Multi-locus Test and Correction for Confounding Effects in Genome-Wide Association Studies
-
Donglai Chen


Abstract
Genome-wide association studies (GWAS) examine a large number of genetic variants, e. g., single nucleotide polymorphisms (SNP), and associate them with a disease of interest. Traditional statistical methods for GWASs can produce spurious associations, due to limited information from individual SNPs and confounding effects. This paper develops two statistical methods to enhance data analysis of GWASs. The first is a multiple-SNP association test, which is a weighted chi-square test derived for big contingency tables. The test assesses combinatorial effects of multiple SNPs and improves conventional methods of single SNP analysis. The second is a method that corrects for confounding effects, which may come from population stratification as well as other ambiguous (unknown) factors. The proposed method identifies a latent confounding factor, using a profile of whole genome SNPs, and eliminates confounding effects through matching or stratified statistical analysis. Simulations and a GWAS of rheumatoid arthritis demonstrate that the proposed methods dramatically remove the number of significant tests, or false positives, and outperforms other available methods.
1 Introduction
Genome-wide association studies (GWAS) contain a large number of genetic variants, i. e., single nucleotide polymorphisms (SNP), for hundreds or thousands of subjects. SNPs are single base differences in DNA sequences among individuals and can serve as genetic markers for identifying disease susceptibility genes. A SNP has three possible genotypes, wild type homozygous, heterozygous, and mutation homozygous. In a GWAS, genotypes of up to millions of SNPs are measured for disease (case) and normal (control) subjects, where differences in SNP genotype frequency between cases and controls imply genetic associations with the disease.
In most situations, statistical analysis of genome-wide association data has been on a single SNP at a time, using simple logistic regression or
Another challenge of GWAS data analysis is the presence of confounding effects. Jakobsdottir et al. [5] examined the viability of using significant SNPs from GWASs as biomarkers for predicting the risk of several diseases, for example, macular degeneration, type II diabetes, Crohn’s disease, and cardiovascular disease. It was found that the predictive power of the significant SNPs was generally poor and using SNPs could not perform better than the commonly known standards for predicting the diseases. Although GWASs tried to follow a randomized case-control design, there are a substantial amount of potential confounding factors in the data and there is often no clear choice of what the control sample should be. Some examples of confounding factors particular to GWASs are 1) genetic artifacts, i. e., systematic “errors” on the SNP array chips, 2) phenotypical and environmental heterogeneity of the subjects such as gender, ancestry, age, etc., and 3) ignorance of disease pathobiology [6–8]. Most confounding factors in GWASs are difficult to quantify as they are not directly observable.
For known confounding factors, e. g., gender, age, known disease conditions etc., multivariate logistic regression is commonly used for disease association of a SNP conditional on the confounding factors. For complicated confounding factors, e. g., latent population structure, Devlin and Roeder [9] developed a method, called Genomic Control, to measure the extent of inflation due to population stratification. Price et al. [10] used principal components analysis to identify principal components of ancestry that may demonstrate population structures. An extensive review of methods that correct for population stratification are in Price et al. [11]. A limitation of these methods is that they mainly work for population stratification but may not account for other types of confounding effects.
In this paper we propose a weighted
We demonstrate our methods on a simulated GWAS and a GWAS of rheumatoid arthritis, which is the GAW16 (Genetic Analysis Workshop 16) data from the North American Rheumatoid Arthritis Consortium. We compare the proposed methods with an existing multi-SNP approach, called Sequence Kernel Association Test (SKAT, Wu et al. [2]), and methods that correct for population stratification, including Genomic Control (Devlin and Roeder [9]) and principal components analysis (EIGENSTRAT, Price et al. [10]). The comparisons show that the proposed method based on the balancing score dramatically removes the number of significant tests, or false positives, and performs superior to the existing methods.
The rest of the paper is organized as follows. In Section 2, we propose a test for a big contingency table when we assess disease association of a block of SNPs. Section 3 introduces the rheumatoid arthritis GWAS and shows the phenomenon of false positive significances. In Section 4, we develop the method of balancing score, to identify confounding effects embedded in GWASs. In Section 5, we conduct simulation studies to compare our methods with SKAT, Genomic Control and EIGENSTRAT. We apply our method in the rheumatoid arthritis GWAS in Section 6. Finally, we conclude with a discussion in Section 7.
