Diagnostic yield and clinical impact of prenatal whole exome sequencing (WES) – four-year single center experience
-
Katleen Janssens


Abstract
Objectives
This study evaluates the diagnostic yield of prenatal whole exome sequencing (WES) and its impact on outcome, such as termination of pregnancy (TOP) or neonatal management.
Methods
A retrospective analysis of more than 4 years of prenatal WES at a single genetic center was performed. Inclusion criteria included normal genome-wide deletion/duplication analysis and ≥1 ultrasound anomaly. Trio analysis was performed, filtering for de novo, compound heterozygous, homozygous, and hemizygous variants, complemented by a genome-wide phenotype-driven analysis.
Results
(Likely) pathogenic variants fitting the phenotype were identified in 36 of 171 cases (21.1 %), of which 19 were de novo, 14 autosomal recessive, one autosomal dominant, and 2 X-linked dominant. Median turnaround time was 16 days. Parents opted for TOP in 21 cases, three resulted in intrauterine death, 11 were carried to term and one was lost to follow-up. Among the neonates, the diagnosis led to optimized postnatal management in 8/11 (72.7 %), abstinence of care in two (18.2 %) and exclusion of syndromic disorders in one (9.1 %).
Conclusions
Our findings indicate that in 1/5 pregnancies with ultrasound anomalies and normal deletion/duplication analysis, a (likely) pathogenic variant explaining the phenotype can be identified. The high proportion (17/36 or 47.2 %) of inherited variants highlights the importance of WES for recurrence risk assessment.
Introduction
Prenatal whole exome sequencing (WES) represents a significant advancement in prenatal diagnostics as it considerably enhances our ability to establish genetic diagnoses in a prenatal setting [1], 2]. When used subsequent to genome-wide deletion/duplication analysis, WES has been shown to increase diagnostic yield by up to 15 % in fetuses with at least one ultrasound anomaly [3], [4], [5], [6] and even up to 21.8 % in fetuses with multiple anomalies [3]. Given the more limited phenotyping options during pregnancy compared to a postnatal setting, early genetic diagnoses are invaluable, as they aid parents in informed decision-making and are essential for optimal postnatal care planning.
However, implementing prenatal WES in a routine diagnostic setting has its challenges, as reported in our previous paper [7], including [1] the quality and quantity of the starting material [2]; the minimization of the turn-around time (TAT) [3]; the interpretation of variants despite limited phenotypic information and [4] the ethical perspective. Despite these hurdles, prenatal WES is an important evolution in the prenatal diagnostic landscape. This study expands on our previous paper which described the implementation and the first year of prenatal WES at our center [7]. Now we report on the diagnostic yield but in particular on the clinical impact of prenatal WES over a period of more than four years.
Materials and methods
The Center of Medical Genetics in Antwerp, one of eight genetic centers in Belgium, annually processes approximately 500 invasive prenatal samples. These samples are collected from the Antwerp University Hospital and several other hospitals in the region. Indications for invasive testing primarily include a known familial genetic disorder, a positive non-invasive prenatal test (NIPT) result or ultrasound anomalies. Ultrasound anomalies qualifying for invasive genetic testing included the presence of at least one major anomaly or more than one minor anomaly, according to institutional guidelines. At our center, the standard procedure for determining the genetic cause of fetal ultrasound anomalies, regardless of gestational age, involves a step-by-step approach. After extraction of genomic DNA from either amniotic fluid or chorion villi, a quantitative fluorescent PCR (QF-PCR) is conducted to rule out common aneuploidies (trisomies 13, 18, 21, sex chromosome aneuploidies) and triploidy. This step also includes checking for maternal cell contamination and confirming fetal identity by comparison of the fetal and maternal profile. Subsequently, CNV (copy number variant) detection is carried out using single nucleotide polymorphism (SNP) array (CMA) or shallow Whole Genome Sequencing (sWGS), both using the CNV Webstore tool [8] with a 400 kb resolution.
Following negative QF-PCR and CNV analysis, further options in fetuses with at least one ultrasound anomaly include single-gene testing (in case of a high suspicion of a particular disorder) or WES. The decision is made on a case-by-case basis during weekly multidisciplinary meetings.
The guidelines for prenatal WES were developed by the Belgian genetic centers and can be found at the website of the Belgian College of Genetics (www.college-genetics.be (V2021)). Variant classification is performed based on the ACMG guidelines [9] and only pathogenic (class V) and likely pathogenic (class IV) variants with a known effect on gene function and matching the fetal phenotype and the inheritance mode are communicated. Variants of uncertain significance (class III) are in principle not communicated, but exceptions can be made for variants for which further clinical exams (ultrasound, MRI, etc.) are recommended to refine variant classification, possibly leading to a genetic diagnosis (upgrade of the variant to class IV/V). The turn-around-time (TAT) was nationally set at eight weeks for ongoing pregnancies.
Library prep is performed on 50 ng of genomic DNA from uncultured or cultured amniocytes or chorion villi using the Twist Human Core Exome kit (Twist Bioscience, South San Francisco, CA, USA) according to the manufacturer’s instructions on a Hamilton STAR robot (Hamilton, Bonaduz, Switzerland). Twenty-four libraries are pooled equimolarly for sequencing on a NextSeq500 or NextSeq550 instrument with a 2 × 75 bp or 2 × 150 bp flow cell (Illumina, San Diego, CA, USA). WES data are analyzed using an in-house developed pipeline which considers only de novo, X-linked and recessive variants, either in a predefined panel (e.g., in case of a well-defined phenotype like skeletal dysplasia) or exome-wide [7], 10]. Additionally, an AI-driven decision-support software is applied to most samples to complement our pipeline with an independent phenotype-driven analysis based on Human Phenotype Ontology (HPO) terms (MOON, Invitae, San Francisco, CA, USA or Franklin, Genoox). This allows the identification of variants outside the panel (if applied) and of dominantly inherited variants. An independent analysis using single-molecule molecular inversion probes (smMIPs) is performed to detect sample swaps and to verify the family relations within each trio.
Results
In the period March 2020-August 2024, 171 WES were performed on prenatal cases that met the national criteria for exome sequencing in an ongoing pregnancy. Eighty-five of these analyses were performed on DNA extracted from uncultured amniotic cells (49.7 %), 58 from cultured amniotic cells (33.9 %) and 28 from chorion villi (16.4 %).
Since 2021, a steady increase in the number of prenatal WES analyses can be observed (see Table 1). This increase is not related to an increase in the total number of invasive tests, which is stable over this five-year period. Rather, it reflects an increase in the percentage of prenatal samples for which WES is requested (from 7.5 to 16.3 %).
Evolution of number of prenatal WES per year.
| Year | Number of invasive tests | Number of prenatal WES | % of invasive tests with WES request |
|---|---|---|---|
| 2020 (from March) | 280 | 21 | 7.5 |
| 2021 | 459 | 24 | 5.2 |
| 2022 | 451 | 29 | 6.4 |
| 2023 | 499 | 47 | 9.4 |
| 2024 (until August) | 326 | 53 | 16.3 |
For the majority of cases (158/171 or 91.8 %), an ‘open’ WES was performed, as the ultrasound anomalies did not allow the selection of a predefined gene panel. For the 13 remaining cases, the analysis was restricted to the skeletal dysplasia gene panel (n=10), the cerebral palsy panel (n=1) or a specific gene (n=2) for which no single gene test was available.
The turnaround time (TAT) was defined as the time between the finalization of the CMA/sWGS report and the WES report. If the request for exome sequencing was made any time after finalizing the CMA/sWGS report, the request date was taken as the start date. For cultured amniocytes, the time required for culturing was included in the calculation of the TAT, as to reflect the impact of this option. The general median TAT for all samples combined was 16 days. For uncultured amniocytes or chorion villi samples the median TAT was 15 and 15.5 days, respectively (range 5–62 and 4–26) (Figure 1). With the exception of two cases, all cases were reported within one month. For one of these cases (#37, not in table since no (likely) pathogenic variant was identified), the urgency of the analysis was no longer applicable since the parents did not consider a termination of pregnancy (TOP), but antenatal reporting of the results ensured optimal postnatal care planning. In the second case (#18), the result, a variant of uncertain significance (VUS) in the FLNA gene, did not align with the prenatal phenotype. Since the parents did not consider TOP, regardless of the genetic diagnosis, it was decided to perform more extensive prenatal phenotyping through a fetal MRI hoping it would allow to upgrade the FLNA variant to class IV in case of periventricular nodular heterotopia (PVNH). However, as the fetal MRI failed to show PVNH, a negative WES report was issued after 62 days. A postnatal brain MRI did reveal PVNH, resulting in the variant being upgraded to class IV. The requirement of culturing the amniocytes in case of insufficient yield increased the median TAT to 19 days (range 8–49); four samples had a TAT that surpassed four weeks. There was no significant difference between the TAT for a panel (median 14 days, range 6–25) or ‘open’ WES (median 16 days, range 4–62, p=0.30).
