A FACS-based screening strategy to assess sequence-specific RNA-binding of Pumilio protein variants in E. coli
-
Stefanie J. Kellermann


Abstract
Sequence-specific and programmable binding of proteins to RNA bears the potential to detect and manipulate target RNAs. Applications include analysis of subcellular RNA localization or post-transcriptional regulation but require sequence-specificity to be readily adjustable to any target RNA. The Pumilio homology domain binds an eight nucleotide target sequence in a predictable manner allowing for rational design of variants with new specificities. We describe a high-throughput system for screening Pumilio variants based on fluorescence-activated cell sorting of E. coli. Our approach should help optimizing variants obtained from rational design regarding folding and stability or identifying new variants with alternative binding modes.
Over the last years there has been an increasing interest not only in the detection of certain RNAs, but also in the manipulation of specific transcripts. RNA-binding proteins with tunable sequence-specificity are of particular interest for these applications, as they can be produced by the cellular machinery and allow binding of endogenous RNAs without the need for additional RNA tags. The family of PUF proteins, named after Drosophila melanogaster Pumilio and Caenorhabditis elegans fem-3 mRNA binding factor (FBF) (Murata and Wharton, 1995; Zhang et al., 1997) seems to be ideally suited to target single-stranded RNA sequence-specifically. Members of the family of PUF proteins contain a conserved motif referred to as the Pumilio homology domain (Pum-HD), which folds into a crescent shaped structure containing eight PUF repeats (Edwards et al., 2001; Wang et al., 2001; Zamore et al., 1997; Zhang et al., 1997). A set of three amino acids from each repeat is responsible for specific binding of one RNA nucleobase (Wang et al., 2002). The underlying binding code could be deciphered and numerous Pumilio variants were designed that bind their target RNAs with nanomolar affinities (Wang et al., 2002; Cheong and Hall, 2006; Dong et al., 2011; Filipovska et al., 2011). The construction of variants with more than one modified PUF repeat has been significantly simplified by the recent development of a repeat module library that can be assembled by Golden Gate cloning (Abil et al., 2014; Adamala et al., 2016). By fusing variants of the Pum-HD to different effector domains, various designer proteins have been generated that combine the sequence-specificity of the RNA-binding domain with either a fluorescent reporter or a specific enzymatic function. Applications of such fusion constructs include RNA detection (Ozawa et al., 2007; Tilsner et al., 2009; Yamada et al., 2011; Yoshimura et al., 2012; Adamala et al., 2016; Kellermann and Rentmeister, 2016), modulation of splicing processes (Wang et al., 2009a; Dong et al., 2011), translation enhancement (Cooke et al., 2011; Campbell et al., 2014; Cao et al., 2014) and inhibition (Cooke et al., 2011; Abil et al., 2014; Cao et al., 2014, 2015), as well as cleavage of RNA (Choudhury et al., 2012).
The growing number of structural datasets available for PUF proteins provides detailed insights into the molecular recognition underlying the protein-RNA interactions. An increasing number of examples reveal deviations from the canonical binding mode, in which eight PUF repeats recognize eight consecutive RNA bases. Alternative binding modes were inter alia identified for Saccharomyces cerevisiae Puf4p and C. elegans FBF, both of which prefer a binding motif of nine nucleotides (Gerber et al., 2004; Opperman et al., 2005). To accommodate the additional nucleotides, up to three of them are flipped-out, pointing away from the protein (Opperman et al., 2005; Miller et al., 2008; Wang et al., 2009b). Another example is a preference for cytosine two bases upstream of the actual binding site. This mode of recognition has been observed for Puf3p, FBF-2, PUF-6 and PUF-11 (Gerber et al., 2004; Zhu et al., 2009; Qiu et al., 2012).
Alternative binding modes could broaden the range of RNA-sequences targetable by Pumilio proteins but at the same time call for thorough validation and testing of engineered Pumilio variants regarding unanticipated sequence-specificity as well as cross reactivity. Furthermore, rationally engineered variants with multiple substitutions are likely to show reduced stability and solubility. These properties can be improved by directed evolution including all residues and not only the three per repeat involved in target recognition, but a suitable high-throughput screening method is required. The yeast hybrid-system is an established method for analysis of protein-RNA interactions and has also been utilized for identifying part of the Pumilio binding code (Dong et al., 2011; Filipovska et al., 2011). However, the method is relatively complex and requires a committed investment into the creation and optimization of its different modules such as molecular cloning and screening strategies – in yeast. We wanted to provide a user-friendly alternative for testing Pumilio-RNA interactions in vivo based on E. coli, which is widely used in laboratories working at the interface of chemistry and biology.
We have previously established tetramolecular fluorescence complementation (TetFC) for sequence-specific detection of RNA in vitro. TetFC uses two variants of the RNA-binding protein Pumilio and a three-partite split-GFP (Figure 1A) (Cabantous et al., 2013; Kellermann et al., 2013). Each of the two Pumilio proteins is tagged with one of the β-strands of GFP (S10 and S11, respectively). TetFC shows a very good signal-to-background ratio (70-fold) and reconstitution of GFP-fluorescence strictly depends on the interaction of both Pumilio fusion proteins with the target RNA and the presence of detector fragment GFP1-9 (containing the remaining nine β-strands, Figure 1A) (Kellermann et al., 2013). Since all of these four components can be genetically encoded, we thought that we could implement TetFC in E. coli. If successful, cells containing a target RNA and two binding Pumilio proteins should show a fluorescent signal discernable from background. In combination with a high-throughput screening technique such as fluorescence-activated cell sorting (FACS), this system could be readily adapted to test libraries of Pumilio proteins or RNA-sequences.
