
Mining DNA Sequence Patterns with Constraints Using Hybridization of Firefly and Group Search Optimization
-
Kuruva Lakshmanna
Kuruva Lakshmanna has received his B-Tech in Computer Science and Engineering from Sri Venkateswara University College of Engineering – Tirupathi, India in the year 2006, M-Tech in Computer Science and Engineering(Information Security) from National Institute of Technology Calicut, Kerala, India in the Year 2009, and currently perusing his PhD from VIT University, India. He is working as an Assistant professor in VIT University, India. His research interests are Data Mining in DNA sequences, algorithms, Knowledge Mining, etc.
 and
Neelu Khare
and
Neelu Khare
Neelu Khare has received her PhD in the year 2011 from NIT Bhopal, India, Masters of Computer Applications in the year 2005 from MP Bhoj University, India, Bachelors in Mathematics and Science in the year 1996 from Barkatullah University, India and Diploma in Electronics and Telecommunications in the year 1994 from MP Board of Technical Education, India. She is currently working as an Associate Professor in VIT University, India. Her research areas are Data Mining, Artificial Intelligence, Soft Computing, Natural Language Processing.

Abstract
DNA sequence mining is essential in the study of the structure and function of the DNA sequence. A few exploration works have been published in the literature concerning sequence mining in information mining task. Similarly, in our past paper, an effective sequence mining was performed on a DNA database utilizing constraint measures and group search optimization (GSO). In that study, GSO calculation was utilized to optimize the sequence extraction process from a given DNA database. However, it is apparent that, occasionally, such an arbitrary seeking system does not accompany the optimal solution in the given time. To overcome the problem, we proposed in this work multiple constraints with hybrid firefly and GSO (HFGSO) algorithm. The complete DNA sequence mining process comprised the following three modules: (i) applying prefix span algorithm; (ii) calculating the length, width, and regular expression (RE) constraints; and (iii) optimal mining via HFGSO. First, we apply the concept of prefix span, which detects the frequent DNA sequence pattern using a prefix tree. Based on this prefix tree, length, width, and RE constraints are applied to handle restrictions. Finally, we adopt the HFGSO algorithm for the completeness of the mining result. The experimentation is carried out on the standard DNA sequence dataset, and the evaluation with DNA sequence dataset and the results show that our approach is better than the existing approach.
1 Introduction
In this digital world where a huge amount of data are available in digital form, a large amount of data contain both significant and non-significant patterns. Here, the main challenge is to find interesting patterns that are helpful to make decisions, which is very tedious and time consuming. Thus, there arises the need for an automated technology that does this job efficiently and effectively. The frequent pattern mining technique is rather useful for this purpose [1]. Various researchers have presented different frequent pattern mining algorithms. These algorithms are categorized into closed sequential pattern mining, constraint-based sequential pattern mining, biological pattern mining, etc. [38]. Sequential pattern mining is an essential task in broad applications. Their functions include analyzing web access patterns, customer purchase patterns, DNA sequences [39], prediction of diseases, etc. [12]. Moreover, sequential pattern mining [11], one of the basic subjects of information mining, is an extra aspect involved in association rule mining [9]. The sequential pattern mining algorithm [2] deals with the problem of determining the frequent sequences in a given database [25]. It also signifies the association among transactions, while association rules describe the intra-transaction relationships. In association rule mining, the mined output is termed as the items that are bought together frequently in a single transaction [36].
The most studied data mining task is classification, whose purpose is forecasting the value (class) of a user-specified goal attribute based on the values of other attributes, called the predictive (feature) attributes. With regard to DNA, clustering is broadly employed in the genome database. Though several techniques were suggested to cluster genome sequences and DNA microarrays [19], there is very minute research in the area of employing DNA computation for clustering. A few plans are put forward to employ DNA computing to work out clustering problems [4]. In addition to this, very few decades have witnessed the individual and joint attempts of data mining and soft computing in the realm of bioinformatics [22]. In DNA sequence mining, soft computing methodologies (involving fuzzy sets, neural networks, genetic algorithms, rough sets, and soft sets), etc., can be broadly employed. There are numerous general classification models, such as naive Bayesian network [7], decision tree, neural networks, and rule learning using evolutionary algorithm, which are put to use here [5]. Moreover, a lot of optimization algorithms are also present to optimize the parameters, such as cuckoo search algorithm [33], chaotic krill herd algorithm [31], stud krill herd algorithm [29], monarch butterfly optimization [28], earthworm optimization algorithm [32], krill herd and cuckoo search [34], group search optimization (GSO) algorithm [8], and firefly algorithm (FA) [37]. In general, the goal of sequential pattern mining algorithms is discovering the sequential patterns from the sequential database. Recently, research work has proved that the frequency is not the best measure to determine the significance of a pattern in different applications [27]. The motive of constraint-based sequential pattern mining is to determine the entire set of sequential patterns that is able to satisfy a regular expression (RE) constraint.
In this paper, we explain a novel approach and efficient DNA sequence mining based on multiple constraints with hybridization of firefly and GSO (HFGSO) algorithm. Initially, we apply the prefix span (PS) algorithm to the dataset. We detect the frequent DNA sequence pattern and eliminate the infrequent pattern. Here, some of the patterns are eliminated, and the size of the dataset is reduced. Then, length and width constraints are applied to the reduced dataset. In order to achieve efficiency and for effective execution of the algorithm, the present study makes use of RE constraint, which saves time and memory. Here, also some of the DNA sequence are removed from the dataset. To improve the efficiency of the mining process, finally we apply the HFGSO algorithm to the mined pattern. It produces the optimal mined DNA sequence. The rest of this paper is organized as follows: Section 2 gives a brief description of the literature survey. Sections 3 and 4 explain the proposed DNA sequence mining, and Section 5 explains the results and discussion part. The conclusion is summed up in Section 6.
2 Literature Survey
For DNA sequence mining, the literature presents several theories. Now, we assess some of the works associated with it. Mallick et al. [20] explained the constraint-based sequential pattern mining. Here, monetary and compactness constraints were included. Moreover, a CFML-PS algorithm was explained by integrating these constraints with the original PS algorithm. This allows discovering all CFML sequential patterns from the sequential database. Kawade and Oza [13] explained the frequent sequential pattern mining with weighted RE and length constraints. The length of the pattern was also an important criterion. Similarly, in multiple biological sequences, Htike and Win [10] clarified the frequent patterns mining. They initially clarified the idea of the primary pattern, which was expanded to form larger patterns in the series. A prefix tree was erected to identify frequent primary patterns. Chen and Liu [6] clarified the problem of frequent pattern mining without user-specified gap constraints. Moreover, they brought in PMBC (namely pattern mining from biological sequences with wildcard constraints) to work out the problem. Similarly, Lin et al. [18] explained the efficient algorithms for mining up-to-date high-utility patterns (UDHUPs). These consider not only utility measure but also the timestamp factor to discover the recent high-utility patterns (HUPs). In Ref. [17], Lin et al. explained the frequent item set mining algorithm based on the principle of inclusion-exclusion and transaction mapping. Moreover, Lakshmanna and Khare [14] have explained the Constraint-Based Measures for DNA Sequence Mining using Group Search Optimization Algorithm. they develop the mining process into three steps such as (1) applying prefix span algorithm, (2) length and width constraints, (3) Optimal mining via group search optimization (GSO).