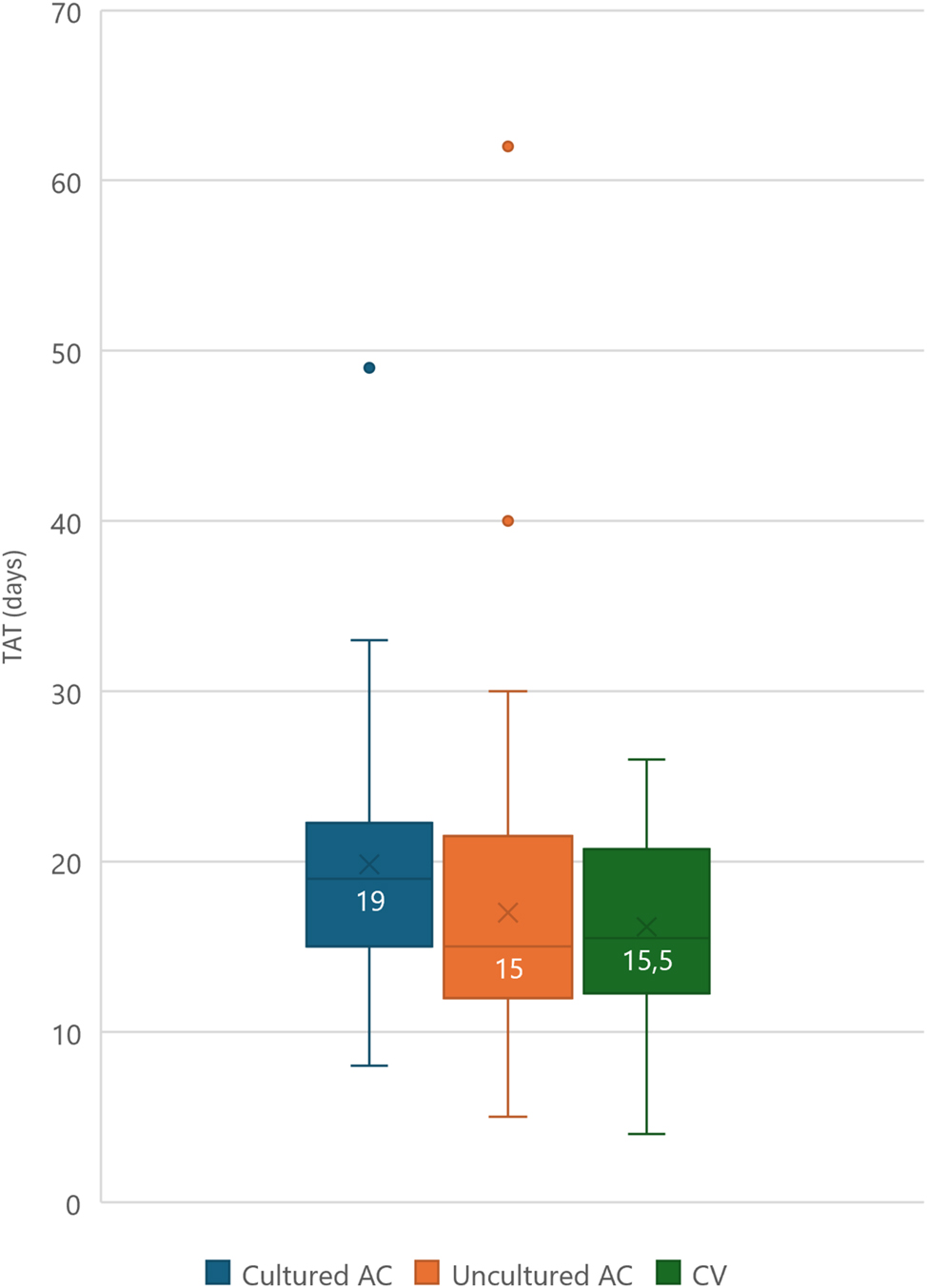
Turnaround time for (un)cultured AC or CV with median. AC, amniocentesis; CV, chorion villi sampling. Horizontal line represents median, x represents average.
Pathogenic or likely pathogenic variants were identified in 36/171 (21.1 %) cases: 19 de novo variants, 14 autosomal recessive, one autosomal dominant inherited from an affected father and two X-linked dominant variants inherited from an (in hindsight) affected mother (Table 2).
Prenatal cases for which WES demonstrated a likely pathogenic or pathogenic variant.
| Case no. | Prenatal phenotype | Gestational age at phenotyping (weeks+days) | Phenotypic group | Gene | Variant | Inheritance | Classification | Associated syndrome | Outcome | Overlap with prenatal phenotype in literature |
| 1a | Abnormal ears, bilateral talipes equinovarus | 22 + 5 | Multisystem | KMT2D | c.450G>A p. (Trp150*) |
AD–de novo | Path | Kabuki syndrome 1 (OMIM# 147920 | TOP | Yes + additional: pes equinovarus |
| 2a | FGR, oligodactyly left hand, hypoplastic ray right hand | 21 | Skeletal | FANCG | c.115C>T p. (Arg39*) |
AR–hom | Path | Fanconi anemia, comple- mentation group G (OMIM# 614082) | TOP | Yes |
| 3a | Edema, rocker bottom foot, retrognathia, abnormal thorax and ribs, increased nuchal translucency | 17 + 6 | Multisystem | CHRNA1 | c.548 A>G p. (Asp183 Gl y) |
AR–hom | Likely path | Multiple pterygium syn- drome, lethal type (OMIM# 253290) | MIU | Yes |
| 4a | Hydrops, ascites, hydrothorax | 29 + 6 | Hydrops | RIT1 | c.297T>A p. (Phe99Leu) |
AD–de novo | Path | Noonan syndrome 8 (OMIM# 615355) | MIU | Yes |
| 5a | Olivopontocerebellar hypoplasia, tetralogy of fallot, hypospadias | 21 + 4 | Multisystem | THOC6 | c.298T>A p. (Trp100Arg) |
AR–comp het (mat) | Path | Beaulieu-boycott-innes syndrome (OMIM# 613680) | TOP | Yes |
| THOC6 | c.700G>C p. (Val234Leu) |
AR–comp het (mat) | Path | |||||||
| THOC6 | c.824G>A p. (Gly275Asp) |
AR–comp het (mat) | Path | |||||||
| THOC6 | c.569G>A p. (Gly190Glu) |
AR–comp het (pat) | Path | |||||||
| 6a | Fetal akinesia, hypotonia, rocker-bottom feet, hydrops, hydrothorax, ascites | 15 + 1 | Multisystem | MUSK | c.2201G>T p. (Gly734Val) |
AR–hom | Likely path | Fetal akinesia defor- mation sequence 1 (OMIM# 208150) | TOP | Yes |
| 7 | Unilateral renal agenesis, cardiomegaly, moderate FGR, unilateral cleft lip, clinodactyly | 24 + 3 | Multisystem | FGFR2 | c.1945A>G p. (Arg649 Gl y) | AD-de novo | Likely path | Lacrimoauriculodentodigital syndrome (OMIM# 149730) | TOP | Yes + additional: FGR and cardiomegaly |
| 8 | Hygroma colli, hydronephrosis, hydrops fetalis, abnormality of the umbilical cord | 11 + 5 | Multisystem | PTPN11 | c.1529 A>C p. (Gln510Pro) | AD-de novo | Path | Rasopathy (OMIM# 163950) | TOP | Yes + additional: hydronephrosis and umbilical cord abnormality |
| 9 | Aplasia of the left hemidiaphragm, bilateral talipes equinovarus, aplasia/hypoplasia of the corpus callosum | 21 + 0 | Multisystem | FLNA | c.437T>C p. (Leu146Pro) | XLD-hemi (mat) | Likely path | Heterotopia, periventricular, (OMIM# 300049) | TOP | Yes + additional: diaphragmatic hernia |
| 10 | Corpus callosum agenesis, multicystic dysplastic kidney, absence of the septum pellucidum | 23 + 4 | Multisystem | SETBP1 | c.2606G>C p. (Ser869 Thr) | AD-de novo | Likely path | Schinzel-giedion midface retraction syndrome (OMIM# 269150) | NN | Yes |
| 11 | FGR, cerebral anomalies, VSD, pes cavus, anal anomalies + maternal symptoms: symmetrical hypomastia, cystic dilation of the 4th ventricle, enlarged posterior fossa, supratentorial hydrocephalus, Dandy-Walker syndrome + history of miscarriage | 26 + 5 | Multisystem | USP9X | c.4086+1G>T | XLD-het (mat) | Likely path | Intellectual developmental disorder, x-linked 99, syndromic, female-restricted (OMIM# 300968) | NN | Yes |
| 12 | Craniosynostosis, FGR, strawberry mark, microcephaly, hypotelorism | 31 + 5 | Multisystem | ADNP | c.3069_3072del p. (Arg1023Serfs*3) | AD-de novo | Path | Helsmoortel-van der Aa syndrome (OMIM# 615873) | NN | Yes |
| 13 | Hydrocephalus, ventriculomegaly, cavum septum pellucidum, low-set ears, edema, single umbilical artery, bilateral talipes equinovarus, aplasia of the phalanges of the toes | 28 | Multisystem | COL27A1 | c.2119C>T p. (Arg707*) | AR-hom | Path | Steel syndrome (OMIM# 615155). | TOP | Yes + additional: hydrocephaly and ventriculomegaly |
| 14 | Generalized edema, ascites, pericardial effusion, severe biventricular dysfunction with bilateral atrioventricular valve insufficiency | 24 + 5 | Cardiac | MYH7 | c.5401G>A p. (Glu1801Lys) | AD-de novo | Path | Complex phenotype characterized by laing Distal myopathy (OMIM# 160500), left ventricular noncompaction 5 (OMIM# 613426) and congenital fiber type disproportion (OMIM# 255310) | TOP | Yes |
| 15 | Bilateral cleft lip and palate, familial | 15 + 5 | Multisystem | ARHGAP29 | c.2330_2331del p. (Gln777Argfs*6) | AD-het (pat) | Path | Nonsyndromic cleft lip with or without cleft palate | NN | Yes |
