Synthesis and structure of [2-((L)-menthoxycarbonyl)ethyl]diphenyltin halides
-
Qingtao Liu


Abstract
Two [2-((L)-menthoxycarbonyl)ethyl]diphenyltin halides, (L)-MenOCOCH2CH2SnXPh2 (X=Cl, 1; I, 2), have been synthesized and characterized by means of elemental analysis, FT-IR, NMR (1H, 13C, and 119Sn) spectroscopy, and X-ray single crystal diffraction. The tin atoms in 1 and 2 are both five-coordinated and possess the [C3SnOX] (X=Cl and I) trigonal bipyramidal environment with the trigonal plane defined by the three carbon atoms and the axial positions occupied by the halogen atom and internal carbonyl oxygen. There is a five-membered chelate ring in 1 and 2 with the Sn-O distance being 2.4781(16) and 2.512(2) Å, respectively.
Introduction
The ability of organotin halides RnSnX(4-n) (n=1–3) to be coordinated effectively by Lewis bases such as N and O, leading to five- and six-coordinated structures, via both inter- and intramolecular interactions, is well established (Davies, 2004). Ester groups are generally weak donors towards organotin acceptors. However, when sited intramolecularly, for example, as in X2Sn(CH2CH2COOR)2 (X=Cl, Br, I) (Harrison et al., 1979; Balasubramanian et al., 1997), X3SnCH2CH2COOR (X=Cl, Br, I) (Harrison et al., 1979; Howie et al., 1986; Howie and Wardell, 2002; Tian et al., 2003, 2005a,b; Lima et al., 2009) and C13SnCH2CH2 CH2COOEt (Howie et al., 1983), they are able to complex strongly to tin centers. The crystal structure determinations (Harrison et al., 1979; Howie et al., 1983, 1986; Howie and Wardell, 2002; Balasubramanian et al., 1997; Tian et al., 2003, 2005a,b; Lima et al., 2009) and spectroscopic data (Hutton et al., 1978, Maughan et al., 1981) showed that the ROCOCH2CH2 unit acts as a chelating ligand by utilizing the carbonyl oxygen as an additional donor center in the solid state and in non-coordinating solvents. Previously, Wardell’s group (Harston et al., 1991) and our group (Tian et al., 2005a,b) reported, respectively, the structures and coordination chemistry of other estertin compounds, ROCOCH2CH2SnXnPh3-n, (n=0, 1, 2; R=Me, Men). So far, for ROCOCH2CH2SnXPh2, only the structure of MeOCOCH2CH2 SnIPh2 (Harston et al., 1991) was reported. In order to continue to investigate the structure and property of the organotin compounds containing the optically active menthyl group, we synthesized and determined the crystal structures of (L)-MenOCOCH2CH2SnXPh2 (X=Cl, 1; I, 2).
Results and discussion
Synthesis
The exchange reaction of compound 1, obtained from the reaction of [2-((L)-menthoxycarbonyl)ethyl]triphenyltin with iodine chloride (Tian et al., 2005a,b), with excess sodium iodide in acetone afforded compound 2 (Scheme 1). In order to react completely, excess of sodium iodide and long reaction time (3 h) were required. Compound 2 is a colorless crystalline solid with a sharp melting point, and soluble in common organic solvents such as benzene, acetone, ethanol, and trichloromethane.
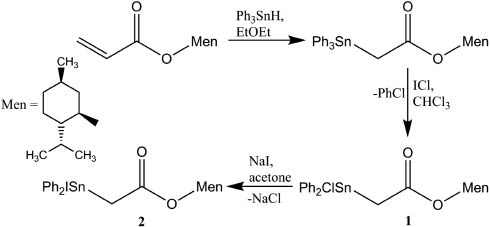
Synthesis of compounds 1 and 2.
