Challenges in data storage and data management in a clinical diagnostic setting
-
Sebastian H. Eck


Abstract
The implementation of next-generation sequencing (NGS) in a clinical diagnostic setting opens vast opportunities through the ability to sequence all genes contributing to a certain morbidity simultaneously at a cost and speed that is superior to traditional sequencing approaches. On the other hand, the practical implementation of NGS in a clinical diagnostic setting involves a variety of new challenges, which need to be overcome. Among these are the generation, analysis and storage of unprecedented amounts of data, strict control of sequencing performance, validation of results, interpretation of detected variants and reporting. In the following sections, key aspects of data management and integration will be discussed. In particular, issues of data storage, data analysis using in-house IT infrastructure vs. data analysis employing cloud computing and the need for data integration from different sources will be covered.
Genetic testing volume
The rise of next-generation sequencing (NGS) methods can be considered as a disruptive technology for genetic testing [1], [2]. The volume of genetic data generated per test rapidly increased. Prior to NGS, the diagnostics of the majority of rare disorders followed a step-wise paradigm where different genes involved in the disorder were sequenced sequentially. NGS enables larger approaches like the sequencing of gene panels (i.e. all genes known to contribute to certain disorders are sequenced simultaneously, typically 5–100 genes) and the sequencing of the clinical exome (i.e. all genes known to be involved in any Mendelian disorder, 2500–6000 genes). Even the sequencing of all protein-coding regions (i.e. whole exome, ~20,000 genes) or the complete genome is possible (Figure 1). Consequently, the amount of data that needs to be pre-processed, analyzed, interpreted and stored per case also dramatically increased (Table 1). There are now several guidelines for the use and quality control of NGS in clinical diagnostics [3], [4], [5], [6], yet the issue of efficient data handling, integration and long-term storage are more rarely covered.

Volume of different genetic tests, size in base pairs (bp).
Output and approximate required disk space to store an instrument run (run folder size, source Illumina bulletin 01/2018).
| Instrument | Run type | Output Gigabases | Run folder size Gigabytes |
|---|---|---|---|
| MiSeq | 2×150 bp | 5 | 16–18 |
| MiSeq | 2×250 bp | 8 | 22–24 |
| MiSeq | 2×300 bp | 15 | 22–26 |
| NextSeq500 | 2×150 bp Mid output | 35 | 22–26 |
| NextSeq500 | 2×150 bp High output | 120 | 60–70 |
| HiSeq2500 | 2×250 bp Rapid run | 150 | 80–100 |
| HiSeq2500 | 2×125 bp High output | 500 | 295–310 |
| NovaSeq | 2×150 bp S2 flowcell | 1000 | 730 |
| NovaSeq | 2×150 bp S4 flowcell | 2500 | 2190 |
Long-term handling and storage of NGS data
The huge amount of raw and processed data from NGS warrants new strategies for the long-term storage and accessibility of these data [4]. There are three possible approaches:
Complete storage of raw data
In this approach, all raw and processed files from the sequencing instrument (Figure 2) and the analysis pipeline are stored in long-term storage. This includes raw base call files (.bcf) or their equivalent for non-Illumina instruments used to generate .fastq files as well as the aligned sequences, preferably in the binary sequence alignment map format (.bam, [7]) and the complete output of the analysis pipeline. This may consist of the detected variants (variant call format, .vcf), quality metrics, coverage analysis, annotations and all parameter settings used to obtain these results. The advantage of this approach is the complete transparency and reproducibility of the results, should circumstances warrant a re-inspection of a particular case. On the other hand, this approach poses a huge burden on the backup and data storage pipeline due to the amount of data and that some data are saved partially redundantly. For example, .bam files may be generated using .fastq files, which in turn can be generated using .bcf files. To reduce the redundancy while maintaining the absolute level of reproducibility, the software versions and settings used when the case was first analyzed need to be saved as well.
 Figure 2:
Figure 2:Illumina raw data folder structure (Illumina bcl2fastq guide v2, page 6).