| 16 | Hydrocephalus, aqueduct stenosis | 27 | CNS | MPDZ | c.5127_5128del p. (Tyr1709*) | AR-hom | Path | Hydrocephalus, congenital, 2, with or without brain or eye anomalies (OMIM# 615219) | TOP | Yes |
| 17 | Bell-shaped thorax, rhizomelia, hydrops | 12 | Skeletal | LBR | c.1748G>A p. (Arg583 Gln) | AR-hom | Path | Greenberg dysplasia (OMIM# 215140) | MIU | Yes |
| 18 | Bilateral clubfeet and arthrogryposis | 20 + 6 | CNS | FLNA | c.208G>A p. (Asp70Asn) | XLD- het (de novo) | Likely path | Heterotopia, periventricular (OMIM# 300049) | NN | Yes + additional: arthrogryposis |
| 19 | Hygroma colli, hydrops | 13 + 1 | Hydrops | PACS1 | c.607C>T p. (Arg203Trp) | AD-de novo | Path | Schuurs-hoeijmakers syndrome (OMIM# 615009) | TOP | Yes |
| 20 | Dolichocephaly, dilatation of the cisterna magna, bilateral kidney dysplasia, small facies, abnormal orbits, anhydramnios, arthrogryposis | 21 + 1 | Multisystem | EXOC3L2 | c.826del p. (Ala276Leufs*39) | AR-hom | Path | Brain malformation renal syndrome (OMIM# 620943) | TOP | Yes + additional: cardiac and nephrological phenotype |
| 21 | Hydrocephaly, polyhydramnnios | 17 + 4 | CNS | CHD7 | c.3422_3423del p. (Val1141Glyfs*27) | AD-de novo | Path | CHARGE syndrome (OMIM# 214800) | NN | Yes + additional: hydrocephaly |
| 22 | FGR and oligohydramnios | 23 + 0 | Multisystem | SAMD9 | c.3841 A>G p. (Arg1281 Gl y) | AD-de novo | Likely path | MIRAGE syndrome (OMIM# 617053) | NN | Yes |
| 23 | Unilateral cleft lip and palate, suspected overriding aorta | 18 + 2 | Multisystem | CHD7 | c.2504_2508del | AD-de novo | Path | CHARGE syndrome (OMIM# 214800) | TOP | Yes |
| 24 | Hydronephrosis, macrosomia, Blake’s pouch or vermis malformation | 29 + 0 | Multisystem | PIGL | c.438C>A p. (Phe146Leu) | AR-hom | Likely path | CHIME syndrome (OMIM# 280000) | NN | Yes |
| 25 | Shortening of long bones, narrow thorax, short ribs, abnormal position of sacrum, abnormal skull, bilateral hydronephrosis | 20 + 0 | Multisystem | FGFR3 | c.1118 A>G | AD-de novo | Path | Thanatophoric dysplasia, type I (OMIM# 187600) | TOP | Yes |
| 26 | Borderline ventriculomegaly (11 mm), abnormal cavum vergae, persistent left superior vena cava, slender aorta | 21 + 6 | Multisystem | SMAD6 | c.706C>T p. (Gln236*) | AR-hom | Likely path | Complex cardiovascular phenotype (PMID: 30,963,242) |
NN | Yes (cardiac phenotype) + additional: CNS malformations |
| 27 | FGR, single umbilical artery, dolichocephaly, underdeveloped left forearm | 23 + 2 | Multisystem | NIPBL | c.5483G>A p. (Arg1828 Gln) | AD-de novo | Path | Cornelia de lange syndrome 1 (OMIM# 122470) | TOP | Yes |
| 28 | FGR, echogenic bowel | 20 + 0 | Multisystem | SKIC3 | c.3486C>A p. (Cys1162*) | AR-hom | Path | Trichohepatoenteric syndrome 1 (OMIM# 222470) | TOP | Yes |
| 29 | Right-sided cleft lip and palate and VSD | 21 + 5 | Multisystem | CHD7 | c.2498+1G>A | AD-de novo | Likely path | CHARGE syndrome (OMIM# 214800) | NN | Yes |
| 30 | FGR, severe bilateral ventriculomegaly, atypical cavum septum pellucidum, corpus callosum agenesis | 28 + 5 | Multisystem | TUBA1A | c.1247G>T p. (Gly416Val) | AD-de novo | Likely path | Lissencephaly 3 (OMIM# 611603) | NN | Yes + additional: FGR |
| 31 | Severe cerebral anomalies | 23 + 0 | CNS | POMGNT2 | c.409C>T p. (Ala137Ser) | AR-hom | Likely path | Muscular dystrophy-dystroglycanopathy (congenital with brain and eye anomalies), type a, 8 (OMIM# 614830) |
TOP | Yes |
| 32 | Unilateral clubfoot, asymmetrical face, deep-set eyes | 20 + 0 | Multisystem | ASPM | c.4795C>T p. (Arg1599*) c.10147dupA p. (Thr3383Asnfs*3) | AR-comp het | Path | Microcephaly 5, primary, autosomal recessive (OMIM# 608716) | TOP | Yes + additional: clubfoot |
| 33 | Short femur, short humerus, thoracic hypoplasia | NA | Skeletal | COL1A1 | c.2498G>A p. (Gly833Asp) | AD-de novo | Likely path | Osteogenesis imperfecta, type 2 (OMIM# 166210) | TOP | Yes |
| 34 | Bowed femur, hypoplasia of lower limb, bilateral clubfeet, thoracic hypoplasia | 20 + 0 | Skeletal | COL1A2 | c.3034G>A p. (Gly1012Ser) | AD-de novo | Path | Osteogenesis imperfecta, type 2 (OMIM# 166210) | TOP | Yes |
| 35 | Hydrocephaly | 27 + 5 | CNS | PLG | c.269 A>T p. (Asp90Val) | AR-hom | Likely path | Plasminogen deficiency, type I (OMIM# 217090) | NN | Yes |
| 36 | Bilateral talipes equinovarus, fetal ascites, hydrothorax, ventriculomegaly | 20 + 3 | Multisystem | PIK3CA | c.1357G>A p. (Glu453Lys) | Somatic-de novo VAF 30 % | Path | Megalencephaly-capillary malformation-polymicrogyria syndrome (OMIM# 602501) | Lost to follow-up | Yes |
-
FGR, fetal growth restriction; VSD, ventricular septal defect; hemi, hemizygous; hom, homozygous; het, heterozygous; mat, maternal; pat: paternal; VAF, variant allele frequency; path, pathogenic; CNS, central nervous system; AD, autosomal dominant; AR, autosomal recessive; TOP, termination of pregnancy; NN, neonate; MIU, mors in utero. aPreviously published [7].
