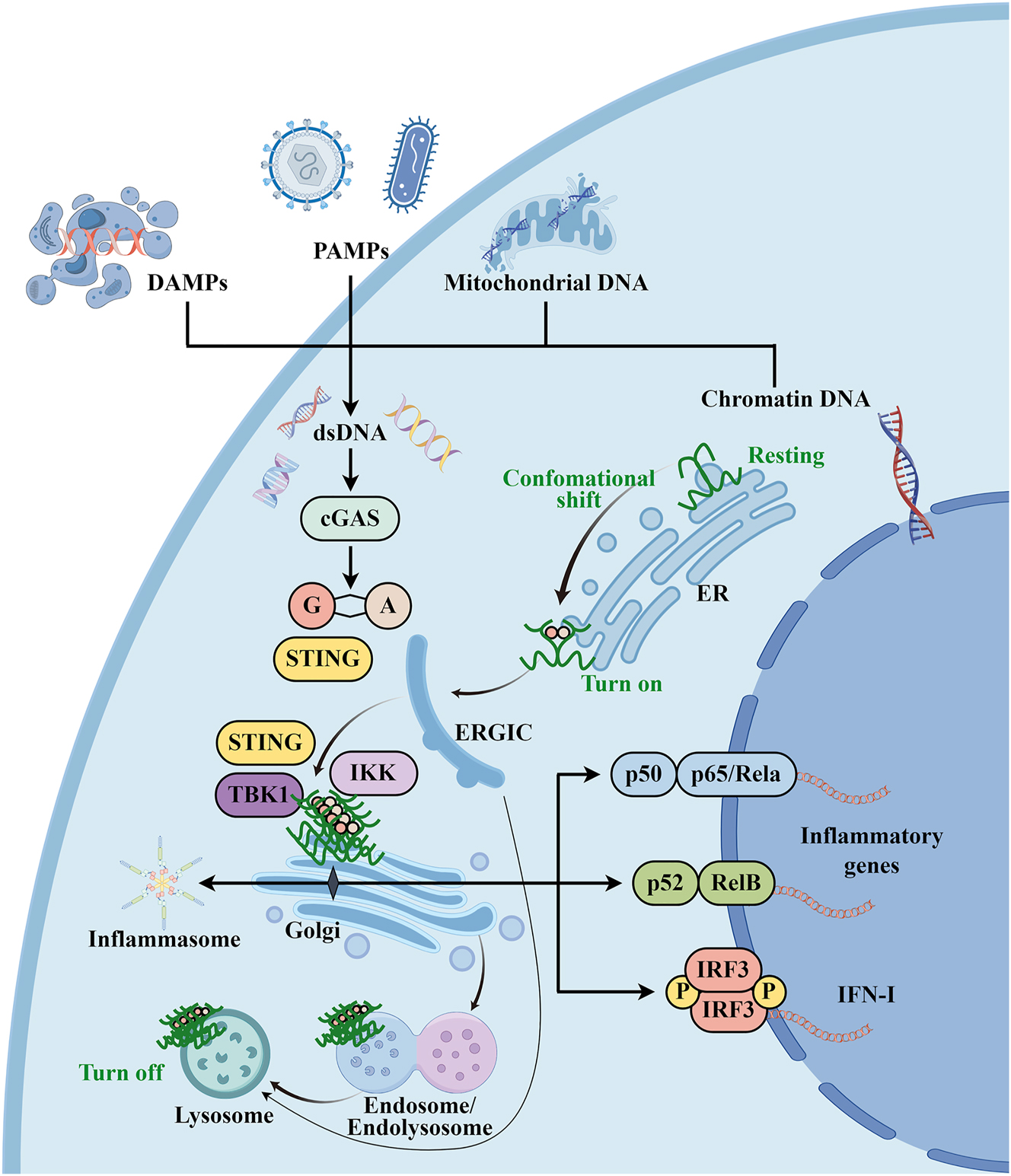
Overview of the cGAS/STING signal transduction pathway. Cytosolic DNA accumulates in response to pathogen infections (e.g., viruses and bacteria) and homeostasis perturbations (e.g., surrounding dead cells). DNA sensors, such cGAS, senses misplaced dsDNA of chromatin, mitochondria, PAMP or DAMP. cGAMP is synthesized by cGAS and then activates its adapter STING, which undergoes a conformational shift and relocates with TBK1 from the ER to perinuclear vesicles containing transcription factors, provoking the transcription activity of IRF3/IFN-I and NF-κB pathways, as well as to initiate a number of cellular processes such as autophagy and NLRP3 inflammasome activation. The degradation of STING occurs in the lysosome, which is accessible by Golgi trafficking or autophagosomes by STING. Figure was illustrated by Figdraw (www.figdraw.com). cGAS, cyclic GMP-AMP synthase; cGAMP, 2′3′-cyclic GMP-AMP; PAMPs, pathogen-associated molecular patterns; DAMPs, damage-associated molecular patterns; STING, stimulator of interferon genes; TBK1, TANK-binding kinase 1; IKK, IκB kinase; IFN-I, type I interferon; ERGIC, endoplasmic reticulum–Golgi intermediate compartment.
STING oligomerization is an essential behavior for its activation
Oligomeric proteins strengthen the selectivity of signal transduction by sequential higher-order assembly of large multimeric protein complex-like inflammasomes, which consist of a cytosolic sensor NOD-like receptor (NLR), the adapter apoptosis-associated speck-like protein with a C-terminal caspase recruitment domain (ASC) and the effector procaspase-1 [10]. Using a scalable approach based on AlphaFold2 to predict homo-oligomeric assemblies across four proteomes, a recent article uncovered that 20 % of eukaryotic, 45 % of bacterial, and 50 % of archeal proteomes form homomers [11]. Thereby, oligomerization represents a prevalent and potentially conserved form of activated functional protein. Oligomerization with a higher-order architecture of STING dimers is an evolutionarily conserved and an early-stage event that happened upon its activation, providing a framework for kinase assembly and signaling transduction [12]. STING oligomerization is overseen by a number of variables.
Elevated rates of reactive oxygen species (ROS) have been detected in the tumor microenvironment of nearly all malignancies, which support many aspects of tumor progression [13]. Activated STING regulates a transcriptional program that promotes the generation of ROS [14]. It has been demonstrated that STING ablation inhibits radiation-induced ROS production and causes resistance to both therapeutic radiation and cisplatin. More importantly, ROS possess the capacity to inactivate DNA, proteins, and lipids and functions as a negative feedback loop. According to a recent study, ROS block IFN production by oxidizing of mouse STING’s cysteine 147, which is equivalent to human STING’s cysteine 148, enabling STING to form disulfide-stabilized oligomers. A recent study observed that STING activation is facilitated by glutathione peroxidase 4 (GPX4) maintained redox equilibrium [15]. Aging is associated with a steady rise in mitochondrial ROS generation and mitochondrial DNA (mtDNA)-triggered cGAS/STING activation [16]. Their link and how STING oligomerization affects inflammaging have not been well-clarified.
In the signaling cascade, intermediate proteins also affect the spatial control of the signaling response, threshold behavior, and signal amplification [10, 17]. Positive cooperativity is characterized by ultrasensitive or switchlike behavior of downstream signalosomes, which can be shown by oligomeric proteins such as activated STING amplified events. Therefore, strategies to modify STING oligomerization, such as chemically induced dimerization or protein engineering, are being investigated (Table 1). Luo et al. developed PC7A, an amphiphilic ultra-pH-sensitive polymeric nanoparticle with a cyclic seven-membered amine ring that directly binds to the STING C-terminal domain and generates biomolecular condensates via polyvalent interactions [18]. PC7A polymer acts as a supramolecular scaffold and directly engages polyvalent interactions, thereby multimerizes STING activation [19]. PC7A assembles into 29-nm nanoparticles with a diameter of less than 50 nm that selectively accumulate inside lymph nodes, priming an intense immunological response without causing considerable systemic damage [19]. A virus study from the same laboratory verified cGAMP-PC7A polymeric nanoparticles effectively inhibited HIV-BaL, HIV-1 (IIIB), and HIV-1 (LAI) reproduction in peripheral blood mononuclear cells (PBMCs), a phenomenon not seen with adjuvants poly(I:C), R848, and CpG. IFN-I appears to be a substantial factor in the effectiveness [20]. A biodegradable block copolymer, PSC7A, was then created by the group due to the accumulation of polymeric PC7A sidechain agonists eventually resulting in dose-limiting toxicity and undesirable consequences [17]. This enhances the therapeutic window of STING activity. Furthermore, small molecule heterocyclic amides can be potent human STING activators [21]. The hydrophobic Compound 53 (C53), for example, is based on an oxindole core structure that allows strong on-target activation of human STING (EC50=185 nM), which leads to the induction of IFN production in human PBMC. C53 is equally potent to at cynomolgus monkey and human STING but inactive at mouse STING. When stimulated with C53 and cGAMP, STING mutations in the binding pocket (H50A, S53L, Y106A, M120L) and the TM3-TM4 loop (V113Q, G114Q, and P115Q) exhibit no or much less high-order oligomerization than wild-type STING [22]. In accordance with the mechanism of C53, sulfated glycosaminoglycans (sGAGs), which are synthesized in the Golgi lumen, induce STING and TBK1 polymerization by binding STING through luminal, positively charged, and polar residues [23]. Remarkably, Li and colleagues discovered that a potent allosteric small molecule, NVS-STG2, functions as the molecular glue to activate human STING via functional screening of a family of chemicals, thereby evokes antitumor activities against established colon tumor MC38 and melanoma B16-SIY via intra-tumoral injection in human STING knock-in mice [24]. Importantly, the aforementioned finding also suggests that NVS-STG2-induced STING activation elicits host antitumor effect instead of intrinsic STING signal in mouse tumor cells. The binding of the substance to a pocket located between the TMDs of nearby STING dimers causes the high-order oligomerization of human STING. The N-terminal TMD R95C mutant is fully capable of being activated by cGAMP except the activity driven by NVS-STG2. The cryo‐electron microscopy (cryo-EM) mapping of the STING tetramer complexed with cGAMP/C53/NVS-STG2 demonstrates that two NVS-STG2 molecules bind to the cavity formed between the TMDs of the two neighboring STING dimers. While orthogonal binding of cGAMP and C53 is respectively formed by side-by-side packing and the overall shape is curled [22]. Excitingly, of recent attention is a non-invasive light-sensitive optogenetic device for reversible control of the cGAS/STING signaling with high spatiotemporal precision. They genetically engineered STING by fusing two copies of the CTT domain (amino acids 341–379) to an optical multimerizer, the N-terminal photolyase-homologous region of Arabidopsis cryptochrome 2 (CRY2) [25]. This region undergoes a monomer-to-oligomer transition after exposure to blue light, forming repeats resembling light-inducible supramolecular organizing centers within dendritic cells (DCs). Upon photo-activation at its primary administration site, mice received adoptive transfer of modified bone marrow-derived DCs with initiated IRF3 and p65 phosphorylation by the CRY2/STING-CTT. It suppresses lung cancer and distant malignancies in a bilateral melanoma model, showing the potential to improve cell-type selectivity and spatiotemporal control, minimizing adverse effects from STING agonist therapy.
Strategies to modify STING oligomerization.
| Properties | PC7A [18], [19], [20]/PSC7A [17] | C53 [22, 89] | sGAGs [23] | NVS-STG2 [22, 24] | CRY2/STING-CTT [25] |
|---|---|---|---|---|---|
| Induction of STING polymers | Yes | Yes | Yes | Yes | Yes |
| Molecular mechanism | An amphiphilic ultra-pH-sensitive polymeric nanoparticle with a cyclic seven-membered amine ring that directly binds to the STING C-terminal domain and generates biomolecular condensates via polyvalent interactions | An oxindole core structure that allows strong on-target activation of human STING | STING binds sGAGs through luminal, positively charged, and polar residues | Two NVS-STG2 molecules bind to the cavity formed between the TMDs of the two neighboring STING dimers. | CRY2/STING-CTT undergoes a monomer-to-oligomer transition after exposure to blue light, forming repeats resembling light-inducible supramolecular organizing centers within DC. |
| Model tested | Colon tumor MC38, melanoma B16-ova and HPV-16 E6/E7-expressing TC-1 tumor model; HIV-BaL, HIV-1 (IIIB), and HIV-1 (LAI) infection. | HEK293T cell; hTert-BJ1 fibroblast cells. | DNA virus infection in vivo. | Colon tumor MC38 and melanoma B16-SIY via intra-tumoral injection in human STING knock-in mice. | Lung cancer LL/2-OVA and distant malignancies in a bilateral B16-OVA melanoma model |
| Other known characteristics | Accumulate inside lymph nodes | sGAG-driven STING polymerization and activation is evolutionally conserved | NVS-STG2-induced STING activation elicits host antitumor effect instead of intrinsic STING signal in mouse tumor cells. | Non-invasive light-sensitive optogenetic device for reversible control of the cGAS/STING signaling with high spatiotemporal precision. |
-
STING, stimulator of interferon genes; C53, compound 53; sGAGs, sulfated glycosaminoglycans; CRY, cryptochrome 2; DC, dendritic cells.