2 Large scale contingency tables for multiple SNPs
In GWAS data analysis, we scan the whole genome and compare disease subjects with normal subjects in terms of genotype frequencies. Significant difference of genotype frequencies at a genomic region implies association with the disease. The commonly used statistical methods analyze SNPs one at a time. We, however, consider a block of consecutive SNPs and analyze combinatorial effects of the SNP block. We use SNP blocks to scan the whole genome and hence refer to blocks as moving windows. For any given window of
There are different ways to model the count data in a
where
For a given window, there are two independent multinomial distributions for SNP genotypes of the control and case populations. We are interested in testing a null hypothesis
which corresponds to no difference between the case and control populations for the given SNP window, or the SNP window is not associated with the disease. This hypothesis is equivalent to
The null hypothesis (1) is equivalent to
Our test for a disease associated SNP window is based on this hypothesis. Let us denote
where
3 False positive significance
We have applied the weighted
We first examine the test statistic
However, when we apply the test statistic
4 Balancing score
Noticing that the false positives are located almost everywhere among all chromosomes, we find it sensible to believe that the case and the control samples are not matched well, at least, not balanced on genotypes that are not associated with the disease. This is a fundamental problem in disease studies. In general, even with case and control samples that have been well balanced on observed phenotypes, there is still a need to balance on genotypes that are not related to the disease of interest. We take a good use of genetic information and consider that unknown confounding factors are embedded in the profile of genome-wide SNPs.
Let
We use the previous notations in large contingency tables, i. e.,
Note that
We define a balancing score for subject
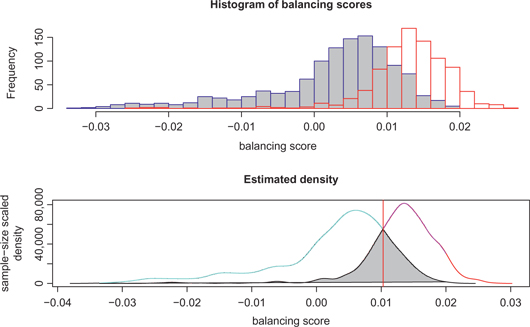
Histograms of the balancing scores (in the logarithm scale) for the case and control samples, where cases are represented in red on the right hand side and controls are represented in blue on the left hand side.
We calculate the balancing scores for all 2,062 subjects in the rheumatoid arthritis GWAS. Figure 1 shows histograms of the balancing scores, in the logarithm scale, from subjects in the case group (red color and on the right hand side) and subjects in the control group (blue color and on the left hand side) respectively. The figures indicate that there are apparent differences between the case and the control groups. In other words, the case and the control groups do not have a balanced distribution in terms of the balancing score. We conclude that confounding effects exist in this rheumatoid arthritis GWAS. In general, less overlap between the case and the control distributions, stronger confounding effects. On the contrary, more overlap between the case and the control distributions, less confounding effects are identified. In Section 6, we will show the latter is the case in the principal components analysis of this data set.
We can interpret the balancing score as a summary feature of all possible confounding factors for the respective subject, where the confounding factors are not limited to population stratification. The confounding effects need to be controlled when we compare the case and the control samples. A simple approach is the sample matching method, which matches a case subject to a control subject with respect to their balancing scores. Without loss of generality, assume control subjects tend to have lower balancing scores. We define a cutoff line at the intersection of the two density curves of the balancing scores for the case and control subjects, as shown by the vertical line in Figure 1. To the left of the cutoff line, where there are more control samples than the cases, every case is matched to a control subject with the nearest balancing score. That is, the control subject most similar to a given case in terms of balancing score is selected for a case-control matching pair. In a rare situation that two control samples have exactly the same balancing score with the case, one control is arbitrarily selected. Analogously, to the right of the cutoff line, where there are more case samples than the controls, every control sample matches a case with the nearest balancing score. This procedure matches subjects only in the intersection of the two balancing score distributions and subjects cannot be matched twice. The weighted
The balancing score can also be used in a stratified analysis. We place subjects into
Each
5 Simulation studies
The simulation is similar to those in Price et al. [10], where confounding effects are simply from population stratification. We demonstrate that the proposed method works very well, although the simulation scenario is in favor of the principal components analysis method.
