Disorders of sex development in children in KwaZulu-Natal Durban South Africa: 20-year experience in a tertiary centre
-
Yasmeen Ganie

 ,
Colleen Aldous
,
Colleen Aldous

Abstract
Background:
The objective of the study was to describe the prevalence, clinical characteristics and aetiological diagnosis in children with disorders of sex development (DSDs) presenting to a tertiary referral centre.
Methods:
This is a retrospective review of all cases of DSD referred to the Paediatric Endocrine Unit in Inkosi Albert Luthuli Central Hospital (IALCH) from January 1995 to December 2014.
Results:
A total of 416 children (15.1%; CI: 13.8%–16.5%) were diagnosed with DSD. The aetiological diagnosis based on the current classification [Lawson Wilkins Paediatric Endocrine Society (LWPES) and European Society for Paediatric Endocrinology (ESPE)] was sex chromosome DSD in 9.5% (n=33), 46 XX DSD in 33% (n=114) and 46 XY DSD in 57.5% (n=199). The most common diagnoses in descending order were a disorder in androgen synthesis and action (not classified) in 53% (n=182), ovotesticular DSD in 22% (n=75) and congenital adrenal hyperplasia (CAH) in 10% (n=36). Overall the median age of presentation was 10 months (IQR: 1 month–4.5 years). There was a significant relationship (p<0.001) between the age of presentation and aetiological diagnosis. The majority (97%) of African patients had a diagnosis of 46 XX DSD. Prematurity was present in 47% (n=83) of children with 46 XY DSD (p<0.001).
Conclusions:
DSD is not an uncommon diagnosis in African patients in sub-Saharan Africa. The most common aetiological diagnosis is 46 XY DSD in androgen synthesis and action, followed by ovotesticular DSD. CAH is only the third most common disorder.
Introduction
Disorders of sex development (DSDs) are a rare heterogeneous group of genetic DSDs. Most are diagnosed at birth; however, DSDs are also diagnosed in childhood and adolescence as a result of a late or missed diagnosis [1].
The prevalence of genital anomalies detected at birth is reported to be as high as one in 300 births [2], but the prevalence of true genital ambiguity is rare, only one in 4500 births [3]. There are few nationally representative data on the prevalence of DSD in Africa and South Africa (SA). A retrospective review published in 2012 of 122 Sudanese children reported that 57% (n=69) of patients had 46 XX DSD and 37% (n=45) had 46 XY DSD. The most common cause of XX DSD was congenital adrenal hyperplasia (CAH) and that of XY DSD was androgen insensitivity syndrome [4]. In contrast in a review published in 2008 of 208 Egyptian children it was reported that 46 XY DSD was more common than 46 XX DSD constituting 65.9% of all cases [5]. Locally Wiersma et al. reported that the vast majority of patients (51%) with DSD presenting to a paediatric surgical unit in KwaZulu Natal had ovotesticular DSD [6]. This is a high incidence compared to global rates of 3%–10% [7].
Despite the advances in genetic and genomic technologies, a genetic diagnosis is made in only 20% of cases of DSD [8]. Only 50% of 46 XY children with DSD will receive a definitive diagnosis and most infants with 46 XX DSD have CAH due to 21 hydroxylase deficiency [9].
A database of DSD in KwaZulu-Natal Durban SA of all patients presenting to the Paediatric Endocrine Unit from 1995 is kept in the unit at Inkosi Albert Luthuli Central Hospital (IALCH). This database was used to retrospectively analyse records of patients and to describe the prevalence, clinical characteristics and aetiological diagnosis of patients with DSD.
Patients and methods
This is a retrospective review of all cases of DSD referred to the Paediatric Endocrine Unit in IALCH from January 1995 to December 2014. A database of DSD in KwaZulu-Natal, Durban, SA of all patients presenting to the unit from 1995 has been maintained. IALCH is a provincial tertiary hospital in KwaZulu-Natal Durban and serves a population of approximately 10 million children from 11 health districts. It is the only referral centre for paediatric patients with endocrine problems in KwaZulu-Natal. Patients are initially evaluated at their district or regional hospitals and referred to IALCH for subspecialty support. All the records of patients with DSD presenting to the unit from 1995 to 2014 were included in the database. This unit was previously situated in King Edward VIII Hospital and relocated to IALCH in 2002. The database has obtained Class Approval from BREC (BE479/14) in 2015.
A total of 416 patients with a diagnosis of DSD were included in the database. Data were extracted from charts and electronic medical records. We included patients with a clinical diagnosis of DSD made by the paediatric endocrinologist and confirmed by biochemistry and or histology. Patients with chromosomal disorders which are included in the current DSD classification were included. The following information was recorded in the database: age at presentation, clinical presentation (ambiguous genitalia or delayed or precocious puberty), demographics including residential area and referral hospital, sex of rearing, gender dysphoria, consanguinity and family history of DSD. Clinical features including anthropometry [height or length, weight and body mass index (BMI)]; assessment of pubertal stage using the Tanner stage, and dysmorphic features was recorded for each patient. The Prader scoring system was used to determine the degree of external virilisation in patients with CAH [10]. The external masculinisation score was used to obtain an objective score of the degree of external masculinisation of the genitalia in males [11]. The karyotype and the results of fluorescence in situ hybridisation (FISH) analysis for SRY gene were also recorded. The patient’s biochemistry and hypothalamic pituitary gonadal axis hormonal assays including gonadotropins, 17-hydroxyprogesterone (17-OHP), dehydroepiandrostendione (DHEA-S), androstenedione (A), testosterone (T) and oestradiol (E2) were included in the database. Sex steroids were measured using the chemiluminescence immunoassay (Siemens Immulite 2000) at a central laboratory in IALCH. Radiological investigations including the findings of the pelvic ultrasound were captured. A short human chorionic gonadotropin (hCG) stimulation test was performed to identify functioning testicular tissue when indicated. However, hCG was not consistently available and the test was performed in a small number of patients. Serum T was measured at baseline and after 3 days of consecutive intramuscular hCG injections. The T response to hCG was normal if the level increased to more than twice the level of the baseline T or to above the upper limit of the prepubertal T levels, depending on the age of the patient [12]. Surgical history, including procedures, histology and outcomes (both short and long-term), including complications, were recorded. Consultations with members of the multidisciplinary team (social worker, clinical psychologist) were also noted.