STING polymerization provides a refined dock for IRF3 dimerization or NF-κB activation. All-or-none behavior is shown in the production of STING puncta and IRF3 nuclear translocation, requiring a certain ratio of STING occupied by cGAMP, signifying a high threshold for STING activation [9, 26, 27]. Strong anticancer or antiviral responses can be elicited by a high threshold of STING polymerization, which helps discriminate between basal levels of self-double stranded DNA (dsDNA) and foreign dsDNA at high acute concentrations. Similarly, polymer formation serves as a ligand discrimination step in other innate immune signaling cascades, including cGAS, the enzyme responsible for producing cGAMP, and the RNA sensing adapter proteins MAVS and MDA5. More research is needed to determine the prerequisites for STING polymerization.
STING is distributed but not restricted on the membranes of organelles along the secretory pathway. In addition to forming the polymer, STING’s membrane association also facilitates its trafficking across endomembranes post ligand-induced oligomerization and initiates a chain reaction of cellular immune pathways in an exceedingly fast, coherent and dynamic process (Figure 1). As a result, STING operations can depend on several trafficking subsectors.
STING signal is governed by the cofactors of the secretion system
The potency and sustenance of STING activities are influenced by the basal flux of the secretory system, as demonstrated by thermoregulation from 37 °C to 20 °C, which slows down post-Golgi vesicle trafficking, causing STING accumulation on the Golgi and boosting sustained STING signaling [28]. Furthermore, Zhang et al. discovered that microtubule depolymerization from the Golgi apparatus to lysosomes is the cause of higher and longer-lasting amounts of active STING [29]. This is due to the interaction of STING with microtubules significantly impacting its intracellular trafficking and distribution. According to these reports, disrupting STING trafficking and keeping it in the Golgi apparatus foster homeostatic cGAS-STING signaling and initiate disease pathology. STING is a highly mobile protein as part of membrane bulk flow. Numerous secretion system regulators also control STING trafficking or STING signaling cascades.
ER retention and ER exit
STING spans the ER membrane under basal conditions. Recent studies uncover that STING interacts with Ca2+ sensor stromal interaction molecule 1 (STIM1) [30] or Toll-interacting protein [31] in ER, preventing either its activation or degradation. In addition, transforming growth factor β-activated kinase 1 (TAK1) activation by STING recruits TAB1 in the ER, which in turn induces STING phosphorylation on S355, thereby facilitates later events of STEEP (STING ER exit protein) mediated STING trafficking [5]. STING(G158E) mutation causes an inability to dimerize and impairs TAK1 activation, indicating that STING dimerization is a prerequisite for TAK1 activation. STING oligomerization and trafficking are eliminated by the deletion of TAK1, STEEP, or a mutation in the CTT domain of human STING (S355A) (mouse STING S348A). Furthermore, ADP-ribosylation factor (ARF) GTPases and the Coat protein complex II complex are required for the translocation of activated STING to the Golgi and the endoplasmic reticulum–Golgi intermediate compartment (ERGIC) [32].
Golgi-to-endosome trafficking
The trans-Golgi network (TGN) acts as a major sorting hub where cargos are sorted into several transport carriers for trafficking to different cell compartments [33]. Numerous investigations have utilized genetic ablation of secretion system cofactors to elucidate the intricate mechanism. Coat complex subunit alpha (COPA) deficiency impairs retrograde COPI vesicle trafficking, leaving activated STING accumulation in Golgi apparatus and triggering IFN mediated tissue pathology in mice [34, 35]. Through a genome-wide CRISPR-Cas9-mediated screening process in HeLa-Cas9 cells that are overexpressed varied STING mutants, STING (N154S/V155M), STING (R281M/R284M) and STING (N154S/V155M/R281M/R284M), Fang et al. have recently shown that lARMH3 interacts with STING in Golgi and recruited phosphatidylinositol 4-kinase beta (PI4KB) to synthesize phosphatidylinositol-4-phosphate (PI4P), which directs STING Golgi-to-endosome trafficking via PI4P-binding proteins adapter protein complex-1 (AP-1) and GGA2 and creates the lipid milieu that is required for STING activation through the reaction of PI4P binding proteins [36]. Similarly, Using the results of a spatiotemporal-resolved proximity labeling screen to define STING interactomes on various organelles along its trafficking route, Tu and his coworkers showed that trans-Golgi coiled-coil protein, GRIP And Coiled-Coil Domain Containing 2 (GCC2), and several RAB GTPases act as key regulators of STING post-Golgi trafficking [28]. The prolonged Golgi-dwell time boosted STING activation is evidenced by the delayed and sustained STING puncta production with a comparable peak observed in the Gcc2-deficiency MEFs. The mechanism is similar to COPA-deficiency while the overall phenotype in Gcc2 deficiency is less severe compared to COPA-deficiency. Importantly, loss of Golgi-to-lysosome STING cofactors, but not ER-to-Golgi cofactors, selectively activates tonic interferon signaling, as evidenced by around twice fold of nuclear phosphorylated IRF3 increases at its peak point. Interestingly, there is hardly any impact on the more ancient NF-κB and a slight change in autophagy. Primordial STING downstream activities, such as NF-κB and autophagy, might be relative earlier events that are perpetuated and occurred before STING Golgi exit, meaning that post-Golgi trafficking stoppage does not affect them [32, 37]. These suggest the uncoordinated perturbations in STING downstream networks and distinct underlying regulatory mechanisms.
STING degradation
Fine‐tuning STING activity is important for mounting a proper immune response. STING signaling is upregulated in the post-ER compartments and rapidly shifts off in the lysosome or proteasome to halt its activity [38] (Figure 1).
Autophagy primarily regulates STING activity negatively, which operates via both canonical and non-canonical pathways [39]. The loss of Atg9a greatly enhances the assembly of STING and TBK1 by dsDNA, leading to aberrant activation of the innate immune response [40]. Furthermore, TBK1 phosphorylated p62/Sequestosome-1 facilitates STING degradation by autophagy‐controlled ubiquitination following DNA stimulation in murine embryonic fibroblasts and THP‐1 monocytes, where p62 knockouts display the bumped IFN responses to cytoplasmic DNA or pathogens. Furthermore, STING expression is negatively regulated by the antioxidant transcription factor NRF2, which transactivates the p62 gene promoter and promotes p62 expression in response to oxidative stress [41]. p62-mediated selective autophagy restricts cGAS-STING signaling by either specifically encouraging the degradation of p62 and STING [42] or by interacting of Beclin1 with cGAS to limit cGAMP synthesis [43]. In addition, AP-1 mediated delivery of phosphorylated STING from TGN46 positive Golgi apparatus into acidic endolysosome via clathrin-coated transport vesicles upon binding a highly conserved dileucine motif and the phosphorylated S366 residue in the STING CTT domain for sequential degradation, complementing the autophagy-mediated degradation of STING. L364F substitution enhances STING binding to AP-1 and speeds STING degradation kinetics, in accordance with a study that found a missense mutation p.LL363LF, located in the EXXXLI motif, which has been linked to human cancer [44]. Seventy-two human STING gene variations are found in a variety of malignancies, the majority of which are dominant-negative mutants. Further proximity-ligation proteomics and unbiased genetic screening reveal that the endosomal sorting complex is required for transport (ESCRT) complex, which includes Hepatocyte growth factor-regulated tyrosine kinase substrate (HGS), Vacuolar protein sorting-associated protein 37A (VPS37A), and Ubiquitin associated protein 1(UBAP1), facilitates the degradation of STING [38]. ESCRT-dependent STING degradation inhibits steady-state and cGAMP-induced signaling. To enable a transient activation of immunity components, a structural mechanism of negative regulation of STING is established as signaling initiation and termination are intricately connected. The ESCRT complex functions as a homeostatic regulator of STING signaling while STING is susceptible to a tonic degradative flux.
While multiple mechanisms are reported to regulate the activated STING, it remains inconclusive how the ER’s nascent STING protein is homeostatically regulated. Recently, Pokatayev et al. revealed that the nascent STING protein level is strictly regulated by a constant tug-of-war between ‘stabilizer’ TOLLIP and ‘degrader’ inositol-requiring enzyme 1α (IRE1α)-lysosome that together preserves tissue immunological homeostasis [31]. In contrast, Ji et al. reported the ER-related degradation of SEL1L–HRD1 (suppressor of lin-12-like–HMG-CoA reductase degradation 1) protein complex ubiquitinates STING protein at the ER for proteasomal degradation [45]. This process uncouples from ER stress or its sensor IRE1α and controls the turnover and amount of the activable STING pool in the baseline state. More intriguingly, a recent study found that Sel1l ablation in CD8+ T-cell reduced the amount of its mature subpopulation and disrupted its peripheral homeostasis, which is worsened by IRE1α deletion and rescued by inhibition of PERK-ATF4-CHOP-Bim signaling [46]. Studying the involved role of STING turnover in controlling CD8+ T-cell biology is worthwhile given the well-documented role that causes T cell death [47], [48], [49], [50]. As a consequence, the maximum activation potential of STING is regulated. Further studies are needed to clarify this.
There are ample documentations on how the secretory system cofactor affects STING activation [38, 51]. Further research is necessary to determine whether the secretory route was responsible for programming and fully furnishing the spatiotemporal dynamics of STING signalosomes. More detailed description on the ratio of STING cellular localizations across organelles in various circumstances is needed.
Posttranslational events, including phosphorylation and dephosphorylation, nitro-alkylation, glycosylation, ubiquitylation and deubiquitylation, SUMOylation, carbonylation, oxidation, and palmitoylation, have been well documented to control STING activity [52]. STING activity is limited by its modification, spatial distribution or substrate. Although there has been a lot of effort in understanding the upstream events including protein modification during STING activation, how the reaction promoted by the posttranslational modification of STING protein proceeds specifically at the organelles remains poorly understood.
The prerequisite and consequence for STING downstream signalosomes
The cGAS-STING signal is a conserved nucleic acid sensing pathway with optimally evolving roles [37]. Based on genetic evidence, the production of IFN-I against DNA viruses and malignancies in most cell types depends on the cytosolic DNA detecting cGAS-STING signal [53, 54]. IFNs production is triggered and restricted by STING-CTT domain-mediated TBK1/IRF3 cascade via STING-LxIS motif for TBK1 phosphorylation and STING-PxPLR motif for TBK1 binding [55]. However, STING’s primary cyclic dinucleotide (CDN)-binding role is extremely ancestral and deeply conserved. The intact cGAS-CDN-STING pathway predates interferon-based immunity. Moreover, the primitive function of the cGAS-STING pathway has been linked to regulating autophagy and NF-κB signal to provide antibacterial immunity, which does not require the CTT [56].