Spectroscopic analysis
Among the stretching modes, the stretch vibration of carbonyl (C=O) in the 1640–1740 cm-1 region is known to depend on the nature of coordination of the carbonyl oxygen to metal (Maughan et al., 1981; Paterson et al., 1984). The ν (C=O) band of 1 and 2 appears at ~1670 cm-1 and exhibits a remarkable red shift compared with that of the free esters (~1740 cm-1), clearly indicating that the carbonyl is coordinated to the tin atom in compounds 1 and 2 (Maughan et al., 1981). The fact that there is almost no variation of the carbonyl frequency between the pure solid and a solution of trichloromethane for 1 and 2 confirms the presence of the C=O→Sn coordination in non-coordination solvents (Maughan et al., 1981; Paterson et al., 1984).
The lH resonance assignments of compounds 1 and 2 are given in the Experimental section. The assignment of the SnCH2 protons was made from the lower 2J(119Sn-1H) (76 Hz) compared with the corresponding 3J(119Sn-1H) (100 Hz) coupling. The lH NMR data also support the presence of carbonyl oxygen to tin coordination in 1 and 2. The δ values of the CHO- proton of the menthoxyl moiety show a downfield shift (Δδ=0.22 for 1, 0.31 for 2) compared with that in free menthyl ester MenOCOCH2CH2SnPh3 (δ 4.60) (Tian et al., 2005a,b) because the coordination of carbonyl to tin (CHOC=O→Sn) causes the deshielding of the CHOC=O proton.
Compared with the 13C NMR of compound 1 reported previously (Tian et al., 2005a), the 13C chemical shift of the SnCH2 carbon atom of 2 increases (from 14.96 ppm for 1 to 17.36 ppm for 2) and the coupling constant 1J(119Sn-13C) decreases (from 550 Hz for 1 to 492 Hz for 2), which is consistent with the 13C NMR of (MeOCOCH2CH2)2SnX2 (X=Cl, I) (Balasubramanian et al., 1997). The 13C δ values of CHO and C=O in 2 are larger (Δδ=4.60 and 5.21, respectively) than those of four-coordinated MenOCOCH2CH2SnPh3 (Tian et al., 2005a), which further prove the coordination of carbonyl to tin (CHOC=O→Sn).
The 119Sn chemical shifts depend on the number and nature of the groups coordinated with the central tin atom (Davies et al., 2008). The 119Sn chemical shift value of compound 2 (-96.6 ppm) is more high field than that of 1 (-74.6 ppm), which is in agreement with the ability of withdrawing electrons of chlorine and iodine. The δ value of 2 is also high field shifted as compared to that of four-coordinated IPh2SnCH2CH2CH2CH2SnPh2I (-55.1 ppm), and similar to that of five-coordinated analog chol-OCOCH2CH2SnPh2I (chol=cholesteryl) (-97.2 ppm) (Buchanan et al., 1998). This indicates that compounds 1 and 2 are five-coordinated in solution.
Structure analysis of 1 and 2
Compounds 1 and 2 both crystallize in the chiral space group P212121. Their molecular structures are shown in Figures 1 and 2, respectively, and selected bond lengths and angles are given in Table 1. In compound 1, the Sn atom is five coordinated and possesses a distorted trigonal bipyramidal geometry with the trigonal plane defined by the C(1), C(14), and C(20) atoms belonging to two phenyl substituents and the menthoxycarbonylethyl group, respectively, and the axial positions occupied by the Cl(1) and O(1) atoms. The Sn(1) atom is 0.155(2) Å out of the C3 trigonal plane of the [C3SnOCl] trigonal bipyramidal polyhedron in the direction of Cl(1), which results in a significant deviation of the C(1)-Sn(1)-Cl(1) (95.00(7)°), C(14)-Sn(1)-Cl(1) (97.90(9)°), and C(20)-Sn(1)-Cl(1) (97.22(7)°) angles from the ideal value of 90°. The three bond angles on the trigonal plane are in the range 115.97(10)°–123.44(10)°, and the bond angle O(1)-Sn(1)-Cl(1) at the axial position is 170.20(4)°. The Sn(1)-O(1) distance of 2.4781(16) Å is in the range of a normal Sn-O coordination bond length (Harston et al., 1991; Buchanan et al., 1996), clearly indicating significant bonding interactions between the Sn atom and the O(1) atom of the carbonyl group. The intramolecular C=O→Sn coordination results in a five-membered chelate ring with a narrow bite angle C(1)-Sn(1)-O(1) (75.21(7)°). The Sn(1)-Cl(1) bond distance (2.4299(7) Å) is longer than that in four-coordinated triorganotin chlorides such as Ph2ClSn(CH2)4SnClPh2 (2.397(3) Å) (Dakternieks et al., 1999) and Ph2ClSnMen (2.396(7) Å) (Hofmann et al., 2008), and comparable with that found in the intramolecularly coordinated diphenyltin chlorides containing five-membered chelates such as Ph2ClSnCH2CH2CH2OMe (2.4412(8) Å) (Lebl et al., 2005) and Ph2ClSnCH2-[19]-crown-6 (2.4323(11) Å) (Kuate et al., 2008).