The aetiological diagnosis was based on the recommended classification in 2006 by the Lawson Wilkins Paediatric Endocrine Society (LWPES) and the European Society for Paediatric Endocrinology (ESPE) [1]. The patients with DSD were divided into three etiologic groups based on their karyotypes: sex chromosome DSD, 46 XY DSD and 46 XX DSD. Sex chromosome DSDs were further subdivided into four categories according to karyotype: 45 XO [Turner syndrome (TS) and variants], 47 XXY (Klinefelter syndrome and variants), 45X/46XY (mixed gonadal dysgenesis) and 46 XX/46 XY (chimerism, ovotesticular DSD).
Statistical methods
Data were analysed using STATA Version 14 (StataCorp., 2015; StataCorp LP, Stata Statistical Software, Release 14, College Station, TX, USA). The descriptive statistical analysis included frequency calculation for categorical data and mean and standard deviations for continuous data. A χ2 test was used to test the homogeneity of proportions as well as independence between categorical variables of interest. Results were considered statistically significant for a p-value < 0.05.
Results
A total number of 2753 patients attended the endocrine clinic over the 20-year study period and 416 (15.1%; CI: 13.8%–16.5%) were diagnosed with DSD.
The aetiological classification of the patient population is reflected in Table 1. Of the total number of patients with DSD (416), data on aetiological diagnosis were missing in 70 patients, hence calculated percentages and numbers are based on 346 patients. Based on the current recommended classification by the LWPES and ESPE, 9.5% (n=33) had an aetiological diagnosis of sex chromosome DSD, 57.5% (n=199) had 46 XY DSD and 33% (n=114) had 46 XX DSD (Table 1). The most common diagnosis in the cohort was a disorder in androgen synthesis and action that could not be further classified in 53% (n=182), followed by ovotesticular DSD in 22% (n=75) patients and CAH in 10% (n=36) patients (Table 1).
Aetiological classification (n=346).
| n | % | |
|---|---|---|
| Sex chromosome | 33 | 9.5 |
| 45 XO | 20 | 61 |
| Trisomy | 4 | 12 |
| Mixed gonadal dysgenesis | 4 | 12 |
| 47 XXY | 3 | 9 |
| 46 XX/46 XY | 1 | 3 |
| Other: 46 XY/XYY | 1 | 3 |
| 46 XY DSD | 199 | 57.5 |
| Disorder of testicular development | 7 | 4 |
| Gonadal dysgenesis | 4 | 57 |
| Ovotesticular | 3 | 43 |
| Disorder of androgen synthesis or action | 192 | 96 |
| Congenital adrenal hyperplasia | 8 | 4 |
| Complete androgen insensitivity | 2 | 1 |
| Not classified | 182 | 95 |
| 46 XX DSD | 114 | 33 |
| Disorder of ovarian development | 76 | 67 |
| Ovotesticular | 69 | 91 |
| 46 XX male | 7 | 9 |
| Androgen excess | 38 | 33 |
| Congenital adrenal hyperplasia | 27 | 71 |
| Cliteromegaly | 9 | 24 |
| Other | 2 | 5 |
The 46 XY DSD was the most common aetiological diagnosis in our cohort and was present in 57.5% (n=199) patients (Table 1). The most frequent aetiology in patients with 46 XY DSD was a disorder in androgen action and synthesis in 96% (n=192) patients. Seven patients (4%) were classified as having a disorder of testicular development. Amongst the patients with disorders in androgen synthesis or action 95% (n=182) did not have a final diagnosis, CAH was diagnosed in 4% (n=8) and complete androgen insensitivity syndrome was diagnosed in only 1% (n=2) patients. Of the seven patients with disorders of testicular development, 57% (n=4) were diagnosed with a disorder of gonadal dysgenesis and 43% (n=3) were diagnosed with ovotesticular DSD.
In the 46 XX DSD group, 67% (n=76) were diagnosed with a disorder of ovarian development and 33% (n=38) had disorders secondary to an excess of androgens (Table 1). Ovotesticular DSD was diagnosed in 61% (n=69), CAH in 24% (n=27) and 46 XX Male in 6% (n=7) of the patients.
Overall CAH was diagnosed in 10% (n=36) patients and 89% (n=15) had severe external virilisation with a Prader score of greater than 3.