Autophagy
The autophagy apparatus is present in all eukaryotes and plays a crucial role in the recycling and degradation of cellular components in response to a variety of stimuli [57, 58]. ERGIC vesicles coated with ligand-binding STING are membrane source for modification by the ubiquitin-like protein LC3, a crucial stage in autophagosome biogenesis [32]. It has been determined that the human STING residues L333, R334, and the corresponding tiny area (residues 330–334) are crucial for autophagy [32]. It is interesting to note that this small region is contained inside the STING unfolded protein response (UPR) motif (residues 322–343), which was first linked to STING-activated ER stress and subsequent T cell death [49]. LC3B lipidation is significantly influenced by the STING-TMD following STING translocation. cGAMP induced LC3 lipidation through a mechanism which is dependent on WIPI2 and ATG5 but independent of the ULK and VPS34–beclin kinase complexes [32]. Further, STING activation causes LC3B lipidation onto single-membrane perinuclear vesicles mediated via non-canonical autophagy factors ATG16L1 via its WD40 domain [59], bypassing the necessity for canonical upstream autophagy machinery. Selective autophagy can be initiated by p62 (mouse S405 and S409, equivalent to human S403 and S407), phosphorylated by CK2 (Casein Kinase II), or TBK1 and the key autophagy-related kinase ULK1 downstream of the STING. Oligomeric p62 detects ubiquitin stress and causes the targets to be degraded by attaching ubiquitinated proteins or intracellular bacterial or viral particles to ATG8 (referred to as LC3 in mammals) presented on the autophagosome membrane. P62-Ub interaction together with p62 phosphorylation and oligomerization are required for the formation and functions of p62-mediated selective autophagy. It is interesting to note that activated ULK1 preferentially suppresses IRF3/IFN signaling and causes immunosuppression by targeting STING on serine 366 after its transcriptional activities [60]. Another autophagy upstream factor CK2 downregulates IRF3 and TBK1 phosphorylation and unidentified intermediate molecules respectively, while it promotes NF-κB activation and proinflammatory cytokine production [61]. Compared to porcine WT STING, IFN-defective mutants such as △CTT, S365A, and pLxIS sub all elicited similar levels of lipid-bound LC3 (LC3-II), indicating that STING-mediated autophagy and p-p62 were not dependent on IFN. Additionally, autophagy positively influenced porcine STING-induced NF-κB activity, suggesting that these two may be inherently linked [62]. These findings imply that autophagy is in opposition to the IRF3 signal while in tandem with the NF-κB signal.
NF-κB
The NF-κB pathway, a crucial transcription factor of STING downstream signalosome, activates a wide range of genes, including inflammatory cytokines (Figure 1). TBK1 and IκB kinase ε (IKKε) act redundantly to elicit NF-κB activation STING downstream in myeloid cells, providing a view that monotherapy inhibiting TBK1 kinase may not entirely block STING-driven pathology [63]. Importantly, IKKb and TAK1/MAP3K7 complexes drive canonical NF-κB responses and mitogen-activated protein kinases (MAPK) p38 and JNK signaling pathway induced proinflammatory cytokines production [5]. STING-activated TAK1 regulates STING trafficking prior to STING dimerization in ER. These findings indicate that NF-κB signaling is an early STING signaling event that occurs before ER exit. In a mouse allograft tumor model, TAK1 activation by monophosphoryl lipid A (MPLA), a TLR4 agonist, boosts cGAMP-induced antitumor immunity reliant on STING phosphorylation, consistent with a study from another individual laboratory. Studies indicates that STING activation is boosted by NF-κB activation caused by a wide range of factors such as Toll-like receptor (TLR), interleukin-1 receptor (IL-1R), tumor necrosis factor receptor (TNFR), growth factor receptor (GF-R), or protein kinase C (PKC) [29]. These highlight a valuable connection between STING activation and other PRRs. Thus, the activation of NF-κB signaling through STING activation is ensured by the sequential action of two molecular machinery in different organelles. Non-canonical NF-κB signaling was associated with higher levels of chromosomal instability and worse clinical prognosis and a lower survival rate by increasing cancer cell fitness [64], [65], [66]. STING activates TRAF3-dependent non-canonical activation of NF-κB (Figure 1), which entails NF-κB-inducing kinase (NIK) stabilization following TRAF3 degradation [67]. In TRAF3-deficient cells, the increase of NIK, a crucial enzyme regulating non-canonical NF-κB activation, leads to deregulation of the canonical NF-κB pathway. A genetic study in human inborn errors of the alternative NF-κB pathway (autosomal-dominant p52LOF/IκBδGOF NF-κB2 disorders, autosomal-recessive NIK, or autosomal-recessive RELB) has shown that these disorders are associated with a higher risk of developing life-threatening COVID-19 pneumonia as well as the development of neutralizing autoantibodies against IFN-I [68]. This suggests that the alternative NF-κB pathway p52-RELB activation is necessary to prevent central T cell tolerance toward IFN-I in human. The coordinated effect of NF-κB signaling and IRF3/IFN-I downstream of STING activation to T cell tolerance remains unclear. Additionally, a study by Hou et al. raises the possibility that radiotherapy dampens the therapeutic benefit by activating non-canonical NF-κB pathways and retarding the IRF3/IFNβ cascade downstream of the STING signal [69]. These results indicate non-canonical NF-κB coordinates perturbation of both canonical NF-κB and IRF3 activation downstream of the STING signal.
Dramatic enhancement of NF-κB signaling was elicited via zebrafish not human STING-CTT [37]. The canonical TRAF6 signaling complex is directly recruited by the zebrafish STING CTT with an additional c-terminal module DPVETTDY, which is absent from human and mammalian STING alleles, to activate NF-κB signaling [37, 70]. STING alleles in humans and mice produced a relatively moderate NF-κB response but an elevated IRF3 response. An evolving substitution the atypical I365 (human STING) with F372 (zebrafish STING) in the IRF3 binding motif obtains IRF3-mediated signaling while a NF-κB hyperactivating module is fine-tuned [37]. These findings show that the STING-CTT is modularly organized and that modifications that modify STING downstream innate immune responses have a low evolutionary threshold. Over the course of evolution, the acquisition of modular motifs in STING-CTT regulates the specificity and intensity of downstream effects, thereby tailoring cGAS-STING immune responses to species-specific pathogen loads. It has been demonstrated that the human STING pathway is more selective and less reactive compared to mice.
Thus, understanding the evolutionary origins of the downstream consequences of STING pathway may provide insight into how this critical pathway has evolved to balance these risks. STING-CTT mediated signaling, including TBK1/IRF3/IFN-I axis and heightened NF-κB responses, is actually a relatively recent innovation. Finding out how pre-existing NF-κB affects latecomer IRF3/IFN-I or the coupling IRF3/IFN-I on top of ancestral NF-κB pathway is of significance, providing a mechanism for how STING accommodates these transcriptional signalosomes.
NF-κB synergizes with IRF3 to induce high amounts of IFN-I and proinflammatory cytokines [29, 71, 72]. At the transcriptional level, the promoter of IFNβ gene is activated by recruiting transcription factors, such as IRF3, NF-κB, and ATF-2-c-Jun [71]. At the posttranslational level, NF-κB activation prevents STING degradation by limiting microtubule-mediated STING trafficking from the Golgi apparatus to lysosomes, prolonging and increasing STING signaling and IFN production [29]. Consequently, this coordinated effect strengthens host antiviral defenses and STING-mediated IFN responses.
IRF3
It is widely known that TBK1 phosphorylates IRF3, which causes it to dimerize and depart to the nucleus, triggering the transcription of IFN-I and other IFN-stimulated genes (ISGs) as well as antigen-specific immune responses against tumor and viral infection [53, 54] (Figure 1). TLR ligands and IL-1β activated TBK1 do not, however, cause IRF3 phosphorylation, suggesting that particular adapter proteins – TRIF for TLR3 and TLR4, MAVS for RIG-I and MDA5, and STING for the DNA-sensing pathways – are involved in signaling pathways that result in IRF3 activation [55, 73]. It has been shown that late endosomes play a significant role as vesicle compartments in STING-mediated IRF3 phosphorylation [5, 74, 75]. Wang et al. and colleagues reported that STING dimerizes upon ligand-binding and interacts with membrane-bound epidermal growth factor receptor (EGFR), which autophosphorylates and provides the platform for the recruitment of cytoplasmic Syk to the signaling complex and its activation. Activated Syk phosphorylates Tyr240 of STING, followed by phosphorylation of Tyr245 by EGFR [75]. The later modification in the ER is required for STING translocation to the late endosome where it binds IRF3. Alternatively, activated STING translocates to the autophagosome in the absence of EGFR with no IRF3 activation, IFN production and antiviral activity in cells and mice. This finding suggests potential therapeutic and experimental strategies for the selective modulation of STING functions [74].
IRF3 phosphorylation is restricted by multiple machineries. According to recent reports from two separate groups, STING ubiquitination has been shown to specifically control IRF3 signal transcriptomes [76, 77]. The results of the mass spectrometry approach in 293T expressing STING reveal that K224 substitution nearly eliminates STING ubiquitination by E3 ligase MUL1 (mitochondrial E3 ubiquitin protein ligase 1) in the presence of cytosolic DNA. Blocking K224 ubiquitination specifically prevents IRF3 but not NF-κB activation [77]. Furthermore, Stempel et al. revealed that m152, a highly conserved protein among various MCMV strains, interacts with murine STING (but not human STING) via their respective ER-luminal domains and traffic together from the ER to the Golgi compartment, leading to a declined STING trafficking and delayed IFN response, however, NF-κB response remains intact [76]. K288R mutation sequesters STING protein in ER after stimulation, the NF-κB pathway is inducible while IFN-I response is invalid. A similar phenomenon is observed in TBK1‐deficient cells. Further study demonstrates that STING activates NF-κB-dependent signaling in ER that is independent of the kinase TBK1, a beneficial outcome for early MCMV transcription [76]. Thus, they uncovered that murine CMV has evolved an attack strategy to specifically antagonize the STING-mediated antiviral IFN response, while preserving its pro-viral NF-κB response, giving it an edge in the establishment of an infection. Importantly, K244 and K288 ubiquitination restricts IRF3 cascade. STING-mediated NF-κB responses appear to be independent of them, indicating that these responses may occur prior to Golgi re-localization. This implies that STING trafficking is not required to stimulate the signaling pathways like NF-κB pathway and uncoupling of IFN-I and NF-κB signaling can occur.
On the opposite to heightened IRF3/IFN-I cascade facilitated by activated NF-κB pathway, IRF3 regulates NF-κB signal by multiple mechanisms at diverse layers in different inflammatory settings. IRF3 restricts kinase IKKβ upstream of the NF-κB signal in the context of metabolic diseases [63], restraining chronic high-fat diet (HFD)-induced hepatic insulin resistance and steatosis through preserving glucose and lipid homeostasis [78]. Furthermore, in cellular and mouse models of Sendai virus infection, IRF3 directly binds to the p65 subunit during nuclear translocation of NF-κB, sequestering it in the cytosol and suppressing lung inflammation [79]. A comparable function of IRF3 in the cytoplasmic sequestration of β-catenin has been documented in relation to colon cancer tumorigenesis [80]. In the context of HFD-induced liver injury, the nontranscriptional function of IRF3-mediated anti-inflammatory protection, as illustrated in Irf3S1/S1 mice with alanine substitutions at Ser-388 and Ser-390 are primarily associated retardation of nuclear translocation of p65 and decreased expression of NF-κB-dependent inflammatory cytokines [78]. The multi-face of IRF3 in the liver is representative as IRF3 activation by viral infection in vivo greatly enhances bile acid- and aspirin-induced hepatotoxicity via stimulating transcriptional suppressor Hes1 to reduce RXRα level [81]. In contrast to this finding of the transcription-independent role of IRF3 on NF-κB repression, two individual groups show that STINGS365A mutants, which substitute the docking site for IRF3, result in precisely abolished transcriptional activity and subsequent IFN-dependent response, also show enhanced NF-κB signal transduction in 293T [62], as well as in primary T cells [82]. Nonetheless, Yan et al. observed that IRF3 promotes MyD88-mediated NF-κB signaling pathway in fish [83]. Thus, uncertainty surrounds the equilibrium between IRF3 and NF-κB transcriptional signalosomes downstream of the STING signal. Further, whether IRF3 regulation of NF-κB signal could reappear in other disease contexts is of interest to examine. TBK1/IRF3 axis can be activated by different adapter proteins downstream of respective innate sensors including RIG-I, cGAS, and TLRs [73]. Given that a range of factors can disrupt both signals, it is noteworthy to investigate the role of STING in maintaining equilibrium between these two downstream signal node molecules. IRF3 also exerts non-transcriptional activation as evidenced by IRF3 with modification of a linear polyubiquitination, activated by the RIG-I pathway recruited TBK1/TRAF2/TRAF6 complex and sequential linear ubiquitin chain assembly complex binding, serves as a proapoptotic factor, as a mechanism to limit virus spread within the host. Thus, the cellular control of IRF3 by posttranslational modifications regulates its activation, activity and stability. IRF3 acts as a network node regulated by multiple mechanisms to be devoid of inflammation and autoimmunity.