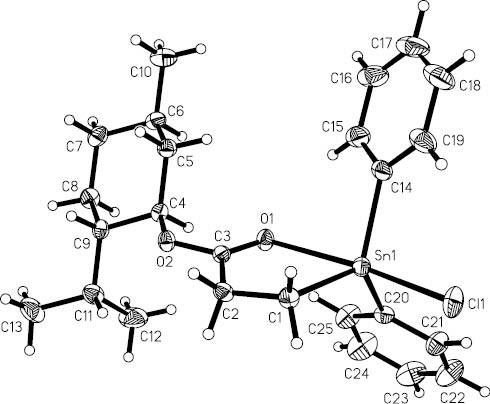
The molecular structure of 1.
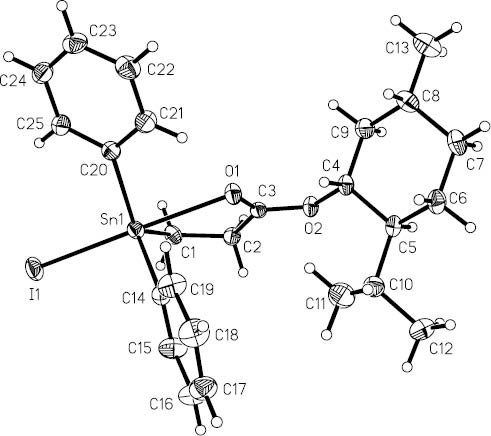
The molecular structure of 2.
Selected bond lengths (Å) and angles (°) for 1 and 2.
| 1 | 2 | 1 | 2 | ||
|---|---|---|---|---|---|
| Sn(1)-O(1) | 2.4781(16) | 2.512(2) | Sn(1)-X(1)a | 2.4299(7) | 2.7938(5) |
| Sn(1)-C(1) | 2.144(2) | 2.139(4) | C(3)-O(1) | 1.205(3) | 1.213(4) |
| Sn(1)-C(14) | 2.135(2) | 2.136(4) | C(3)-O(2) | 1.311(3) | 1.310(4) |
| Sn(1)-C(20) | 2.142(2) | 2.140(4) | C(4)-O(2) | 1.471(3) | 1.475(4) |
| C(1)-Sn(1)-O(1) | 75.21(7) | 73.54(12) | C(1)-Sn(1)-X(1) | 95.00(7) | 96.16(10) |
| C(14)-Sn(1)-O(1) | 86.49(10) | 84.62(13) | O(1)-Sn(1)-X(1) | 170.20(4) | 169.60(6) |
| C(20)-Sn(1)-O(1) | 88.63(8) | 84.32(12) | C(14)-Sn(1)-X(1) | 97.90(9) | 101.42(11) |
| C(1)-Sn(1)-C(14) | 116.58(10) | 114.52(15) | C(20)-Sn(1)-X(1) | 97.22(7) | 100.49(10) |
| C(1)-Sn(1)-C(20) | 123.44(10) | 122.75(15) | C(3)-O(1)-Sn(1) | 110.08(16) | 109.2(2) |
| C(14)-Sn(1)-C(20) | 115.97(10) | 115.00(15) | C(2)-C(1)-Sn(1) | 114.46(16) | 113.8(2) |
aX=Cl for 1, I for 2.