Storage of data needed to repeat analysis
The second approach tries to omit saving data directly from the instruments, and instead focuses on all data needed to repeat the analysis. This typically means that the .fastq files and/or .bam files along all parameter settings and pipeline components are archived, while primary data from the instruments like .bcf files are not. This approach relaxes the conditions of storage requirements somewhat, while still maintaining the ability to repeat all primary analysis steps if necessary. The only part that is not reproducible is the actual base calling, i.e. the translation of instrument output into continuous reads.
Storage of results and parameter settings
In the last approach, only processed results like detected variants and the parameter setting with which they were obtained are stored, all intermediate files used to generate the results are deleted after analysis. This includes .fastq and .bam files. As a consequence, results cannot be easily reproduced. Instead, the sample has to be re-sequenced in an additional instrument run, potentially requiring new primary material ethylenediaminetetraacetic acid (EDTA) blood. While this sounds like a disadvantageous strategy, it could be more attractive for large-scale data sets like whole genome and exome data. Due to further declining sequencing cost, the financial burden of storage space for all samples can outweigh the cost for re-sequencing of few selected cases where re-analysis is necessary.
All approaches share the requirement of recording pipeline versions and parameter settings of how the results were obtained.
Cloud vs. in-house data analysis
Cloud computing, by definition, is a way to organize computing and storage resources so that multiple users may rent certain amounts of CPU hours and storage according to their specific need [8]. Since the inception, cloud computing concepts have extended to additionally provide platforms, i.e. pre-configured computing environments, as well as already installed and ready-to-use software. Major cloud computing providers include Amazon, Google and Microsoft, but also data resources and large scale data sets such as GenBank, the 1000 Genomes project [9] and the Exome Aggregation Consortium data (ExAC [10]) are stored in clouds. Different cloud concepts may be classified by services that are offered; broad categories are Data-as-a-Service, Infrastructure-as-a-Service, Software-as-a-Service and Platform-as-a-Service. An overview with example providers for each category and possible services can be seen in Table 2. The defining feature of cloud computing models is “elasticity”, which in this context describes the ability to access exactly the amounts of resources needed for a specific task. Two typical problems when considering investments in IT infrastructure are under-provisioning, i.e. the lack of computational power and/or storage to efficiently handle a task, and problems of over-provisioning, i.e. mostly idle resources not needed routinely. These problems may be solved by employing cloud solutions as only the resources specifically needed are rented and paid for.
Cloud computing models, example services and providers [8].
| Cloud service | Acronym | Example services | Example providers |
|---|---|---|---|
| Data-as-a-Service | DaaS | Access to large data sets | GenBank ExaC 1000 Genomes |
| Infrastructure-as-a-Service | IaaS | Virtualized, computational infrastructure (compute power and/or storage) | Amazon Web Service AWS Amazon EC2 Amazon S3 Google Cloud Platform IBM Cloud |
| Software-as-a-Service | SaaS | Software applications, open source analysis pipelines | Illumina Base Space DNAnexus |
| Platform-as-a-Service | PaaS | Programming language environments, web servers, databases | Amazon Web Services Software Development Kit (AWS SDK) Google App Engine |
In genomics research, cloud computing solutions are not only used for primary analysis in large-scale projects like the 1000 Genomes Project [9], the Exome Aggregation Consortium [10] or the Cancer Genome Atlas [11], they also enable global access to these data sets. This allows re-analysis of the data which can assure reproducibility and exploitation for different projects by, for example, providing reference data sets.
The main reasoning for choosing a cloud computing approach is to reduce the burden of storage and computing requirements that need to be maintained in-house, and to have access to a sufficient resource when needed.
On the other hand, maintaining an in-house server structure for primary data analysis has the advantage of having full control over the computing systems. New software may easily be deployed and utilized, which may be difficult depending on the chosen cloud computing model. Another advantage is that all data remain in an in-house network and no large data sets need to be transferred, reducing the cost and requirements for the cable connection. This comes at the price of having to install, update and maintain the in-house servers. This approach is also not readily scalable so that an increase in sequencing capacity often warrants new investments in the IT infrastructure for storage and analysis.