Moreover, the recognition of promoters in DNA sequences using weightily averaged one-dependence estimators was clarified by Wu et al. [35]. They have also elucidated the growing interest in the process of gene finding and gene recognition from DNA sequences. Park et al. [24] clarified the protein function forecast in view of gap constraints. Also, Lin et al. [18] clarified the proficient calculations for mining UDHUP. It considers utility measure as well as timestamp variable to find the late HUPs. The UDHUP-apriori was initially acquainted with mine UDHUPs in a level-wise manner. In Ref. [3], Aloysius and Binu explained an approach for products placement in supermarkets using PS algorithm. An approach was put forward to mine user buying patterns using PS algorithm and to place the products on shelves based on the order of mined purchasing patterns. In [16], Lakshmanna et al. have explained Enhanced Algorithm for Frequent Pattern Mining from Biological Sequences. Masseglia et al. [21] explained the efficient mining of sequential patterns with time constraints. They introduced an algorithm, GTC (graph for time constraints), for mining such patterns in very large databases. Moreover, Nakamura et al. [23] explained the mining approximate patterns with frequent locally optimal occurrences. Here, candidate patterns were generated without duplication using the suffix tree of a given string. They further define a k-gap-constrained setting, in which the number of gaps in the alignment between a pattern and an occurrence is limited to at most k. Moreover; Lakshmanna and Khare [15] have explained Frequent DNA Sequence Mining Using FBSB and Optimization. Here, to optimize the pattern, the Prefix Span with Group Search Optimization (PSGSO) was hybrid.
3 Technical Preliminaries
In this section, we explain the algorithm presented in the paper. After that, we go to the proposed DNA sequence pattern mining process.
3.1 Sequential Pattern Mining
Sequential pattern mining aims to mine a complete set of sequential patterns with respect to a given sequence database DS . Let DS be a DNA sequential database where each transaction T contains the ID and a set of the item involved in the transaction. Let P={p1, p2, pm } be a unique set of items. An item set is a non-empty subset of items, and an item set with k is called the k-item set. A sequence S is an ordered list of item sets based on the timestamp. It is denoted as <s1,s2,…,sn , where, sj , j∈1,2,…, n is an item set that is also called an element of the sequence S and sj ∈I. A sequence of K items (or of length k) is called k-sequence. For example, <(a)(c)(e)>, <(b)(c,d)>, and <(a)(b)(a)> are all three sequences. A sequence <s1, s2,…, sn > is called a subsequence of another sequence
3.2 PS Algorithm
PS is the most promising pattern-growth method. It is based on the recursive construction of the patterns and a simultaneous restriction of the search to projected databases [26]. A database is a set of subsequences. They are suffixes of the sequences that have a prefix. In each step, the algorithm looks for frequent sequences with prefix a, in the correspondent projected database. Let us consider the DNA sequence database DS having n-numbers of sequence. For this, the sequential patterns are mined from this database by using PS algorithm. Let our running database be DNA sequence database DS specified in Figure 2 and min_support=2.
The set of the items in the database in Table 1 is {p, q, r, s, t, u, v}. The sequence <p (p q r) (p r) s (r u)> has five elements: (p), (p q r), (p r), (s), and (r u), where item p and r appear more than once, respectively, in different elements. It is also a nine-sequence set, as there are nine instances appearing in that sequence. Item p happens three times in this sequence, so it contributes 3 to the length of the sequence. The step-by-step process is explained below.
DNA Sequence Database.
| ID | Sequences |
|---|---|
| 10 | <p(p q r) (p r) s (r u)> |
| 20 | <(p s) r ( q r) (p t)> |
| 30 | <(t u) (p q) (s u) r q> |
| 40 | <t v (p u) r q r> |
Step 1: Find length-1 sequential pattern
Find the length of sequence patterns for the DNA sequence database DB considering the minimum support that has been given. Initially, the scanning process is prepared on the database once to find all the frequent items in sequences. Each of these frequent items is a length-1 sequential pattern (Table 2), as revealed in Figure 3. They are 〈P〉:4,〈Q〉:4,〈R〉:4,〈S〉:3,〈T〉:3,〈U〉:3 and 〈V〉:1, where the notation of “<pattern>: count” symbolizes the pattern and its related support count [30].
Obtained Length-1 Frequent Item Sets.
| <P> | <Q> | <R> | <S> | <T> | <U> | <V> |
|---|---|---|---|---|---|---|
| 4 | 4 | 4 | 3 | 3 | 3 | 1 |
| <P><Q><R><S><T><U> |
Step 2: Divide the search space
Divide the search space into the prefixes whose support is greater than the minimum support. That is, the complete patterns can be divided into the subsequent four prefixes: the ones with prefix <P><Q><R><S><T><U>.
Step 3: Find subsets of sequential patterns
The subsets of sequential patterns can be mined by constructing corresponding projected databases and mining each recursively. The projected databases, as well as sequential patterns found in them, are listed in Table 3, while the mining process is explained as follows.
Projected Database and Mined Sequence.
| Prefix | Projected database | Sequential patterns |
|---|---|---|
| <p> | <(p q r) (p r) s (r u)>, <(_s) r (q r) (p t)>, <(_ q) (s u) r q>, <(_u) r q r> | <p>, <pp>,<pq>, <p(qr)>, <p(qr)p>, <pqp>, <pqr>, <(pq)>, <(pq)r>, <(pq)s>, <(pq)u>, <(pq)sr>, <pr>, <prp>, <prq>, <prr>, <ps>, <psr>, <pu> |
| <q> | <(_r) (p r) s (r u)>, <(_r) (p t)>, <(s u) r q>, <r> | <q>, <qp>, <qr>, <(qr)>, <(qr)p>, <qs>, <qsr>, <qu> |
| <r> | <(p r) s (r u)>, <(q r) (p t)>, <q>, <q r> | <r>, <rp>, <rq>, <rr> |
| <s> | <(r u)>, <r (q r) (p t)>, <(_u) r q>, | <s>, <sq>,<sr>, <srq> |
| <t> | <(_u) (p q) (s u) r q>, <-v (p u) r q r> | <t>, <tp>, <tpq>, <tpr>, <tprq>, <tq>, <tqr>, <tr>, <trq>, <tu>, <tuq>, <tur>, <turq> |
| <u> | <(p q) (s u) r q><r q r> | <u>, <uq>, <uqr>, <ur>, <urq> |
3.3 RE Constraints
An RE constraint is the constraint that is used to mine the sequential pattern present in the database. An RE constraint R is indicated as an RE over the alphabet of sequence elements utilizing the setup set of the RE operator, for example disjunction (|) and Kleeneclosure (*). Thus, an RE constraint Reither specifies a language of strings over the element alphabet, or equivalently, it specifies a regular family of sequential patterns that is of interest to the user. Thus, given any RE R, it is possible to build a deterministic finite automaton AR such that AR extracts the language generated by R. Informally, a deterministic finite automaton is a finite-state machine with (i) a well-defined start state (denoted by a) and one or more accept states, and (ii) deterministic transitions across states on symbols of the input alphabet (in our case, sequence elements). A transition from state b to state c on the element si is denoted by
4 Proposed Sequential Pattern Mining
The basic idea of our proposed methodology is to mine DNA sequential patterns with constraints using the HFGSO algorithm. Normally, the DNA sequence database has a big number of items. The long sequence creates a great dispute for presented sequential pattern discovery algorithms. According to this, we mine the frequent pattern. Basically, this paper consists of three modules: (i) PS module, (ii) constraint module, and (iii) hybrid optimization module. In each module, the repeated DNA sequences are mined. The overall diagram of the proposed DNA sequence mining is shown in Figure 1.