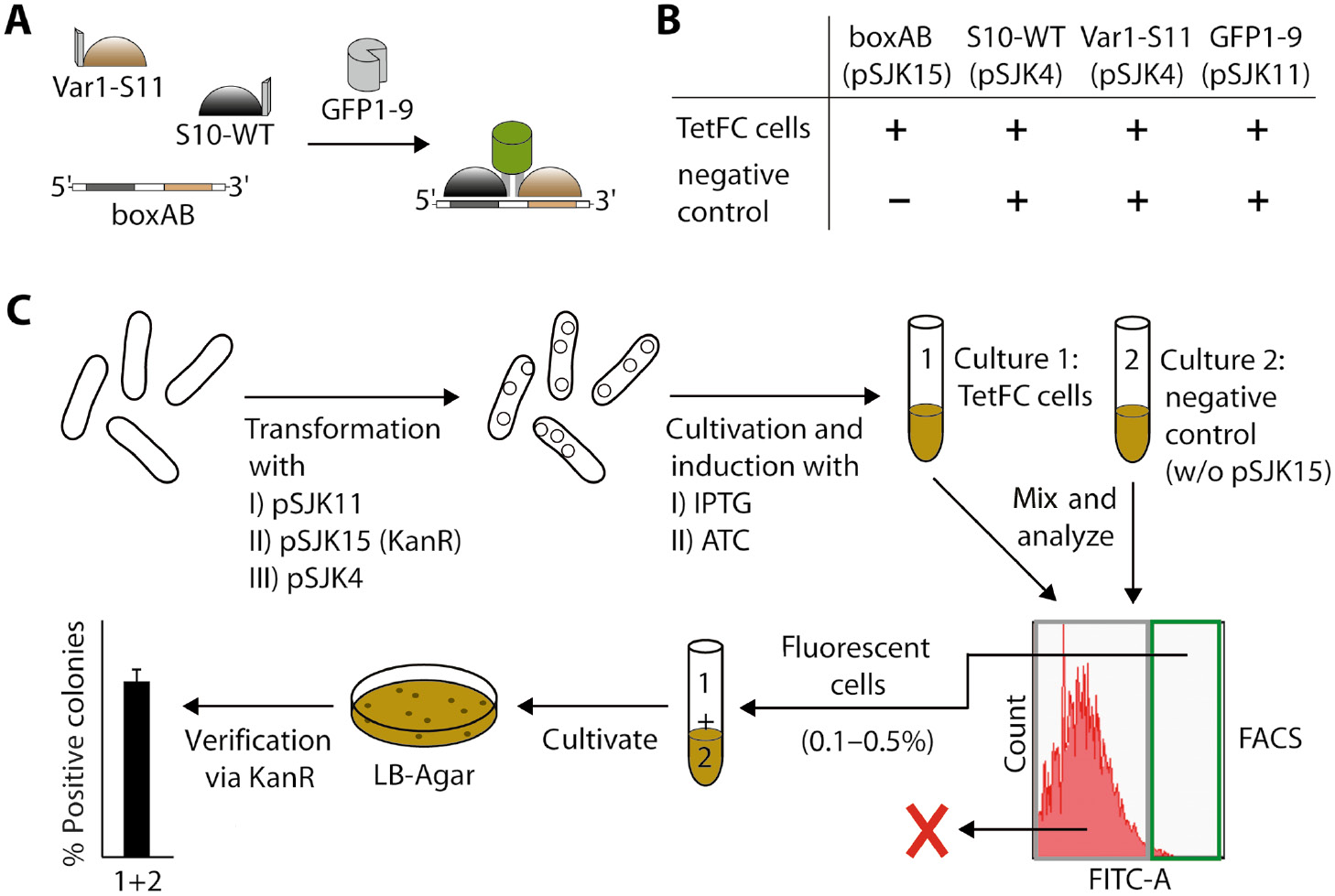
FACS-based screening strategy for analysis of sequence-specific RNA-protein interaction using tetramolecular fluorescence complementation (TetFC).
(A) General design of TetFC. Pumilio fusion proteins S10-WT and Var1-S11 come into close proximity only if the target RNA (here boxAB) is present. Upon addition of non-fluorescent reporter fragment GFP1-9, GFP can reconstitute, resulting in a fluorescence signal. (B) Comparison of TetFC cells and negative control cells. Plasmids for production of each component of TetFC are indicated in brackets. (C) Flow scheme of the FACS-based screening strategy. First, E. coli cells were transformed with I) pSJK11 for production of GFP1-9, then with II) pSJK15 for target RNA boxAB and lastly with III) pSJK4 for simultaneous production of Pumilio fusion proteins S10-WT and Var1-S11. Cells were cultivated and gene expression was induced by addition of IPTG. Production of RNA boxAB was induced independently using anhydrotetracycline (ATC). For validation, TetFC cells and negative control cells (without plasmid pSJK15 but induced with ATC) were mixed and sorted using FACS. The 0.1–0.5% strongest fluorescing cells were collected, cultivated and subsequently tested for kanamycin resistance.
For in vivo application, the four individual TetFC components, namely S10-WT, Var1-S11, GFP1-9 and the RNA of interest, had to be produced by the cellular machinery. We established and tested four different approaches to introduce the different genes into E. coli: (i) chromosomal integration of gfp1-9 into the E. coli genome, (ii) a two plasmid approach, (iii) stabilization of the target RNA, and (iv) target RNA production decoupled from the production of the TetFC proteins (supplementary Figure S1).
The simplest system was based on chromosomal integration of the detector fragment GFP1-9. This is the only TetFC component that never has to be changed and therefore integration into the genome seemed reasonable. The gfp1-9 was placed under the control of the constitutive Lambda phage promoter pL (λpL) (see supplementary Figure S2 for the expression system and the cloning cassette). We chose three different integration sites that had previously been described for the generation of E. coli knockout strains or the insertion of gfp into the genome of E. coli (supplementary Figure S2B) (Pinheiro et al., 2008; Miao et al., 2009; Gerstle et al., 2012). The resulting strains BL21(DE3) (ΔrybA::FRT-KanR-FRT-gfp1-9), BL21(DE3) (ΔznuA::FRT-KanR-FRT-gfp1-9) and BL21(DE3) (283526–284439 FRT-KanR-FRT-gfp1-9) were termed ACR001, ACR002 and ACR003, respectively. All three strains were equally vital (supplementary Figure S3B). Comparable amounts of soluble GFP1-9 could be detected in ACR001, ACR002 and ACR003 using bimolecular fluorescence complementation (BiFC) with the previously described fusion protein Pum-S10-S11 (containing WT HsPumHD and the remaining two GFP strands S10 and S11 (Rath et al., 2014; supplementary Figure S3C,D). However, in strains with only one copy of gfp1-9 integrated into the genome, the protein concentration might be relatively low and mainly depend on the promoter strength. Indeed, when testing lysate of E. coli BL21(DE3) ACR002 cells for BiFC, we observed a relatively low fluorescence signal compared to cells containing a medium copy plasmid with the same promoter/gene combination (Figure 2E). The difference in fluorescence intensity correlated with the estimated copy number of the pET-plasmid used (15–40) (Sambrook and Russell, 2001; Novagen, 2004), suggesting that using an entirely plasmid-based integration of the TetFC system might be advantageous.