To summarize, both approaches have their respective advantages and caveats. Smaller scale data sets like primarily multi-gene panel sequencing can be efficiently handled on in-house servers. The capital investment for the necessary IT infrastructure is manageable and all data are generated, processed and stored in the same place. On the other hand, cloud computing solutions are more suited for the analysis of larger scale data sets, ranging from primarily exome sequencing to whole genome sequencing data analysis. As cloud solutions are readily scalable, the risk of wrong IT infrastructure investments is eliminated.
Data analysis and integration
Efficient data handling goes far beyond the problems of which data need to be stored. In state-of-the art diagnostics, the integration of different data sources, both external and internal, from different tests, workflows and clinical specialties are vital for optimal patient care. In the following paragraphs two examples of integrated diagnostics will be discussed.
The first example is the diagnostic routing for non-small cell lung carcinoma (NSCLC, Figure 3) where data from different tests and the cooperation between different clinical specialties are combined for an optimal performance [12]. The workflow is as follows:

Diagnostic routing and interplay of different tests in the diagnosis of NSCLC.
In pathology, tumor diagnosis and classification of the tumor tissue is performed on the basis of the primary or re-biopsy material. After morphological examination, multi-gene panel sequencing is performed to identify driver and passenger mutations. Based on the results, a molecular profile is created with the aim of identifying therapeutic options. The molecular profile can be used in a follow-up by liquid biopsy or liquid profiling from peripheral blood. Targeted ctDNA analysis is performed for the monitoring of minimal residual disease or therapy resistance. Tumor markers complement and increase the sensitivity of the follow-up. In case of clinical deterioration, therapy/drug resistance or conspicuous imaging results, a re-biopsy with a morphological examination and molecular tumor profile is recommended.
The second example constitutes the integration of systematic phenotype information in the diagnostics of rare Mendelian disorders, in particular unexplained intellectual disability and developmental delay (ID/DD) [13], [14]. Rare disorders often manifest during infancy or early childhood, yet are difficult to diagnose due to high phenotypic variability and masking by auxiliary symptoms. The majority of rare disease patients remain without diagnosis or has to endure a diagnostic odyssey before the diagnosis could be obtained. Current routine diagnostic testing for patients with ID/DD of unclear etiology consists of karyotyping and array comparative genomic hybridization (array CGH) analysis. However, the major cause of severe ID/DD are de novo point mutations [15], which cannot be detected by these genome-wide low-resolution tests. A further approach in routine diagnostics is targeted sequencing of single genes with disease association, which is best suited for highly distinct clinical conditions that are caused by just one or a few genes. Many rare pediatric conditions including neurodevelopmental disorders are characterized by a large genetic heterogeneity, so testing each candidate gene individually is not feasible, whereas sequencing all genes simultaneously, such as in a whole exome sequencing (WES) approach, enables a fast and comprehensive genetic analysis [16]. Moreover, trio analysis of the WES data of the affected child and both unaffected parents enables benign familial genetic variants to be filtered out and de novo variants that are only present in the child to be identified.
By broad NGS-based diagnostic approaches like WES, the amount of clinical data increases tremendously, which necessitates the integration of phenotype and genotype data for fast and precise data interpretation and generation of conclusive genetic reports. To record phenotypic information in a standardized way, the Human Phenotype Ontology (HPO, [17], [18], [19]) was developed and is now widely employed. The HPO provides a hierarchic system of standardized vocabulary of phenotypic abnormalities encountered in human diseases and their semantic relationships, which can be stored in a relational database for easy query. Patient’s clinical features can thereby be used for filtering and evaluating genetic variants as described in the Multiple Integration and Data Annotation Study (MIDAS, [20]), which aims to accelerate WES data analysis by automated variant prioritization based on the patients’ phenotype. Besides, it would be beneficial for diagnostic purposes to integrate and correlate NGS data with additional genetic data or further resources like results from biochemical testing or medical imaging to support or exclude certain diagnoses (conf. MIDAS concept, Figure 4).