The structure of compound 2 is similar to that of 1, and the tin atom is also five-coordinated and has distorted trigonal bipyramidal geometry with an axial bond angle O(1)-Sn(1)-I(1) being 169.60(6)°. The Sn(1) atom is displaced by 0.346(3) Å from the C3 trigonal plane of the [C3SnOI] trigonal bipyramidal polyhedron in the direction of I(1). The intramolecular Sn(1)-O(1) distance (2.512(2) Å) is longer than that of 1 (2.4781(16) Å), which is consistent with the increase of the tin atom Lewis acidity in the sequence I<Cl (Spencer et al., 1990), and shorter than that of an analog MeOCOCH2CH2SnIPh2 (2.55(2) Å) (Harston et al., 1991).
Conclusion
Two optically active [2-((L)-menthoxycarbonyl)ethyl]diphenyltin halides have been synthesized and characterized. The tin atoms in 1 and 2 are both five-coordinated and possess trigonal bipyramidal geometry with a five-membered chelate ring formed by the intramolecular C=O→Sn coordination in the solid state as well as in non-coordinating solvents.
Experimental section
Materials and physical measurements
[2-((L)-menthoxycarbonyl)ethyl]triphenyltin was prepared according to the reported method (Tian et al., 2005a). The other chemicals (Sinopharm Chemical Reagent Company Limited, Shanghai, China) were of reagent grade and were used without further purification. Carbon and hydrogen analyses were obtained using a Perkin Elmer 2400 Series II elemental analyzer (Perkin Elmer, Waltham, MA, USA). Melting points were measured on an XR4 microscopic melting point apparatus (Shanghai Optical Instrument Factory, Shanghai, China). IR spectra were recorded on a Nicolet 470 FT-IR spectrophotometer using KBr disks in the range 4000–400 cm-1 (Thermo Nicolet Corporation, Madison, WI, USA). 1H and 13C NMR spectroscopic data were collected using a Bruker Avance 300 FT-NMR spectrometer (Bruker Corporation, Switzerland) with CDCl3 as solvent and Me4Si as internal standard. 119Sn NMR spectra were recorded in CDCl3 on a Varian Mercury Vx300 spectrometer (Varian Corporation, USA) using Me4Sn external reference.
Synthesis of [2-((L)-menthoxycarbonyl)ethyl]diphenyltin chloride (1)
This compound was synthesized according to the literature procedure (Tian et al., 2005a). The oily product 1 was dissolved in dichloromethane-hexane (1:1, v/v), and the solution obtained was left in the refrigerator for 2 days. The resulting colorless crystals were filtered and dried in vacuum. M.p. 48–49°C [literature: oil (Tian et al., 2005a)], yield 58%. Anal. Found: C, 57.80; H, 6.37. Calc. for C25H33ClO2Sn: C, 57.78; H, 6.40%. IR (KBr, ν, cm-1): 1667 (C=O), 1229 (C-O). 1H NMR (CDCl3, δ, ppm): 0.65 (d, J=6.9 Hz, 3H, CH3), 0.82–1.89 (m, 15H, Men), 1.75 (t, J=7.5 Hz, 2J(119Sn-1H)=78 Hz, 2H, CH2Sn), 2.82 (t, J=7.5 Hz, 3J(119Sn-1H)=100 Hz, 2H, COCH2), 4.82 (dt, Jae=4.3 Hz, Jaa=11.1 Hz, 1H, HCO), 7.33–7.41 (m, 6H, (p-, m-H)-C6H5), 7.79–7.84 (m, 4H, 2J(119Sn-1H)=62 Hz, o-H-C6H5).