We generate genotype data of 100,000 SNPs for 500 cases and 500 controls. We consider a mixture of two populations in the samples. For the 500 cases, a proportion
We generate ten simulation data sets for both scenarios of no population stratification and population substructure. We then compare a variety of association tests under each scenario in Table 1. The association tests include the standard Armitage trend
Comparison of association tests. Average number of significant SNPs from 10 simulations among true causal SNPs (True Positive) and non-causal SNPs (False Positive) are reported.
| No stratification | Population stratification | |||
|---|---|---|---|---|
| Causal | Non-causal | Causal | Non-causal | |
| SNPs | SNPs | SNPs | SNPs | |
| Armitage | 10 | 5.6 | 9 | 39,357 |
| EIGENSTRAT | 10 | 8.8 | 10 | 9.5 |
| SKAT | 12.6 | 13.4 | – | – |
| SKAT with PCs | – | – | 14.4 | 18.8 |
| Unmatched | 18 | 13 | 18 | 6,878 |
| Balancing score | 18 | 12.1 | 17.7 | 12.7 |
Table 1 shows the average numbers of significant SNPs or SNP windows by different methods in ten simulations. Note that the Armitage and EIGENSTRAT tests are for SNPs one by one, but SKAT and our method test for multiple SNPs. There are 10 causal SNPs but 18 causal SNP windows, where a causal window is referred to as a window of size 5 that contains at least two causal SNPs. For the scenario of no population stratification, the standard SKAT is used, which does not account for confounding effects. For the scenario of population stratification, we include the first two principal components of the genotype data in the SKAT regression model, so that it also corrects for the population substructure. Our simulations indicate that when there is no population confounding effect, all methods work well. However, when there is population stratification, the methods that do not consider confounding effect, i. e., Armitage and unmatched
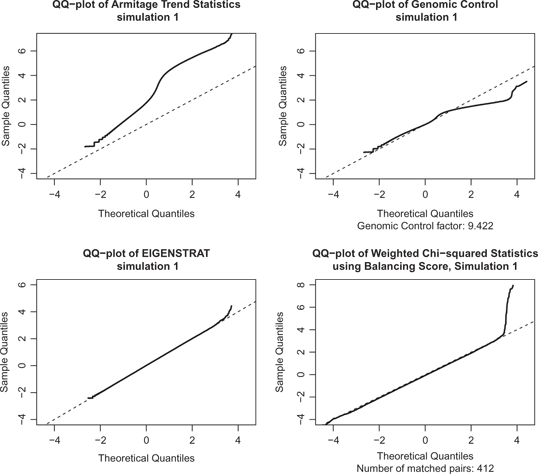
QQ-plots of four methods of association tests for the simulation data.
Figure 2 shows QQ-plots of four associate tests for one simulation. Besides Armitage, EIGENSTRAT, and our balancing score method, we also illustrate the Genomic Control adjusted test, which divides the Armitage test by the Genomic Control factor
6 Application in a GWAS
We have applied the Armitage
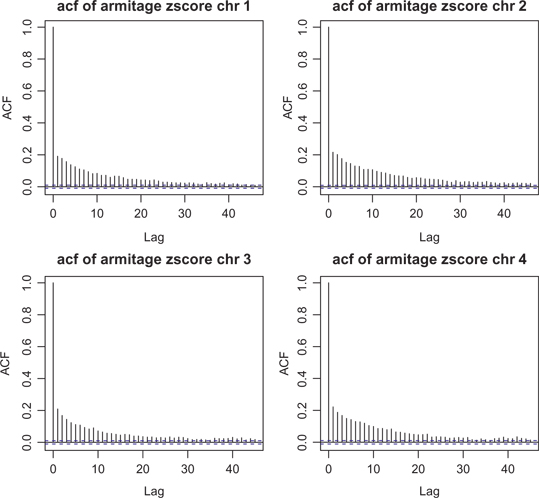
The autocorrelation function (acf) of the Armitage statistic for the first four chromosomes of the GWAS data of rheumatoid arthritis. The reduction of SNP autocorrelation over lag values indicates that the SNP window size 10 is a fine choice.
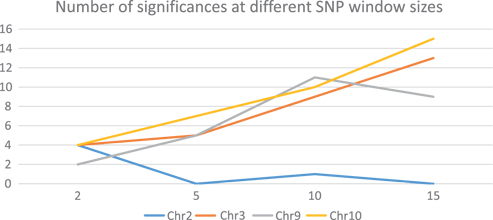
Effect of SNP window size as indicated by the number of significances over different window sizes. Most chromosomes are similar to the lines of Chromosome 2 at the bottom, with very few significances.
Applying our method, we have previously showed that there is substantial difference in the distributions of the balancing scores between the case subjects and the control subjects (Figure 1). Hence, our method correctly identifies the confounding effect. On the other hand, Figure 5 demonstrates that the first two principal components from EIGENSTRAT have great overlaps between the distributions of the case subjects and the control subjects. As the distributions are similar between the case and control groups, the principal components from EIGENSTRAT do not find the confounding effects that are indicated by the balancing score.