TS was diagnosed in 61% (n=20) of patients with sex chromosome DSD. Mixed gonadal dysgenesis and trisomy disorders were present in 12% (n=4) of patients. There were three (9%) patients with Klinefelter syndrome. Chimerism of 46 XX/XY and 46 XY/XYY was diagnosed in one patient (3%) each. Ovotesticular DSD was diagnosed in 50% (n=2) of the patients with mixed gonadal dysgenesis and in the single patient with 46 XX/XY chimerism (Table 2).
Ovotesticular DSD – Karyotype (n=75).
| Chromosome | Number of patients, % |
|---|---|
| XY | 3 |
| XX | 69 |
| Mixed gonadal dysgenesis | 2 |
| 46 XX/46 XY | 1 |
The 45XO karyotype was confirmed in 85% of patients with TS. The median age of presentation of patients with TS was 8 years (IQR: 7 months–11 years). Amongst the patients with TS, 45% (n=9) presented after 10 years of age with short stature and delayed puberty, 30% presented between 5 and 10 years of age with short stature and dysmorphology and 25% (n=5) presented in infancy with dysmorphology and lymphoedema.
Of the 416 patients, data on both age and aetiological diagnosis were available in 337 patients. The age of presentation of patients with DSD was widely varied and ranged from 1 day to 22 years with a median age of presentation of 10 months (IQR: 1 month–4.5 years). Overall 42% of patients were assessed before 6 months of age, 28% between 1 and 5 years of age, 15% between 6 and 10 years of age and 6% after 10 years of age, respectively.
The age of presentation also varied significantly by aetiological diagnosis (p<0.001) and is represented in Figure 1. The median age of presentation of patients with sex chromosome disorders, 46 XY DSD and 46 XX DSD was 8 years (IQR: 7 months–11 years); 11 months (IQR: 2 months–3 years) and 5 months (IQR: 15 days–5 years) (Figure 2). Of the patients with sex chromosome DSD, 33% (n=11) presented after 10 years of age and 15% (n=5) presented before the age of 6 months (Figure 1). In the 46 XY DSD group, 43% (n=83) of patients presented before 6 months of age and a significant number of patients (n=69, 35%) presented between 1 and 5 years of age (Figure 1). In the 46 XX DSD group, 50% (n=55) of patients presented before 6 months of age, 19% (n=21) presented between 6–10 years of age and 1–5 years of age, respectively (Figure 1).
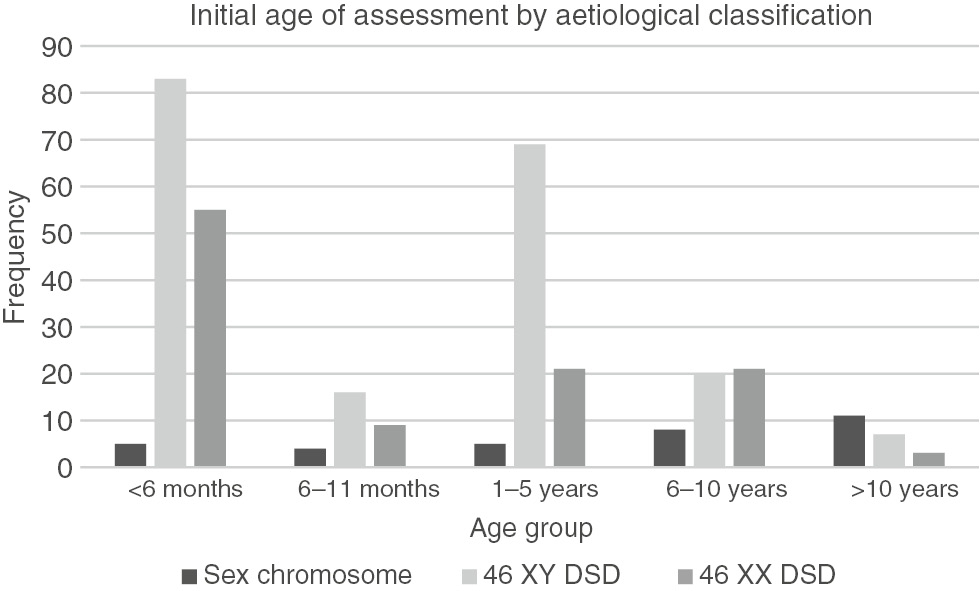
Bar graph of age of presentation by aetiological classification (n=337).