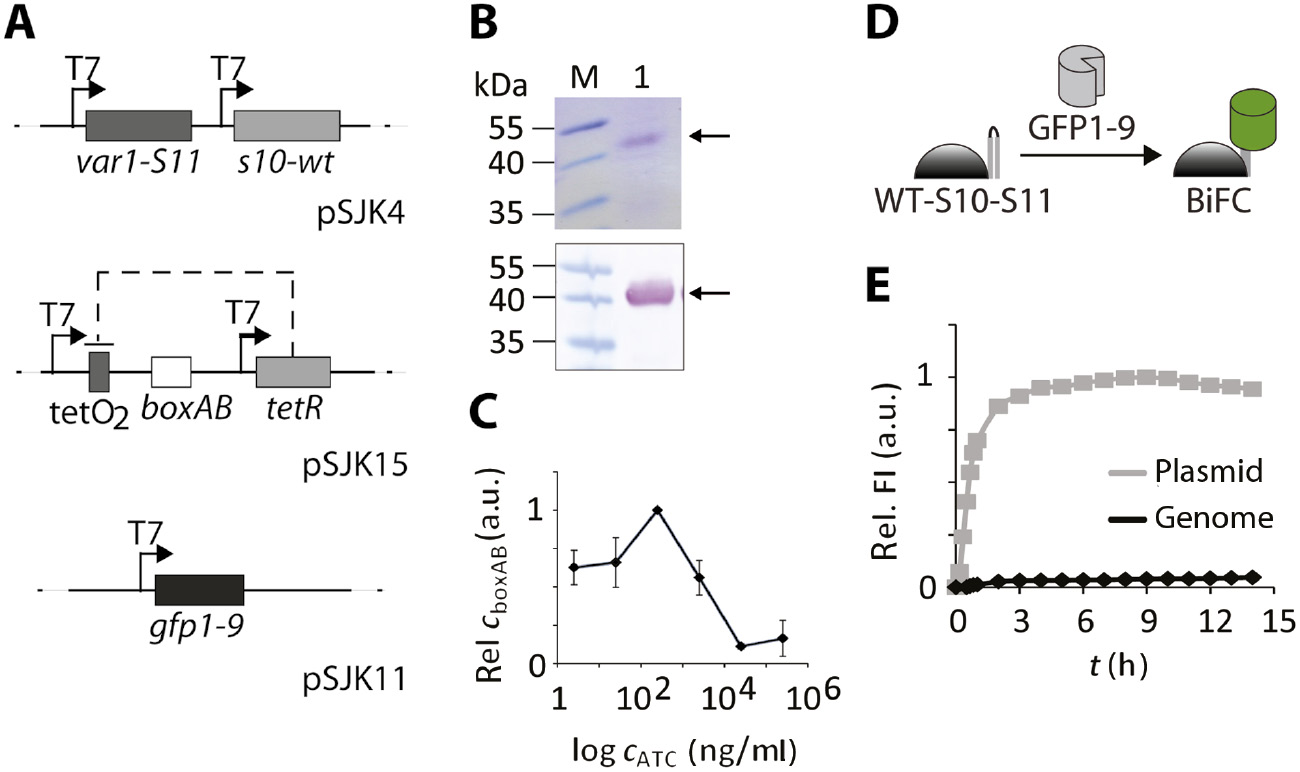
In vitro characterization of the expression systems used for TetFC in E. coli cells.
(A) Schematic of an expression system in E. coli, in which boxAB RNA production was decoupled from recombinant protein production. GFP1-9 was encoded on a pUC plasmid (pSJK11), pACYCDuet™-1 was used for production of fusion proteins Var1-S11 and S10-WT (pSJK4). Vector pRSDuet™-1 was chosen for the target RNA boxAB (pSJK15). For independent production of RNA boxAB, a TetR/tetracycline system was used. Genes tetR, as well as var1-S11, S10-wt and gfp1-9, were each placed under the control of an individual T7 promoter. For plasmid pSJK15, two TetR binding sites (tetO2) were inserted between the first T7 promoter and boxAB. (B) Production of TetFC fusion proteins in the expression system shown in (A). E. coli BL21(DE3) pSJK4 cells were harvested after recombinant protein production (0.16 mm IPTG at OD600=0.4, 3 h, 37°C). Purified proteins were analyzed using SDS-PAGE and Coomassie-staining (top) and Western blot using anti-his antibody (bottom). (M) Marker PageRuler™ Prestained Protein Ladder, (1) purified proteins. (C) Validation boxAB RNA production at different ATC concentrations. E. coli BL21(DE3) pSJK11+pSJK15+pSJK4 cells were harvested after recombinant protein and RNA production (0.16 mm IPTG at OD600=0.4 for 2 h, 2.5–2.5×105 ng/ml ATC for 1.5 h, 37°C). The qRT-PCRs were performed with isolated total RNA and normalized to the housekeeping gene IdnT. Standard deviation of three independent experiments is shown. (D) Schematic illustration of the bimolecular fluorescence complementation (BiFC) using Pumilio-WT-S10-S11 as a reporter of soluble GFP1-9 levels. (E) Comparison of chromosomal integration (black line) and vector-based integration of gfp1-9 (gray line) by BiFC with WT-S10-S11. For chromosomally integrated gfp1-9E. coli ACR002 cells were used. Cell lysate of E. coli BL21(DE3) pSJK1 was applied for plasmid-based gfp1-9 production. λex=488 nm, λem=530 nm. Data were normalized to the maximum fluorescence of the plasmid-based integration. Data points are average values of a typical experiment performed in duplicate.