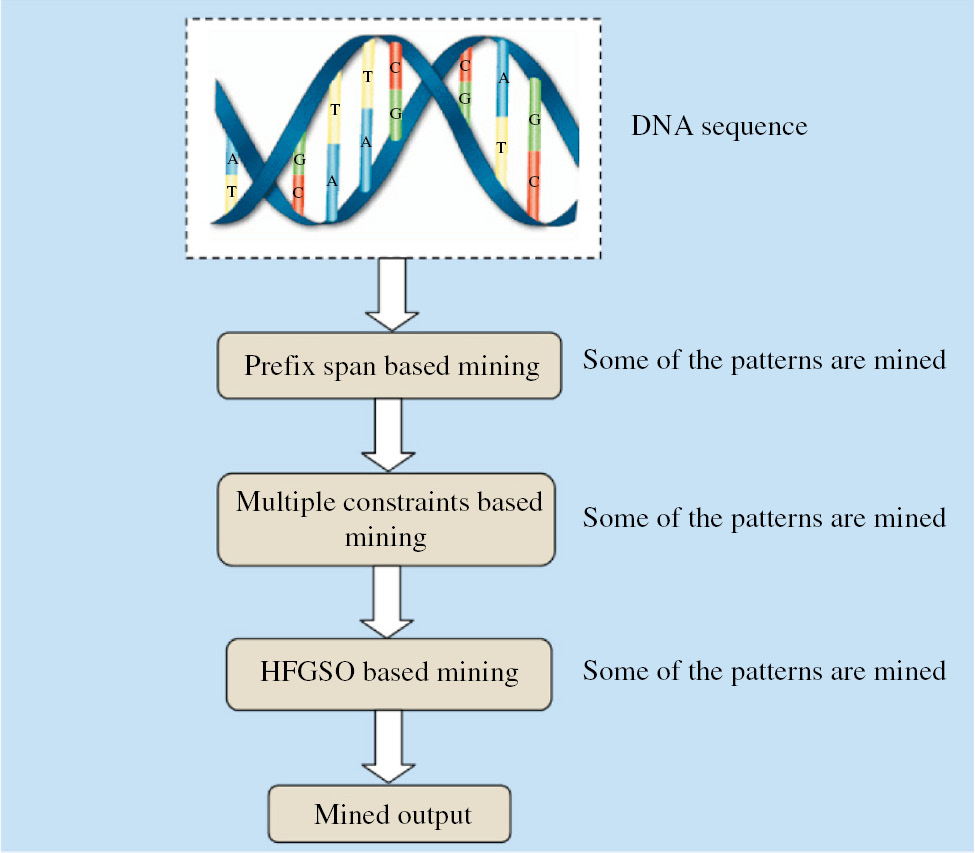
Overall Diagram of the Proposed DNA Sequence Mining.
Module 1: Mining based on PS algorithm
The PS algorithm is one of the important algorithms to mine the DNA sequence. The DNA sequence database consists of five types of items, such as <A><G><C><T>. This algorithm was carried out to perform the initial-level mining process. The detailed explanation of sequential pattern mining is given in Section 4.
Module 2: DNA sequential pattern mining based on constraints
The mined DNA sequence obtained from PS algorithm is given in this section. In the first module, some of the sequences are mined. Here, we use three types of constraints, such as length, weight, and RE constraints, for discovering the frequent patterns. This algorithm takes as inputs the weight of each item obtained from the PS algorithm, an RE, and min_length and max_length. This algorithm scans the database only once and finds frequent sequential patterns that satisfy the given min_weight, min_length, and max_length. The RE constraint is explained in Section 4. In this work, we take the RE as <A*C*G*T>. Moreover, the sequence [ACGT], [ACG], [GT] is the valid sequence. Here, we also find out the total number of patterns that satisfy the RE. After that, we check whether these obtained sequences satisfy the length and weight constraints. Finally, we check which sequences satisfy all the three constraints, which are taken as the mined pattern. All the other patterns are eliminated.
Module 3: Optimal mining via the HFGSO algorithm
The fullness of the mining process is prepared through the HFGSO algorithm after length, width, and RE constraints. The optimization is prepared to decrease the redundancy and duplication of sequences from the DNA sequence data. Here, at first, we apply the GSO algorithm to the DNA sequence pattern to mine the pattern. GSO [8] algorithms have three operators, such as producer, scrounger, and ranger. To improve the effectiveness of the system, we hybridized the FA [33, 37] and the GSO algorithm [8]. By hybridizing these two classifiers, we assume that the mining performance will be increased, which will help improve the accuracy of the mined pattern. By our assumptions, the results section shows that the proposed optimization algorithm of HFGSO achieved better performance than the individual optimization algorithm. The step-by-step process is explained below.
Step 1: Initialize the search solution as well as the head angle
Solution encoding is the important stage of the optimization algorithm. Here, we create the solution for the hybrid of the GSO and FA algorithms. The solution was based on the DNA sequences obtained from module 2. HFGSO considers the extracted sequences as the first population. At first, the search solution is initialized and, in the case of the novel technique, the solution characterizes the DNA sequence obtained from module 2. HFGSO considers the extracted sequence as the first population. Let us reflect on the first population, as follows:
The set PS represents the population of the extracted sequence, and the individuals in the population are represented with P1 to Pn . In respect of each and every individual population, the head angle can be expressed as shown in Eq. (2), and the direction of the member is given in Eq. (3):
The polar and Cartesian coordinate transformations are effectively deployed to appraise the direction of search based on the head angle.
Step 2: Fitness calculation
Once we have created the solution, then we calculate the fitness of the population. The fitness for the functions is planned based on the support, confidence, frequency, and lift parameters of the suggested approach. The support of the sequence is described as the relevance of a specific sequence in the DNA sequence database, and the support is the ratio of presence of a specific sequence in the transactions to the total number of transactions in the DNA sequence database. The minimum support is the support necessary to maintain a sequence regarding the DNA sequence database. The minimum support is symbolized as main support and is described as
where T(X,Y) is the number of transactions that enclose the sequences and Tn is the total number of transactions. The other characteristics that are referred to the fitness function are confidence and lift parameters. The parameters confidence and lift are obtained from the parameter support. The parameter can be obtained as
Hence, based on these parameters, we expand a fitness function for the suggested method for optimizing the sequences. The fitness is described by relating the confidence, support, and the lift value.