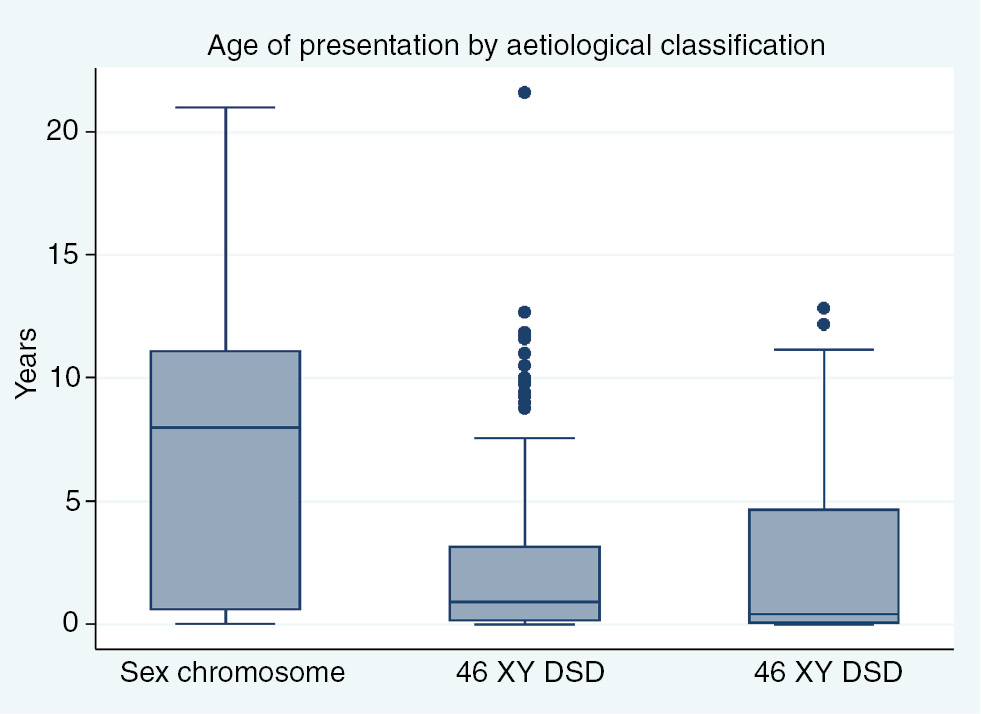
Age of presentation by aetiological classification.
Data on both age and race were available in 346 patients. The majority of patients (86%, n=299) were of African ethnicity. Thirty (9%) patients were Indian, ten (3%) White and seven (2%) were classified as mixed ancestry (Figure 3). A Pearson χ2 test showed a statistically significant association between population group and aetiological diagnosis (p<0.001). Substantially more than expected Indian (18%) and White (21%) patients were diagnosed with sex chromosome DSD. There were fewer than expected African (61%) and no mixed ancestry patients diagnosed with sex chromosome DSD. More Indian (11%) patients were diagnosed with 46 XY DSD than expected. Fewer than expected Indian (2%), mixed ancestry (1%) and no White patients were diagnosed with 46 XX DSD. A significant number of African (97%) patients had 46 XX DSD.
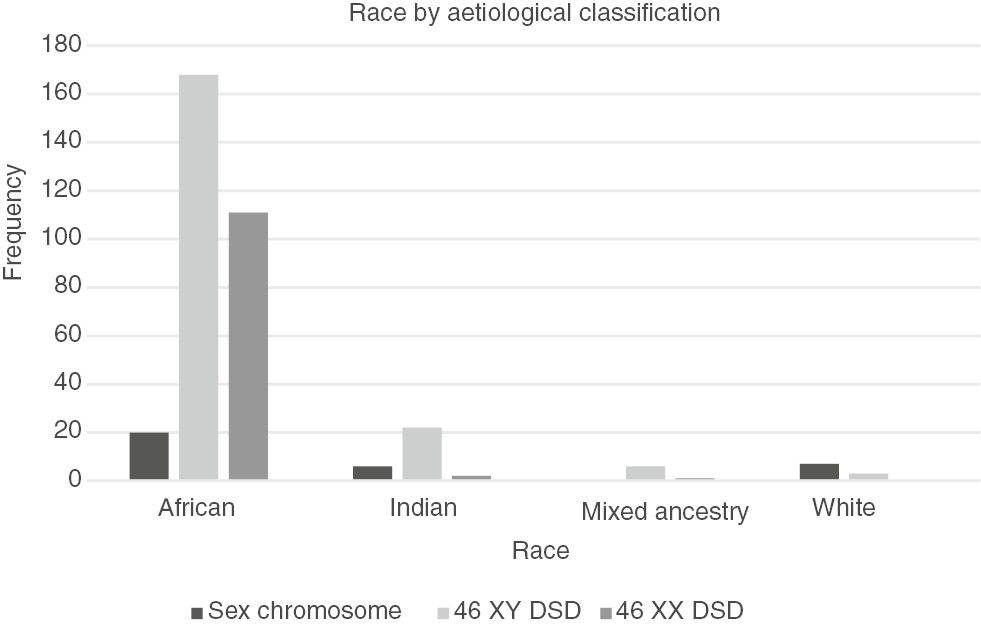
Race by aetiological classification (n=346).
Overall, 37% (n=155) of patients were initially evaluated at the endocrine clinic and 34% (n=141) were evaluated at the urogenital clinics, respectively.
Associated congenital malformations were present in 14% (n=58) patients. Overall dysmorphology was present in 4% (n=18) of patients, whilst 4% of patients also had an underlying neurological or gastrointestinal problem. Renal pathology was present in only 2% of patients in the cohort.
Information on both birth weight and the aetiological diagnosis was available in 286 patients. A significant association (p<0.001) between birth weight and the aetiological diagnosis was found in these patients. Of the 286 patients, 64% (n=182) of the patients with DSD were delivered at term and the remaining 36% (n=104) were preterm. More patients with 46 XY DSD 47% (n=83) were premature at birth compared with patients with 46 XX DSD and chromosomal DSD.
A positive family history of DSD was obtained in 5% (n=21) of patients. In this group, 10 patients had an aetiological diagnosis of CAH and nine patients were diagnosed with 46 XY DSD. The remaining two patients had a diagnosis of ovotesticular DSD. There were two sibling pairs with a diagnosis of CAH in the cohort. There was no positive history of consanguinity reported amongst any of the patients in the study.