NLRP3 inflammasome
NLRP3 functions as a guardian of intracellular homeostasis and monitors a range of endogenous cues [84], such as the efflux of potassium ions (K+) or chloride ions (Cl−), the flux of calcium ions (Ca2+), ROS, cytosolic mtDNA and cardiolipin, mitochondrial damage, lysosomal disruption, ER stress, trans-Golgi disassembly, intracellular kinase signaling, and lipid uptake and accumulation. Recently, the role of STING in perturbing NLRP3 inflammasome has gained increasing attention (Figure 1). Inflammasome activation requires two signals, priming and activation, both of which have differential requirements for STING [85]. A recent report described the histone methylation mediated by WDR5/DOT1L plays a crucial role in augmenting IRF3 binding to the Nlrp3 promoter and facilitating STING-induced Nlrp3 transcription [86]. NF-κB primes the NLRP3-inflammasome for activation by promoting pro-IL-1β and NLRP3 expression. However, it also hinders NLRP3-inflammasome activation by causing a delayed accumulation of the autophagy receptor p62/SQSTM1[87]. The “NF-κB-p62-mitophagy” pathway is an intrinsic regulatory loop in macrophages that allows NF-κB to control its own proinflammatory activity and triggers a self-limiting host response that preserves homeostasis and promotes tissue repair [87]. During HSV-1 infection, STING recruits and interacts with NLRP3 on ER, attenuating K48- and K63-linked polyubiquitination of NLRP3, thereby facilitating NLRP3 inflammasome formation [88]. It has been discovered recently that NLRP3 inflammasome activation in Golgi vesicles is triggered by STING’s proton channel activity [85, 89]. The mechanism remains unclear. It is noteworthy that two individual groups reported PI4P-binding proteins mediate STING or NLRP3 Golgi-to-endosome trafficking and the importance of PI4P in mediating STING activation and NLRP3 inflammasome aggregation [90]. It is unknown if the coupling protein contact occurred during trafficking. The coordinated effect mediates hepatocyte pyroptosis involving oxidative stress and metabolic reprogramming [86]. cGAMP induction of IFN-I precedes inflammasome activation, which then occurs when IFN-I is waning. cGAMP activates the inflammasome in addition to IFN-I, and both are required to prevent DNA virus infection.
Cellular heterogeneity skews downstream consequences of STING activation in cancer
By sensing accumulated cytosolic DNA, tumor infiltrated cells secret a second messenger cGAMP into the surrounding as the tumors grow. STING holds the potential to enhance tumor rejection by activating IFN-I dependent responses in tumor cells and infiltrated immune cell types including DCs, and monocyte [91, 92]. Selective STING stimulation in DCs promotes their maturation and primes antitumor cytotoxic T cell responses [93, 94] (Figure 2). Many natural and synthetic STING agonists have been created since the discovery of STING and have been used in preclinical and clinical settings for various malignancies, either with or without immune checkpoint blockade therapy [1]. However, there has been little success with STING therapy. Thereby, the opposing effects of STING activation have recently attracted more attention (Figure 2).

Diverse outcomes arise from cell preference of STING activation. Tumor infiltrated cells secret a second messenger cGAMP into the surrounding as the tumors grow. STING is universally expressed in a variety of cell subsets. Thereby the surrounding cells exhibit preference to STING downstream effects. STING activation in dendritic cells (DC) and cancer cells promotes IFN-I production and anti-tumor responses. STING activation in adaptive immune cells result in T cell death and regulatory B cell induction. Intrinsic STING signal in cancer cells also promote its survival and metastasis. Thus, cytocial delivery system, such as LNPs, ADC, and exosomes, allows precise spatiotemporal and control over the STING signaling. Figure was illustrated by Figdraw. cGAS, cyclic GMP-AMP synthase; STING, stimulator of interferon genes; DC, dendritic cells; IFN-I, type I interferon; LNPs, lipid nanoparticles; ADC, antibody-drug conjugates.
STING retards cytotoxic cells mediated immune surveillance to tumors. T cells with intrinsic STING signal activation experience substantial cell death, which is consistently observed by several individual labs [47], [48], [49], [50]. Further, sustained activation of the STING/IRF3 axis is partially counteracted with TCR-mediated mTORC1 activation, leading to the suppression of T-cell proliferation [95], which requires a distinct C-terminal domain of STING that activates NF-κB and its re-localization to the Golgi apparatus [50]. In parallel, STING agonists have the ability to simultaneously induce the expression of inhibitory molecules [96, 97], such as indoleamine 2, 3-dioxygenase 1 (IDO1), PD-L1, IL-10, IL-35, and regulatory immune cell types accumulated in the tumor local site [96, 98, 99], which counteract the tumor-suppressive effects [100]. The molecular mechanisms of STING’s disparate functions in different immune cell populations remain unconclusive (Figure 2).
The role of intrinsic STING signal in cancer cells is also debatable. STING mitigates tumorigenesis [101], clears reawakened metastatic tumor cells and prevents spontaneous outbreaks in a T cell- and natural killer (NK) cell-dependent manner [102]. Previous research showed the presence of missense mutations and epigenetic silencing of endogenous cGAS/STING in the tumor microenvironment [44]. However, renal cell carcinoma (RCC) exerts an increased STING expression pattern [103]. STING depletion weakens mitochondrial function, inhibits mTORC1/S6K signaling, which results in RCC growth retardation, and amplifies mitochondrial voltage-dependent anion channel VDAC2/GRP75-mediated mitochondria-ER contact formation to raise mitochondrial ROS/calcium levels. Contrary to its established tumor-suppressive function [101, 102, 104], additional research has revealed that the STING system may also be involved in increasing tumor burden and worsening illness outcomes in mouse tumor models [65, 105] (Figure 2). In cancer cells, constitutive activation of NF-κB can drive the development and spread of tumors while also enhancing cell survival proliferation [106]. STING activation promotes the proliferation and chemoresistance of cancer cells by triggering the release of inflammatory cytokines that are TBK1/NF-κB dependent. The secretion of TNF led to the disruption of blood vessels following the STING agonist treatment [107]. Chronic STING activation and induction of the STING-dependent non-canonical NF-κB pathway are pro-tumorigenic and warrant further studies to clearly delineate the mechanisms. Furthermore, brain metastatic cancer cells engage the Cx43/PCDH7 gap-junctional network to transfer cGAMP to astrocytes, which in turn causes paracrine signals by astrocytes to secrete the inflammatory cytokines TNFα and IFNα. This leads to the activation of the NF-κB and STAT1 pathways in brain metastatic cells, further supporting tumor growth and chemoresistance [108]. More remarkably, autophagy can impede CD8-mediated immune surveillance by downregulated MHC-I on the surface of pancreatic cancer cells [109]. In a radiation pneumonitis model, autophagy also facilitates NKG2D internalization and reduces NK cell function [110]. Chronic inflammatory cytokine secretions by p65’s transcription activity, such as IL-1 and IL-6, led to tumor progression [111]. Furthermore, p65 activation-mediated CXCL14 promoted angiogenesis and tumor growth [112]. HIF1-a and VEGFa are also regulated by p65 affecting epithelial-mesenchymal transition and angiogenesis [113]. As a result, highly aggressive, unstable tumors have evolved to co-opt these programs to drive tumorigenic behaviors (Figure 2).
Potentiate selective STING signalosomes for cancer therapeutics
The use of STING agonists raises a serious concern regarding the potential induction of systemic inflammation due to their pleiotropic effects on various immune cell types [114] (Figure 2). Different forms of STING agonists may bypass particular cell types, which would modify how the immune system responds. Moreover, substantial dosages are frequently required to achieve efficient concentration and retention within tumors, which might increase side effects [114]. These findings highlight the critical need to develop a technique that would allow precise spatiotemporal and control over the STING signaling, particularly in antigen-presenting cells (APCs) such as DCs, to lessen the previously described undesirable side effects. Exosomes, antibody-drug conjugates (ADC), and lipid nanoparticles (LNPs) have all demonstrated encouraging advancements in the targeted in vivo delivery of desired genes to particular cell types (Figure 2).
STING activation by intercellular transmission
STING traffics across organelles through the secretory pathway. Recently, a study revealed that STING is a secreted protein. RAB22A-mediated non-canonical autophagosome, which originated from the ER and is released as an extracellular vesicle, allows activated STING to shuttle across cells to spread antitumor immunity [115]. These studies provided a rationale for adopting extracellular vesicles as a form of delivery for activated STING. Natural STING agonist cGAMP shuttles through gap junctions, receptor-based transport, membrane fusion, and viral particles. Mice receiving immune checkpoint inhibition saw increased tumor-specific T cell responses and antitumor effects when combined with virus-like particles delivering cGAMP. Nanotechnology has been employed to solve the aforementioned issues by altering the pharmacokinetics, biodistribution and cytosolic delivery of CDN therapy, resulting in moderate impact in preclinical models [18]. Apart from the cGAMP-PC7A polymeric nanoparticles above [18], CDN–Mn2+ nanoparticle particle, a self-assembled Mn2+ coordinated with CDN STING agonists, effectively delivers STING agonists to immune cells and offers a new platform for local and systemic cancer treatments [116, 117]. A similar nanoscale coordination polymer encapsulated with CDA, ZnCDA [118], also mediates robust antitumor effects in a diverse set of preclinical cancer models. These investigations highlight the enormous potential of coordinated nanomedicine in the field of metalloimmunotherapy. A formulation of CDN-conjugated poly (β-amino ester) nanoparticles that targets TME and secondary lymphoid organs was recently developed [119]. The proximal immune cells in the TME were gradually stimulated by the drug internalized by cancer cells. The differentiation of immunological memory is aided by nanoparticles that gathered in the spleen. These results demonstrate that APCs may benefit more from the selectively organ-targeted STING agonists, particularly in poorly immunogenic tumors. ADC exert with highly specific targeting capabilities is leading a new era of targeted cancer therapy. CRD5500 (Takeda) and XMT-2056 (Mersana), the leading candidates of HER2-targeted STING-agonist ADC [92], are undergoing clinical research, awaiting the effect results of efficacy and estimated toxicity.
By utilizing the delivery routes, the encapsulated STING agonists are able to overcome the barriers of poor stability, deliverability, and unwanted effects of cell heterogeneity consequences after systemic delivery. However, tumors can easily evade this suppression by downregulation or loss of endogenous STING due to epigenetic silencing or missense mutations in TME [44].
Established mimics imitate STING actions independent of endogenous expression
Take advantage of ultra-light-sensitive CRY2-clustering system CRY2clust as STING oligomerization inducible inducer, further conjunction with STING-CTT as the scaffold for interactions with downstream TBK1 and IRF3, which together constitutively recapitulate STING activation [25]. The platform confers antitumor immune response with high spatiotemporal precision and minimizes adverse effects. The innovation relies on and is restricted to the adoptive cell transfer of DC expressing CRY2clust/STING (Table 2). In an investigation regarding to a ribonucleoprotein STING agonist [120], STING lacking the TMD complexed with cGAMP efficiently initiates STING signaling even in STING-deficient cell lines, which is striking since the STING-TMD associated oligomerization is prerequisite for its activation [22] (Table 2). This ribonucleoprotein self-assembled a tetrameric form under physiological conditions. The molecular mechanism is still unknown. It requires cGAMP presence for the preassembly of the STING/cGAMP protein complex, and therefore represents a challenge for intracellular delivery. In 2023, the groundbreaking work on the mRNA vaccines against COVID-19 by Drs. Katalin Karikó and Drew Weissman is recognized by the Nobel Prize, sparking a new area of medicine with broad therapeutic possibilities. An mRNA-encoded constitutively active STING mutant (V155M) mobilizes immune response without dependence of endogenous STING expression in TME [121] (Table 2). This mutant, localizing STING to the ERGIC, activates downstream signaling including IRF3/IFN-I and pro-inflammatory NF-κB/p65/IL-6 that consequently initiates undesired dose-limiting side toxicity as described above. To tackle this, it is necessary to consider uncoupling beneficial and detrimental signalosomes for cancer therapeutics design.
Established mimics imitates STING’ actions independent of endogenous expression.
| Properties | mRNA-encoded STINGV155M [121] | Self-assembled cGAMP-STING△TM [120] | CRY2clust/STING-CTT [25] |
|---|---|---|---|
| Independent of endogenous STING expression | Yes | Yes | Yes |
| Prerequisite | Constitutively active | Need cGAMP presence for preassembly of the STING/cGAMP ribonucleoprotein complex | Need adoptive cell transfer of DC expressing CRY2clust/STING |
| Consequence | Activated IRF3/IFN-I and NF-κB/p65 pathways | Activated IRF3/IFN-I and increases TNF | Activated IRF3/IFN-I and NF-κB/p65 pathways |
| Tumor types tested | HPV+TC-1 lung primary and metastatic cancer, MC38 colon adenocarcinoma | B16-OVA melanoma; MC38 murine colon adenocarcinoma | LL/2 lung cancer; B16-F10 melanoma |
-
STING, stimulator of interferon genes; TM, transmembrane; CRY2, cryptochrome 2; CTT, C-terminal tail; DC, dendritic cells.