Now, ileft and iright are the item sets in the left side and right side of a sequence correspondingly. Once all the fitness values are computed, the fitness values are supplied to a fitness set, which encloses the fitness of the sequences.
Step 3: Find the producerZpof the group
The member with the top fitness of Zi is known as the producer and indicated as Zp .
Producer performance
In the course of the functioning of the GSO technique, the action of the producer Zp at the sth iteration may be described as given below.
It carries out the scanning assignment at zero degree:
(12)where dmax denotes the maximum search distance.
It accomplishes the scanning function at the right-hand-side hypercube:
(13)It executes the scanning task at the left-hand side hypercube:
where ε1 points to a normally distributed random numbers with zero mean and unity standard deviation and ε2 stands for a uniformly distributed random sequence, which has values within the range 0 and 1.
The maximum search angle Φmax is effectively represented as
The evaluation of maximum search distance dmax includes the ensuing equations:
Here, dUi and dLi represent the upper and lower limits of the ith dimension, correspondingly. The best location consisting of the most beneficial resource may be achieved by means of Eqs. (4), (5), and (6). The existing best location will give way for a new best location if its existing resource is found to be inferior to that in the new location. Otherwise, the producer preserves its location and turns its head as per the head angle direction, which is randomly produced by Eq. (19):
When the producer is unable to identify a better position even after the completion of m iterations, its head would then assume its initial position as given in Eq. (21):
Step 4: Scrounger performance
In all the iterations, many members other than the producer are selected and they are termed as scroungers. The scrounging action of the GSO generally includes the area copying task. During the sth iteration, the function of area copying, which the ith scrounger carries out, may be shaped as a movement to inch toward the producer in an intimate manner, which is illustrated as
Here, ᴏ specifies the Hadamard product that determines the product of the two vectors in an entry-wise manner and ε3 denotes a uniform random sequence lying in the interval of (0, 1). The ith scrounger continues to be in its searching task so as to make a selection of the superior chance for the purpose of linking.
Step 5: Solution update via firefly operator
The FA works based on the brightness of the birds. The firefly update is based on Eq. (23):
where B0 is the degree of attractiveness of the firefly at distance r=0, r is the distance between any two fireflies, and γ is the coefficient of light absorption.
Once the iteration is over, the fitness between the old sequence and novel sequence are compared, and the one with higher fitness is maintained. If the novel sequence has better fitness, it will be substituted with the old sequence. Alternatively, if the old sequence has higher fitness, it will be subjected to development in the next iteration of HFGSO. Likewise, the processes are prolonged until each sequence is revised. The last step of the HFGSO algorithm is optimizing the sequences based on the fitness threshold. A set for the optimized sequence is produced for storing the optimized sequences from the extorted sequences based on the fitness, defined by Sop . Let the set of sequences be S and Sd be the rejected sequences:
The above expressions specify that the set of sequences si in S is passed to either the set of optimized sequences or the set of discarded sequences.
Step 6: Termination criteria
The algorithm discontinues its execution only if a maximum number of iterations is achieved and the solution that is holding the best fitness value is selected. Once the best fitness is attained by means of the HFGSO algorithm, the selected sequences are mined DNA sequences.
5 Results and Discussion
In this section, the experimental results of the proposed approach for DNA sequence mining are explained. We evaluate the efficiency and performance of our proposed approach by comparing it with the traditional algorithm PS. In this approach, we use two sets of DNA sequence datasets, such as AF008216.1 (dataset 1) and AF348525.1 (dataset 2) [26]. The DNA to be sequenced is prepared as a single strand. The DNA sequence presents the dideoxy nucleotides (A, G, C, and T). The proposed approach has been programmed using JAVA (jdk 1.6), and the experimentation is performed on a 3.0 GHz Pentium PC machine with 2 GB main memory.
5.1 Experimental results of analysis
The basic idea of our research is to mine DNA sequence patterns with constraints using HFGSO. The following Figures 3 and 4 show our proposed approach experimental result outputs. Table 4 shows the sample data sequences. The parameter used in this HFGSO algorithm is shown in Table 5. To test the scalability of the algorithms, we choose 3000 sequences from the DNA sequence dataset and divide the selected sequences into five groups. In each group, we use protein sequences with similar length to form one or two test data sets. In different groups, data sets have different numbers of sequences from 100 to 500. Table 6 shows the number of data sets and the number of sequences in each data set.
Sample DNA Sequence Database.
| ID | Sequence |
|---|---|
| 10 | ACTATTGTAGAGTA |
| 20 | AGTATTAATCGAT |
| 30 | ACTAGTCGATCG |
| 40 | CTAGTGCGATCTATGCTTAA |
| 50 | GAGTGCTTAATCG |
Parameters of the HFGSO Algorithm.
| Step size factor α | Population size, N | Absorption coefficient, γ | Initial head angle | Producer | Scrounger | Ranger | φmax |
|---|---|---|---|---|---|---|---|
| 0–1 | 100 | 1 | 45 | 1 | 16 | 3 | π/6 |
Groups of Sequences Tested.
| Group ID | Total number of sequences | Number of datasets | Number of sequences in each dataset |
|---|---|---|---|
| 1 | 200 | 2 | 100 |
| 2 | 400 | 2 | 200 |
| 3 | 600 | 2 | 300 |
| 4 | 800 | 2 | 400 |
| 5 | 1000 | 2 | 500 |
The experimental results obtained from the proposed approach with the two types of DNA sequence datasets are described in Figure 2. Initially, the input dataset are given to the proposed approach of three-module DNA sequence mining (DNASM) algorithm to mine the sequence. The mining performance with respect to the mined sequence is given in the graphs shown in Figures 2 and 3. Figure 2A shows the total number of mined patterns by varying the length of the sequence. If the length of the sequence is 3, we obtain the mined pattern of 22. Likewise, Figure 2B shows the performance of the proposed method for dataset 1 by varying the width of the sequence. In our work, we use three constraints, such as length, width, and RE. Here, the algorithm uses an RE for discovering user-interested patterns. Weights are used to discover the pattern according to the importance of items, and length constraint is used to restrict the length of the pattern so as to reduce search space and find user-interested pattern efficiently and effectively. Figure 2C shows that we obtain the total number of mined patterns by varying the iteration. From the figure, we understand that the number of iterations increases as the total number of mined sequences also increases. Similarly, Figure 4 shows the experimental result based on dataset 2. Moreover, Figure 4 shows the number of patterns mined using different constraints. In this work, we utilized a hybridization of the three constraints of length, width, and RE. From Figure 5, we clearly understand that our proposed hybrid approach is better than the individual constraints such as length, width, and RE.