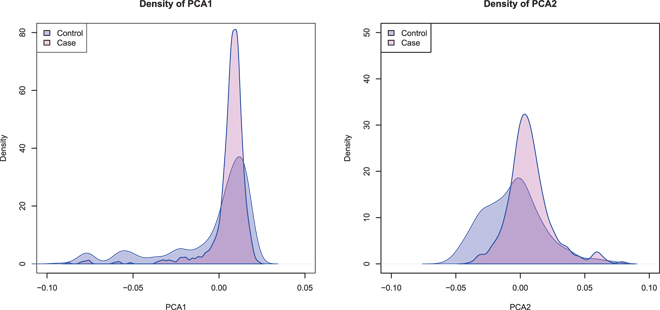
Distributions of the top two principal components from EIGENSTRAT in the case and control groups. The great overlaps indicate the principal components analysis is not able to identify the confounding effect in the GWAS data set.
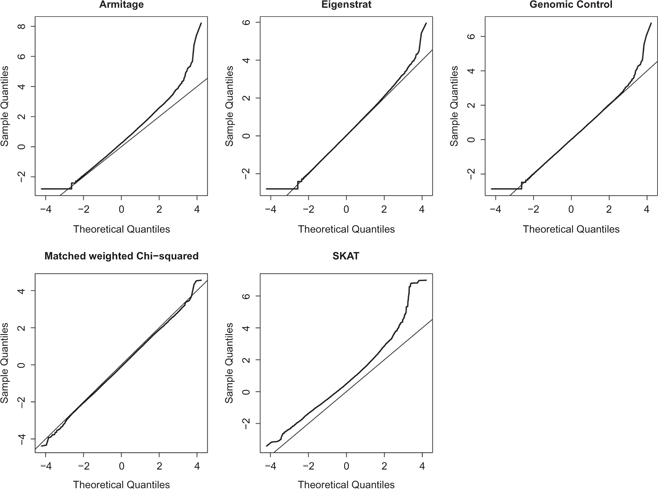
QQ-plots of five methods of association tests for Chromosome 1 in the rheumatoid arthritis GWAS. Tails on the top of the plots are likely false positives.
Figure 6 displays QQ-plots of five association tests for Chromosome 1, which comprises 40,929 SNPs. We use p-value
The QQ-plots of our method for all chromosomes are shown on the second panel in Figure 7. The number of significant test statistics is greatly reduced for all chromosomes except for Chromosome 6. The excessive number of false positives on the first panel in Figure 7 are removed by adjusting for the balancing score. The significant SNP windows in Chromosome 6 correspond to the known disease associated gene HLA. Most other chromosomes do not have any significant SNP windows. The significant SNP windows in Chromosome 3, 9, 10, and 22 suggest possible genetic variants that may associate with the disease rheumatoid arthritis.
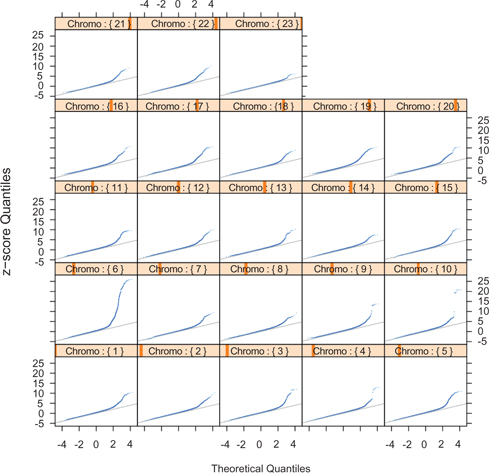
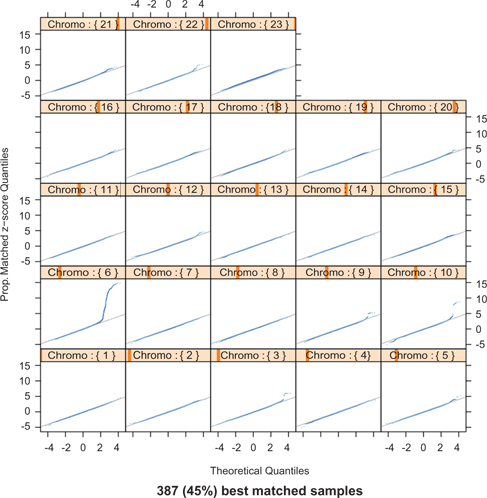
Comparison of the amount of false significant test statistics (the up tails of the QQ plots) for the raw data (the first panel) and the balancing score matched data (the second panel). The first panel: QQ-plots of the proposed test statistic Y for SNP blocks in each chromosome from the GWAS of rheumatoid arthritis. The test statistic is converted to a z-score,
To study the issue of automatic reduction of false positives due to the smaller sample size of about 400 matched pairs, we calculate the proposed test statistic
Alternatively, we stratify all 2,062 subjects in the rheumatoid arthritis GWAS into
7 Discussion
We define a balancing score that represents potential confounding factors from the genome-wide SNP profile. Different distributions of the balancing scores between the case and the control groups indicate confounding effects. We propose the matching and stratification methods that emulate a more balanced design prior to analysis. Our methods are specially useful when confounding factors are unknown and not limited to population stratification. Although the balancing score is particularly defined for blocks of multiple SNPs, it can also be calculated for single-SNP analysis. In fact, we may simply consider a SNP window size 1 with three genotypes of the single SNP. The calculation as in Section 4 follows automatically.