To increase the concentration of GFP1-9 within the cell while keeping the system simple and stress to cells as low as possible, we next designed a two-plasmid-based approach (supplementary Figure S1B). Plasmid pSJK7 has a medium copy number and contains genes for fusion proteins S10-WT and Var1-S11, as well as the sequence for target RNA boxAB, each under control of an individual T7 promoter. For the second plasmid a pUC vector was chosen, as it contains a derivative of the pMB1 origin of replication for a higher copy number of up to several hundred (Sambrook and Russell, 2001). The gfp1-9 was placed under control of a fourth T7 promoter (pSJK11). This way, all four components should be produced simultaneously upon addition of IPTG. We could validate presence of both target RNA and fusion proteins in the lysate of respective E. coli cells at different optical densities (supplementary Figure S4), but did not observe an increase in fluorescence intensity during flow cytometric analysis compared to a negative control producing only GFP1-9 (supplementary Figure S5).
One reason for the lack of fluorescence could be the low stability of the RNA produced. Depending on their function, prokaryotic mRNAs have a mean half-life of less than 7 min (Selinger et al., 2003). To prolong the availability of target RNA boxAB for TetFC we tried to stabilize boxAB by placing stem loop structures on both the 5′- and 3′-end of the transcript (5F-boxAB-3F, supplementary Figures S1C and S6) (Blind et al., 1999; Hunsicker et al., 2009). Production of target RNA was confirmed in Northern blots (supplementary Figure S6E). TetFC performed in vitro with 5F-boxAB-3F RNA revealed that GFP complementation was still possible in the presence of two flanking stem loops. In fact, applying construct 5F-boxAB-3F RNA resulted in a higher fluorescence signal compared to non-stabilized RNA, suggesting that the RNA is either more stable or more accessible (supplementary Figure S6D). To implement this system in vivo,E. coli cells were transformed with three plasmids: one plasmid for GFP1-9 production, a second plasmid for production of S10-WT and Var1-S11, and a third plasmid for production of stabilized RNA 5F-boxAB-3F (supplementary Figure S6A). Cells were analyzed using flow cytometry after recombinant protein and target RNA production at different conditions (i.e. temperature, concentration of IPTG), but no increase in fluorescence intensity was observed (supplementary Figure S7).
An alternative approach to address the drawback of a short mRNA half-life is to decouple RNA production from the production of the remaining TetFC components. We therefore introduced a second promoter that is controlled by tetracycline, as described for the two-partite split-GFP (Valencia-Burton et al., 2009). Here, the tetracycline repressor TetR is produced under control of a T7 promoter and binds to tetO sites upstream of the DNA segment of interest, blocking transcription. Upon addition of a suitable inducer such as tetracycline or one of its derivatives, TetR-tetO-binding is interrupted and transcription occurs (Figure 2A). With this in mind, we generated pSJK15, a pRSFDuet™-1-derived plasmid compatible with both pSJK4 (for production of fusion proteins S10-WT and Var1-S11) and pSJK11 (for production of detector GFP1-9). Again, we could validate in vivo availability of fusion proteins S10-WT and Var1-S11 in E. coli pSJK4 cells using Western blots (Figure 2B). Next, E. coli BL21-Gold(DE3) cells were consecutively transformed with all three plasmids (Figure 2A). Bacterial growth was not affected by transformation of cells with a third plasmid, compared to respective cells containing only plasmids pSJK11 and pSJK15 (supplementary Figure S8A). Expression of the protein coding genes was induced by adding IPTG, whereas RNA production could be induced at a later stage by addition of anhydrotetracycline (ATC). Within the available tetracycline derivatives, ATC was favored due to its low toxicity (Rasmussen et al., 1991). A negative effect on cell growth was only observed in presence of a very high ATC concentration (25 000 ng/ml, supplementary Figure S8B). We performed quantitative real-time (qRT)-PCR from total RNA of E. coli BL21-Gold(DE3) pSJK11+pSJK15+pSJK4 cells after ATC induction at different concentrations (final concentrations of 2.5–2.5×105 ng/ml). We found that the highest concentration of boxAB RNA was available if 250 ng/ml ATC was used for induction (Figure 2C), which is in accordance with previous studies (Valencia-Burton et al., 2009).
Next, we analyzed whether this three-plasmid system that allows for late induction of RNA expression can be used for tetramolecular fluorescence complementation in E. coli BL21-Gold(DE3) cells. This strain can be very efficiently transformed – an important point because expression from several plasmids is required to obtain a functional system. Another crucial point to consider during data evaluation is fluorescence caused by ATC. Hence, we always applied ATC at the indicated concentrations to both TetFC cells and control cells that do not produce target RNA (Figure 1C). Only experiments at identical concentrations of ATC were compared (ranging from 2.5 to 2.5×105 ng/ml). To determine whether RNA-dependent fluorescence complementation can be detected in vivo, cells were analyzed by flow cytometry regarding their green fluorescence (Figure 3A).