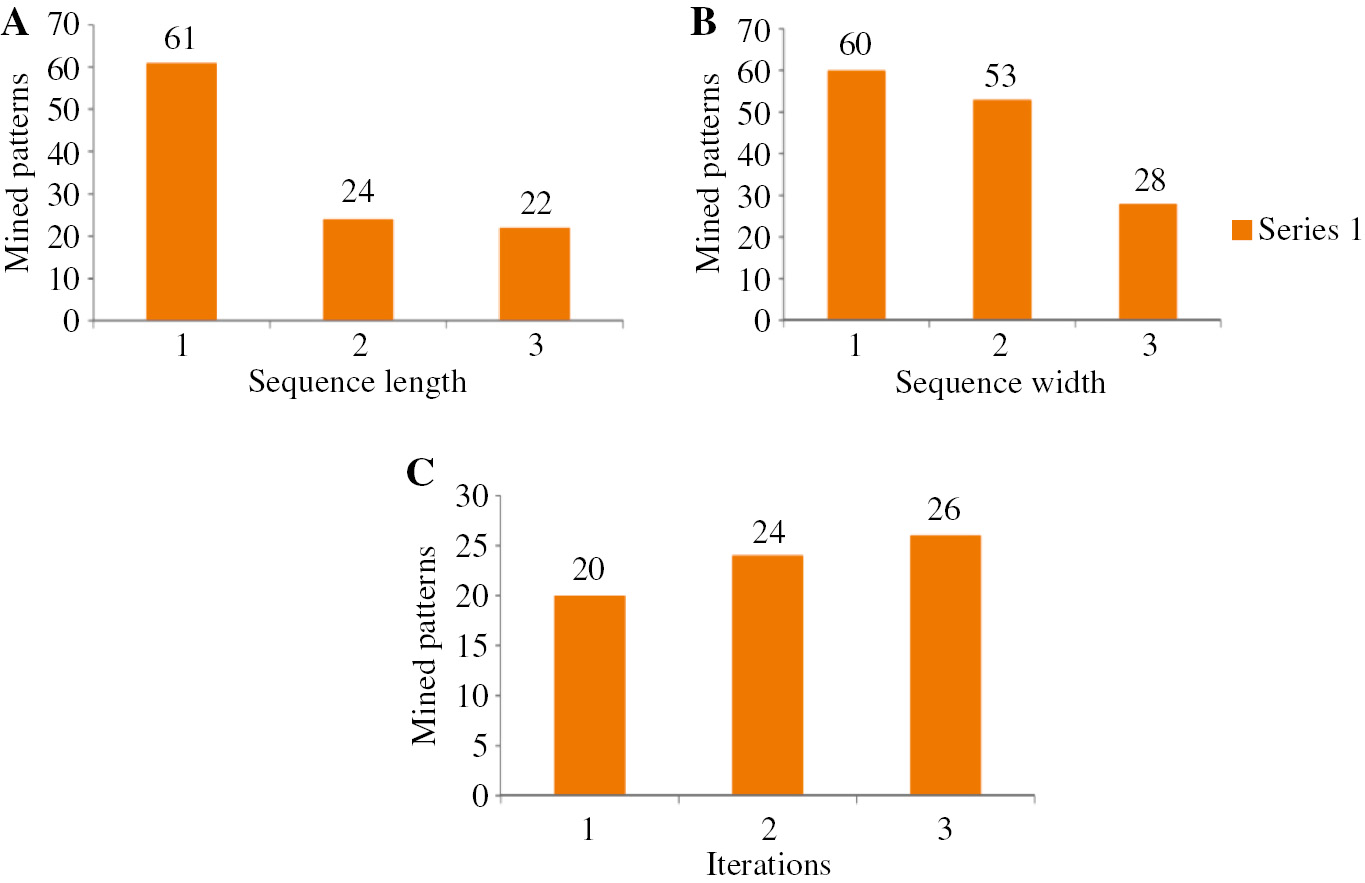
Number of Patterns Mined from Dataset 1.
(A) By varying length. (B) By varying width. (C) By varying iterations.
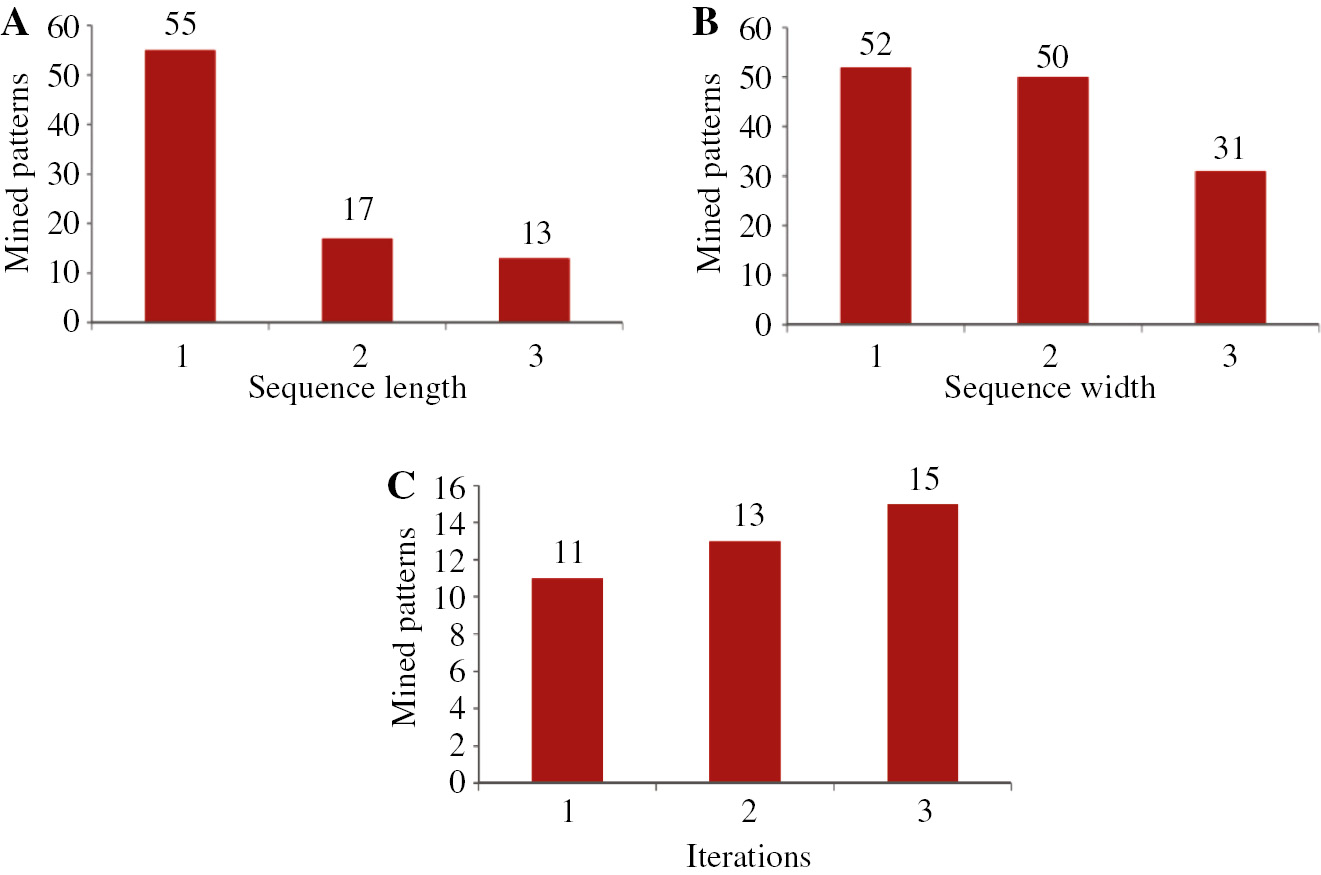
Number of Patterns Mined from Dataset 2.