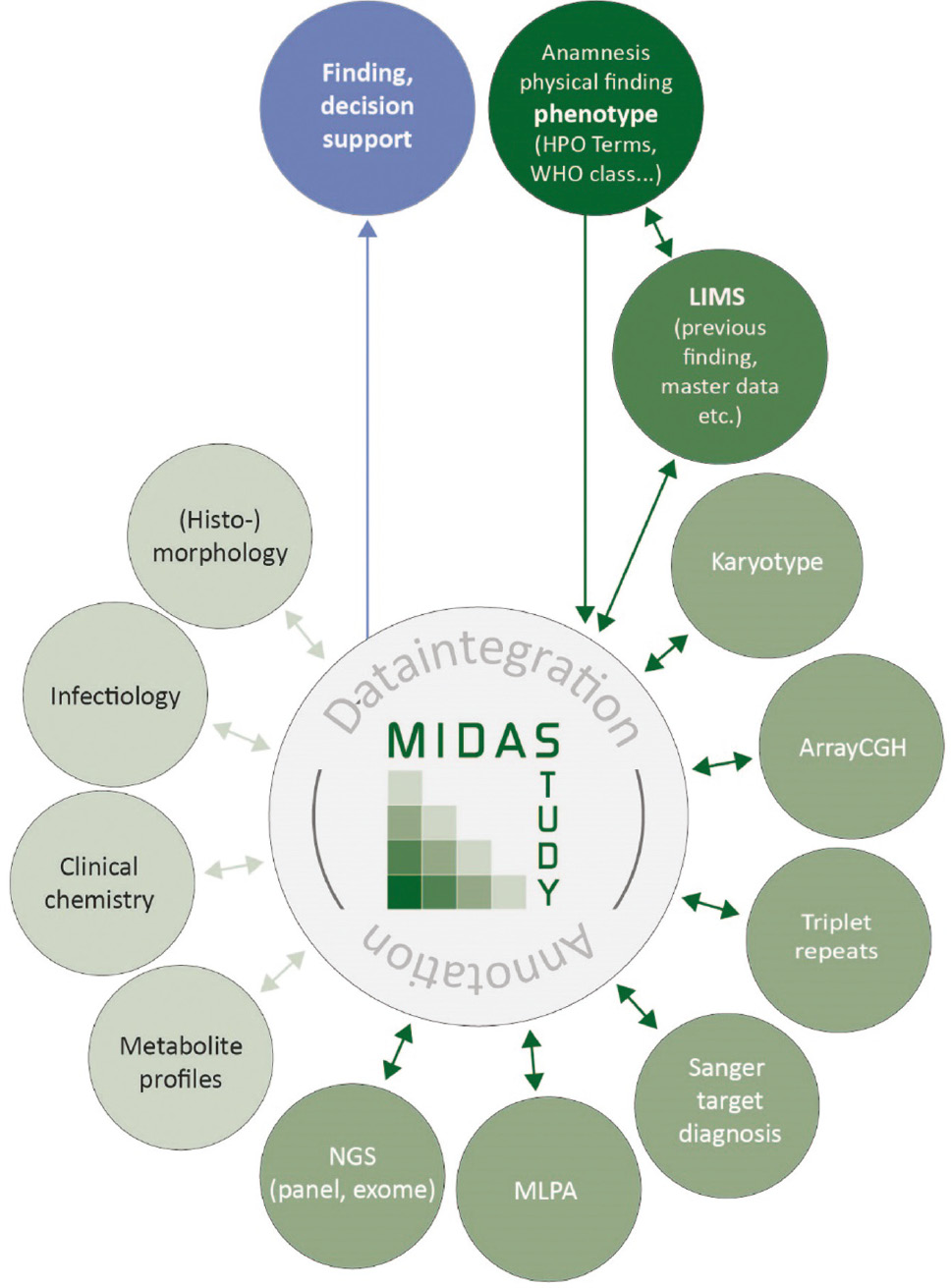
MIDAS data integration concept.
These examples show that strategies for long-term data storage, data handling and integration need to be developed, as these points are crucial for the success of future genetic testing in a clinical laboratory. The ever-expanding amounts of data generated per test warrant a more and more sophisticated approach to the data to extract the most useful information and exploit synergies of different data sets and clinical specialties by means of integrative diagnostic concepts.