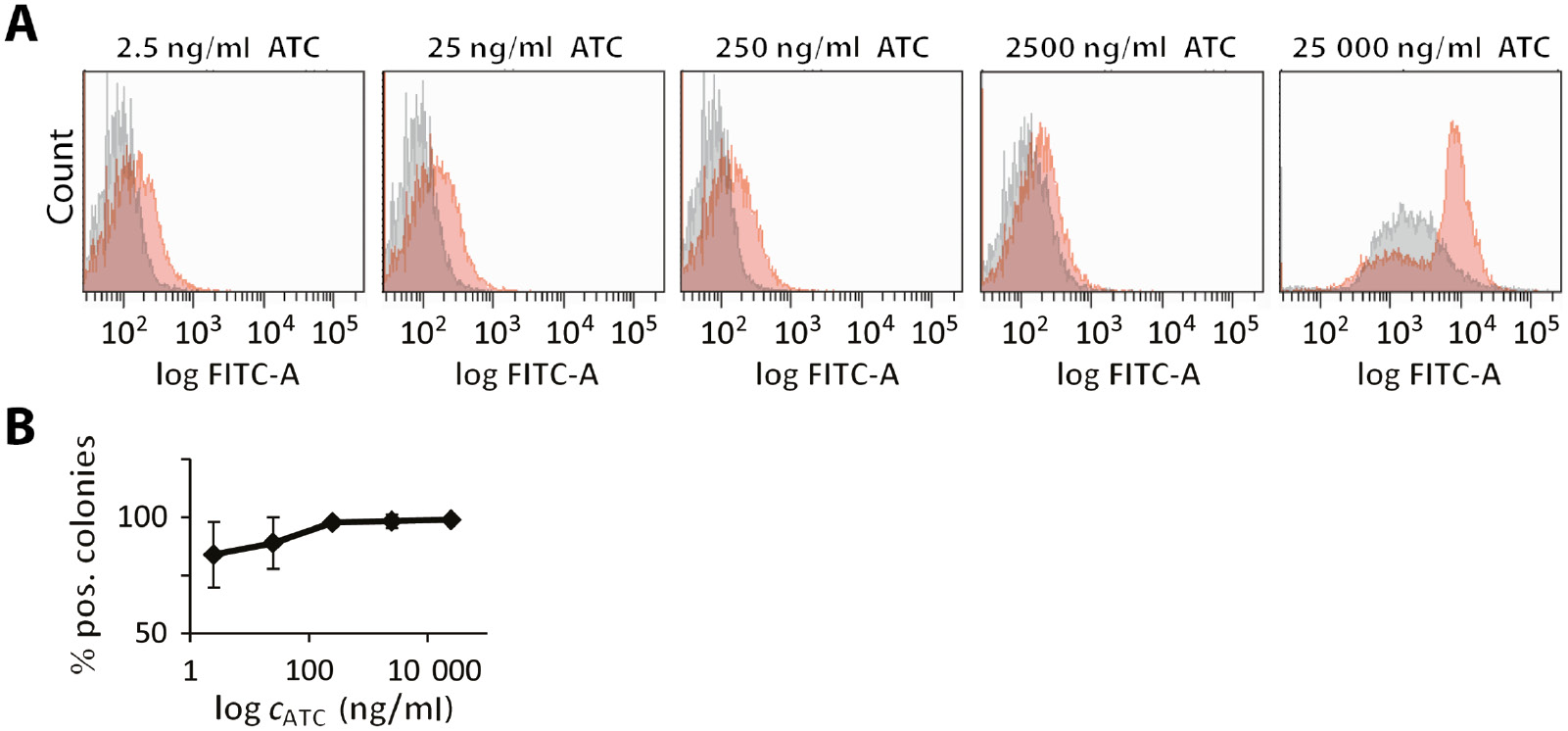
Fluorescence-activated sorting of TetFC and control E. coli cells.
(A) Different ATC concentrations (0–25 000 ng/ml) were used in both TetFC (red) and negative control (gray) cells treated and grown under the same conditions. (B) Equal numbers of TetFC and negative control cells were mixed and then sorted. The 0.1–0.5% strongest fluorescing cells were collected, regrown and tested for kanamycin resistance indicative of TetFC cells (plasmid encoding boxAB RNA). Error bars indicate the standard deviation of three independent experiments.
All experiments showed an ATC-dependent increase in green fluorescence. To elucidate whether there is a target RNA-dependent increase in fluorescence, we therefore directly compared TetFC and control cells grown in parallel under identical conditions – including ATC addition. These experiments were carried out at different ATC concentrations and revealed that TetFC cells showed consistently higher fluorescence than control cells (Figure 3A). The difference in fluorescence between TetFC and control cells seemed to be most pronounced at a concentration of 250 ng ATC/ml medium, in line with qRT-PCR data that showed the highest RNA expression at this ATC concentration (Figure 2C). These conditions were thus chosen to test whether TetFC causes fluorescence changes sufficient to discern cells expressing boxAB RNA in a mixture. To do so, TetFC and negative control cells were grown in parallel and mixed in a 1/1 ratio before sorting as schematically illustrated in Figure 1C. The top 0.1–0.5% of green fluorescing cells were sorted and regrown. Positive colonies (i.e. TetFC cells) were identified via a simple selection using antibiotics, since only TetFC cells contain plasmid pSJK15, which mediates kanamycin resistance. Regrowth was thus performed both in the presence and absence of kanamycin and the fraction of correctly sorted cells was calculated. Starting from a 1/1 mixture, we observed a higher fraction of ATC-resistant cells after sorting (Figure 3B). Importantly, the amount of ATC-resistant cells depended on the ATC concentration, reaching 99% at 250 ng ATC/ml medium and above. Taken together, these data demonstrate that TetFC was successfully implemented in E. coli and can be used to distinguish cells expressing the target RNA from control cells by FACS. Experiments have to be performed in a controlled manner to rule out fluorescence background originating from ATC. Nevertheless, the option to induce RNA production at a later timepoint and independent of recombinant protein expression proved essential to make the system functional, most likely due to the amount of RNA available for Pumilio binding.
We reason that TetFC in E. coli could provide a viable alternative to current yeast-based screening strategies for RNA-protein interactions. Based on our in vitro data, we know that TetFC is not prone to self-complementation, reducing the number of false positives. It has to be stated, however, that our in vivo TetFC shows a rather low signal increase compared to related work on RNA-dependent fluorescence complementation systems in E. coli analyzed via flow cytometry (Valencia-Burton et al., 2007, 2009; Yiu et al., 2011). This is most likely due to the number of components required for GFP complementation. We think that in combination with FACS, which can efficiently sort large numbers of cells, it will be beneficial to have a low number of false positives and test large libraries under stringent conditions. Experiments to use TetFC for directed evolution of Pumilio proteins are currently underway.