At the time of presentation 29% (n=119) of patients had a female karyotype and 71% (n=297) had a male karyotype. Only 20 patients in the cohort were tested for the SRY gene and one patient tested positive. With respect to sex assignment, all patients were assigned a gender after evaluation by the multidisciplinary team.
In the 46 XY DSD group, most patients had gonads which were located normally in both the scrotal folds. However, 46% presented with varying degrees of cryptorchidism. The mean external masculinisation score was seven with a standard deviation 2.62 (range 1–12). The biochemical investigations in the patients with 46 XY DSD were performed mainly after 6 months of age and were in the pre-pubertal range. A substantial number of patients had biochemistry suggestive of a partial androgen insensitivity disorder with elevated luteinizing hormone (LH) and T levels but this could not be confirmed. The hCG stimulation was only performed in 61 patients with undetectable basal levels of steroids. The T levels increased from basal confirming functioning Leydig cells in 49% (n=30) of these patients.
Discussion
There are limited published data on DSD in developing countries, specifically in Africa and sub-Saharan Africa. To our knowledge, this review represents the largest cohort of patients with DSD in sub-Saharan Africa and is the first study evaluating the prevalence, clinical characteristics and aetiological diagnosis of DSD using the new LWPES/ESPE classification system.
The prevalence of DSD in the general population of SA is unknown; however, based on the number of patients recorded in our database over the last two decades, it appears that it is not uncommon. A total number of 416 patients met the criteria for DSD and were included in this retrospective descriptive analysis. The prevalence of DSD was 15.1% in our cohort.
The frequency of DSD will also vary depending on the underlying aetiology. Based on the current LWPES/ESPE recommended classification for DSD, the most common aetiology of DSD in our cohort was 46 XY DSD (57.5%). The second commonest aetiology was 46 XX DSD (33%) and only 9.5% of patients had sex chromosome DSD (Table 1). When compared to studies in Africa, these results are similar to the findings by Mazen et al. on Egyptian children published in 2008, where 65.9% of patients with DSD had a diagnosis of 46 XY DSD [5]. Our findings are in contrast to that of Abdulla et al. where the majority (57%) of Sudanese children had a diagnosis of 46 XX DSD [4].
The most common diagnosis in our cohort of 416 patients was 46 XY disorder in androgen synthesis or action that could not be classified (53%). The second most common diagnosis overall was ovotesticular DSD (22%). CAH was the third most common diagnosis and was present in only 10% of patients. In the Egyptian study, the majority of patients with 46 XY DSD had a diagnosis of 5 α-reductase deficiencies, and in Sudan the most common diagnosis in the patients with 46 XY DSD was androgen insensitivity syndrome [4, 5]. In our unit in KwaZulu-Natal, we are unable to perform specific hormonal assays such as an anti-mullerian hormone, inhibin B and dihydrotestosterone which can assist in reaching a specific diagnosis in patients with 46 XY DSD. Urine steroid profiles measuring metabolites of steroidgenesis are also not available. β-hCG has also not been consistently available hence stimulation tests are not routinely performed on patients. A significant number of patients had biochemistry suggestive of a partial androgen insensitivity disorder with elevated LH and T levels but this could not be confirmed. Hence, the final diagnosis in this large group of patients with 46 XY DSD was compromised and was largely dependent on clinical assessment and basic investigations.
Molecular genetic analyses of specific genes for patients with DSD are also currently not available in KwaZulu-Natal and in SA. The wide availability of new genomic technologies including chromosomal microarray and whole exome sequencing can detect mutations in known DSD genes and can identify variants in novel DSD genes. This has changed the diagnostic approach of patients with DSD as genetic investigations can be undertaken early in the diagnosis of DSD [12]. The lack of these advanced molecular techniques is likely to alter the classification of our cohort of patients with DSD when these techniques become available in future. However, it has previously been reported, that in patients with 46 XY DSD, a genetic mutation is identified in less than 50% of patients [9].
Ovotesticular DSD (22%) was the second most common aetiological diagnosis in our cohort; this rate is high compared with global rates of 3%–10% [7]. The diagnosis of ovotesticular DSD is based on histology confirming the presence of ovarian follicles and testicular tissue in the same patient. A high incidence of ovotesticular DSD has previously been reported in SA [10, 13, 14]. Wiersma et al. reported that ovotesticular DSD was the single most common diagnosis (51%) amongst Black African patients with ambiguous genitalia presenting to a local surgical unit [7]. Our numbers were lower and this could be explained by the fact that the new DSD classification includes a range of disorders including chromosomal disorders and medically managed DSD’s which would attenuate the final number of patients in each aetiological category.
The most common cause of DSD worldwide is CAH and is reported to occur in 1:15,000 live births [15]. In our cohort, however, ovotesticular DSD and 46 XY DSD defects in androgen synthesis and action were more common than CAH. Newborns are not routinely screened for CAH in SA and this missed diagnosis will result in a high mortality especially in boys who present with electrolyte disturbances and no genital ambiguity. This can be improved through education and the training of paediatricians to recognise these patients early and refer timeously for specialist treatment.
There was a wide variation in the age of presentation in patients with DSD. Overall, the majority of the patients (42%) were evaluated early, before 6 months of age and only 6% were evaluated after 10 years (Figure 1). However, overall the median age of presentation was 10 months. There is a postnatal surge of gonadotropins and sex steroids that peak between 1 week and 3 months of age which affords a window of opportunity to investigate and make a diagnosis in patients with DSD [16]. This opportunity is missed when patients present after 6 months of age and limits making a specific diagnosis.