(A) By varying length. (B) By varying width. (C) By varying iteration.
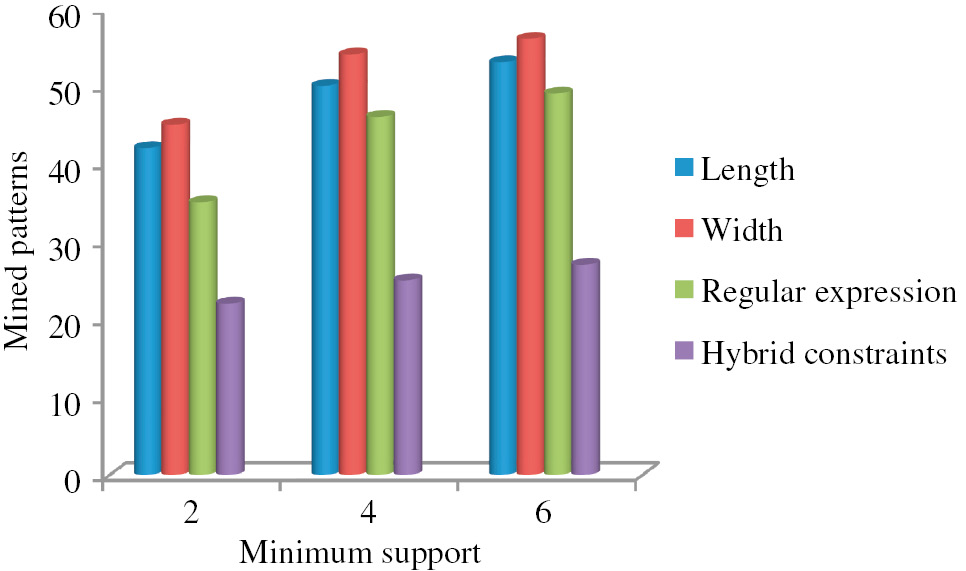
Number of Patterns Mined Using Different Constraints.
5.2 Comparative analysis of the proposed approach
This section describes the comparative analysis of the proposed approach to PS, PS+GSO, and PS+FA. The comparative result clearly ensures that the proposed approach provides an optimal order of sequential patterns compared to the existing algorithm. In the proposed approach, we use length 3 and width 2.
In this DNA sequence mining method, we use the hybrid optimization algorithm, which increases the effectiveness of the approach. We use HFGSO for the mined pattern. To prove the effectiveness of the proposed approach, we use two types of data sets for the comparison. Actually, the second dataset is synthetically generated with the view of an installation that will be done with respect to the first patterns. The support of the mined patterns from the first dataset is relatively high in the second dataset as per the data given in Tables 7 and 8. Table 7 shows the comparative analysis of the proposed approach for dataset 1. When the minimum support is 2, we obtain the mined pattern of 58 for the proposed approach, 13,118 for the PS approach, 5098 for the PS+GSO approach, and 7854 for PS+FA, which are associated with dataset 1. Similarly, we choose the minimum support of 4, which means that our proposed approach achieves the mined pattern of 63. Moreover, Table 8 shows the comparative analysis of the proposed approach for dataset 2. Here, our proposed approach achieves the minimum mined pattern of 56. In this work, we use three types of constrains that increase the effectiveness of the system. The RE constraints used to select the RE of the sequence of other patterns are eliminated. Moreover, Figure 5A and B shows the comparative analysis based on computation time. Here, we compare our proposed work with PS, PS+GSO, and PS+FA. When analyzing Figure 5A, our proposed work takes minimum time compared to other approaches. When analyzing Figure 5B, compared to other techniques, the PS-based DNA sequence approach takes maximum time. From Tables 7 and 8, we clearly understand that our proposed approach achieves better performance compared to the existing approach.
Comparative Analysis of the Proposed Approach for Dataset 1.
| Min_support | Mined patterns | |||
|---|---|---|---|---|
| PS+HFGSO | PS | PS+GSO | PS+FA | |
| 2 | 58 | 13118 | 5098 | 7854 |
| 3 | 61 | 6142 | 4321 | 6543 |
| 4 | 63 | 3768 | 2567 | 5247 |
Comparative Analysis of the Proposed Approach for Dataset 2.
| Min_support | Mined patterns | |||
|---|---|---|---|---|
| PS+HFGSO | PS | PS+GSO | PS+FA | |
| 2 | 56 | 8072 | 6543 | 7932 |
| 3 | 55 | 3636 | 5643 | 6893 |
| 4 | 53 | 2168 | 4562 | 5342 |
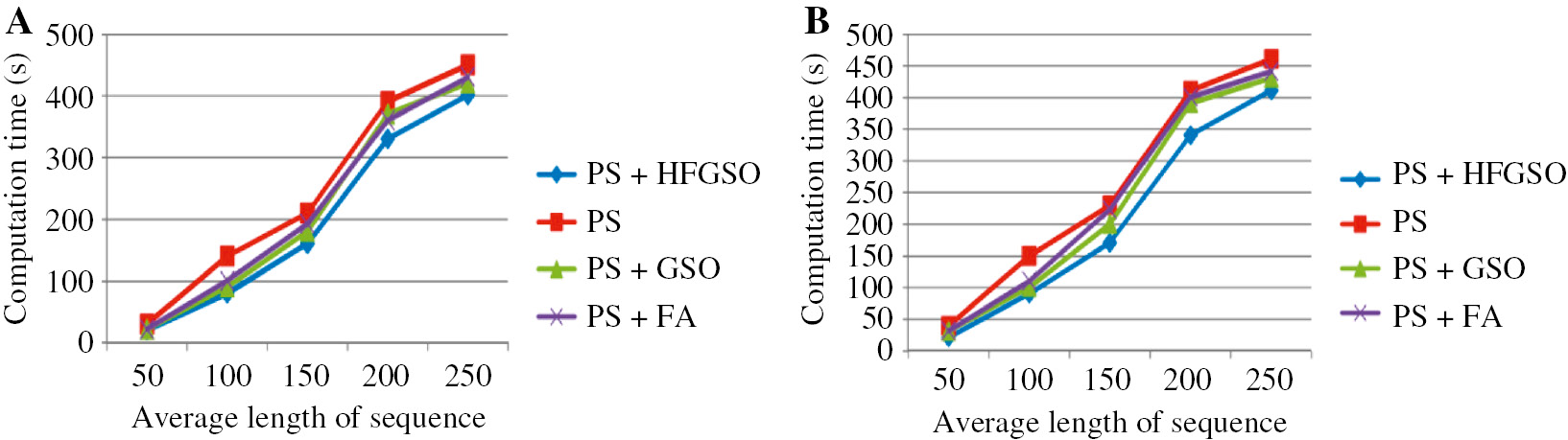
Performance Comparison Based on Computation Time.