In conclusion, elucidation of the Pumilio binding code in combination with the recent development of elegant cloning strategies now provides access to tailor-made Pumilio variants for sequence-specific RNA-binding (Wang et al., 2002; Cheong and Hall, 2006; Dong et al., 2011; Filipovska et al., 2011; Abil et al., 2014; Adamala et al., 2016). However, introduction of several substitutions can be expected to compromise stability and solubility of the resulting variants. Furthermore, alternative binding modes cannot be excluded and are likely to become more prominent as variants deviate more from the original RNA consensus sequence. Therefore, validation and optimization of Pumilio proteins will be required to optimize variants, including substitutions at positions far away from the sequence-recognition code. We showed that TetFC can be implemented in E. coli and is compatible with FACS-based screening. As a proof of concept we showed that E. coli cells can be distinguished from negative control cells that do not express the target RNA. TetFC thus holds promise to screen libraries of Pumilio proteins for variants with improved or new binding affinities. Additionally, we propose that our system could be easily tailored to screen other RNA-binding proteins. In particular, the pentatricopeptide repeat proteins (PPRs) will be promising starting points for directed evolution, because they exhibit a ‘one repeat/one base’ binding mode similar to that of the Pum-HD (Wang et al., 2002; Yagi et al., 2013; Barkan et al., 2012), but have neither been as extensively studied nor utilized as binding domains for sequence-specific RNA modification.
Acknowledgments
We are grateful to Prof. Dr. Ulrich Hahn for continuous access to the FACSAria™ III and laboratory space during flow cytometric/FACS experiments. S. J. K. would also like to express her gratitude towards Dr. Florian Mittelberger and Thorsten Mix for training and technical support at the FACS. We thank Prof. Beatrix Süß for suggesting and providing plasmid pWH610L3F5F. We also thank Dr. Cheom-Gil Cheong and Dr. Traci M. T. Hall for plasmids encoding Pumilio WT and Pumilio Variant 1, Dr. Geoffrey S. Waldo for the GFP1-9, and pTet-SpecR constructs, Dr. Natalia E. Broude for plasmid pMB38 and Dr. Anna K. Rath for plasmid pSK17-Hb-boxAB. This work was supported by the Emmy-Noether-Programme of the Deutsche Forschungsgemeinschaft (DFG) (RE2796/2-1) and partly supported by the DFG EXC 1003 Cells in Motion Cluster of Excellence, Münster, Germany. A. R. thanks the Fonds der Chemischen Industrie for financial support (Dozentenstipendium).
References
Abil, Z., Denard, C.A., and Zhao, H. (2014). Modular assembly of designer PUF proteins for specific post-transcriptional regulation of endogenous RNA. J. Biol. Eng. 8, 7.10.1186/1754-1611-8-7Search in Google Scholar PubMed PubMed Central
Adamala, K.P., Martin-Alarcon, D.A., and Boyden, E.S. (2016). Programmable RNA-binding protein composed of repeats of a single modular unit. Proc. Natl. Acad. Sci. USA 113, E2579–E2588.10.1073/pnas.1519368113Search in Google Scholar PubMed PubMed Central
Barkan, A., Rojas, M., Fujii, S., Yap, A., Chong, Y. S., Bond, C. S., and Small, I. (2012). A combinatorial amino acid code for RNA recognition by pentatricopeptide repeat proteins. PLoS Genet 8, e1002910.10.1371/journal.pgen.1002910Search in Google Scholar PubMed PubMed Central
Blind, M., Kolanus, W., and Famulok, M. (1999). Cytoplasmic RNA modulators of an inside-out signal-transduction cascade. Proc. Natl. Acad. Sci. USA 96, 3606–3610.10.1073/pnas.96.7.3606Search in Google Scholar PubMed PubMed Central
Cabantous, S., Nguyen, H.B., Pedelacq, J.D., Koraichi, F., Chaudhary, A., Ganguly, K., Lockard, M.A., Favre, G., Terwilliger, T.C., and Waldo, G.S. (2013). A new protein-protein interaction sensor based on tripartite split-GFP association. Sci. Rep. 3, 2854.10.1038/srep02854Search in Google Scholar PubMed PubMed Central
Campbell, Z.T., Valley, C.T., and Wickens, M. (2014). A protein-RNA specificity code enables targeted activation of an endogenous human transcript. Nat. Struct. Mol. Biol. 21, 732–738.10.1038/nsmb.2847Search in Google Scholar PubMed PubMed Central
Cao, J., Arha, M., Sudrik, C., Schaffer, D.V., and Kane, R.S. (2014). Bidirectional regulation of mRNA translation in mammalian cells by using PUF domains. Angew. Chem. Int. Ed. 53, 4900–4904.10.1002/anie.201402095Search in Google Scholar PubMed
Cao, J., Arha, M., Sudrik, C., Mukherjee, A., Wu, X., and Kane, R.S. (2015). A universal strategy for regulating mRNA translation in prokaryotic and eukaryotic cells. Nucleic Acids Res. 43, 4353–4362.10.1093/nar/gkv290Search in Google Scholar PubMed PubMed Central
Cheong, C.G. and Hall, T.M. (2006). Engineering RNA sequence specificity of Pumilio repeats. Proc. Natl. Acad. Sci. USA 103, 13635–13639.10.1073/pnas.0606294103Search in Google Scholar PubMed PubMed Central
Choudhury, R., Tsai, Y.S., Dominguez, D., Wang, Y., and Wang, Z. (2012). Engineering RNA endonucleases with customized sequence specificities. Nat. Commun. 3, 1147.10.1038/ncomms2154Search in Google Scholar PubMed PubMed Central
Cooke, A., Prigge, A., Opperman, L., and Wickens, M. (2011). Targeted translational regulation using the PUF protein family scaffold. Proc. Natl. Acad. Sci. USA 108, 15870–15875.10.1073/pnas.1105151108Search in Google Scholar