Uncoupling STING signalosomes for cancer therapeutics
Sustained activation of inflammatory autophagy and NF-κβ signal or suppressed IFN are hallmarks of immunosuppressive tumors [44, 122]. Respective mechanisms described above or unknown machinery regulate these STING downstream signalosomes. A recent study revealed an attempt for uncoupling STING signalosomes to treat cancer. GB1275, a CD11b agonist, enters the phase I clinical trial of advanced solid tumors (NCT040603420), elicits robust T cell specific responses by uncoupling two signaling pathways in macrophages, including NF-κB p65/IL-1 inhibition and STING/IRF3/IFN-I/CXCL axis activation [123]. Specifically, through an axis known as focal adhesion kinase (FAK)/sirtuin-3 (SIRT3)/ROS, GB1275 promotes STING-dependent IFN-I production. Overproduction of ROS causes damage to the mitochondria, which allows mtDNA to be released into the cytosol and activate STAT1-dependent antitumor immunity and cGAS/STING signaling. More importantly, CD11b agonists induce p65 degradation to repress NF-κB/IL-1 signaling dependent on proteasome activity, which lessens detrimental STING effects. Further research and drug development that potentially uncouple the STING downstream pathways are needed. Recent reports implicate poly (ADP-ribose) polymerase-1 (PARP-1) in the activation of NF-κB [124, 125]. Cytosolic PARP1 suppresses antiviral immunity through PARylating human cGAS on D191 or mouse cGAS on E176 to terminate its DNA binding. Furthermore, the transglutaminase TG2 expression has been linked to constitutive activation of NF-κB, autophagy, and chemotherapy resistance in lymphoma cells, while inhibition of its transamidating activity facilitates the TBK1-IRF3 interaction in the cytosol correlated with an increase in the IFN-I transcripts in human melanoma [126]. Repressing TG2 may enhance the effect of STING pathway-based cancer therapeutics. These initiatives, such as targeting PARP-1 or TG2, provide a theoretical basis for the proposal to decouple STING signalosomes for cancer therapeutics.
Discussion
cGAS-STING pathway impacts on infectious disease [8], aging [16], neurological diseases [16], autoinflammatory disease [100], and cancer [127]. The cGAS/STING pathway is one of the most promising targets in immuno-oncology. In fact, the related research interest is growing tremendously, as seen by the number of publications in the past decade (Figure 3). Multiple cancer therapies exposing tumor cells to radiation, DNA-damaging agents, or mitotic inhibitors can induce chromosome instability (CIN) or mitochondria damage, which results in nucleic acid release and recognition. The recognition of DNA as an immune-stimulatory molecule is a mechanism to initiate rapid innate immune reactions and cultivate the antigen-specific responses. The foundation for cellular network transmission is laid by the second messenger, cGAMP and STING protein, which can transit the surrounding in the TME. Activated STING enhances immunological surveillance against tumors yet encourages the tumorigenesis, survival and metastasis by perturbing beneficial and detrimental downstream signalosomes in tumor cells or infiltrating immune cell types with diverse intrinsic sensory machinery. As its versatile consequences in physiological and pathological context, the cGAS-STING pathway is evolutionarily conserved with high-threshold activated by meticulous machinery such as a restricted amount of apo-STING in ER, multiple degradation mechanisms, conformational changes, cofactors throughout the secretory route, posttranslational modification of STING protein and redundant prerequisite conditions for diverse downstream pathways. Importantly, STING pathway has been shown difference on substation and strengthen across species, evidencing its adaptations through modular optimization on the STING-CTT domain with a low evolutionary bar to modify STING downstream innate immune responses.
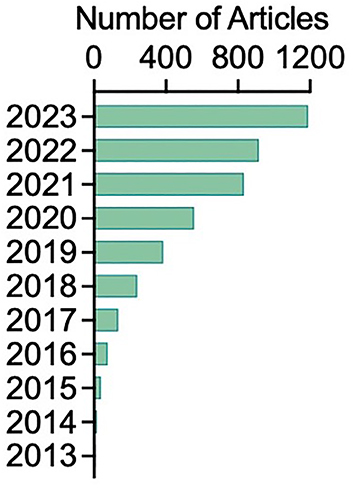
The number of articles retrieved in Google Scholar with key words of “stimulator of interferon genes” and “cancer immunotherapy”.
There are increasing attention in defining STING’s innovative function. In addition to regulating cellular metabolism by limiting glycolysis regardless of its innate role [128], activated STING also acts as ion channel [90, 129] and regulates platelet activation [130] and wound healing [131]. Moreover, STING plays a nuclear role in stimulating the transcription factor AHR’s activation, which opposes the regular signaling route and is not dependent on DNA sensing [132]. Together with STING downstream processes including ER stress and cell death, their function and effect on cancer outcomes may change the immunological landscape in cancer, which merits more research. Precise positioning of STING characteristics is necessary to ensure immunological balance and avoid detrimental inflammation and tissue damage. The disease setting and time course also matters in activated STING-mediated immune response outcomes. There are unsolved issues of STING based cancer therapeutics, future studies are needed to solve it. It is necessary to develop a better STING activator for cancer immunotherapy that enhances tumor clearance while maintaining superior safety profiles.
Funding source: National Natural Science Foundation of China
Award Identifier / Grant number: 82000003
Funding source: China Postdoctoral Science Foundation
Award Identifier / Grant number: 2023M743039
Funding source: National Key Research and Development Program of China
Award Identifier / Grant number: 2022YFC3401400
Acknowledgments
We acknowledge all the authors of the reference we cited.
-
Research ethics: Not applicable.
-
Informed consent: Not applicable.
-
Author contributions: All authors have accepted responsibility for the entire content of this manuscript and approved its submission.
-
Competing interests: Authors state no conflict of interest.
-
Research funding: National Natural Science Foundation of China (82000003);China Postdoctoral Science Foundation (2023M743039); National Key Research and Development Program of China (2022YFC3401400).
-
Data availability: Not applicable.
References
1. Zhong, B, Yang, Y, Li, S, Wang, YY, Li, Y, Diao, F, et al.. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity 2008;29:538–50. https://doi.org/10.1016/j.immuni.2008.09.003.Search in Google Scholar PubMed
2. Sun, W, Li, Y, Chen, L, Chen, H, You, F, Zhou, X, et al.. ERIS, an endoplasmic reticulum IFN stimulator, activates innate immune signaling through dimerization. Proc Natl Acad Sci USA 2009;106:8653–8. https://doi.org/10.1073/pnas.0900850106.Search in Google Scholar PubMed PubMed Central
3. Jin, L, Waterman, PM, Jonscher, KR, Short, CM, Reisdorph, NA, Cambier, JC. MPYS, a novel membrane tetraspanner, is associated with major histocompatibility complex class II and mediates transduction of apoptotic signals. Mol Cell Biol 2008;28:5014–26. https://doi.org/10.1128/mcb.00640-08.Search in Google Scholar
4. Ishikawa, H, Barber, GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008;455:674–8. https://doi.org/10.1038/nature07317.Search in Google Scholar PubMed PubMed Central
5. Ma, M, Dang, Y, Chang, B, Wang, F, Xu, J, Chen, L, et al.. TAK1 is an essential kinase for STING trafficking. Mol Cell 2023;83:3885–903.e5. https://doi.org/10.1016/j.molcel.2023.09.009.Search in Google Scholar PubMed
6. Zhao, B, Du, F, Xu, P, Shu, C, Sankaran, B, Bell, SL, et al.. A conserved PLPLRT/SD motif of STING mediates the recruitment and activation of TBK1. Nature 2019;569:718–22. https://doi.org/10.1038/s41586-019-1228-x.Search in Google Scholar PubMed PubMed Central
7. Liu, S, Yang, B, Hou, Y, Cui, K, Yang, X, Li, X, et al.. The mechanism of STING autoinhibition and activation. Mol Cell 2023;83:1502–18.e10. https://doi.org/10.1016/j.molcel.2023.03.029.Search in Google Scholar PubMed
8. Hopfner, KP, Hornung, V. Molecular mechanisms and cellular functions of cGAS–STING signalling. Nat Rev Mol Cell Biol 2020;21:501–21. https://doi.org/10.1038/s41580-020-0244-x.Search in Google Scholar PubMed
9. Ergun, SL, Fernandez, D, Weiss, TM, Li, L. STING polymer structure reveals mechanisms for activation, hyperactivation, and inhibition. Cell 2019;178:290–301.e10. https://doi.org/10.1016/j.cell.2019.05.036.Search in Google Scholar PubMed
10. Wu, H. Higher-order assemblies in a new paradigm of signal transduction. Cell 2013;153:287–92. https://doi.org/10.1016/j.cell.2013.03.013.Search in Google Scholar PubMed PubMed Central
11. Schweke, H, Pacesa, M, Levin, T, Goverde, CA, Kumar, P, Duhoo, Y, et al.. An atlas of protein homo-oligomerization across domains of life. Cell 2024;187:999–1010.e15. https://doi.org/10.1016/j.cell.2024.01.022.Search in Google Scholar PubMed
12. Ko, TP, Wang, YC, Yang, CS, Hou, MH, Chen, CJ, Chiu, YF, et al.. Crystal structure and functional implication of bacterial STING. Nat Commun 2022;13:26. https://doi.org/10.1038/s41467-021-26583-3.Search in Google Scholar PubMed PubMed Central
13. Storz, P. Reactive oxygen species in tumor progression. Front Biosci 2005;10:1881. https://doi.org/10.2741/1667.Search in Google Scholar PubMed
14. Hayman, TJ, Baro, M, MacNeil, T, Phoomak, C, Aung, TN, Cui, W, et al.. STING enhances cell death through regulation of reactive oxygen species and DNA damage. Nat Commun 2021;12:2327. https://doi.org/10.1038/s41467-021-22572-8.Search in Google Scholar PubMed PubMed Central
15. Jia, M, Qin, D, Zhao, C, Chai, L, Yu, Z, Wang, W, et al.. Redox homeostasis maintained by GPX4 facilitates STING activation. Nat Immunol 2020;21:727–35. https://doi.org/10.1038/s41590-020-0699-0.Search in Google Scholar PubMed
16. Gulen, MF, Samson, N, Keller, A, Schwabenland, M, Liu, C, Glück, S, et al.. cGAS–STING drives ageing-related inflammation and neurodegeneration. Nature 2023;620:374–80. https://doi.org/10.1038/s41586-023-06373-1.Search in Google Scholar PubMed PubMed Central
17. Bennett, ZT, Li, S, Sumer, BD, Gao, J. Polyvalent design in the cGAS-STING pathway. Semin Immunol 2021;56:101580. https://doi.org/10.1016/j.smim.2021.101580.Search in Google Scholar PubMed PubMed Central
18. Luo, M, Wang, H, Wang, Z, Cai, H, Lu, Z, Li, Y, et al.. A STING-activating nanovaccine for cancer immunotherapy. Nat Nanotechnol 2017;12:648–54. https://doi.org/10.1038/nnano.2017.52.Search in Google Scholar PubMed PubMed Central
19. Li, S, Luo, M, Wang, Z, Feng, Q, Wilhelm, J, Wang, X, et al.. Prolonged activation of innate immune pathways by a polyvalent STING agonist. Nat Biomed Eng 2021;5:455–66. https://doi.org/10.1038/s41551-020-00675-9.Search in Google Scholar PubMed PubMed Central