Acknowledgments
The author would like to thank Oliver Wachter for the input to the NSCLC workflow and Yasemin Dinçer for the input to the integration of HPO terms and critical revision of the manuscript.
Author contributions: The author has accepted responsibility for the entire content of this submitted manuscript and approved submission.
Research funding: Bayerisches Staatsministerium für Wirtschaft, Infrastruktur, Verkehr und Technologie: Funder Id: 10.13039/501100005017, Grant Number: MED-1603-0001.
Employment or leadership: None declared.
Honorarium: None declared.
Competing interests: The funding organization(s) played no role in the study design; in the collection, analysis, and interpretation of data; in the writing of the report; or in the decision to submit the report for publication.
References
1. Wheeler DA, Srinivasan M, Egholm M, Shen Y, Chen L, McGuire A, et al. The complete genome of an individual by massively parallel DNA sequencing. Nature 2008;452:872–6.http://gateway.webofknowledge.com/gateway/Gateway.cgi?GWVersion=2&SrcApp=PARTNER_APP&SrcAuth=LinksAMR&KeyUT=WOS:000255026000048&DestLinkType=FullRecord&DestApp=ALL_WOS&UsrCustomerID=b7bc2757938ac7a7a821505f8243d9f310.1038/nature06884Search in Google Scholar PubMed
2. Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 2008;456:53–9.10.1038/nature07517http://gateway.webofknowledge.com/gateway/Gateway.cgi?GWVersion=2&SrcApp=PARTNER_APP&SrcAuth=LinksAMR&KeyUT=WOS:000260674000039&DestLinkType=FullRecord&DestApp=ALL_WOS&UsrCustomerID=b7bc2757938ac7a7a821505f8243d9f3Search in Google Scholar PubMed PubMed Central
3. Rehm HL, Bale SJ, Bayrak-Toydemir P, Berg JS, Brown KK, Deignan JL, et al. ACMG clinical laboratory standards for next-generation sequencing. Genet Med 2013;15:733–47.10.1038/gim.2013.92http://gateway.webofknowledge.com/gateway/Gateway.cgi?GWVersion=2&SrcApp=PARTNER_APP&SrcAuth=LinksAMR&KeyUT=WOS:000324172000010&DestLinkType=FullRecord&DestApp=ALL_WOS&UsrCustomerID=b7bc2757938ac7a7a821505f8243d9f3Search in Google Scholar PubMed PubMed Central
4. Matthijs G, Souche E, Alders M, Corveleyn A, Eck S, Feenstra I, et al. Guidelines for diagnostic next-generation sequencing. Eur J Hum Genet 2016;24:2–5.10.1038/ejhg.2015.226http://gateway.webofknowledge.com/gateway/Gateway.cgi?GWVersion=2&SrcApp=PARTNER_APP&SrcAuth=LinksAMR&KeyUT=WOS:000366615000002&DestLinkType=FullRecord&DestApp=ALL_WOS&UsrCustomerID=b7bc2757938ac7a7a821505f8243d9f3Search in Google Scholar PubMed PubMed Central
5. Vogl I, Eck SH, Benet-Pagès A, Greif PA, Hirv K, Kotschote S, et al. Diagnostic applications of next generation sequencing: working towards quality standards. J Lab Med 2012;36: 227–39.Search in Google Scholar
6. Bauer P, Wildhardt G, Gläser D, Müller-Reible C, Bolz HJ, Klein HG, et al. German guidelines for molecular genetic diagnostic testing using high-throughput technology, such as next-generation sequencing. OBM Genet 2018;2:019.10.21926/obm.genet.1802019Search in Google Scholar
7. Li Y, Vinckenbosch N, Tian G, Huerta-Sanchez E, Jiang T, Jiang H, et al. Resequencing of 200 human exomes identifies an excess of low-frequency non-synonymous coding variants. Nat Genet 2010;42:969–72.http://gateway.webofknowledge.com/gateway/Gateway.cgi?GWVersion=2&SrcApp=PARTNER_APP&SrcAuth=LinksAMR&KeyUT=WOS:000283540500013&DestLinkType=FullRecord&DestApp=ALL_WOS&UsrCustomerID=b7bc2757938ac7a7a821505f8243d9f310.1038/ng.680Search in Google Scholar PubMed