There was a statistically significant difference in the age of presentation amongst the different aetiological groups (Figure 1). The majority (33%) of patients with sex chromosome DSD presented late, after 10 years of age and only 15% presented early, before 6 months of age. Two-thirds of patients (61%) in the sex chromosome group had TS. The median age of presentation of girls with TS was 8 years (IQR: 7 months–11 years). TS is a common sex chromosome disorder and has a reported incidence of 1:2000–1:5000 live births [17]. Girls with TS have dysmorphic features and normal external genitalia but abnormal chromosomes and gonadal function. In our cohort, the diagnosis of TS was missed by clinicians in infancy and childhood, and girls were referred in late childhood or adolescence when they presented with short stature and delayed puberty. This would impact on the treatment outcome of girls with TS as final height can be improved with growth hormone therapy that is initiated in childhood [11, 18, 19]. A diagnosis of Klinefelter syndrome was made in only three patients in the cohort. This is low compared to the reported incidence of 1:500–1:1000 live births [20]. The dysmorphic features in boys with Klinefelter syndrome are subtle and it is likely that the diagnosis is missed until patients present in adolescence with delayed puberty.
In the 46 XY DSD group, the median age of presentation was 11 months (IQR: 2 months–3 years). Most of the patients (43%) presented early, before 6 months of age but a significant number (35%) presented in early childhood between 1 and 5 years of age. Gonadotropins and sex steroids including DHEAS, androstenedione, T and dihydrotestosterone measured during the mini pubertal surge before 6 months of age assists with making a specific diagnosis of T biosynthetic defects or action in patients with 46 XY DSD. This late presentation once again impacts on making a specific diagnosis in patients with 46 XY DSD. It will also impact on medical management, surgical procedures and long-term outcomes.
Half of the patients with 46 XX DSD presented before 6 months of age. The median age of presentation was 5 months (IQR: 15 days–5 years). The most common diagnosis in this group was ovotesticular DSD confirmed on histopathology of the gonads, followed by CAH. This is in contrast to most studies where the commonest diagnosis in patients with 46 XX DSD is CAH 21 hydroxylase deficiency [9]. The external genitalia in patients with ovotesticular DSD are ambiguous and a spectrum of abnormalities is described. The clinical features described include the abnormal combination of the phallus, labio-scrotal folds and gonads, generally, results in an earlier referral [21]. Girls with CAH present with virilisation of the genitalia and or electrolyte abnormalities and the clinical suspicion results in an earlier referral. However, boys with CAH may be missed as their genitalia appear normal and the electrolyte disturbances may be associated with gastroenteritis which is more common in our setting. Neonatal screening programs can prevent this life-threatening diagnosis from being missed [22].
The distribution of population by population groups in our cohort reflects the socio-demographic status of the population in KwaZulu-Natal with 85% of the patients being of African origin, 11% Indian and 2% mixed ancestry and White, respectively. There was a statistically significant relationship between the different population groups and aetiological diagnosis (Figure 3). The majority of African patients (97%) had an aetiological diagnosis of 46 XX DSD. In the 46 XX DSD group, 61% (n=69) of patients were diagnosed with ovotesticular DSD (Table 2). Further analysis is necessary to confirm this high prevalence of ovotesticular DSD in the African population.
Approximately two-thirds of patients were referred appropriately to the endocrine and urogenital clinics for assessment, but in the remaining one-third of patients, the diagnosis of DSD was missed resulting in a late presentation. More health personnel have to be educated and trained to recognise and refer children with DSD early. The health infrastructure should ensure that this condition is recognised at a primary level and that these children are channelled appropriately for early expert management.
Associated congenital anomalies are reported to occur in 27% of patients with DSD and renal anomalies are reported to occur in 30% patients with DSD [23]. In our cohort, only 14% of patients had congenital abnormalities and only 2% of patients had renal pathology.
Birth weight was statistically significant between the different aetiological groups. The majority of patients were term deliveries. Almost half of the patients with 46 XY DSD were preterm and small for gestational age compared with 46 XX DSD and chromosomal DSDs. This has previously been reported by Cox et al. from data from the I-DSD registry, where the most common associated condition in 46 XY DSD patients was small for gestational age present in 23% of their patients [23].
A history of consanguinity is reported to be high in endogamous communities like Saudi Arabia and Egypt and low in Western communities [24]. It is noted that none of the patients in our cohort reported a history of consanguinity which reflects the social, cultural and religious background of our population. A positive family history was reported in 5% of patients and most of these patients had a diagnosis of either CAH which is an autosomal recessive disorder or 46 XY DSD. Interestingly, there were two sibling pairs in our cohort with CAH. In the one family, both female siblings had classic salt wasting CAH whilst in the second family, one sibling had 46XX CAH classic salt wasting syndrome and the second sibling had 46 XY CAH classic non-salt wasting syndrome.
Conclusions
In this large retrospective review describing patients with DSD over two decades, we have shown that DSD is not an uncommon diagnosis in African patients in sub-Saharan Africa. The most common aetilogical diagnosis is 46 XY DSD disorder in androgen synthesis and action, followed by ovotesticular DSD. CAH is only the third most common disorder. In resource-poor settings, an accurate diagnosis of patients with 46 XY DSD is compromised due to the limited availability of investigations. There is an unusually high incidence of ovotesticular DSD amongst Black South Africans that warrants further investigation. The majority of the patients are referred late, limiting the diagnostic workup and management. A history of consanguity is rare in African patients with DSD. The 46 XY DSD is more common in preterm babies than term babies.