Synthesis of [2-((L)-menthoxycarbonyl)ethyl]diphenyltin iodide (2)
The solutions of [2-((L)-menthoxycarbonyl)ethyl]diphenyltin chloride (1.04 g, 2 mmol) in acetone (30 mL) and sodium iodide (0.60 g, 4 mmol) in acetone (30 mL) were mixed and heated at reflux for 3 h under stirring, and then cooled to room temperature and filtered. The filtrate was evaporated under reduced pressure by a rotary evaporator. The solid residue was extracted into dichloromethane (20 mL) and filtered. A white solid product was obtained by removal of solvent under reduced pressure, and recrystallized from ethanol and dried in vacuum. Yield 0.98 g (80%), m.p. 72–73°C,
Crystal structure determination of compounds 1 and 2
The colorless single crystals were obtained from chloroform-hexane (1:2, v/v) by slow evaporation at room temperature. The intensity data for crystals of compounds were measured at 291(2) K on a Bruker Smart Apex area detector fitted with graphite monochromatized MoKα radiation (0.71073 Å) using the φ and ω scan technique. The structures were solved by a direct method and refined by a full-matrix least-squares procedure based on F2 using SHELXL-97 (Sheldrick, 2008). The non-hydrogen atoms were refined anisotropically, and hydrogen atoms were placed at calculated positions (C-H=0.93 Å for aromatic H atoms, C-H=0.96 Å for methyl H atoms, C-H=0.97 Å for methylene H atoms, and C-H=0.98 Å for methine H atoms) and were included in the refinement in the riding-model approximation. The crystallographic parameters and refinements are summarized in Table 2. Crystallographic data have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication numbers CCDC 904059 and 904060.
Crystallographic data and structure refinements of 1 and 2.
| 1 | 2 | |
|---|---|---|
| Empirical formula | C25H33ClO2Sn | C25H33IO2Sn |
| Formula weight | 519.65 | 611.10 |
| Crystal system | Orthorhombic | Orthorhombic |
| Temperature (K) | 291(2) | 291(2) |
| Space group | P212121 | P212121 |
| a (Å) | 10.7964(7) | 10.6619(16) |
| b (Å) | 13.7389(9) | 15.083(2) |
| c (Å) | 17.2450(12) | 16.467(3) |
| Volume (Å3) | 2558.0(3) | 2648.1(7) |
| Z | 4 | 4 |
| Dc (g cm-3) | 1.349 | 1.533 |
| μ (mm-1) | 1.120 | 2.147 |
| F(000) | 1064 | 1208 |
| θ range (°) | 1.90–25.99 | 1.83–26.00 |
| Crystal size (mm) | 0.40×0.25×0.18 | 0.14×0.14×0.12 |
| Total reflections | 20010 | 20931 |
| Uniq. reflections, Rint | 5010, 0.0207 | 5214, 0.0296 |
| Reflections with I>2σ(I) | 4623 | 4804 |
| GOF on F2 | 1.024 | 1.032 |
| Flack parameter | -0.03(2) | -0.03(2) |
| R1, wR2 [I>2σ(I)] | 0.0226, 0.0555 | 0.0282, 0.0643 |
| R1, wR2 (all data) | 0.0255, 0.0571 | 0.0316, 0.0659 |
| Δρmin, Δρmax (e Å-3) | -0.249, 0.306 | -0.345, 0.441 |
Acknowledgments
This work was supported by Shandong Provincial Natural Science Foundation, China (ZR2013BM007), the National Natural Science Foundation of China (21302110), and the Undergraduate Innovation Project in Qufu Normal University (2015).
References
Balasubramanian, R.; Chohan, Z. H.; Doidge-Harrison, S. M. S. V.; Howie, R. A.; Wardell, J. L. Some further studies of estertin compounds: crystal structures of [NEt4][MeO2CCH2CH2Sn(dmio)2] (dmio=1,3-dithiole-2-one-4,5-dithiolato) and (MeO2CCH2CH2)2SnX2 [X2=I2, (NCS)2 or Cl, Br]. Polyhedron1997, 16, 4283–4295.Search in Google Scholar