When grouped based on the organ system(s) involved (see also Mellis et al. 2022 [4]), the highest diagnostic yield was obtained in fetuses with skeletal anomalies (4/14 cases, 28.6 %), hydrops (2/7, 28.6 %), multisystem anomalies (24/87, 27.6 %) or central nervous system anomalies (5/28, 17.9 %). In 20 fetuses with a cardiac anomaly, only one diagnosis was obtained. No diagnosis was found in one case with a neoplasm and in 14 cases with isolated severe fetal growth restriction (Figure 2).
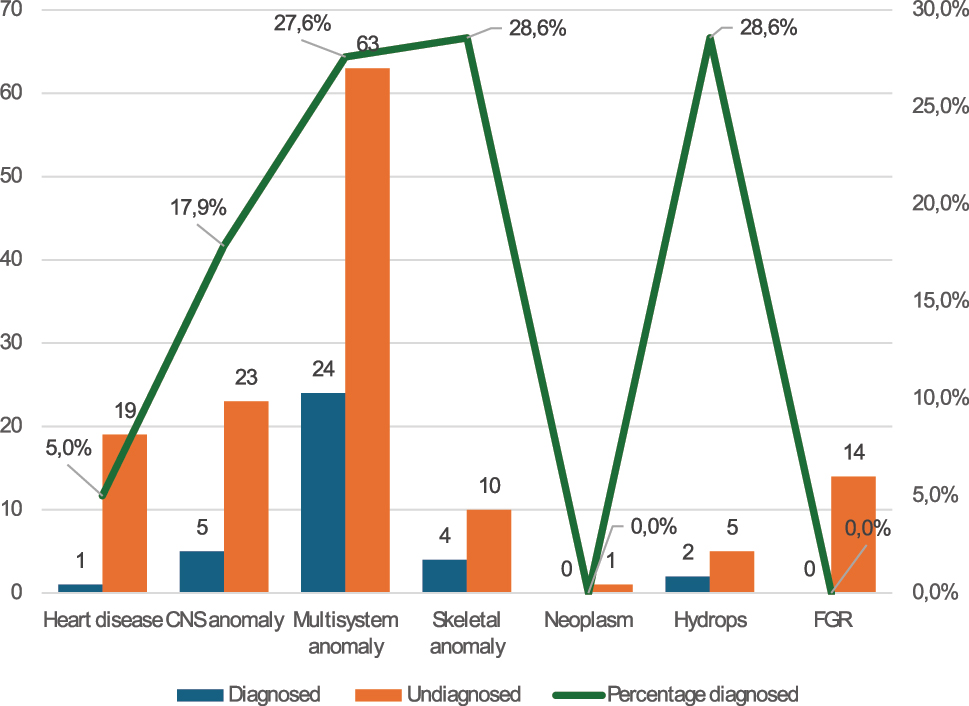
Distribution of cases according to involved organ system(s) and the results of the WES analysis. CNS, central nervous system; FGR, fetal growth restriction.
In the majority of cases the molecular diagnosis was made prenatally. In three cases (#11,#12 and #22) fetal distress or maternal complications led to the obstetric decision to induce a premature delivery, and the WES result was issued in the perinatal period. This eliminated the parental choice for TOP based on the genetic diagnosis.
The prenatal phenotype was consistent with the diagnosis and published prenatal phenotype in all 36 cases. However, in 11 fetuses (cases #1,#7,#8,#9,#13,#18,#20,#21,#26,#30 and #32), the ultrasound abnormalities remained partially unexplained, which might represent unreported or new prenatal presentations of the diagnosed syndrome or could be phenotypic presentations of a second (genetic) cause (Table 2).
Of the 36 cases in which a (likely) pathogenic variant was identified, parents chose to terminate the pregnancy in 21 cases (58.3 %). In three cases (8.3 %), intrauterine death occurred which could be explained by the genetic diagnoses: Greenberg dysplasia (case #17) [11], Noonan syndrome with hydrops (RIT1, case #4) [12] and lethal type multiple pterygium syndrome (case #3) [13], [14], [15], [16]. 11 cases (30.6 %) were brought to term and one case was lost to follow-up (2.8 %). Table 3 provides an overview of the prenatal and postnatal phenotypes of all cases brought to term. For these cases, having a genetic diagnosis was relevant in several ways. In one case (#15), we identified a non-syndromic cause of cleft lip (ARHGAP29 mutation). The exclusion of a syndromic disorder associated with the observed bilateral cleft lip and palate and the fact that it was paternally inherited, gave the parents confidence to proceed with the pregnancy. In 8 cases (72.7 %; cases #10,#11,#12,#18,#21,#26,#29 and #36, details in table 3) the diagnosis facilitated optimal postnatal management, enabling immediate screening and treatment of health problems associated with the diagnosed syndrome. In two cases (18.2 %, cases #22 and #24), the decision to abstain from care was made based on a genetic diagnosis with poor prognoses, i.e. MIRAGE and CHIME syndrome. MIRAGE syndrome is characterized by myelodysplasia, infections, growth restriction, adrenal insufficiency, genital abnormalities and enteropathy. Most patients die in infancy or early childhood, with a median age at death of three years [17], [18], [19], [20]. CHIME syndrome is a rare ectodermal dysplasia syndrome characterized by ocular colobomas, heart defects, ichthyosiform dermatosis, intellectual disability, ear anomalies (e.g. conductive hearing loss) and epilepsy, with a reported poor prognosis [21], 22].
Prenatal and postnatal phenotypes of cases brought to term.