20. Aroh, C, Wang, Z, Dobbs, N, Luo, M, Chen, Z, Gao, J, et al.. Innate immune activation by cGMP-AMP nanoparticles leads to potent and long-acting antiretroviral response against HIV-1. J Immunol 2017;199:3840–8. https://doi.org/10.4049/jimmunol.1700972.Search in Google Scholar PubMed PubMed Central
21. Basu, S, Middya, S, Banerjee, M, Ghosh, R, Pryde, DC, Yadav, DB, et al.. The discovery of potent small molecule cyclic urea activators of STING. Eur J Med Chem 2022;229:114087. https://doi.org/10.1016/j.ejmech.2021.114087.Search in Google Scholar PubMed
22. Lu, D, Shang, G, Li, J, Lu, Y, Bai, XC, Zhang, X. Activation of STING by targeting a pocket in the transmembrane domain. Nature 2022;604:557–62. https://doi.org/10.1038/s41586-022-04559-7.Search in Google Scholar PubMed PubMed Central
23. Fang, R, Jiang, Q, Guan, Y, Gao, P, Zhang, R, Zhao, Z, et al.. Golgi apparatus-synthesized sulfated glycosaminoglycans mediate polymerization and activation of the cGAMP sensor STING. Immunity 2021;54:962–75.e8. https://doi.org/10.1016/j.immuni.2021.03.011.Search in Google Scholar PubMed
24. Li, J, Canham, SM, Wu, H, Henault, M, Chen, L, Liu, G, et al.. Activation of human STING by a molecular glue-like compound. Nat Chem Biol 2024;20:365–72. https://doi.org/10.1038/s41589-023-01434-y.Search in Google Scholar PubMed PubMed Central
25. Dou, Y, Chen, R, Liu, S, Lee, YT, Jing, J, Liu, X, et al.. Optogenetic engineering of STING signaling allows remote immunomodulation to enhance cancer immunotherapy. Nat Commun 2023;14:5461. https://doi.org/10.1038/s41467-023-41164-2.Search in Google Scholar PubMed PubMed Central
26. Mukai, K, Konno, H, Akiba, T, Uemura, T, Waguri, S, Kobayashi, T, et al.. Activation of STING requires palmitoylation at the Golgi. Nat Commun 2016;7:11932. https://doi.org/10.1038/ncomms11932.Search in Google Scholar PubMed PubMed Central
27. Almine, JF, O’Hare, CAJ, Dunphy, G, Haga, IR, Naik, RJ, Atrih, A, et al.. IFI16 and cGAS cooperate in the activation of STING during DNA sensing in human keratinocytes. Nat Commun 2017;8:14392. https://doi.org/10.1038/ncomms14392.Search in Google Scholar PubMed PubMed Central
28. Tu, X, Chu, TT, Jeltema, D, Abbott, K, Yang, K, Xing, C, et al.. Interruption of post-Golgi STING trafficking activates tonic interferon signaling. Nat Commun 2022;13:6977. https://doi.org/10.1038/s41467-022-33765-0.Search in Google Scholar PubMed PubMed Central
29. Zhang, L, Wei, X, Wang, Z, Liu, P, Hou, Y, Xu, Y, et al.. NF-κB activation enhances STING signaling by altering microtubule-mediated STING trafficking. Cell Rep 2023;42:112185. https://doi.org/10.1016/j.celrep.2023.112185.Search in Google Scholar PubMed
30. Srikanth, S, Woo, JS, Wu, B, El-Sherbiny, YM, Leung, J, Chupradit, K, et al.. The Ca2+ sensor STIM1 regulates the type I interferon response by retaining the signaling adaptor STING at the endoplasmic reticulum. Nat Immunol 2019;20:152–62. https://doi.org/10.1038/s41590-018-0287-8.Search in Google Scholar PubMed PubMed Central
31. Pokatayev, V, Yang, K, Tu, X, Dobbs, N, Wu, J, Kalb, RG, et al.. Homeostatic regulation of STING protein at the resting state by stabilizer TOLLIP. Nat Immunol 2020;21:158–67. https://doi.org/10.1038/s41590-019-0569-9.Search in Google Scholar PubMed PubMed Central
32. Gui, X, Yang, H, Li, T, Tan, X, Shi, P, Li, M, et al.. Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature 2019;567:262–6. https://doi.org/10.1038/s41586-019-1006-9.Search in Google Scholar PubMed PubMed Central
33. Di Martino, R, Sticco, L, Luini, A. Regulation of cargo export and sorting at the trans‐Golgi network. FEBS Lett 2019;593:2306–18. https://doi.org/10.1002/1873-3468.13572.Search in Google Scholar PubMed
34. Steiner, A, Hrovat-Schaale, K, Prigione, I, Yu, CH, Laohamonthonkul, P, Harapas, CR, et al.. Deficiency in coatomer complex I causes aberrant activation of STING signalling. Nat Commun 2022;13:2321. https://doi.org/10.1038/s41467-022-29946-6.Search in Google Scholar PubMed PubMed Central
35. Deng, Z, Chong, Z, Law, CS, Mukai, K, Ho, FO, Martinu, T, et al.. A defect in COPI-mediated transport of STING causes immune dysregulation in COPA syndrome. J Exp Med 2020;217:e20201045. https://doi.org/10.1084/jem.20201045.Search in Google Scholar PubMed PubMed Central
36. Fang, R, Jiang, Q, Jia, X, Jiang, Z. ARMH3-mediated recruitment of PI4KB directs Golgi-to-endosome trafficking and activation of the antiviral effector STING. Immunity 2023;56:500–15.e6. https://doi.org/10.1016/j.immuni.2023.02.004.Search in Google Scholar PubMed
37. De Oliveira Mann, CC, Orzalli, MH, King, DS, Kagan, JC, Lee, ASY, Kranzusch, PJ. Modular architecture of the STING C-terminal tail allows interferon and NF-κB signaling adaptation. Cell Rep 2019;27:1165–75.e5. https://doi.org/10.1016/j.celrep.2019.03.098.Search in Google Scholar PubMed PubMed Central
38. Gentili, M, Liu, B, Papanastasiou, M, Dele-Oni, D, Schwartz, MA, Carlson, RJ, et al.. ESCRT-dependent STING degradation inhibits steady-state and cGAMP-induced signalling. Nat Commun 2023;14:611. https://doi.org/10.1038/s41467-023-36132-9.Search in Google Scholar PubMed PubMed Central
39. Prabakaran, T, Bodda, C, Krapp, C, Zhang, B, Christensen, MH, Sun, C, et al.. Attenuation of cGAS-STING signaling is mediated by a p62/SQSTM1-dependent autophagy pathway activated by TBK1. EMBO J 2018;37:e97858.Search in Google Scholar
40. Lu, Q, Chen, Y, Li, J, Zhu, F, Zheng, Z. Crosstalk between cGAS-STING pathway and autophagy in cancer immunity. Front Immunol 2023;14:1139595. https://doi.org/10.3389/fimmu.2023.1139595.Search in Google Scholar PubMed PubMed Central
41. Olagnier, D, Brandtoft, AM, Gunderstofte, C, Villadsen, NL, Krapp, C, Thielke, AL, et al.. Nrf2 negatively regulates STING indicating a link between antiviral sensing and metabolic reprogramming. Nat Commun 2018;9:3506. https://doi.org/10.1038/s41467-018-05861-7.Search in Google Scholar PubMed PubMed Central
42. Prabakaran, T, Bodda, C, Krapp, C, Zhang, B, Christensen, MH, Sun, C, et al.. Attenuation of cGAS-STING signaling is mediated by a p62/SQSTM1-dependent autophagy pathway activated by TBK1. EMBO J 2018;37:e97858. https://doi.org/10.15252/embj.201797858.Search in Google Scholar PubMed PubMed Central
43. Liang, Q, Seo, GJ, Choi, YJ, Kwak, MJ, Ge, J, Rodgers, MA, et al.. Crosstalk between the cGAS DNA sensor and Beclin-1 autophagy protein shapes innate antimicrobial immune responses. Cell Host Microbe 2014;15:228–38. https://doi.org/10.1016/j.chom.2014.01.009.Search in Google Scholar PubMed PubMed Central
44. Konno, H, Yamauchi, S, Berglund, A, Putney, RM, Mulé, JJ, Barber, GN. Suppression of STING signaling through epigenetic silencing and missense mutation impedes DNA damage mediated cytokine production. Oncogene 2018;37:2037–51. https://doi.org/10.1038/s41388-017-0120-0.Search in Google Scholar PubMed PubMed Central
45. Ji, Y, Luo, Y, Wu, Y, Sun, Y, Zhao, L, Xue, Z, et al.. SEL1L–HRD1 endoplasmic reticulum-associated degradation controls STING-mediated innate immunity by limiting the size of the activable STING pool. Nat Cell Biol 2023;25:726–39. https://doi.org/10.1038/s41556-023-01138-4.Search in Google Scholar PubMed PubMed Central
46. Gao, Y, Li, W, Wang, Z, Zhang, C, He, Y, Liu, X, et al.. SEL1L preserves CD8+ T-cell survival and homeostasis by fine-tuning PERK signaling and the IL-15 receptor-mediated mTORC1 axis. Cell Mol Immunol 2023;20:1232–50. https://doi.org/10.1038/s41423-023-01078-x.Search in Google Scholar PubMed PubMed Central
47. Kuhl, N, Linder, A, Philipp, N, Nixdorf, D, Fischer, H, Veth, S, et al.. STING agonism turns human T cells into interferon-producing cells but impedes their functionality. EMBO Rep 2023;24:e55536. https://doi.org/10.15252/embr.202255536.Search in Google Scholar PubMed PubMed Central
48. Gulen, MF, Koch, U, Haag, SM, Schuler, F, Apetoh, L, Villunger, A, et al.. Signalling strength determines proapoptotic functions of STING. Nat Commun 2017;8:427. https://doi.org/10.1038/s41467-017-00573-w.Search in Google Scholar PubMed PubMed Central
49. Wu, J, Chen, YJ, Dobbs, N, Sakai, T, Liou, J, Miner, JJ, et al.. STING-mediated disruption of calcium homeostasis chronically activates ER stress and primes T cell death. J Exp Med 2019;216:867–83. https://doi.org/10.1084/jem.20182192.Search in Google Scholar PubMed PubMed Central
50. Cerboni, S, Jeremiah, N, Gentili, M, Gehrmann, U, Conrad, C, Stolzenberg, MC, et al.. Intrinsic antiproliferative activity of the innate sensor STING in T lymphocytes. J Exp Med 2017;214:1769–85. https://doi.org/10.1084/jem.20161674.Search in Google Scholar PubMed PubMed Central
51. Jeltema, D, Abbott, K, Yan, N. STING trafficking as a new dimension of immune signaling. J Exp Med 2023;220:e20220990. https://doi.org/10.1084/jem.20220990.Search in Google Scholar PubMed PubMed Central
52. Kang, J, Wu, J, Liu, Q, Wu, X, Zhao, Y, Ren, J. Post-translational modifications of STING: a potential therapeutic target. Front Immunol 2022;13:888147. https://doi.org/10.3389/fimmu.2022.888147.Search in Google Scholar PubMed PubMed Central
53. Diamond, MS, Kinder, M, Matsushita, H, Mashayekhi, M, Dunn, GP, Archambault, JM, et al.. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med 2011;208:1989–2003. https://doi.org/10.1084/jem.20101158.Search in Google Scholar PubMed PubMed Central
54. Li, XD, Wu, J, Gao, D, Wang, H, Sun, L, Chen, ZJ. Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science 2013;341:1390–4. https://doi.org/10.1126/science.1244040.Search in Google Scholar PubMed PubMed Central
55. Tanaka, Y, Chen, ZJ. STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci Signal 2012;5:ra20. https://doi.org/10.1126/scisignal.2002521.Search in Google Scholar PubMed PubMed Central
56. Margolis, SR, Wilson, SC, Vance, RE. Evolutionary origins of cGAS-STING signaling. Trends Immunol 2017;38:733–43. https://doi.org/10.1016/j.it.2017.03.004.Search in Google Scholar PubMed
57. Levine, B, Mizushima, N, Virgin, HW. Autophagy in immunity and inflammation. Nature 2011;469:323–35. https://doi.org/10.1038/nature09782.Search in Google Scholar PubMed PubMed Central
58. Feng, Y, He, D, Yao, Z, Klionsky, DJ. The machinery of macroautophagy. Cell Res 2014;24:24–41. https://doi.org/10.1038/cr.2013.168.Search in Google Scholar PubMed PubMed Central
59. Fischer, TD, Wang, C, Padman, BS, Lazarou, M, Youle, RJ. STING induces LC3B lipidation onto single-membrane vesicles via the V-ATPase and ATG16L1-WD40 domain. J Cell Biol 2020;219:e202009128. https://doi.org/10.1083/jcb.202009128.Search in Google Scholar PubMed PubMed Central