Buchanan, H.; Howie, R. A.; Khan, A.; Spencer, G. M.; Wardell, J. L. Preparations and crystal structures of Sn(CH2CH2CO2Me)2(C3S5) and [Q][Sn(CH2CH2CO2Me)(C3S5)2] (Q=NEt4 or 1,4-dimethylpyridinium, C3S5=4,5-disulfanyl-1,3-dithiole-2-thionate). J. Chem. Soc., Dalton Trans. 1996, 541–548.10.1039/dt9960000541Search in Google Scholar
Buchanan, H. J.; Cox, P. J., Wardell, J. L. Organotin substituted cholesteryl ethers and esters. Main Group Metal Chem. 1998, 21, 751–764.Search in Google Scholar
Dakternieks, D.; Lim, A. E. K.; Jurkschat, K.; Tiekink, E. R. T. Crystal structure of chloro[4-(1-chloro-1,1-diphenylstannyl)butyl]diphenylstannane, Ph2ClSn(CH2)4SnPh2Cl. Z. Kristallogr. – New Crystal Structures1999, 214, 515–516.10.1515/ncrs-1999-0458Search in Google Scholar
Davies, A. G. Organotin Chemistry, 2nd Edition. Wiley-VCH: Weinheim, 2004; p. 174.10.1002/3527601899Search in Google Scholar
Davies, A. G.; Gielen, M.; Pannell, K. H.; Tiekink, E. R. T. Tin Chemistry: Fundamentals, Frontiers, and Applications; John Wiley & Sons: Chichester, UK, 2008, p. 17.10.1002/9780470758090Search in Google Scholar
Harrison, P. G.; King, T. J.; Healy, M. A. Structural studies in main group chemistry: XXIII. Estertin derivatives, structural and spectroscopic studies. J. Organometal. Chem. 1979, 182, 17–36.Search in Google Scholar
Harston, P.; Howie, R. A.; McQuillan, G. P.; Wardell, J. L.; Zanetti, E.; Doidge-Harrison, S. M. S. V.; Stewart, N. S.; Cox, P. J. Crystal structure and coordination chemistry of (2-carbomethoxyethyl)iododiphenylstannane, IPh2SnCH2CH2CO2Me. Polyhedron1991, 10, 1085–1090.Search in Google Scholar
Hofmann, M.; Clark, T.; Heinemann, F. W.; Zenneck, U. Rock around the ring: an experimental and theoretical study of the molecular dynamics of stannyltriphospholes with chiral tin substituents. Eur. J. Inorg. Chem. 2008, 2225–2237.10.1002/ejic.200701321Search in Google Scholar
Howie, R. A.; Wardell, S. M. S. V. (2-Carbomethoxyethyl)triiodotin at 120 K. Acta Cryst. Section E2002, 58, m220–m222.10.1107/S1600536802007080Search in Google Scholar
Howie, R. A.; Paterson, E. S.; Wardell J. L.; Burley, J. W. Crystal structure and coordination chemistry of Cl3SnCH2CH2CH2CO2Et. J. Organomet. Chem. 1983, 259, 71–78.Search in Google Scholar
Howie, R. A.; Paterson, E. S.; Wardell, J. L.; Burley, J. W. Further study of estertin trichlorides, Cl3SnCH2CH2CO2R, Lewis acidity towards acetonitrile. Crystal structure of Cl3SnCH2CH2CO2Pr-i. J. Organomet. Chem. 1986, 304, 301–308.Search in Google Scholar
Hutton, R. E.; Burley, J. W.; Oakes, V. β-Substituted alkyltin halides: I. Monoalkyltin trihalides: synthetic, mechanistic and spectroscopic aspects. J. Organometal. Chem. 1978, 156, 369–382.Search in Google Scholar
Kuate, A. C. T.; Reeske, G.; Schurmann, M.; Costisella, B.; Jurkschat, K. Organotin compounds Ph2XSnCH2-[19]-crown-6 (X=Ph, F, Cl, Br, I, SCN) and Ph2ISnCH2Sn(I)PhCH2-[19]-crown-6 as ditopic receptors for potassium salts. Organometallics2008, 27, 5577–5587.Search in Google Scholar
Lebl, T.; Zoufala, P.; Bruhn, C. A comparative assessment of the effect of the Lewis acidity of the central tin atom on intramolecular coordination of (3-methoxypropyl)stannanes. Eur. J. Inorg. Chem. 2005, 2536–2544.10.1002/ejic.200400877Search in Google Scholar
Lima, G. M. D.; Milne, B. F.; Pereira, R. P.; Rocco, A. M.; Skakle, J. M. S.; Travis, A. J.; Wardell, J. L.; Wardell, S. M. S. V. Experimental and ab initio structural study of estertin compounds, X3SnCH2CH2CO2Me: crystal structures of Cl3SnCH2 CH2CO2Me at 120 K and Br3SnCH2CH2CO2Me at 120 and 291 K. J. Mol. Struct. 2009, 921, 244–250.Search in Google Scholar