Dong, S., Wang, Y., Cassidy-Amstutz, C., Lu, G., Bigler, R., Jezyk, M.R., Li, C., Hall, T.M., and Wang, Z. (2011). Specific and modular binding code for cytosine recognition in Pumilio/FBF (PUF) RNA-binding domains. J. Biol. Chem. 286, 26732–26742.10.1074/jbc.M111.244889Search in Google Scholar
Edwards, T.A., Pyle, S.E., Wharton, R.P., and Aggarwal, A.K. (2001). Structure of Pumilio reveals similarity between RNA and peptide binding motifs. Cell 105, 281–289.10.1016/S0092-8674(01)00318-XSearch in Google Scholar
Filipovska, A., Razif, M.F., Nygard, K.K., and Rackham, O. (2011). A universal code for RNA recognition by PUF proteins. Nat. Chem. Biol. 7, 425–427.10.1038/nchembio.577Search in Google Scholar PubMed
Gerber, A.P., Herschlag, D., and Brown, P.O. (2004). Extensive association of functionally and cytotopically related mRNAs with Puf family RNA-binding proteins in yeast. PLoS Biol. 2, E79.10.1371/journal.pbio.0020079Search in Google Scholar PubMed PubMed Central
Gerstle, K., Klatschke, K., Hahn, U., and Piganeau, N. (2012). The small RNA RybA regulates key-genes in the biosynthesis of aromatic amino acids under peroxide stress in E. coli. RNA Biol. 9, 458–468.10.4161/rna.19065Search in Google Scholar PubMed
Hunsicker, A., Steber, M., Mayer, G., Meitert, J., Klotzsche, M., Blind, M., Hillen, W., Berens, C., and Suess, B. (2009). An RNA aptamer that induces transcription. Chem. Biol. 16, 173–180.10.1016/j.chembiol.2008.12.008Search in Google Scholar PubMed
Kellermann, S.J. and Rentmeister, A. (2016). A genetically encodable system for sequence-specific detection of RNAs in two colors. Chembiochem 17, 895–899.10.1002/cbic.201500705Search in Google Scholar PubMed
Kellermann, S.J., Rath, A.K., and Rentmeister, A. (2013). Tetramolecular fluorescence complementation for detection of specific RNAs in vitro. Chembiochem 14, 200–204.10.1002/cbic.201200734Search in Google Scholar PubMed
Miao, H., Ratnasingam, S., Pu, C.S., Desai, M.M., and Sze, C.C. (2009). Dual fluorescence system for flow cytometric analysis of Escherichia coli transcriptional response in multi-species context. J. Microbiol. Methods 76, 109–119.10.1016/j.mimet.2008.09.015Search in Google Scholar PubMed
Miller, M.T., Higgin, J.J., and Hall, T.M. (2008). Basis of altered RNA-binding specificity by PUF proteins revealed by crystal structures of yeast Puf4p. Nat. Struct. Mol. Biol. 15, 397–402.10.1038/nsmb.1390Search in Google Scholar
Murata, Y. and Wharton, R.P. (1995). Binding of pumilio to maternal hunchback mRNA is required for posterior patterning in Drosophila embryos. Cell 80, 747–756.10.1016/0092-8674(95)90353-4Search in Google Scholar
Novagen. (2004). User Protocol TB340 Rev. D 0304.Search in Google Scholar
Opperman, L., Hook, B., DeFino, M., Bernstein, D.S., and Wickens, M. (2005). A single spacer nucleotide determines the specificities of two mRNA regulatory proteins. Nat. Struct. Mol. Biol. 12, 945–951.10.1038/nsmb1010Search in Google Scholar PubMed
Ozawa, T., Natori, Y., Sato, M., and Umezawa, Y. (2007). Imaging dynamics of endogenous mitochondrial RNA in single living cells. Nat. Methods 4, 413–419.10.1038/nmeth1030Search in Google Scholar PubMed
Pinheiro, L.B., Gibbs, M.D., Vesey, G., Smith, J.J., and Bergquist, P.L. (2008). Fluorescent reference strains of bacteria by chromosomal integration of a modified green fluorescent protein gene. Appl. Microbiol. Biotechnol. 77, 1287–1295.10.1007/s00253-007-1253-9Search in Google Scholar PubMed
Qiu, C., Kershner, A., Wang, Y., Holley, C.P., Wilinski, D., Keles, S., Kimble, J., Wickens, M., and Hall, T.M. (2012). Divergence of Pumilio/fem-3 mRNA binding factor (PUF) protein specificity through variations in an RNA-binding pocket. J. Biol. Chem. 287, 6949–6957.10.1074/jbc.M111.326264Search in Google Scholar PubMed PubMed Central
Rasmussen, B., Noller, H.F., Daubresse, G., Oliva, B., Misulovin, Z., Rothstein, D.M., Ellestad, G.A., Gluzman, Y., Tally, F.P., and Chopra, I. (1991). Molecular basis of tetracycline action: identification of analogs whose primary target is not the bacterial ribosome. Antimicrob. Agents Chemother. 35, 2306–2311.10.1128/AAC.35.11.2306Search in Google Scholar PubMed PubMed Central
Rath, A.K., Kellermann, S.J., and Rentmeister, A. (2014). Programmable design of functional ribonucleoprotein complexes. Chem. Asian J. 9, 2045–2051.10.1002/asia.201402220Search in Google Scholar PubMed
Sambrook, J. and Russell, D.W. (2001). Molecular Cloning: A Laboratory Manual (Cold Spring Harbor, NY, USA: Cold Spring Harbor Laboratory Press).Search in Google Scholar
Selinger, D.W., Saxena, R.M., Cheung, K.J., Church, G.M., and Rosenow, C. (2003). Global RNA half-life analysis in Escherichia coli reveals positional patterns of transcript degradation. Genome Res. 13, 216–223.10.1101/gr.912603Search in Google Scholar PubMed PubMed Central
Tilsner, J., Linnik, O., Christensen, N.M., Bell, K., Roberts, I.M., Lacomme, C., and Oparka, K.J. (2009). Live-cell imaging of viral RNA genomes using a Pumilio-based reporter. Plant J. 57, 758–770.10.1111/j.1365-313X.2008.03720.xSearch in Google Scholar PubMed