8. Langmead B, Nellore A. Cloud computing for genomic data analysis and collaboration. Nat Rev Genet 2018;19:208–19.http://gateway.webofknowledge.com/gateway/Gateway.cgi?GWVersion=2&SrcApp=PARTNER_APP&SrcAuth=LinksAMR&KeyUT=WOS:000427488600006&DestLinkType=FullRecord&DestApp=ALL_WOS&UsrCustomerID=b7bc2757938ac7a7a821505f8243d9f310.1038/nrg.2017.113Search in Google Scholar PubMed PubMed Central
9. 1000 Genomes Project Consortium, Abecasis GR, Altshuler D, Brooks LD, Durbin RM, Gibbs RA, et al. A map of human genome variation from population-scale sequencing. Nature 2010;467:1061–73.10.1038/nature09534http://gateway.webofknowledge.com/gateway/Gateway.cgi?GWVersion=2&SrcApp=PARTNER_APP&SrcAuth=LinksAMR&KeyUT=WOS:000283548600039&DestLinkType=FullRecord&DestApp=ALL_WOS&UsrCustomerID=b7bc2757938ac7a7a821505f8243d9f3Search in Google Scholar PubMed PubMed Central
10. Exome Aggregation Consortium, Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016;536:285–91.10.1038/nature19057http://gateway.webofknowledge.com/gateway/Gateway.cgi?GWVersion=2&SrcApp=PARTNER_APP&SrcAuth=LinksAMR&KeyUT=WOS:000381804900026&DestLinkType=FullRecord&DestApp=ALL_WOS&UsrCustomerID=b7bc2757938ac7a7a821505f8243d9f3Search in Google Scholar PubMed PubMed Central
11. Cancer Genome Atlas Research Network, Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA, et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet 2013;45:1113–20.10.1038/ng.2764http://gateway.webofknowledge.com/gateway/Gateway.cgi?GWVersion=2&SrcApp=PARTNER_APP&SrcAuth=LinksAMR&KeyUT=WOS:000324989600005&DestLinkType=FullRecord&DestApp=ALL_WOS&UsrCustomerID=b7bc2757938ac7a7a821505f8243d9f3Search in Google Scholar PubMed PubMed Central
12. Hagemann IS, Devarakonda S, Lockwood CM, Spencer DH, Guebert K, Bredemeyer AJ, et al. Clinical next-generation sequencing in patients with non-small cell lung cancer. Cancer 2015;121:631–9.10.1002/cncr.29089http://gateway.webofknowledge.com/gateway/Gateway.cgi?GWVersion=2&SrcApp=PARTNER_APP&SrcAuth=LinksAMR&KeyUT=WOS:000349395200020&DestLinkType=FullRecord&DestApp=ALL_WOS&UsrCustomerID=b7bc2757938ac7a7a821505f8243d9f3Search in Google Scholar PubMed
13. Rauch A, Wieczorek D, Graf E, Wieland T, Endele S, Schwarzmayr T, et al. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet 2012;380:1674–82.http://gateway.webofknowledge.com/gateway/Gateway.cgi?GWVersion=2&SrcApp=PARTNER_APP&SrcAuth=LinksAMR&KeyUT=WOS:000310951400029&DestLinkType=FullRecord&DestApp=ALL_WOS&UsrCustomerID=b7bc2757938ac7a7a821505f8243d9f310.1016/S0140-6736(12)61480-9Search in Google Scholar PubMed
14. de Ligt J, Willemsen MH, van Bon BW, Kleefstra T, Yntema HG, Kroes T, et al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med 2012;367:1921–9.http://gateway.webofknowledge.com/gateway/Gateway.cgi?GWVersion=2&SrcApp=PARTNER_APP&SrcAuth=LinksAMR&KeyUT=WOS:000311029300008&DestLinkType=FullRecord&DestApp=ALL_WOS&UsrCustomerID=b7bc2757938ac7a7a821505f8243d9f310.1056/NEJMoa1206524Search in Google Scholar PubMed
15. Vissers LE, de Ligt J, Gilissen C, Janssen I, Steehouwer M, de Vries P, et al. A de novo paradigm for mental retardation. Nat Genet 2010;42:1109–12.http://gateway.webofknowledge.com/gateway/Gateway.cgi?GWVersion=2&SrcApp=PARTNER_APP&SrcAuth=LinksAMR&KeyUT=WOS:000284578800014&DestLinkType=FullRecord&DestApp=ALL_WOS&UsrCustomerID=b7bc2757938ac7a7a821505f8243d9f310.1038/ng.712Search in Google Scholar PubMed
16. Wright CF, FitzPatrick DR, Firth HV. Paediatric genomics: diagnosing rare disease in children. Nat Rev Genet 2018; 19:253–68.10.1038/nrg.2017.116http://gateway.webofknowledge.com/gateway/Gateway.cgi?GWVersion=2&SrcApp=PARTNER_APP&SrcAuth=LinksAMR&KeyUT=WOS:000430048800006&DestLinkType=FullRecord&DestApp=ALL_WOS&UsrCustomerID=b7bc2757938ac7a7a821505f8243d9f3Search in Google Scholar PubMed