60. Konno, H, Konno, K, Barber, GN. Cyclic dinucleotides trigger ULK1 (ATG1) phosphorylation of STING to prevent sustained innate immune signaling. Cell 2013;155:688–98. https://doi.org/10.1016/j.cell.2013.09.049.Search in Google Scholar PubMed PubMed Central
61. Du, M, Liu, J, Chen, X, Xie, Y, Yuan, C, Xiang, Y, et al.. Casein kinase II controls TBK1/IRF3 activation in IFN response against viral infection. J Immunol 2015;194:4477–88. https://doi.org/10.4049/jimmunol.1402777.Search in Google Scholar PubMed
62. Jiang, S, Xia, N, Luo, J, Zhang, Y, Cao, Q, Zhang, J, et al.. The porcine cyclic GMP-AMP synthase-STING pathway exerts an unusual antiviral function independent of interferon and autophagy. Williams, BRG, editor. J Virol 2022;96:e01476–22. https://doi.org/10.1128/jvi.01476-22.Search in Google Scholar PubMed PubMed Central
63. Balka, KR, Louis, C, Saunders, TL, Smith, AM, Calleja, DJ, D’Silva, DB, et al.. TBK1 and IKKε act redundantly to mediate STING-induced NF-κB responses in myeloid cells. Cell Rep 2020;31:107492. https://doi.org/10.1016/j.celrep.2020.03.056.Search in Google Scholar PubMed
64. Hoong, BYD, Gan, YH, Liu, H, Chen, ES. cGAS-STING pathway in oncogenesis and cancer therapeutics. Oncotarget 2020;11:2930–55. https://doi.org/10.18632/oncotarget.27673.Search in Google Scholar PubMed PubMed Central
65. Hong, C, Schubert, M, Tijhuis, AE, Requesens, M, Roorda, M, Van Den Brink, A, et al.. cGAS–STING drives the IL-6-dependent survival of chromosomally instable cancers. Nature 2022;607:366–73. https://doi.org/10.1038/s41586-022-04847-2.Search in Google Scholar PubMed
66. Li, J, Hubisz, MJ, Earlie, EM, Duran, MA, Hong, C, Varela, AA, et al.. Non-cell-autonomous cancer progression from chromosomal instability. Nature 2023;620:1080–8. https://doi.org/10.1038/s41586-023-06464-z.Search in Google Scholar PubMed PubMed Central
67. Mukherjee, T, Kumar, N, Chawla, M, Philpott, DJ, Basak, S. The NF-κB signaling system in the immunopathogenesis of inflammatory bowel disease. Sci Signal 2024;17:eadh1641. https://doi.org/10.1126/scisignal.adh1641.Search in Google Scholar PubMed
68. Le Voyer, T, Parent, AV, Liu, X, Cederholm, A, Gervais, A, Rosain, J, et al.. Autoantibodies against type I IFNs in humans with alternative NF-κB pathway deficiency. Nature 2023;623:803–13. https://doi.org/10.1038/s41586-023-06717-x.Search in Google Scholar PubMed PubMed Central
69. Hou, Y, Liang, H, Rao, E, Zheng, W, Huang, X, Deng, L, et al.. Non-canonical NF-κB antagonizes STING sensor-mediated DNA sensing in radiotherapy. Immunity 2018;49:490–503.e4. https://doi.org/10.1016/j.immuni.2018.07.008.Search in Google Scholar PubMed PubMed Central
70. Balka, KR, De Nardo, D. Understanding early TLR signaling through the Myddosome. J Leukoc Biol 2019;105:339–51. https://doi.org/10.1002/jlb.mr0318-096r.Search in Google Scholar
71. Fan, J, Li, Q, Liang, J, Chen, Z, Chen, L, Lai, J, et al.. Regulation of IFNβ expression: focusing on the role of its promoter and transcription regulators. Front Microbiol 2023;14:1158777. https://doi.org/10.3389/fmicb.2023.1158777.Search in Google Scholar PubMed PubMed Central
72. Smale, ST. Selective transcription in response to an inflammatory stimulus. Cell 2010;140:833–44. https://doi.org/10.1016/j.cell.2010.01.037.Search in Google Scholar PubMed PubMed Central
73. Liu, S, Cai, X, Wu, J, Cong, Q, Chen, X, Li, T, et al.. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 2015;347:aaa2630. https://doi.org/10.1126/science.aaa2630.Search in Google Scholar PubMed
74. Wang, C, Wang, X, Veleeparambil, M, Kessler, PM, Willard, B, Chattopadhyay, S, et al.. EGFR‐mediated tyrosine phosphorylation of STING determines its trafficking route and cellular innate immunity functions. EMBO J 2020;39:e104106. https://doi.org/10.15252/embj.2019104106.Search in Google Scholar PubMed PubMed Central
75. Wang, C, Sharma, N, Veleeparambil, M, Kessler, PM, Willard, B, Sen, GC. STING-mediated interferon induction by herpes simplex virus 1 requires the protein tyrosine kinase Syk. Horner, SM, editor. mBio 2021;12:e03228–21. https://doi.org/10.1128/mbio.03228-21.Search in Google Scholar PubMed PubMed Central
76. Stempel, M, Chan, B, Juranić Lisnić, V, Krmpotić, A, Hartung, J, Paludan, SR, et al.. The herpesviral antagonist m152 reveals differential activation of STING-dependent IRF and NF-κB signaling and STING’s dual role during MCMV infection. EMBO J 2019;38:e100983. https://doi.org/10.15252/embj.2018100983.Search in Google Scholar PubMed PubMed Central
77. Ni, G, Konno, H, Barber, GN. Ubiquitination of STING at lysine 224 controls IRF3 activation. Sci Immunol 2017;2:eaah7119. https://doi.org/10.1126/sciimmunol.aah7119.Search in Google Scholar PubMed PubMed Central
78. Sanz‐Garcia, C, McMullen, MR, Chattopadhyay, S, Roychowdhury, S, Sen, G, Nagy, LE. Nontranscriptional activity of interferon regulatory factor 3 protects mice from high‐fat diet‐induced liver injury. Hepatol Commun 2019;3:1626–41. https://doi.org/10.1002/hep4.1441.Search in Google Scholar PubMed PubMed Central
79. Popli, S, Chakravarty, S, Fan, S, Glanz, A, Aras, S, Nagy, LE, et al.. IRF3 inhibits nuclear translocation of NF-κB to prevent viral inflammation. Proc Natl Acad Sci USA 2022;119:Article no. e2121385119. https://doi.org/10.1073/pnas.2121385119.Search in Google Scholar PubMed PubMed Central
80. Tian, M, Wang, X, Sun, J, Lin, W, Chen, L, Liu, S, et al.. IRF3 prevents colorectal tumorigenesis via inhibiting the nuclear translocation of β-catenin. Nat Commun 2020;11:5762. https://doi.org/10.1038/s41467-020-19627-7.Search in Google Scholar PubMed PubMed Central
81. Chow, EK, Castrillo, A, Shahangian, A, Pei, L, O’Connell, RM, Modlin, RL, et al.. A role for IRF3-dependent RXRα repression in hepatotoxicity associated with viral infections. J Exp Med 2006;203:2589–602. https://doi.org/10.1084/jem.20060929.Search in Google Scholar PubMed PubMed Central
82. Wu, J, Dobbs, N, Yang, K, Yan, N. Interferon-independent activities of mammalian STING mediate antiviral response and tumor immune evasion. Immunity 2020;53:115–26.e5. https://doi.org/10.1016/j.immuni.2020.06.009.Search in Google Scholar PubMed PubMed Central
83. Yan, X, Zhao, X, Huo, R, Xu, T. IRF3 and IRF8 regulate NF-κB signaling by targeting MyD88 in teleost fish. Front Immunol 2020;11:606. https://doi.org/10.3389/fimmu.2020.00606.Search in Google Scholar PubMed PubMed Central
84. Swanson, KV, Deng, M, Ting, JPY. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol 2019;19:477–89. https://doi.org/10.1038/s41577-019-0165-0.Search in Google Scholar PubMed PubMed Central
85. Gaidt, MM, Ebert, TS, Chauhan, D, Ramshorn, K, Pinci, F, Zuber, S, et al.. The DNA inflammasome in human myeloid cells is initiated by a STING-cell death program upstream of NLRP3. Cell 2017;171:1110–24.e18. https://doi.org/10.1016/j.cell.2017.09.039.Search in Google Scholar PubMed PubMed Central
86. Xiao, Y, Zhao, C, Tai, Y, Li, B, Lan, T, Lai, E, et al.. STING mediates hepatocyte pyroptosis in liver fibrosis by epigenetically activating the NLRP3 inflammasome. Redox Biol 2023;62:102691. https://doi.org/10.1016/j.redox.2023.102691.Search in Google Scholar PubMed PubMed Central
87. Zhong, Z, Umemura, A, Sanchez-Lopez, E, Liang, S, Shalapour, S, Wong, J, et al.. NF-κB restricts inflammasome activation via elimination of damaged mitochondria. Cell 2016;164:896–910. https://doi.org/10.1016/j.cell.2015.12.057.Search in Google Scholar PubMed PubMed Central
88. Wang, W, Hu, D, Wu, C, Feng, Y, Li, A, Liu, W, et al.. STING promotes NLRP3 localization in ER and facilitates NLRP3 deubiquitination to activate the inflammasome upon HSV-1 infection. In: Zhu, F, editor. PLoS Pathog; 2020;16:e1008335. https://doi.org/10.1371/journal.ppat.1008335.Search in Google Scholar PubMed PubMed Central
89. Liu, B, Carlson, RJ, Pires, IS, Gentili, M, Feng, E, Hellier, Q, et al.. Human STING is a proton channel. Science 2023;381:508–14. https://doi.org/10.1126/science.adf8974.Search in Google Scholar PubMed PubMed Central
90. Chen, J, Chen, ZJ. PtdIns4P on dispersed trans-Golgi network mediates NLRP3 inflammasome activation. Nature 2018;564:71–6. https://doi.org/10.1038/s41586-018-0761-3.Search in Google Scholar PubMed PubMed Central
91. Cai, Y, Zhang, A, Lu, C. Precision intervention of cell type-specific targeting is required for future cancer immunotherapy. Med Rev 2023;2:553–4. https://doi.org/10.1515/mr-2022-0037.Search in Google Scholar PubMed PubMed Central
92. Amouzegar, A, Chelvanambi, M, Filderman, J, Storkus, W, Luke, J. STING agonists as cancer therapeutics. Cancers 2021;13:2695. https://doi.org/10.3390/cancers13112695.Search in Google Scholar PubMed PubMed Central
93. Pan, BS, Perera, SA, Piesvaux, JA, Presland, JP, Schroeder, GK, Cumming, JN, et al.. An orally available non-nucleotide STING agonist with antitumor activity. Science 2020;369:eaba6098. https://doi.org/10.1126/science.aba6098.Search in Google Scholar PubMed
94. Jneid, B, Bochnakian, A, Hoffmann, C, Delisle, F, Djacoto, E, Sirven, P, et al.. Selective STING stimulation in dendritic cells primes antitumor T cell responses. Sci Immunol 2023;8:eabn6612. https://doi.org/10.1126/sciimmunol.abn6612.Search in Google Scholar PubMed
95. Imanishi, T, Unno, M, Kobayashi, W, Yoneda, N, Matsuda, S, Ikeda, K, et al.. Reciprocal regulation of STING and TCR signaling by mTORC1 for T-cell activation and function. Life Sci Alliance 2019;2:e201800282. https://doi.org/10.26508/lsa.201800282.Search in Google Scholar PubMed PubMed Central
96. Li, S, Mirlekar, B, Johnson, BM, Brickey, WJ, Wrobel, JA, Yang, N, et al.. STING-induced regulatory B cells compromise NK function in cancer immunity. Nature 2022;610:373–80. https://doi.org/10.1038/s41586-022-05254-3.Search in Google Scholar PubMed PubMed Central
97. Lemos, H, Ou, R, McCardle, C, Lin, Y, Calver, J, Minett, J, et al.. Overcoming resistance to STING agonist therapy to incite durable protective antitumor immunity. J Immunother Cancer 2020;8:e001182. https://doi.org/10.1136/jitc-2020-001182.Search in Google Scholar PubMed PubMed Central
98. Ding, L, Huang, XF, Dong, GJ, Hu, EL, Chen, S, Wang, TT, et al.. Activated STING enhances Tregs infiltration in the HPV-related carcinogenesis of tongue squamous cells via the c-jun/CCL22 signal. Biochim Biophys Acta (BBA) – Mol Basis Dis 2015;1852:2494–503. https://doi.org/10.1016/j.bbadis.2015.08.011.Search in Google Scholar PubMed