Maughan, D.; Wardell, J. L.; Burley, J. W. Lewis acidity of carboxyethyltin chlorides, Cl3SnCH2CH2CO2R and Cl2Sn(CH2CH2CO2R)2. J. Organomet. Chem. 1981, 212, 59–70.Search in Google Scholar
Paterson, E. S.; Wardell, J. L.; Burley, J. W. Acidity of Cl3SnCH2CH2 CO2H. J. Organometal. Chem. 1984, 273, 313–318.10.1016/0022-328X(84)80545-8Search in Google Scholar
Sheldrick, G. M. A short history of SHELX. Acta Crystallogr. Section A2008, 64, 112–122.10.1107/S0108767307043930Search in Google Scholar PubMed
Spencer, J. N.; Ganunis, T.; Zafar, A.; Eppley, H.; Otter, J. C.; Coley, S. M.; Yoder, C. H. The effect of the halide on the Lewis acidity of organotin halides. J. Organomet. Chem. 1990, 389, 295–300.Search in Google Scholar
Tian, L.-J.; Yu, Q.-S.; Liu, X.-J.; Shang, Z.-C.; Wang, T.; Zhao, W.-N. Synthesis and structural characterization of 3-trichlorostannylpropionates and their complexes. ChineseJ. Org. Chem. 2003, 23, 441–446.Search in Google Scholar
Tian, L.-J.; Sun, Y.-X.; Liu, X.-J.; Yang, G.-M.; Shang, Z.-C. Synthesis and structural characterization of 2-(-)-menthoxycarbonylethyltin compounds. Polyhedron2005a, 24, 2027–2034.10.1016/j.poly.2005.05.025Search in Google Scholar
Tian, L.-J.; Yu, Q.-S.; Zhang, L.-P.; Sun, Y.-X. Structure and transesterification reaction of methyl 3-(phenyldihalostannyl) propionates. Chin. Chem. Lett. 2005b, 16, 1010–1012.Search in Google Scholar
©2015 by De Gruyter
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Articles in the same Issue
- Frontmatter
- Research Articles
- Amino-functionalised metal xanthates
- The disilanes Cp*SiCl2SiH3 and Cp*SiH2SiH2Cp*
- Synthesis and characterization of gallium(III) dithiocarbamates as suitable nano-gallium(III) sulfide precursors
- Synthesis, structure and in vitro cytotoxic activity of two organotin complexes of 2-phenyl-1,2, 3-triazole-4-carboxylic acid
- Synthesis and structure of [2-((L)-menthoxycarbonyl)ethyl]diphenyltin halides
- A low-temperature, environment-friendly approach to the synthesis of magnesium borates using magnesium waste scraps
- Short Communications
- Synthesis and crystal structure of a new 1D Pb(II) coordination polymer containing salicylate and 2,2′-bipyridine ligands
- Molecular structure of novel heterobimetallic thiolate [Cu(PPh3)]4(ZnCl)2(SEt)6.6THF
- Mercury(I) chloride in vivo oxidation: a thermodynamic study
Articles in the same Issue
- Frontmatter
- Research Articles
- Amino-functionalised metal xanthates
- The disilanes Cp*SiCl2SiH3 and Cp*SiH2SiH2Cp*
- Synthesis and characterization of gallium(III) dithiocarbamates as suitable nano-gallium(III) sulfide precursors
- Synthesis, structure and in vitro cytotoxic activity of two organotin complexes of 2-phenyl-1,2, 3-triazole-4-carboxylic acid
- Synthesis and structure of [2-((L)-menthoxycarbonyl)ethyl]diphenyltin halides
- A low-temperature, environment-friendly approach to the synthesis of magnesium borates using magnesium waste scraps
- Short Communications
- Synthesis and crystal structure of a new 1D Pb(II) coordination polymer containing salicylate and 2,2′-bipyridine ligands
- Molecular structure of novel heterobimetallic thiolate [Cu(PPh3)]4(ZnCl)2(SEt)6.6THF
- Mercury(I) chloride in vivo oxidation: a thermodynamic study