17. Köhler S, Doelken SC, Mungall CJ, Bauer S, Firth HV, Bailleul- Forestier I, et al. The Human Phenotype Ontology project: linking molecular biology and disease through phenotype data. Nucleic Acids Res 2014;42:D966–74.http://gateway.webofknowledge.com/gateway/Gateway.cgi?GWVersion=2&SrcApp=PARTNER_APP&SrcAuth=LinksAMR&KeyUT=WOS:000331139800142&DestLinkType=FullRecord&DestApp=ALL_WOS&UsrCustomerID=b7bc2757938ac7a7a821505f8243d9f310.1093/nar/gkt1026Search in Google Scholar PubMed PubMed Central
18. Köhler S, Vasilevsky NA, Engelstad M, Foster E, McMurry J, Aymé S, et al. The Human Phenotype Ontology in 2017. Nucleic Acids Res 2017;45:D865–76.10.1093/nar/gkw1039http://gateway.webofknowledge.com/gateway/Gateway.cgi?GWVersion=2&SrcApp=PARTNER_APP&SrcAuth=LinksAMR&KeyUT=WOS:000396575500121&DestLinkType=FullRecord&DestApp=ALL_WOS&UsrCustomerID=b7bc2757938ac7a7a821505f8243d9f3Search in Google Scholar PubMed PubMed Central
19. Köhler S, Schulz MH, Krawitz P, Bauer S, Dölken S, Ott CE, et al. Clinical diagnostics in human genetics with semantic similarity searches in ontologies. Am J Hum Genet 2009;85:457–64.http://gateway.webofknowledge.com/gateway/Gateway.cgi?GWVersion=2&SrcApp=PARTNER_APP&SrcAuth=LinksAMR&KeyUT=WOS:000270836000003&DestLinkType=FullRecord&DestApp=ALL_WOS&UsrCustomerID=b7bc2757938ac7a7a821505f8243d9f310.1016/j.ajhg.2009.09.003Search in Google Scholar PubMed PubMed Central
20. Dincer Y, Schulz J, Wilson SC, Marschall C, Cohen MY, Mall V, et al. Multiple Integration and Data Annotation Study (MIDAS): improving next-generation sequencing data analysis by genotype-phenotype correlations. J Lab Med 2018;42:1–8.http://gateway.webofknowledge.com/gateway/Gateway.cgi?GWVersion=2&SrcApp=PARTNER_APP&SrcAuth=LinksAMR&KeyUT=WOS:000429039300001&DestLinkType=FullRecord&DestApp=ALL_WOS&UsrCustomerID=b7bc2757938ac7a7a821505f8243d9f310.1515/labmed-2017-0072Search in Google Scholar
©2018 Walter de Gruyter GmbH, Berlin/Boston
Articles in the same Issue
- Frontmatter
- Editorial
- Shaping the digital transformation of laboratory medicine
- Shaping the digital transformation of laboratory medicine
- Mini Review
- Challenges in data storage and data management in a clinical diagnostic setting
- Challenges in data storage and data management in a clinical diagnostic setting
- Review
- Continuous glucose monitoring: data management and evaluation by patients and health care professionals – current situation and developments
- Continuous glucose monitoring: data management and evaluation by patients and health care professionals – current situation and developments
- Opinion Paper
- Long-term medical data storage: challenges with test results obtained by direct-to-consumer testing
- Long-term medical data storage: challenges with test results obtained by direct-to-consumer testing
- Legal aspects of storage and transmission of laboratory results
- Legal aspects of storage and transmission of laboratory results
- Review
- Interoperability of laboratory data in Switzerland – a spotlight on Bern
- Interoperability of laboratory data in Switzerland – a spotlight on Bern
- Mini Review
- Using HL7 CDA and LOINC for standardized laboratory results in the Austrian electronic health record
- Using HL7 CDA and LOINC for standardized laboratory results in the Austrian electronic health record