| Case no. | Prenatal phenotype | Timing of diagnosis | Gene/syndrome | Gestational age at birth | Biometry (W, L, HC) | Postnatal phenotype | Impact of diagnosis | Current status |
| 10 | Corpus callosum agenesis, multicystic dysplastic kidney, absence of the septum pellucidum, later in the pregnancy also bilateral hydronephrosis and polyhydramnios | Prenatal |
SETBP1
Schinzel-giedion midface retraction syndrome (OMIM# 269150) |
37 w+3 d induction due to unstable position and polyhydramnios | 3,300 g, 49 cm, 33.5 cm | Consistent with diagnosis: Dysmorphisms (prominent forehead, temporal narrowing, large eyes, infraorbital crease, upturned nasal tip, wide mouth, low-set ears, single palmar crease on the right hand and mild rocker-bottom feet), pelvic-ureteropelvic junction obstruction, hydronephrosis with recurrent urinary tract infections, chronic kidney disease stage III, blindness, bilateral sensorineural hearing loss, ASD with PDA and peripheral pulmonary stenosis, epilepsy, severe GDD[23] | Planning of optimal postnatal management: Immediate postnatal screening for associated health problems and follow-up (as recommended for the syndrome) [24] Advanced care planning with DNR decision (DNR2). | >2 y |
| 11 | FGR, cerebral anomalies (moderate hydrocephalus with the presence of cavum vergae), VSD, pes cavus, anal anomalies + maternal symptoms: Symmetrical hypomastia, cystic dilation of the 4th ventricle, enlarged posterior fossa, supratentorial hydrocephalus, Dandy-Walker syndrome, arterial hypertension | Postnatal due to prematurity |
USP9X
Intellectual developmental disorder, X-linked 99, syndromic, female-restricted (OMIM# 300968) |
30 w+3 d section due to suboptimal CTG and maternal hypertension | 1,050 g, 35 cm, 27 cm | Consistent with diagnosis: GDD, visual disturbances (congenital nystagmus, electroretinogram shows signs of retinal dysfunction), ventriculomegaly, apical VSD and PDA + maternal symptoms | Early diagnosis of underlying genetic cause of health problems in neonate and mother. Immediate postnatal screening for associated health problems and follow-up (as recommended for the syndrome) | >2 y |
| 12 | FGR, strawberry mark of the skull, microcephaly, hypotelorism, suspicion of craniosynostosis [25] | Postnatal due to prematurity |
ADNP
Helsmoortel-van der Aa syndrome (OMIM# 615873) |
35 w+4 d section due to premature contractions and fetal distress | 1,575 g, 40.5 cm, 28.6 cm | Consistent with diagnosis: Autism spectrum disorder, GDD, dysmorphisms (high prominent forehead, deep-set eyes, small nose with small alae nasi) | Postnatal screening for associated health problems and follow-up by pediatric neurologist (as recommended for the syndrome) [26] | >2 y |
| 15 | Bilateral cleft lip and palate, familial | Prenatal |
ARHGAP29
Nonsyndromic cleft lip with or without cleft palate |
37 w+3 d | 3,335 g, 52 cm, 37.4 cm | Consistent with diagnosis: Bilateral cleft lip and palate | Exclusion of syndromic cause, prenatal counseling on surgery. | 20 m |
| 18 | Bilateral clubfeet and arthrogryposis | Prenatal: class 3 Postnatal based on phenotype: Class 4 |
FLNA
Heterotopia, periventricular (OMIM# 300049) |
39 w+1 d section due to multiple congenital anomalies | 2,870 g, 48 cm, 34.5 cm | Consistent with diagnosis: Pulmonary valve stenosis, partially absent corpus callosum, periventricular nodular heterotopia, megacisterna magna, GDD, bilateral clubfeet, hyperlaxity, hip dysplasia, fragile skin Additional finding: Arthrogryposis and hypotonia |
Planning of optimal postnatal management: immediate postnatal screening for associated health problems and follow-up (as recommended for the syndrome) [27], 28] | 18 m |
| 21 | Hydrocephaly, polyhydramnios | Prenatal |
CHD7
CHARGE syndrome (OMIM# 214800) |
39 w+3 d section due to non-progressing labor | 3,170 g, 52 cm, 39 cm | Consistent with diagnosis: asymmetrical face, PDA for which ductus clipping was performed, biventricular hypertrophy, hearing loss, hypoPTH and hypocalcemia, small cerebellum, small internal auditory canal, small corpus callosum, small brainstem, bilateral deafness due to abnormal anatomy, acquired vocal cord paralysis with absent swallowing function, continuous tube feeding via jejunostomy, arytenoid hypertrophy with obstructive breathing, requiring continuous positive airway pressure | Prenatal counseling with pediatric neurologist and immediate post-natal screening for associated health problems and follow-up (as recommended for the syndrome) [29], 30] | 9 m |
| 22 | FGR and oligohydramnios | Postnatal due to prematurity |
SAMD9
MIRAGE syndrome (OMIM# 617053) |
28 w, section due to FGR, abnormal CTG and oligohydramnios | 670 g, 34.5 cm, 23 cm | Consistent with diagnosis: prematurity, adrenal insufficiency, dysmorphisms (low-set and posteriorly rotated ears, camptodactyly, micrognathia, genital abnormality), thrombocytopenia | DNR3 decision due to instability and poor prognosis | Died after 4 d |
| 24 | Hydronephrosis, macrosomia, Blake’s pouch or vermis malformation | Prenatal: class 3 Postnatal based on phenotype: Class 4 |
PIGL
CHIME syndrome (OMIM# 280000) |
38+3 d, section due to macrosomia | 4,025 g, NA, 35.2 cm | Consistent with diagnosis: vermis hypoplasia, macrosomia, heart defect, cleft palate, dysplastic ears, puffy hands and feet, hypoplasia of the corpus callosum and olfactory sulci, epilepsy, tetralogy of fallot, suspected Hirschsprung’s disease, bilateral hydronephrosis, short thorax, widely spaced nipples, low implanted ears, short and thickened neck | DNR3 decision after increasing cardiorespiratory failure, poor expected neurological outcome, and the need for at least four surgical interventions for long-term survival | Died<1 m |
| 26 | Borderline ventriculomegaly (11 mm), abnormal cavum vergae, persistent left superior vena cava, slender aorta | Prenatal |
SMAD6
Complex cardiovascular phenotype [31], 32] |
39 w+1 d | 3,020 g, 48 cm, 32.5 cm | Consistent with diagnosis: Pulmonary valve stenosis, ASD type 2, PDA, GDD, peripheral hypertonia, central hypotonia, partial corpus callosum dysgenesis, white matter abnormalities with bilateral periventricular diffusion restriction and bilateral (limited) subdural hematoma, dysmorphisms (brachycephaly, downslanting palpebral fissures, low-set ears, long philtrum, thin upperlip) | Planning of optimal postnatal management: Immediate postnatal screening for associated health problems incl. Craniosynostosis or radio-ulnar synostosis with cardiac, transfontanellar ultrasound and renal ultrasound [31], 32] | 7 m |
| 29 | Right-sided cleft lip and palate and VSD | Prenatal |
CHD7
CHARGE syndrome (OMIM# 214800) |
39 w | 2,670 g, 50 cm, 34.5 cm | Consistent with diagnosis: Severe cardiac abnormality | Prenatal planning of follow-up and immediate post-natal screening for associated health problems according to guidelines [29], 30], DNR decision after determining that the heart defect was too complex for surgery. | Died after 3 weeks |
| 36 | Hydrocephaly | Prenatal: Class 3 Postnatal based on phenotype: Class 4 |
PLG
Plasminogen deficiency, type I (OMIM# 217090) |
39 w | 3,080 g, NA, 34.5 cm | Consistent with diagnosis: Supratentorial hydrocephalus and hypoplastic aspect of the cerebellum without a classic Dandy-Walker malformation, ligneous conjunctivitis bilateral, recurrent respiratory infections | Immediate screening for associated health problems and optimal treatment (as recommended for the syndrome), screening for inclusion in clinical trial for plasminogen deficiency [33] | >4 y |
-
ASD, atrial septal defect; CTG, cardiotocography; DNR, do not resuscitate; EEG, electroencephalogram; GDD, global developmental delay; FGR, fetal growth restriction; PDA, patent ductus arteriosus; VSD, ventricular septal defect; NA, not available; d, days; w, weeks; m, months; y, years.
Discussion
This study reports on the experience with prenatal WES over a period of more than four years, analyzing 171 cases and yielding 36 (21.1 %) (likely) diagnoses. This yield is higher than what is generally reported in the literature [3], [4], [5], [6], which can probably be explained by the fact that the majority of the included cases had multiple anomalies. Our results confirm that the diagnostic yield varies greatly depending on the organ system involved; it is highest in fetuses with hydrops and skeletal or multisystem anomalies [3], 4], 6] and lowest in case of cardiac anomalies or isolated fetal growth restriction. While this information can assist clinicians in deciding whether to recommend an exome analysis, it should be kept in mind that the fetal phenotype is subject to change throughout pregnancy, and that prenatal phenotyping has its limitations. In this context, one could advocate for performing prenatal WES in all pregnancies exhibiting ultrasound anomalies, even isolated minor ones, as the selection of candidates for prenatal WES relies on the limited information available from the ultrasound at a specific gestational age [4]. However, given the cost of WES and depending on the healthcare system, information on the diagnostic yield associated with different types of ultrasound anomalies is of great help to determine which pregnancies would benefit the most from WES. One could also argue that exome sequencing in fetuses without ultrasound anomalies is warranted, as a phenotype might develop later in pregnancy or even only postnatally (in case of e.g. neurodevelopmental disorders). Although this has become common practice in some countries [34], we do not foresee this to happen in Belgium in the near future, nor is it currently supported by the International Society for Prenatal Diagnosis [1]. First, there is a risk of a procedure-related miscarriage. Second, there is no such thing as the (genetically) perfectly healthy baby. Even if we screen for all known variants in currently known genes associated with early-onset severe disorders, we will still miss numerous diagnoses since [1] the causal variant can reside in a non-coding or other region inaccessible with exome sequencing [2]; not all genes that have been associated with disease are known [3]; not all types of variants can be detected with the current prenatal diagnostic tests (e.g. inversions or balanced translocations interrupting a gene, copy number variants between ∼50 bp and 400 kb) [35]. Therefore, we deem it more useful to improve the currently available technologies or implement new technologies to increase the diagnostic yield in fetuses with ultrasound anomalies that are likely to be genetic in origin. We do observe a steady increase in the percentage of prenatal samples for which WES is requested in our center (from 7.5 to 16.3 %). This growth is largely attributed to the broader spectrum of indications for prenatal WES.