Limitations
This is a retrospective descriptive study which is accompanied by the usual issues of data integrity such as missing data. Given that we operate in a resource-poor setting where there is limited availability of biochemical, molecular and cytogenetic investigations, we are restricted in making a final aetiological diagnosis and classification in children with DSD. The aetiological diagnosis and classification of the patients in our cohort is likely to change with the availability of genetic studies at a reduced cost in future.
Author contributions: All the authors have accepted responsibility for the entire content of this submitted manuscript and approved submission.
Research funding: None declared.
Employment or leadership: None declared.
Honorarium: None declared.
Competing interests: The funding organization(s) played no role in the study design; in the collection, analysis, and interpretation of data; in the writing of the report; or in the decision to submit the report for publication.
References
1. Hughes IA. Disorders of sex development: a new definition and classification. Best Pract Res Clin Endocrinol Metab 2008;22:119–34.10.1016/j.beem.2007.11.001Search in Google Scholar PubMed
2. Ahmed SF, Dobbie R, Finlayson AR, Gilbert J, Youngson G, et al. Regional and temporal variation in the occurrence of genital anomalies amongst singleton births, 1988–1997, Scotland. Arch Dis Child Fetal Neonatal Ed 2004;89:F149–F51.10.1136/adc.2002.024034Search in Google Scholar PubMed PubMed Central
3. Hamerton JL, Canning N, Ray M, Smith S. A cytogenetic survey of 14,069 newborn infants. Incidence of chromosome abnormalities. Clin Genet 1975;4:223–43.10.1097/00006254-197606000-00015Search in Google Scholar
4. Abdullah MA, Saeed U, Abass A, Lubna K, Weam A, et al. Disorder of Sex Development among Sudanese children: 5-year experience of a Paediatric Endocrinology Clinic. J Pediatr Endocrinol Metab 2012;25:1065–72.10.1515/jpem-2011-0467Search in Google Scholar PubMed
5. Mazen I, Hiort O, Bassiouny R, El Gammal M. Differential diagnosis of disorders of sex development in Egypt. Horm Res 2008;70:118–23.10.1159/000137657Search in Google Scholar PubMed
6. Wiersma R, Ramdial PK. The gonads of 111 South African patients with Ovotesticular Disorder of Sex Differentiation. J Paediatr Surg 2009;44:556–60.10.1016/j.jpedsurg.2008.08.013Search in Google Scholar PubMed
7. Krstic ZD, Smoljanic Z, Vukanic D, Janjic G. True Hermaphroditism: 10-year experience. Paediatr Surg Int 2000;16:580–3.10.1007/s003830000415Search in Google Scholar PubMed
8. Grumbach MM, Hughes IA, Conte FA. Disorders of sex differentiation. In: Larsen PR, Kronenberg HM, Melmed S, Polonsky KS, editors. Williams Textbook of Endocrinology, 10th ed. Heidelberg Germany: Saunders, 2003:842–1002.Search in Google Scholar
9. Ahmed SF, Cheng A, Dovey L, Hawkins JR, Martin H, et al. Phenotypic features, androgen receptor binding and mutational analysis in 278 clinical cases reported as androgen insensitivity syndrome. J Clin Endocrinol Metab 2000;85:658–65.10.1210/jc.85.2.658Search in Google Scholar
10. Wiersma R, Constantinides CG. True Hermaphroditism. A common cause of intersex in Africa. Abstract in S. Afr J Surg 1993;31:36.Search in Google Scholar
11. Bolar K, Hoffman AR, Maneatis T, Lippe B. Long-term safety of recombinant human growth hormone in turner syndrome. J Clin Endocrinol Metab 2008;93:344–51.10.1210/jc.2007-1723Search in Google Scholar PubMed
12. Ahmed SF, Acherman JC, Arlt W, Balen A, Conway G, et al. Society for Endocrinology UK Guidance on the initial evaluation of an infant or an adolescent with a suspected disorder of sex development (Revised 2015). Clin Endocrinol 2016;84:771–88.10.1111/cen.12857Search in Google Scholar PubMed PubMed Central
13. Aarinson IA. True Hermaphroditism. A review of 41 cases with observations on testicular histology and function. Br J Urol 1985;57:775–9.10.1111/j.1464-410X.1985.tb07052.xSearch in Google Scholar
14. Van Niekerk WA. True Hermaphroditism: an analytical review with a report of 3 new cases. Am J Obstet Gynecol 1976;126:890–907.10.1016/0002-9378(76)90677-3Search in Google Scholar
15. Speiser P. Congenital adrenal hyperplasia owing to 21 hydroxylase deficiency. Endocrinol Metab Clin North Am 2001;30:31–59.10.1016/S0889-8529(08)70018-5Search in Google Scholar
16. Kuiri-Hanninen T, Sankilampi U, Dunkel L. Activation of the hypothalamic-pituitary-gonadal axis in infancy: MiniPuberty. Horm Res Paediatr 2014;82:73–80.10.1159/000362414Search in Google Scholar
17. Donaldson MD, Gault EJ, Tan KW, Dunger DB. Optimising management in Turner syndrome: from infancy to adult transfer. Arch Dis Child 2006;91:513–20.10.1136/adc.2003.035907Search in Google Scholar
18. Davenport ML, Crowe BJ, Travers SH, Rubin K, Ross JL, et al. Growth hormone treatment of early growth failure in toddlers with Turner syndrome: a randomized, controlled, multicenter trial. J Clin Endocrinol Metab 2007;92:3406–16.10.1210/jc.2006-2874Search in Google Scholar