(A) Dataset 1. (B) Dataset 2.
6 Conclusion
In this paper, we explained an algorithm that enables to efficiently manage the constant-based DNA sequence mining task. The detailed DNA sequence mining process contains mainly three modules: (i) mining based on PS algorithm, (ii) constrain-based mining, and (iii) optimal mining via HFGSO. In the first step, the concept of PS is presented, which detects frequent DNA sequence patterns using a prefix tree. After that, we apply constraint-based mining. We use three types of constraints, such as weight, length, and RE. The proposed RE constraints in the pattern mining process are used to generate a frequent pattern of user interest. Also, we used the weight of each item, which is an important indicator of each item. Similarly, our proposed algorithm uses length constraints, which restrict the length of the pattern. This algorithm generates a frequent pattern that satisfies the minimum weight, length, and RE constraints. Finally, the optimized mining result is obtained through the HFGSO algorithm. The experimentation results demonstrated that the proposed system achieved higher-quality results compared with other methods.
About the authors

Kuruva Lakshmanna has received his B-Tech in Computer Science and Engineering from Sri Venkateswara University College of Engineering – Tirupathi, India in the year 2006, M-Tech in Computer Science and Engineering(Information Security) from National Institute of Technology Calicut, Kerala, India in the Year 2009, and currently perusing his PhD from VIT University, India. He is working as an Assistant professor in VIT University, India. His research interests are Data Mining in DNA sequences, algorithms, Knowledge Mining, etc.

Neelu Khare has received her PhD in the year 2011 from NIT Bhopal, India, Masters of Computer Applications in the year 2005 from MP Bhoj University, India, Bachelors in Mathematics and Science in the year 1996 from Barkatullah University, India and Diploma in Electronics and Telecommunications in the year 1994 from MP Board of Technical Education, India. She is currently working as an Associate Professor in VIT University, India. Her research areas are Data Mining, Artificial Intelligence, Soft Computing, Natural Language Processing.
Bibliography
[1] R. Agrawal and R. Srikant, Fast algorithms for mining association rules, in: Proc. of the 20th Int’l Conference on Very Large Databases, Santiago, Chile, September 1994, expanded version available as IBM Research Report RJ9839, June 1994.Search in Google Scholar
[2] R. Agrawal and R. Srikant, Mining sequential patterns, In: Proceedings of the 11th International Conference on Data Engineering, pp. 3–14, Taiwan, 1995.Search in Google Scholar
[3] G. Aloysius and D. Binu, An approach to products placement in supermarkets using Prefix Span algorithm, J. King Saud Univ. Comput. Inform. Sci.25 (2013), 77–87.10.1016/j.jksuci.2012.07.001Search in Google Scholar
[4] R. B. A. Bakar, J. Watada and W. Pedrycz, A DNA computing approach to data clustering based on mutual distance order, In: Proceedings 9th Czech-Japan Seminar, pp. 139–145, 2006.Search in Google Scholar
[5] W. Banzaf, P. Nordin, R. Keller and F. Francone, Genetic Programming – An Introduction, Morgan Kaufmann, San Francisco, CA, 1997.Search in Google Scholar
[6] L. Chen and W. Liu, Frequent patterns mining in multiple biological sequences, Comput. Biol. Med.43 (2013), 1444–1452.10.1016/j.compbiomed.2013.07.009Search in Google Scholar PubMed
[7] C. Eugene, Bayesian network without tears, AI Mag.12 (1991), 50–63.Search in Google Scholar
[8] S. He, Q. H. Wu and J. R. Saunders, A group search optimizer for neural network training, Lect. Notes Comput. Sci.3982 (2006), 934–943.10.1007/11751595_98Search in Google Scholar
[9] S. Hou and X. Zhang, Alarms association rules based on sequential pattern mining algorithm, In: Proceedings of the 5th International Conference on Fuzzy Systems and Knowledge Discovery, Shandong, 2, 556–560, 2008.10.1109/FSKD.2008.11Search in Google Scholar
[10] Z. Z. Htike and S. L. Win, Recognition of promoters in DNA sequences using weightily averaged one-dependence estimators, Proc. Comput. Sci.23 (2013), 60–67.10.1016/j.procs.2013.10.009Search in Google Scholar
[11] K. Julisch, Data Mining for Intrusion Detection – A Critical Review, Application of Data Mining in Computer Security, Kluwer Academic Publisher, Boston, MA, 2002.10.1007/978-1-4615-0953-0_2Search in Google Scholar
[12] D. R. Kawade and K. S. Oza, Exploration of DNA sequences using pattern mining, Int. J. Emerg. Technol. Comput. Appl. Sci.2 (2013), 144–148.Search in Google Scholar
[13] D. R. Kawade and K. S. Oza, Frequent sequential pattern mining with weighted regular expression and length constraint, Int. J. Sci. Res.4 (2015), 3–7.Search in Google Scholar
[14] K. Lakshmanna and N. Khare, Constraint-based measures for DNA sequence mining using group search optimization algorithm, IJIES9 (2016), 91–100.10.22266/ijies2016.0930.09Search in Google Scholar
[15] K. Lakshmanna and N. Khare, FDSMO: frequent DNA sequence mining using FBSB and optimization, IJIES9 (2016), 157–166.10.22266/ijies2016.1231.17Search in Google Scholar
[16] K. Lakshmanna, K. Rajesh, G. Thippa Reddy, G. Nagaraja and D. V. Subramanian, An enhanced algorithm for frequent pattern mining from biological sequences. Int. J. Pharm. Technol. 8 (2016), 12776–12784.Search in Google Scholar
[17] K. C. Lin, I. E. Liao, T. P. Chang and S. F. Lin, A frequent itemset mining algorithm based on the principle of inclusion-exclusion and transaction mapping, J. Inform. Sci.276 (2014), 278–289.10.1016/j.ins.2014.02.060Search in Google Scholar
[18] J. C.W. Lin, W. Gan, T. P. Hong and V. S. Tseng, Efficient algorithms for mining up-to-date high-utility patterns, J. Adv. Eng. Inform.29 (2015), 648–661.10.1016/j.aei.2015.06.002Search in Google Scholar
[19] X. Lu, Y. Lin, X. Li, Y. Yi, L. Cai and H. Wang, Gene cluster algorithm based on most similarity tree, In: Proceedings of the Eighth International Conference on High-Performance Computing in Asia-Pacific Region, 2005.Search in Google Scholar