Prenatal WES is performed to allow informed decision-making for expecting parents. In 21/36 (58.3 %) cases the parents opted for a TOP because of ultrasound anomalies in combination with the genetic diagnosis, while in 11/36 cases the parents and medical team had the opportunity to prepare optimal postnatal care. This corresponds to the results of a similar study in which in 48.9 % of cases with a molecular diagnosis parents decided to terminate the pregnancy [3].
In 2023, we reported on our experience of the first year of prenatal WES in our center [7]. In that paper, we referred to the many hurdles that had to be overcome to offer WES in the prenatal setting including [1] the quality and quantity of the starting material [2]; the short TAT [3]; the interpretation of variants and [4] the ethical perspective. Well into our fifth year of offering prenatal WES, we are in the position to comment on these hurdles [1]. In our hands, DNA extracted directly from amniotic fluid or chorion villi is of high quality and maternal cell contamination is rarely an issue. As the input for our library prep is only 50 ng, the majority (66,1 %) of our exome analyses can be performed on DNA extracted from uncultured cells, reducing the TAT with a couple of days (median 19 vs. 15 days) [2]. With biweekly sequencing runs, the median turnaround time (TAT) is 16 days, significantly shorter than the national threshold of 8 weeks. This enables parents to make timely decisions regarding the continuation or termination of their pregnancy. Even when WES is requested in the third trimester, results are available before birth, facilitating the implementation of appropriate neonatal management [3]. Our variant detection and filtering pipeline has been optimized to minimize the number of variants that need manual curation. This again reflects positively on the TAT and drastically reduces the number of variants of unknown significance and incidental findings that require discussion in a multidisciplinary team meeting. On a total of 171 cases, only 2 ‘hot’ class 3 variants (1.2 %; data not shown) and two incidental findings (1.2 %; data not shown) have been reported, even though most analyses were exome-wide and not panel-based. While variant interpretation remains challenging due to the limited prenatal phenotypic information, our pipeline ensures that clinically relevant variants are accurately identified [4]. From an ethical perspective, it is paramount to balance the benefits of prenatal diagnosis against the potential psychological impact on expectant parents. The discovery of incidental findings, in particular, can add complexity to the counseling. However, with our low number of incidental findings and adequate pre- and post-test counseling, we demonstrate prenatal WES to be beneficial to parents in case of ultrasound anomalies.
Obtaining a diagnosis is not only of relevance in the context of the ongoing pregnancy, but also for future pregnancies. In case of a de novo variant, the recurrence risk is low, but not zero, as low-level parental mosaicism cannot be excluded; an invasive test followed by targeted sequencing of the familial variant can bring reassurance to parents. In case of an inherited variant, the recurrence risk can be as high as 50 % and preimplantation genetic testing can be offered, reducing the risk tremendously. In our cohort, there was a high recurrence risk (25–50 %) in 17/36 (47.2 %) of diagnoses, underscoring the impact of WES on family planning.
In three out of 36 positive cases (8.3 %), the diagnosis would have been missed by using a panel-based approach like the Genomics England Fetal Anomalies panel (2,187 genes; version v5.0), demonstrating that a) it remains difficult to keep panels up to date, as new gene-disease correlations are published on a regular basis and b) the described postnatal phenotype might not warrant the inclusion of a particular gene in the Fetal Anomalies panel, since no fetal phenotype has been described thus far. For the disorders detected in these three cases, there was low or no evidence for a prenatal phenotype, explaining why the genes (COL27A1, SMAD6 and SKIC3) are not included in the Genomics England Fetal Anomalies panel (Supplementary Table S1). By opting for an open WES, we contribute in defining the fetal phenotype of known disorders while, with optimized bioinformatic pipelines, keeping the number of incidental findings and variants of unknown significance low.
The use of Moon, Franklin or any other AI-based software for HPO-based prioritization of variants is complementary to our own pipelines for two reasons [1]; it can prioritize variants in novel genes in case of a panel-based approach and [2] it will return dominantly inherited variants that fit the phenotype but can have a variable presentation. In our cohort of 36 positive cases, three (8.3 %) carried a dominantly inherited variant, which would have been missed in 2/3 cases when using the standard filtering only (for de novo, compound heterozygous, homozygous and hemizygous variants).
The interpretation and classification of variants requires detailed prenatal phenotyping. Achieving this, demands highly trained gynecologists and excellent communication between the genetics and gynecology departments. In our center, all cases are discussed during a weekly multidisciplinary prenatal meeting, ensuring the best diagnostic approach and facilitating the interpretation and discussion of genetic testing results when necessary.
A limitation of our study is that neonates born from pregnancies without a genetic diagnosis are not routinely re-evaluated by a geneticist after birth [1], 2]. The decision to do so is left to the discretion of the parents or pediatrician at the hospital where the baby is born, which is often different from the hospital where pregnancy follow-up and the request for genetic testing took place. This lack of routine re-evaluation prevents us from identifying additional phenotypic features and determining if reanalysis of WES data is necessary. The strength of this study lies in the reporting on the diagnostic yield of a comprehensive, unbiased cohort from a single institution over an extended period of time, enabling a broad and thorough evaluation of the performance of prenatal WES.
Conclusions
Our findings indicate that in one out of five pregnancies with ultrasound anomalies and normal deletion/duplication analysis, a (likely) pathogenic variant explaining the phenotype can be identified. The short turnaround time allows for timely decisions regarding the ongoing pregnancy and neonatal management. In addition, we demonstrated an 8.3 % higher diagnostic yield by performing an ‘open’ WES compared to a gene panel like the Genomics England Fetal Anomalies panel. Furthermore, 47.2 % (17/36) of the identified variants were inherited, highlighting the importance of this analysis for future family planning, as the recurrence risk can be as high as 50 %.
Acknowledgments
The authors wish to thank the patients for their willingness to participate in this study. In addition, we would like to acknowledge our colleagues from the Center for Medical Genetics, Gynecology and Pediatrics department of the Antwerp University Hospital for sharing their expertise. Lastly, we would like to thank all laboratory staff for their dedicated work, ensuring the short turnaround times.
-
Research ethics: Ethical approval for retrospective studies is not required by the responsible Ethics Review Committee.
-
Informed consent: Written and signed informed consent was obtained from the participating patients/their legal guardian and are stored in their medical files.
-
Author contributions: Lab work and interpretation of WES data K.J.2; conceptualization, methodology, writing original draft preparation, writing review and editing, K.J.1, M.D.R., J.M., B.B. and K.J.2; data curation, K.J.1, M.D.R., J.M., B.B. and K.J.2. All authors have read and agreed to the published version of the manuscript. All authors have accepted responsibility for the entire content of this manuscript and approved its submission.
-
Use of Large Language Models, AI and Machine Learning Tools: None declared.
-
Conflict of interest: The authors state no conflict of interest.
-
Research funding: None declared.
-
Data availability: Data available on request due to privacy/ethical restrictions.