99. Ni, H, Zhang, H, Li, L, Huang, H, Guo, H, Zhang, L, et al.. T cell-intrinsic STING signaling promotes regulatory T cell induction and immunosuppression by upregulating FOXP3 transcription in cervical cancer. J Immunother Cancer 2022;10:e005151. https://doi.org/10.1136/jitc-2022-005151.Search in Google Scholar PubMed PubMed Central
100. Decout, A, Katz, JD, Venkatraman, S, Ablasser, A. The cGAS–STING pathway as a therapeutic target in inflammatory diseases. Nat Rev Immunol 2021;21:548–69. https://doi.org/10.1038/s41577-021-00524-z.Search in Google Scholar PubMed PubMed Central
101. He, L, Xiao, X, Yang, X, Zhang, Z, Wu, L, Liu, Z. STING signaling in tumorigenesis and cancer therapy: a friend or foe? Cancer Lett 2017;402:203–12. https://doi.org/10.1016/j.canlet.2017.05.026.Search in Google Scholar PubMed
102. Hu, J, Sánchez-Rivera, FJ, Wang, Z, Johnson, GN, Ho, YJ, Ganesh, K, et al.. STING inhibits the reactivation of dormant metastasis in lung adenocarcinoma. Nature 2023;616:806–13. https://doi.org/10.1038/s41586-023-05880-5.Search in Google Scholar PubMed PubMed Central
103. Zhu, Z, Zhou, X, Du, H, Cloer, EW, Zhang, J, Mei, L, et al.. STING suppresses mitochondrial VDAC2 to Govern RCC growth independent of innate immunity. Adv Sci 2023;10:2203718. https://doi.org/10.1002/advs.202203718.Search in Google Scholar PubMed PubMed Central
104. Han, C, Liu, Z, Zhang, Y, Shen, A, Dong, C, Zhang, A, et al.. Tumor cells suppress radiation-induced immunity by hijacking caspase 9 signaling. Nat Immunol 2020;21:546–54. https://doi.org/10.1038/s41590-020-0641-5.Search in Google Scholar PubMed
105. Bakhoum, SF, Ngo, B, Laughney, AM, Cavallo, JA, Murphy, CJ, Ly, P, et al.. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 2018;553:467–72. https://doi.org/10.1038/nature25432.Search in Google Scholar PubMed PubMed Central
106. Taniguchi, K, Karin, M. NF-κB, inflammation, immunity and cancer: coming of age. Nat Rev Immunol 2018;18:309–24. https://doi.org/10.1038/nri.2017.142.Search in Google Scholar PubMed
107. Corrales, L, Glickman, LH, McWhirter, SM, Kanne, DB, Sivick, KE, Katibah, GE, et al.. Direct activation of STING in the tumor microenvironment leads to potent and systemic tumor regression and immunity. Cell Rep 2015;11:1018–30. https://doi.org/10.1016/j.celrep.2015.04.031.Search in Google Scholar PubMed PubMed Central
108. Chen, Q, Boire, A, Jin, X, Valiente, M, Er, EE, Lopez-Soto, A, et al.. Carcinoma–astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 2016;533:493–8. https://doi.org/10.1038/nature18268.Search in Google Scholar PubMed PubMed Central
109. Yamamoto, K, Venida, A, Yano, J, Biancur, DE, Kakiuchi, M, Gupta, S, et al.. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature 2020;581:100–5. https://doi.org/10.1038/s41586-020-2229-5.Search in Google Scholar PubMed PubMed Central
110. Wang, R, Ma, X, Zhang, X, Jiang, D, Mao, H, Li, Z, et al.. Autophagy-mediated NKG2D internalization impairs NK cell function and exacerbates radiation pneumonitis. Front Immunol 2023;14:1250920. https://doi.org/10.3389/fimmu.2023.1250920.Search in Google Scholar PubMed PubMed Central
111. Dougan, M, Li, D, Neuberg, D, Mihm, M, Googe, P, Wong, KK, et al.. A dual role for the immune response in a mouse model of inflammation-associated lung cancer. J Clin Invest 2011;121:2436–46. https://doi.org/10.1172/jci44796.Search in Google Scholar PubMed PubMed Central
112. Wente, MN, Mayer, C, Gaida, MM, Michalski, CW, Giese, T, Bergmann, F, et al.. CXCL14 expression and potential function in pancreatic cancer. Cancer Lett 2008;259:209–17. https://doi.org/10.1016/j.canlet.2007.10.021.Search in Google Scholar PubMed
113. Pramanik, K, Makena, M, Bhowmick, K, Pandey, M. Advancement of NF-κB signaling pathway: a novel target in pancreatic cancer. Int J Mol Sci 2018;19:3890. https://doi.org/10.3390/ijms19123890.Search in Google Scholar PubMed PubMed Central
114. Garland, KM, Sheehy, TL, Wilson, JT. Chemical and biomolecular strategies for STING pathway activation in cancer immunotherapy. Chem Rev 2022;122:5977–6039. https://doi.org/10.1021/acs.chemrev.1c00750.Search in Google Scholar PubMed PubMed Central
115. Gao, Y, Zheng, X, Chang, B, Lin, Y, Huang, X, Wang, W, et al.. Intercellular transfer of activated STING triggered by RAB22A-mediated non-canonical autophagy promotes antitumor immunity. Cell Res 2022;32:1086–104. https://doi.org/10.1038/s41422-022-00731-w.Search in Google Scholar PubMed PubMed Central
116. Wang, C, Guan, Y, Lv, M, Zhang, R, Guo, Z, Wei, X, et al.. Manganese increases the sensitivity of the cGAS-STING pathway for double-stranded DNA and is required for the host defense against DNA viruses. Immunity 2018;48:675–87.e7. https://doi.org/10.1016/j.immuni.2018.03.017.Search in Google Scholar PubMed
117. Sun, X, Zhang, Y, Li, J, Park, KS, Han, K, Zhou, X, et al.. Amplifying STING activation by cyclic dinucleotide–manganese particles for local and systemic cancer metalloimmunotherapy. Nat Nanotechnol 2021;16:1260–70. https://doi.org/10.1038/s41565-021-00962-9.Search in Google Scholar PubMed PubMed Central
118. Yang, K, Han, W, Jiang, X, Piffko, A, Bugno, J, Han, C, et al.. Zinc cyclic di-AMP nanoparticles target and suppress tumours via endothelial STING activation and tumour-associated macrophage reinvigoration. Nat Nanotechnol 2022;17:1322–31. https://doi.org/10.1038/s41565-022-01225-x.Search in Google Scholar PubMed
119. Dosta, P, Cryer, AM, Dion, MZ, Shiraishi, T, Langston, SP, Lok, D, et al.. Investigation of the enhanced antitumour potency of STING agonist after conjugation to polymer nanoparticles. Nat Nanotechnol 2023;18:1351–63. https://doi.org/10.1038/s41565-023-01447-7.Search in Google Scholar PubMed
120. He, Y, Hong, C, Yan, EZ, Fletcher, SJ, Zhu, G, Yang, M, et al.. Self-assembled cGAMP-STINGΔTM signaling complex as a bioinspired platform for cGAMP delivery. Sci Adv 2020;6:eaba7589. https://doi.org/10.1126/sciadv.aba7589.Search in Google Scholar PubMed PubMed Central
121. Tse, SW, McKinney, K, Walker, W, Nguyen, M, Iacovelli, J, Small, C, et al.. mRNA-encoded, constitutively active STINGV155M is a potent genetic adjuvant of antigen-specific CD8+ T cell response. Mol Ther 2021;29:2227–38. https://doi.org/10.1016/j.ymthe.2021.03.002.Search in Google Scholar PubMed PubMed Central
122. Lawson, KA, Sousa, CM, Zhang, X, Kim, E, Akthar, R, Caumanns, JJ, et al.. Functional genomic landscape of cancer-intrinsic evasion of killing by T cells. Nature 2020;586:120–6. https://doi.org/10.1038/s41586-020-2746-2.Search in Google Scholar PubMed PubMed Central
123. Liu, X, Hogg, GD, Zuo, C, Borcherding, NC, Baer, JM, Lander, VE, et al.. Context-dependent activation of STING-interferon signaling by CD11b agonists enhances anti-tumor immunity. Cancer Cell 2023;41:1073–90. https://doi.org/10.1016/j.ccell.2023.04.018.Search in Google Scholar PubMed PubMed Central
124. Kim, C, Wang, XD, Yu, Y. PARP1 inhibitors trigger innate immunity via PARP1 trapping-induced DNA damage response. Elife 2020;9:e60637. https://doi.org/10.7554/elife.60637.Search in Google Scholar PubMed PubMed Central
125. Wang, F, Zhao, M, Chang, B, Zhou, Y, Wu, X, Ma, M, et al.. Cytoplasmic PARP1 links the genome instability to the inhibition of antiviral immunity through PARylating cGAS. Mol Cell 2022;82:2032–49.e7. https://doi.org/10.1016/j.molcel.2022.03.034.Search in Google Scholar PubMed
126. Occhigrossi, L, D’Eletto, M, Vecchio, A, Piacentini, M, Rossin, F. Transglutaminase type 2-dependent crosslinking of IRF3 in dying melanoma cells. Cell Death Discov 2022;8:498. https://doi.org/10.1038/s41420-022-01278-w.Search in Google Scholar PubMed PubMed Central
127. Ritchie, C, Carozza, JA, Li, L. Biochemistry, cell biology, and pathophysiology of the innate immune cGAS–cGAMP–STING pathway. Annu Rev Biochem 2022;91:599–628. https://doi.org/10.1146/annurev-biochem-040320-101629.Search in Google Scholar PubMed
128. Zhang, L, Jiang, C, Zhong, Y, Sun, K, Jing, H, Song, J, et al.. STING is a cell-intrinsic metabolic checkpoint restricting aerobic glycolysis by targeting HK2. Nat Cell Biol 2023;25:1208–22. https://doi.org/10.1038/s41556-023-01185-x.Search in Google Scholar PubMed PubMed Central
129. Xun, J, Zhang, Z, Lv, B, Lu, D, Yang, H, Shang, G, et al.. A conserved ion channel function of STING mediates noncanonical autophagy and cell death. EMBO Rep 2024;25:544–69. https://doi.org/10.1038/s44319-023-00045-x.Search in Google Scholar PubMed PubMed Central
130. Yang, M, Jiang, H, Ding, C, Zhang, L, Ding, N, Li, G, et al.. STING activation in platelets aggravates septic thrombosis by enhancing platelet activation and granule secretion. Immunity 2023;56:1013–26.e6. https://doi.org/10.1016/j.immuni.2023.02.015.Search in Google Scholar PubMed
131. Geng, K, Ma, X, Jiang, Z, Huang, W, Gu, J, Wang, P, et al.. High glucose-induced STING activation inhibits diabetic wound healing through promoting M1 polarization of macrophages. Cell Death Discov 2023;9:136. https://doi.org/10.1038/s41420-023-01425-x.Search in Google Scholar PubMed PubMed Central
132. Zhang, R, Yu, C, Zeh, HJ, Wang, H, Kroemer, G, Klionsky, DJ, et al.. Nuclear localization of STING1 competes with canonical signaling to activate AHR for commensal and intestinal homeostasis. Immunity 2023;56:2736–54.e8. https://doi.org/10.1016/j.immuni.2023.11.001.Search in Google Scholar PubMed PubMed Central
© 2024 the author(s), published by De Gruyter, Berlin/Boston
This work is licensed under the Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License.
Articles in the same Issue
- Frontmatter
- Reviews
- Understanding autoimmune response after SARS-CoV-2 infection and the pathogenesis/mechanisms of long COVID
- Improving lung allograft function in the early post-operative period through the inhibition of pyroptosis
- The occurrence mechanism, assessment, and non-pharmacological treatment of dyspnea
- Regenerative rehabilitation: a novel multidisciplinary field to maximize patient outcomes
- Advances in the prerequisite and consequence of STING downstream signalosomes
- Complement factor H in molecular regulation of angiogenesis
Articles in the same Issue
- Frontmatter
- Reviews
- Understanding autoimmune response after SARS-CoV-2 infection and the pathogenesis/mechanisms of long COVID
- Improving lung allograft function in the early post-operative period through the inhibition of pyroptosis
- The occurrence mechanism, assessment, and non-pharmacological treatment of dyspnea
- Regenerative rehabilitation: a novel multidisciplinary field to maximize patient outcomes
- Advances in the prerequisite and consequence of STING downstream signalosomes
- Complement factor H in molecular regulation of angiogenesis