[20] B. Mallick, D. Garg and P. S. Grover, Constraint-based sequential pattern mining: a pattern growth algorithm incorporating compactness, length and monetary, Int. Arab J. Inform. Technol.11 (2014), 33–42.Search in Google Scholar
[21] F. Masseglia, Poncelet and M. Teisseire, Efficient mining of sequential patterns with time constraints: reducing the combinations, Expert Syst. Appl.36 (2009), 2677–2690.10.1016/j.eswa.2008.01.021Search in Google Scholar
[22] S. Mitra and T. Acharya, Data Mining: Multimedia, Soft Computing, and Bioinformatics, John Wiley & Sons, New York, 2003.Search in Google Scholar
[23] A. Nakamura, I. Takigawa, H. Tosaka, M. Kudo and H. Mamitsuka, Mining approximate patterns with frequent locally optimal occurrences, J. Discr. Appl. Math.200 (2016), 123–152.10.1016/j.dam.2015.07.002Search in Google Scholar
[24] H. A. Park, T. Kim, M. Li, H. S. Shon, J. S. Park and K. H. Ryu, Application of gap-constraints given sequential frequent pattern mining for protein function prediction, Sci. Direct6 (2015), 12–120.10.1016/j.phrp.2015.01.006Search in Google Scholar PubMed PubMed Central
[25] J. Parmar and S. Garg, Modified web access pattern (mWAP) approach for sequential pattern mining, J. Comput. Sci.6 (2007), 46–54.Search in Google Scholar
[26] J. Pei, J. Han, B. Mortazavi-Asl, H. Pinto, Q. Chen, U. Dayal and M. Hsu, Prefixspan: mining sequential patterns by prefix-projected growth, In: Proceedings of the 17th International Conference on Data Engineering, pp. 215–224, IEEE Computer Society, Washington, DC, 2001.Search in Google Scholar
[27] T. Slimani and A. Lazzez, Efficient analysis of pattern and association rule mining approaches, Int. J. Inform. Technol. Comput. Sci. (IJITCS)6 (2014), 70–81.10.5815/ijitcs.2014.03.09Search in Google Scholar
[28] G. G. Wang and S. D. Z. Cui, Monarch butterfly optimization, Neural Comput. Appl. (2015), 1–20.10.1007/s00521-015-1923-ySearch in Google Scholar
[29] G. G. Wang, A. H. Gandomi and A. H. Alavi, Stud krill herd algorithm, J. Neuro Comput.128 (2014), 223–370.10.1016/j.neucom.2013.08.031Search in Google Scholar
[30] G. G. Wang, L. Guo, H. Duan and H. Wang, A new improved firefly algorithm for global numerical optimization, J. Comput. Theor. Nano.11 (2014), 477–485.10.1166/jctn.2014.3383Search in Google Scholar
[31] G. G. Wang, L. Guo, A. H. Gandomi and H. Wang, Chaotic krill herd algorithm, Inf. Sci.274 (2015), 17–34.10.1016/j.ins.2014.02.123Search in Google Scholar
[32] G. G. Wang, S. Deb and L. D. S. Coelho, Earthworm optimization algorithm: a bio-inspired metaheuristic algorithm for global optimization problems, Int. J. Bio-Inspired Comput. (2015).10.1504/IJBIC.2015.10004283Search in Google Scholar
[33] G. G. Wang, S. Deb, A. H. Gandomi, Z. Zhang and A. H. Alavi, Chaotic cuckoo search, Soft Comput.20 (2015), 3349–3362.10.1007/s00500-015-1726-1Search in Google Scholar
[34] G. G. Wang, A. H. Gandomi, X. S. Yang and A. H. Alavi, A new hybrid method based on krill herd and cuckoo search for global optimization tasks, Int. J. Bio-Inspired Comput.8 (2016), 286–299.10.1504/IJBIC.2016.079569Search in Google Scholar
[35] X. Wu, X. Zhu, Y. He and A. N. Arslan, PMBC: pattern mining from biological sequences with wildcard constraints, Comput. Biol. Med.43 (2013), 481–492.10.1016/j.compbiomed.2013.02.006Search in Google Scholar PubMed
[36] E. Yafi, A. A. Hegami, A. Afsar and B. Ranjit, YAMI: incremental mining of interesting association patterns, Int. Arab J. Inform. Technol.9 (2012), 504–510.Search in Google Scholar
[37] X. S. Yang and X. He, Firefly algorithm: recent advances and applications, Int. J. Swarm Intell.1 (2013), 36–50.10.1504/IJSI.2013.055801Search in Google Scholar
[38] U. Yun, WIS: weighted interesting sequential pattern mining with a similar level of support and/or weight, ETRI J.29 (2007), 336–352.10.4218/etrij.07.0106.0067Search in Google Scholar
[39] U. Yun and K. H. Ryu, Discovering important sequential patterns with length-decreasing weighted support constraints, Int. J. Inform. Technol. Decis. Making9 (2010), 575–599.10.1142/S0219622010003968Search in Google Scholar
©2018 Walter de Gruyter GmbH, Berlin/Boston
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Articles in the same Issue
- Frontmatter
- ADOFL: Multi-Kernel-Based Adaptive Directive Operative Fractional Lion Optimisation Algorithm for Data Clustering
- A Multiple Criteria-Based Cost Function Using Wavelet and Edge Transformation for Medical Image Steganography
- Mining DNA Sequence Patterns with Constraints Using Hybridization of Firefly and Group Search Optimization
- An Approach to Detect Vehicles in Multiple Climatic Conditions Using the Corner Point Approach
- Adaptive Algorithm for Solving the Load Flow Problem in Distribution System
- Modeling and Optimizing Boiler Design using Neural Network and Firefly Algorithm
- Models for Predicting Development Effort of Small-Scale Visualization Projects
- An Effective Technique for Reducing Total Harmonics Distortion of Multilevel Inverter
- Neural Network Classifier for Fighter Aircraft Model Recognition
- Feature Selection Using Harmony Search for Script Identification from Handwritten Document Images
- A Novel Hybrid ABC-PSO Algorithm for Effort Estimation of Software Projects Using Agile Methodologies
Articles in the same Issue
- Frontmatter
- ADOFL: Multi-Kernel-Based Adaptive Directive Operative Fractional Lion Optimisation Algorithm for Data Clustering
- A Multiple Criteria-Based Cost Function Using Wavelet and Edge Transformation for Medical Image Steganography
- Mining DNA Sequence Patterns with Constraints Using Hybridization of Firefly and Group Search Optimization
- An Approach to Detect Vehicles in Multiple Climatic Conditions Using the Corner Point Approach
- Adaptive Algorithm for Solving the Load Flow Problem in Distribution System
- Modeling and Optimizing Boiler Design using Neural Network and Firefly Algorithm
- Models for Predicting Development Effort of Small-Scale Visualization Projects
- An Effective Technique for Reducing Total Harmonics Distortion of Multilevel Inverter
- Neural Network Classifier for Fighter Aircraft Model Recognition
- Feature Selection Using Harmony Search for Script Identification from Handwritten Document Images
- A Novel Hybrid ABC-PSO Algorithm for Effort Estimation of Software Projects Using Agile Methodologies