A slight and known tradeoff to the matching approach is the possible reduction of sensitivity due to the loss of subjects in the analysis. We argue however that the removal of confounding effects greatly outweighs the loss in sensitivity, especially as we were able to still declare significance of the association between SNPs in Chromosome 6 and the prevalence of rheumatoid arthritis in the given GWAS.
Although we recommend our technique for any GWAS study, it is not the end-all-be-all solution to controlling confounding effects. Because the balancing score is a summarized measure it may not be able to adjust for some specific confounding factors as the effect can be “lost” within the summarization. Thus we recommend our technique be used not as a substitute for current methods that adjust GWAS studies for known confounding variables but rather as a technique to augment analysis. If a known confounding factor affects analysis, it may be prudent to adjust analysis prior to estimating the balancing score. For example if gender is known to alter the likelihood of a subject being affected by disease we suggest stratifying subjects by gender and then calculating the balancing score within each stratum. Adjusting for a known confounding variable will likely trump the effect of the balancing score but the advantage of the balancing score is more specifically to adjust for unknown confounding variables which can potentially exist in any study. As such our method may be seen as an exploratory method as well to identify the degree of confound effects among the subjects. Additionally, this technique is not limited to GWAS studies but can be employed for any case-control study involving genomic sequence data.
Funding source: National Science Foundation
Award Identifier / Grant number: DMS-1007678
Funding source: National Institutes of Health
Award Identifier / Grant number: R21GM101504
Funding statement: This work is supported by the National Science Foundation Grant DMS-1007678 and the National Institutes of Health Grant R21GM101504.
Acknowledgments
The authors thank the Editor and the anonymous referees for their helpful comments and suggestions that enhanced the paper. The authors also thank Kelvin Ma for assistance in an early draft and implementation of the stratification method in Section 6.
References
1. McCarthy M, Abecasis G, Cardon L, Goldstein D, Little J, Ioannidis J, et al. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev Genet 2008;9(5):356–69.10.1038/nrg2344Suche in Google Scholar PubMed
2. Wu MC, Lee S, Cai T, Li Y, Boehnke M, Lin X. Rare-variant association testing for sequencing data with the sequence Kernel association test. Am J Human Genet 2011;89:82–93.10.1016/j.ajhg.2011.05.029Suche in Google Scholar PubMed PubMed Central
3. Zhang Y. A novel bayesian graphical model for genome-wide multi-snp association mapping. Genet Epidemiol 2012;36:36–47.10.1002/gepi.20661Suche in Google Scholar PubMed PubMed Central
4. Qiao D, Cho MH, Fier H, Bakke PS, Gulsvik A, Silverman EK, et al. On the simultaneous association analysis of large genomic regions: a massive multi-locus association test. Bioinformatics 2014;30:157–64.10.1093/bioinformatics/btt654Suche in Google Scholar PubMed PubMed Central
5. Jakobsdottir J, Gorin MB, Conley YP, Ferrell RE, Weeks DE. Interpretation of genetic association studies: Markers with replicated highly significant odds ratios may be poor classifiers. PLoS Genet 2009;5:e10000337.10.1371/journal.pgen.1000337Suche in Google Scholar PubMed PubMed Central
6. Begum F, Ghosh D, Tseng GC, Feingold E. Comprehensive literature review and statistical considerations for gwas meta-analysis. Nucleic Acids Res 2012;40:3777–84.10.1093/nar/gkr1255Suche in Google Scholar PubMed PubMed Central
7. Moore JH, Asselbergs FW, Williams SM. Bioinformatics challenges for genome-wide association studies. Bioinformatics 2010;26:445–55.10.1093/bioinformatics/btp713Suche in Google Scholar PubMed PubMed Central