References
1. Van den Veyver, IB, Chandler, N, Wilkins-Haug, LE, Wapner, RJ, Chitty, LS, ISPD Board of Directors. International society for prenatal diagnosis updated position statement on the use of genome-wide sequencing for prenatal diagnosis. Prenat Diagn 2022;42:796–803. https://doi.org/10.1002/pd.6157.Search in Google Scholar PubMed PubMed Central
2. Monaghan, KG, Leach, NT, Pekarek, D, Prasad, P, Rose, NC. The use of fetal exome sequencing in prenatal diagnosis: a points to consider document of the American college of medical genetics and genomics (ACMG). Genet Med 2020;22:675–80. https://doi.org/10.1038/s41436-019-0731-7.Search in Google Scholar PubMed
3. Diderich, KEM, Bruggenwirth, HT, Joosten, M, Thurik, F, Mijalkovic, J, Polak, M, et al.. The high diagnostic yield of prenatal exome sequencing followed by 3400 gene panel analysis in 629 ongoing pregnancies with ultrasound anomalies. Prenat Diagn 2024;44:1444–50. https://doi.org/10.1002/pd.6676.Search in Google Scholar PubMed
4. Mellis, R, Oprych, K, Scotchman, E, Hill, M, Chitty, LS. Diagnostic yield of exome sequencing for prenatal diagnosis of fetal structural anomalies: a systematic review and meta-analysis. Prenat Diagn 2022;42:662–85. https://doi.org/10.1002/pd.6115.Search in Google Scholar PubMed PubMed Central
5. Lord, J, McMullan, DJ, Eberhardt, RY, Rinck, G, Hamilton, SJ, Quinlan-Jones, E, et al.. Prenatal exome sequencing analysis in fetal structural anomalies detected by ultrasonography (PAGE): a cohort study. Lancet 2019;393:747–57. https://doi.org/10.1016/s0140-6736(18)31940-8.Search in Google Scholar PubMed PubMed Central
6. Petrovski, S, Aggarwal, V, Giordano, JL, Stosic, M, Wou, K, Bier, L, et al.. Whole-exome sequencing in the evaluation of fetal structural anomalies: a prospective cohort study. Lancet 2019;393:758–67. https://doi.org/10.1016/s0140-6736(18)32042-7.Search in Google Scholar
7. Janicki, E, De Rademaeker, M, Meunier, C, Boeckx, N, Blaumeiser, B, Janssens, K. Implementation of exome sequencing in prenatal diagnostics: chances and challenges. Diagnostics 2023;13. https://doi.org/10.3390/diagnostics13050860.Search in Google Scholar PubMed PubMed Central
8. Vandeweyer, G, Reyniers, E, Wuyts, W, Rooms, L, Kooy, RF. CNV-WebStore: online CNV analysis, storage and interpretation. BMC Bioinf 2011;12:4. https://doi.org/10.1186/1471-2105-12-4.Search in Google Scholar PubMed PubMed Central
9. Richards, S, Aziz, N, Bale, S, Bick, D, Das, S, Gastier-Foster, J, et al.. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med 2015;17:405–24. https://doi.org/10.1038/gim.2015.30.Search in Google Scholar PubMed PubMed Central
10. Vandeweyer, G, Van Laer, L, Loeys, B, Van den Bulcke, T, Kooy, RF. VariantDB: a flexible annotation and filtering portal for next generation sequencing data. Genome Med 2014;6:74. https://doi.org/10.1186/preaccept-1622561303131056.Search in Google Scholar
11. Gregersen, PA, McKay, V, Walsh, M, Brown, E, McGillivray, G, Savarirayan, R. A new case of Greenberg dysplasia and literature review suggest that Greenberg dysplasia, dappled diaphyseal dysplasia, and astley-kendall dysplasia are allelic disorders. Mol Genet Genom Med 2020;8:e1173. https://doi.org/10.1002/mgg3.1173.Search in Google Scholar PubMed PubMed Central
12. Miceikaite, I, Bak, GS, Larsen, MJ, Kristiansen, BS, Torring, PM. Prenatal cases with rare RIT1 variants causing severe fetal hydrops and death. Clin Case Rep 2021;9:e04507. https://doi.org/10.1002/ccr3.4507.Search in Google Scholar PubMed PubMed Central
13. de Die-Smulders, CE, Vonsée, HJ, Zandvoort, JA, Fryns, JP. The lethal multiple pterygium syndrome: prenatal ultrasonographic and postmortem findings; a case report. Eur J Obstet Gynecol Reprod Biol 1990;35:283–9. https://doi.org/10.1016/0028-2243(90)90175-z.Search in Google Scholar PubMed
14. Zhuang, J, Wang, J, Luo, Q, Zeng, S, Chen, Y, Jiang, Y, et al.. Case report: novel compound heterozygous variants in CHRNA1 gene leading to lethal multiple pterygium syndrome: a case report. Front Genet 2022;13:964098. https://doi.org/10.3389/fgene.2022.964098.Search in Google Scholar PubMed PubMed Central
15. Vogt, J, Harrison, BJ, Spearman, H, Cossins, J, Vermeer, S, ten Cate, LN, et al.. Mutation analysis of CHRNA1, CHRNB1, CHRND, and RAPSN genes in multiple pterygium syndrome/fetal akinesia patients. Am J Hum Genet 2008;82:222–7. https://doi.org/10.1016/j.ajhg.2007.09.016.Search in Google Scholar PubMed PubMed Central
16. de Die-Smulders, CE, Schrander-Stumpel, CT, Fryns, JP. The lethal multiple pterygium syndrome: a nosological approach. Genet Counsel 1990;1:13–23.Search in Google Scholar
17. Narumi, S, Amano, N, Ishii, T, Katsumata, N, Muroya, K, Adachi, M, et al.. SAMD9 mutations cause a novel multisystem disorder, MIRAGE syndrome, and are associated with loss of chromosome 7. Nat Genet 2016;48:792–7. https://doi.org/10.1038/ng.3569.Search in Google Scholar PubMed
18. Tanase-Nakao, K, Olson, TS, Narumi, S. 2020 Nov 25. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from: https://www.ncbi.nlm.nih.gov/books/NBK564655/.Search in Google Scholar
19. Suntharalingham, JP, Ishida, M, Del Valle, I, Stalman, SE, Solanky, N, Wakeling, E, et al.. Emerging phenotypes linked to variants in SAMD9 and MIRAGE syndrome. Front Endocrinol 2022;13:953707. https://doi.org/10.3389/fendo.2022.953707.Search in Google Scholar PubMed PubMed Central
20. Panaitescu, AM, Huluță, I, Gorecki, GP, Cima, LN, Voiculescu, VM, Nedelea, FM, et al.. Prenatal features of MIRAGE syndrome-case report and review of the literature. Children 2024;11. https://doi.org/10.3390/children11030310.Search in Google Scholar PubMed PubMed Central
21. Winter-Paquette, LM, Al Suwaidi, HH, Sajjad, Y, Bricker, L. Congenital diaphragmatic hernia and early lethality in PIGL-Related disorder. Eur J Med Genet 2022;65:104501. https://doi.org/10.1016/j.ejmg.2022.104501.Search in Google Scholar PubMed
22. Zunich, J, Esterly, N. Chime syndrome (zunich syndrome). In: Ruggieri, M, Pascual-Castroviejo, I, Di Rocco, C, editors. Neurocutaneous Disorders Phakomatoses and Hamartoneoplastic Syndromes. Vienna: Springer Vienna; 2008:949–55 pp.10.1007/978-3-211-69500-5_64Search in Google Scholar
23. Lehman, AM, McFadden, D, Pugash, D, Sangha, K, Gibson, WT, Patel, MS. Schinzel-giedion syndrome: report of splenopancreatic fusion and proposed diagnostic criteria. Am J Med Genet 2008;146a:1299–306. https://doi.org/10.1002/ajmg.a.32277.Search in Google Scholar PubMed
24. Duis, J, van Bon, BWM. 2024 Mar 7. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2024. Available from: https://www.ncbi.nlm.nih.gov/books/NBK601394/ Search in Google Scholar
25. Rosenblum, J, Van der Veeken, L, Aertsen, M, Meuwissen, M, Jansen, AC. Abnormal fetal ultrasound leading to the diagnosis of ADNP syndrome. Eur J Med Genet 2023;66:104855. https://doi.org/10.1016/j.ejmg.2023.104855.Search in Google Scholar PubMed
26. Van Dijck, A, Vandeweyer, G, Kooy, F. ADNP-related disorder. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 2016:1993–2025 pp. Available from: https://www.ncbi.nlm.nih.gov/books/NBK355518/.Search in Google Scholar
27. Lange, M, Kasper, B, Bohring, A, Rutsch, F, Kluger, G, Hoffjan, S, et al.. 47 patients with FLNA associated periventricular nodular heterotopia. Orphanet J Rare Dis 2015;10:134. https://doi.org/10.1186/s13023-015-0331-9.Search in Google Scholar PubMed PubMed Central
28. Chen, MH, Walsh, CA. FLNA deficiency. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 2002:1993–2025 pp. Available from: https://www.ncbi.nlm.nih.gov/sites/books/NBK1213/.Search in Google Scholar
29. Trider, CL, Arra-Robar, A, van Ravenswaaij-Arts, C, Blake, K. Developing a CHARGE syndrome checklist: health supervision across the lifespan (from head to toe). Am J Med Genet 2017;173:684–91. https://doi.org/10.1002/ajmg.a.38085.Search in Google Scholar PubMed
30. de Geus, CM, Free, RH, Verbist, BM, Sival, DA, Blake, KD, Meiners, LC, et al.. Guidelines in CHARGE syndrome and the missing link: cranial imaging. Am J Med Genet C Semin Med Genet 2017;175:450–64. https://doi.org/10.1002/ajmg.c.31593.Search in Google Scholar PubMed PubMed Central
31. Kloth, K, Bierhals, T, Johannsen, J, Harms, FL, Juusola, J, Johnson, MC, et al.. Biallelic variants in SMAD6 are associated with a complex cardiovascular phenotype. Hum Genet 2019;138:625–34. https://doi.org/10.1007/s00439-019-02011-x.Search in Google Scholar PubMed
32. Luyckx, I, Walton, IS, Boeckx, N, Van Schil, K, Pang, C, De Praeter, M, et al.. Homozygous SMAD6 variants in two unrelated patients with craniosynostosis and radioulnar synostosis. J Med Genet 2024;61:363–8. https://doi.org/10.1136/jmg-2023-109151.Search in Google Scholar PubMed PubMed Central
33. Shapiro, AD, Menegatti, M, Palla, R, Boscarino, M, Roberson, C, Lanzi, P, et al.. An international registry of patients with plasminogen deficiency (History). Haematologica 2020;105:554–61. https://doi.org/10.3324/haematol.2019.241158.Search in Google Scholar PubMed PubMed Central
34. Vaknin, N, Azoulay, N, Tsur, E, Tripolszki, K, Urzi, A, Rolfs, A, et al.. High rate of abnormal findings in prenatal exome trio in low risk pregnancies and apparently normal fetuses. Prenat Diagn 2022;42:725–35. https://doi.org/10.1002/pd.6077.Search in Google Scholar PubMed
35. Brabbing-Goldstein, D, Bazak, L, Ruhrman-Shahar, N, Lidzbarsky, GA, Orenstein, N, Lifshiz-Kalis, M, et al.. Potentially missed diagnoses in prenatal versus postnatal exome sequencing in the lack of informative phenotype: lessons learned from a postnatal cohort. Prenat Diagn 2024;44:1423–34. https://doi.org/10.1002/pd.6659.Search in Google Scholar PubMed
Supplementary Material
This article contains supplementary material (https://doi.org/10.1515/jpm-2025-0302).