19. Backeljauw P. Does growth hormone therapy before 4 years of age enhance the linear growth of girls with Turner’s syndrome? Nat Clin Pract Endocrinol Metab 2008;4:78–9.10.1038/ncpendmet0678Search in Google Scholar
20. Paduch DA, Bolyakov A, Cohen P, Travis A. Reproduction in men with Klinefelter syndrome: the past, the present, and the future. Semin Reprod Med 2009;27:137–48.10.1055/s-0029-1202302Search in Google Scholar
21. Wiersma R. Ovotesticular disorders of sex development in Southern Africa: the clinical picture. Pediatr Surg Int 2004;20:363–8.10.1007/s00383-004-1200-0Search in Google Scholar
22. Therrell BL. Newborn screening for congenital adrenal hyperplasia. Endocrinol Metab Clin North Am 2001;30:15–30.10.1016/S0889-8529(08)70017-3Search in Google Scholar
23. Cox K, Bryce J, Jiang J, Rodie M, Sinnott R, et al. Novel associations in disorders of sex development: findings from the I-DSD Registry. J Clin Endocrinol Metab 2014;99:E348–E55.10.1210/jc.2013-2918Search in Google Scholar PubMed PubMed Central
24. Bashamboo A, McElreavy K. Consanguinity and disorders of sex development. Hum Hered 2014;77:108–17.10.1159/000360763Search in Google Scholar PubMed
©2017 Walter de Gruyter GmbH, Berlin/Boston
Articles in the same Issue
- Frontmatter
- Editorial
- Disorders of sex development
- Original Articles
- Fertility and sexual function: a gap in training in pediatric endocrinology
- Disorders of sex development in children in KwaZulu-Natal Durban South Africa: 20-year experience in a tertiary centre
- Novel mutations of the SRD5A2 and AR genes in Thai patients with 46, XY disorders of sex development
- Sensitivity and specificity of different methods for cystic fibrosis-related diabetes screening: is the oral glucose tolerance test still the standard?
- Anti-hyperglycemic activity of Aegle marmelos (L.) corr. is partly mediated by increased insulin secretion, α-amylase inhibition, and retardation of glucose absorption
- Risk factors that affect metabolic health status in obese children
- Nocturnal levels of chemerin and progranulin in adolescents: influence of sex, body mass index, glucose metabolism and sleep
- Elevated endogenous secretory receptor for advanced glycation end products (esRAGE) levels are associated with circulating soluble RAGE levels in diabetic children
- Food exchange estimation by children with type 1 diabetes at summer camp
- Usefulness of non-fasting lipid parameters in children
- Monitoring steroid replacement therapy in children with congenital adrenal hyperplasia
- Clinical and molecular characterization of Beckwith-Wiedemann syndrome in a Chinese population
- An analysis of the sequence of the BAD gene among patients with maturity-onset diabetes of the young (MODY)
- Case Reports
- Hypogonadotropic hypogonadism in a female patient with congenital arhinia
- Transdermal testosterone gel for induction and continuation of puberty in adolescent boys with hepatic dysfunction
- Long-term response to growth hormone therapy in a patient with short stature caused by a novel heterozygous mutation in NPR2
- Three cases of Japanese acromicric/geleophysic dysplasia with FBN1 mutations: a comparison of clinical and radiological features
Articles in the same Issue
- Frontmatter
- Editorial
- Disorders of sex development
- Original Articles
- Fertility and sexual function: a gap in training in pediatric endocrinology
- Disorders of sex development in children in KwaZulu-Natal Durban South Africa: 20-year experience in a tertiary centre
- Novel mutations of the SRD5A2 and AR genes in Thai patients with 46, XY disorders of sex development
- Sensitivity and specificity of different methods for cystic fibrosis-related diabetes screening: is the oral glucose tolerance test still the standard?
- Anti-hyperglycemic activity of Aegle marmelos (L.) corr. is partly mediated by increased insulin secretion, α-amylase inhibition, and retardation of glucose absorption
- Risk factors that affect metabolic health status in obese children
- Nocturnal levels of chemerin and progranulin in adolescents: influence of sex, body mass index, glucose metabolism and sleep
- Elevated endogenous secretory receptor for advanced glycation end products (esRAGE) levels are associated with circulating soluble RAGE levels in diabetic children
- Food exchange estimation by children with type 1 diabetes at summer camp
- Usefulness of non-fasting lipid parameters in children
- Monitoring steroid replacement therapy in children with congenital adrenal hyperplasia
- Clinical and molecular characterization of Beckwith-Wiedemann syndrome in a Chinese population
- An analysis of the sequence of the BAD gene among patients with maturity-onset diabetes of the young (MODY)
- Case Reports
- Hypogonadotropic hypogonadism in a female patient with congenital arhinia
- Transdermal testosterone gel for induction and continuation of puberty in adolescent boys with hepatic dysfunction
- Long-term response to growth hormone therapy in a patient with short stature caused by a novel heterozygous mutation in NPR2
- Three cases of Japanese acromicric/geleophysic dysplasia with FBN1 mutations: a comparison of clinical and radiological features