8. Tian C, Gregersen PK, Seldin MF. Accounting for ancestry: population substructure and genome-wide association studies. Human Mol Genet 2008;17:R143–R150.10.1093/hmg/ddn268Suche in Google Scholar PubMed PubMed Central
9. Devlin B, Roeder K. Genomic control for association studies. Biometrics 1999;55(4):997–1004.10.1111/j.0006-341X.1999.00997.xSuche in Google Scholar
10. Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 2006;38:904–9.10.1038/ng1847Suche in Google Scholar PubMed
11. Price AL, Zaitlen NA, Reich D, Patterson N. New approaches to population stratification in genome-wide association studies. Nat Rev Genet 2010;11:459–63.10.1038/nrg2813Suche in Google Scholar
12. Liu C, Xie J. Large scale two sample multinomial inferences and its applications in genome wide association studies. Int J Approximate Reasoning 2013. doi:10.1016/j.ijar.2013.04.010Suche in Google Scholar
13. Moschopoulos P, Canada WB. The distribution function of a linear combination of chi-squares. Comput Math Appl 1984;10:383–6.10.1016/0898-1221(84)90066-XSuche in Google Scholar
14. Balding D, Nichols R. A method for quantifying differentiation between populations at multi-allelic loci and its implications for investigating identify and paternity. Genetica 1995;96:3–12.10.1007/BF01441146Suche in Google Scholar PubMed
15. Armitage P. Tests for linear trends in proportions and frequencies. Biometrics 1955;11:375–86.10.2307/3001775Suche in Google Scholar
© 2016 Walter de Gruyter GmbH, Berlin/Boston
Artikel in diesem Heft
- Research Articles
- A Comparison of Some Approximate Confidence Intervals for a Single Proportion for Clustered Binary Outcome Data
- Effect of Smoothing in Generalized Linear Mixed Models on the Estimation of Covariance Parameters for Longitudinal Data
- Adaptive Design for Staggered-Start Clinical Trial
- A Binomial Integer-Valued ARCH Model
- Testing Equality in Ordinal Data with Repeated Measurements: A Model-Free Approach
- Mendelian Randomization using Public Data from Genetic Consortia
- Tree Based Method for Aggregate Survival Data Modeling
- Multi-locus Test and Correction for Confounding Effects in Genome-Wide Association Studies
- Semiparametric Regression Estimation for Recurrent Event Data with Errors in Covariates under Informative Censoring
- Joint Model for Mortality and Hospitalization
- Effect Estimation in Point-Exposure Studies with Binary Outcomes and High-Dimensional Covariate Data – A Comparison of Targeted Maximum Likelihood Estimation and Inverse Probability of Treatment Weighting
- Sample Size for Assessing Agreement between Two Methods of Measurement by Bland−Altman Method
- Using Relative Statistics and Approximate Disease Prevalence to Compare Screening Tests
- Multiple Comparisons Using Composite Likelihood in Clustered Data
Artikel in diesem Heft
- Research Articles
- A Comparison of Some Approximate Confidence Intervals for a Single Proportion for Clustered Binary Outcome Data
- Effect of Smoothing in Generalized Linear Mixed Models on the Estimation of Covariance Parameters for Longitudinal Data
- Adaptive Design for Staggered-Start Clinical Trial
- A Binomial Integer-Valued ARCH Model
- Testing Equality in Ordinal Data with Repeated Measurements: A Model-Free Approach
- Mendelian Randomization using Public Data from Genetic Consortia
- Tree Based Method for Aggregate Survival Data Modeling
- Multi-locus Test and Correction for Confounding Effects in Genome-Wide Association Studies
- Semiparametric Regression Estimation for Recurrent Event Data with Errors in Covariates under Informative Censoring
- Joint Model for Mortality and Hospitalization
- Effect Estimation in Point-Exposure Studies with Binary Outcomes and High-Dimensional Covariate Data – A Comparison of Targeted Maximum Likelihood Estimation and Inverse Probability of Treatment Weighting
- Sample Size for Assessing Agreement between Two Methods of Measurement by Bland−Altman Method
- Using Relative Statistics and Approximate Disease Prevalence to Compare Screening Tests
- Multiple Comparisons Using Composite Likelihood in Clustered Data