- NPU, LOINC, and SNOMED CT: a comparison of terminologies for laboratory results reveals individual advantages and a lack of possibilities to encode interpretive comments
- NPU, LOINC, and SNOMED CT: a comparison of terminologies for laboratory results reveals individual advantages and a lack of possibilities to encode interpretive comments
- Review
- Laboratory information system and necessary improvements in function and programming
- Laboratory information system and necessary improvements in function and programming
- Percentiler and Flagger – low-cost, on-line monitoring of laboratory and manufacturer data and significant surplus to current external quality assessment
- Percentiler and Flagger – low-cost, on-line monitoring of laboratory and manufacturer data and significant surplus to current external quality assessment
- Review
- Informatics External Quality Assurance (IEQA) Down Under: evaluation of a pilot implementation
- Informatics External Quality Assurance (IEQA) Down Under: evaluation of a pilot implementation
- Acknowledgment
- Acknowledgment
Articles in the same Issue
- Frontmatter
- Editorial
- Shaping the digital transformation of laboratory medicine
- Shaping the digital transformation of laboratory medicine
- Mini Review
- Challenges in data storage and data management in a clinical diagnostic setting
- Challenges in data storage and data management in a clinical diagnostic setting
- Review
- Continuous glucose monitoring: data management and evaluation by patients and health care professionals – current situation and developments
- Continuous glucose monitoring: data management and evaluation by patients and health care professionals – current situation and developments
- Opinion Paper
- Long-term medical data storage: challenges with test results obtained by direct-to-consumer testing
- Long-term medical data storage: challenges with test results obtained by direct-to-consumer testing
- Legal aspects of storage and transmission of laboratory results
- Legal aspects of storage and transmission of laboratory results
- Review
- Interoperability of laboratory data in Switzerland – a spotlight on Bern
- Interoperability of laboratory data in Switzerland – a spotlight on Bern
- Mini Review
- Using HL7 CDA and LOINC for standardized laboratory results in the Austrian electronic health record
- Using HL7 CDA and LOINC for standardized laboratory results in the Austrian electronic health record
- NPU, LOINC, and SNOMED CT: a comparison of terminologies for laboratory results reveals individual advantages and a lack of possibilities to encode interpretive comments
- NPU, LOINC, and SNOMED CT: a comparison of terminologies for laboratory results reveals individual advantages and a lack of possibilities to encode interpretive comments
- Review
- Laboratory information system and necessary improvements in function and programming
- Laboratory information system and necessary improvements in function and programming
- Percentiler and Flagger – low-cost, on-line monitoring of laboratory and manufacturer data and significant surplus to current external quality assessment
- Percentiler and Flagger – low-cost, on-line monitoring of laboratory and manufacturer data and significant surplus to current external quality assessment
- Review
- Informatics External Quality Assurance (IEQA) Down Under: evaluation of a pilot implementation
- Informatics External Quality Assurance (IEQA) Down Under: evaluation of a pilot implementation
- Acknowledgment
- Acknowledgment