© 2025 the author(s), published by De Gruyter, Berlin/Boston
This work is licensed under the Creative Commons Attribution 4.0 International License.
Articles in the same Issue
- Frontmatter
- Commentary
- The IAPM New Delhi 2025 Declaration on Svadharma and Professional Responsibility
- Reviews
- The use of laser therapy in fetal medicine: a narrative review
- Nutriepigenomics in perinatal medicine: maternal nutrition as a modulator of fetal gene expression and long-term health
- Original Articles – Obstetrics
- Community births in the United States, 2016–2024: post-pandemic patterns across racial and ethnic groups
- Preeclampsia is associated with increased NT-proBNP and altered lipid profiles in umbilical cord blood: a propensity score-matched analysis
- Ureaplasma parvum detected in umbilical cord tissues diagnosed with funisitis associated with adverse pregnancy outcomes and neonatal pneumonia
- Implementation of a universal low-dose aspirin protocol for the prevention of preeclampsia in a federally qualified health center
- Comparative evaluation of manual and VOCAL methods for amniotic sac volume measurement in early pregnancy
- McDonald vs. Shirodkar cerclage for the prevention of preterm birth in patients with obesity
- The incidence and outcomes of perinatal asphyxia in spontaneous extreme preterm birth: a retrospective cohort study
- Comparative effectiveness of oxytocin, carbetocin, and tranexamic acid for postpartum hemorrhage prevention in cesarean deliveries: a prospective cohort analysis
- Risk factors for primary cesarean delivery in women with gestational diabetes mellitus: a predictive model for clinical risk assessment
- Evaluation of Doppler parameters and obstetric outcomes in intrahepatic cholestasis of pregnancy
- Diagnostic yield and clinical impact of prenatal whole exome sequencing (WES) – four-year single center experience
- Associations between previous cesarean section and maternal and neonatal complications: the modification of long inter-pregnancy interval
- Maternal and neonatal outcomes of ultrasound-guided percutaneous nephrostomy for symptomatic hydronephrosis in pregnancy: a retrospective cohort study
- Original Articles – Fetus
- Abnormal Doppler and perinatal outcomes according to the placental lesions of maternal and fetal vascular malperfusion in preterm fetal growth restriction
- The additional value of fetal MRI to ultrasonography in prenatal diagnosis: an evaluation based on postnatal confirmation
- The fetal exposome and preterm birth: a systematic synthesis of environmental exposures and multi-omics evidence
- Original Articles – Neonates
- Pregnancy-associated anemia and its effects in term neonates
- Longitudinal changes of left ventricular hypoplasia and ventricular disproportion in congenital diaphragmatic hernia neonates
- Outcomes in early term neonates requiring extracorporeal membrane oxygenation
- Letters to the Editor
- Therapeutic trials of maternal position therapy are needed to clarify the causes of abnormal maternal hemodynamic profiles and reduced umbilical vein flow in fetal growth restriction
- Let’s use diverse technologies to assess maternal hemodynamics-- both before and during labor and to both predict and prevent bad outcomes!
- Corrigendum
- The FAIR framework: ethical hybrid peer review
Articles in the same Issue
- Frontmatter
- Commentary
- The IAPM New Delhi 2025 Declaration on Svadharma and Professional Responsibility
- Reviews
- The use of laser therapy in fetal medicine: a narrative review
- Nutriepigenomics in perinatal medicine: maternal nutrition as a modulator of fetal gene expression and long-term health
- Original Articles – Obstetrics
- Community births in the United States, 2016–2024: post-pandemic patterns across racial and ethnic groups
- Preeclampsia is associated with increased NT-proBNP and altered lipid profiles in umbilical cord blood: a propensity score-matched analysis
- Ureaplasma parvum detected in umbilical cord tissues diagnosed with funisitis associated with adverse pregnancy outcomes and neonatal pneumonia
- Implementation of a universal low-dose aspirin protocol for the prevention of preeclampsia in a federally qualified health center
- Comparative evaluation of manual and VOCAL methods for amniotic sac volume measurement in early pregnancy
- McDonald vs. Shirodkar cerclage for the prevention of preterm birth in patients with obesity
- The incidence and outcomes of perinatal asphyxia in spontaneous extreme preterm birth: a retrospective cohort study
- Comparative effectiveness of oxytocin, carbetocin, and tranexamic acid for postpartum hemorrhage prevention in cesarean deliveries: a prospective cohort analysis
- Risk factors for primary cesarean delivery in women with gestational diabetes mellitus: a predictive model for clinical risk assessment
- Evaluation of Doppler parameters and obstetric outcomes in intrahepatic cholestasis of pregnancy
- Diagnostic yield and clinical impact of prenatal whole exome sequencing (WES) – four-year single center experience
- Associations between previous cesarean section and maternal and neonatal complications: the modification of long inter-pregnancy interval
- Maternal and neonatal outcomes of ultrasound-guided percutaneous nephrostomy for symptomatic hydronephrosis in pregnancy: a retrospective cohort study
- Original Articles – Fetus
- Abnormal Doppler and perinatal outcomes according to the placental lesions of maternal and fetal vascular malperfusion in preterm fetal growth restriction
- The additional value of fetal MRI to ultrasonography in prenatal diagnosis: an evaluation based on postnatal confirmation
- The fetal exposome and preterm birth: a systematic synthesis of environmental exposures and multi-omics evidence
- Original Articles – Neonates
- Pregnancy-associated anemia and its effects in term neonates
- Longitudinal changes of left ventricular hypoplasia and ventricular disproportion in congenital diaphragmatic hernia neonates
- Outcomes in early term neonates requiring extracorporeal membrane oxygenation
- Letters to the Editor
- Therapeutic trials of maternal position therapy are needed to clarify the causes of abnormal maternal hemodynamic profiles and reduced umbilical vein flow in fetal growth restriction
- Let’s use diverse technologies to assess maternal hemodynamics-- both before and during labor and to both predict and prevent bad outcomes!
- Corrigendum
- The FAIR framework: ethical hybrid peer review