Valencia-Burton, M., McCullough, R.M., Cantor, C.R., and Broude, N.E. (2007). RNA visualization in live bacterial cells using fluorescent protein complementation. Nat. Methods 4, 421–427.10.1038/nmeth1023Search in Google Scholar
Valencia-Burton, M., Shah, A., Sutin, J., Borogovac, A., McCullough, R.M., Cantor, C.R., Meller, A., and Broude, N.E. (2009). Spatiotemporal patterns and transcription kinetics of induced RNA in single bacterial cells. Proc. Natl. Acad. Sci. USA 106, 16399–16404.10.1073/pnas.0907495106Search in Google Scholar
Wang, X., Zamore, P.D., and Hall, T.M. (2001). Crystal structure of a Pumilio homology domain. Mol. Cell 7, 855–865.10.1016/S1097-2765(01)00229-5Search in Google Scholar
Wang, X., McLachlan, J., Zamore, P.D., and Hall, T.M. (2002). Modular recognition of RNA by a human pumilio-homology domain. Cell 110, 501–512.10.1016/S0092-8674(02)00873-5Search in Google Scholar
Wang, Y., Cheong, C.G., Hall, T.M., and Wang, Z. (2009a). Engineering splicing factors with designed specificities. Nat. Methods 6, 825–830.10.1038/nmeth.1379Search in Google Scholar PubMed PubMed Central
Wang, Y., Opperman, L., Wickens, M., and Hall, T.M. (2009b). Structural basis for specific recognition of multiple mRNA targets by a PUF regulatory protein. Proc. Natl. Acad. Sci. USA 106, 20186–20191.10.1073/pnas.0812076106Search in Google Scholar PubMed PubMed Central
Yagi, Y., Hayashi, S., Kobayashi, K., Hirayama, T., and Nakamura, T. (2013). Elucidation of the RNA recognition code for pentatricopeptide repeat proteins involved in organelle RNA editing in plants. PLoS One 8, e57286.10.1371/journal.pone.0057286Search in Google Scholar PubMed PubMed Central
Yamada, T., Yoshimura, H., Inaguma, A., and Ozawa, T. (2011). Visualization of nonengineered single mRNAs in living cells using genetically encoded fluorescent probes. Anal. Chem. 83, 5708–5714.10.1021/ac2009405Search in Google Scholar PubMed
Yiu, H.-W., Demidov, V.V., Toran, P., Cantor, C.R., and Broude, N.E. (2011). RNA detection in live bacterial cells using fluorescent protein complementation triggered by interaction of two RNA aptamers with two RNA-binding peptides. Pharmaceuticals 4, 494–508.10.3390/ph4030494Search in Google Scholar
Yoshimura, H., Inaguma, A., Yamada, T., and Ozawa, T. (2012). Fluorescent probes for imaging endogenous b-actin mRNA in living cells using fluorescent protein-tagged pumilio. ACS Chem. Biol. 7, 999–1005.10.1021/cb200474aSearch in Google Scholar PubMed
Zamore, P.D., Williamson, J.R., and Lehmann, R. (1997). The Pumilio protein binds RNA through a conserved domain that defines a new class of RNA-binding proteins. RNA 3, 1421–1433.Search in Google Scholar
Zhang, B., Gallegos, M., Puoti, A., Durkin, E., Fields, S., Kimble, J., and Wickens, M.P. (1997). A conserved RNA-binding protein that regulates sexual fates in the C. elegans hermaphrodite germ line. Nature 390, 477–484.10.1038/37297Search in Google Scholar PubMed
Zhu, D., Stumpf, C.R., Krahn, J.M., Wickens, M., and Hall, T.M. (2009). A 5′ cytosine binding pocket in Puf3p specifies regulation of mitochondrial mRNAs. Proc. Natl. Acad. Sci. USA 106, 20192–20197.10.1073/pnas.0812079106Search in Google Scholar PubMed PubMed Central
Supplemental Material:
The online version of this article (DOI: 10.1515/hsz-2016-0214) offers supplementary material, available to authorized users.
©2016, Andrea Rentmeister, published by De Gruyter.
This work is licensed under the Creative Commons Attribution-NonCommercial-NoDerivatives 3.0 License.
Articles in the same Issue
- Frontmatter
- Guest Editorial
- Highlight issue: protein design
- HIGHLIGHT: MOSBACH COLLOQUIUM 2016 “PROTEIN DESIGN”
- Multifunctional biomaterial coatings: synthetic challenges and biological activity
- Advances in the design and engineering of peptide-binding repeat proteins
- Fusion proteins of an enoate reductase and a Baeyer-Villiger monooxygenase facilitate the synthesis of chiral lactones
- Anticalins directed against vascular endothelial growth factor receptor 3 (VEGFR-3) with picomolar affinities show potential for medical therapy and in vivo imaging
- Development of a screening system for inteins active in protein splicing based on intein insertion into the LacZα-peptide
- A FACS-based screening strategy to assess sequence-specific RNA-binding of Pumilio protein variants in E. coli
- Review
- Alpha-synuclein at the intracellular and the extracellular side: functional and dysfunctional implications
- Research Articles/Short Communications
- Protein Structure and Function
- A novel plant enzyme with dual activity: an atypical Nudix hydrolase and a dipeptidyl peptidase III
- Comparison of the ability of mammalian eEF1A1 and its oncogenic variant eEF1A2 to interact with actin and calmodulin
- Structural analysis and interaction studies of acyl-carrier protein (acpP) of Staphylococcus aureus, an extraordinarily thermally stable protein
- Characterization of a new toxin from the entomopathogenic fungus Metarhizium anisopliae: the ribotoxin anisoplin
Articles in the same Issue
- Frontmatter
- Guest Editorial
- Highlight issue: protein design
- HIGHLIGHT: MOSBACH COLLOQUIUM 2016 “PROTEIN DESIGN”
- Multifunctional biomaterial coatings: synthetic challenges and biological activity
- Advances in the design and engineering of peptide-binding repeat proteins
- Fusion proteins of an enoate reductase and a Baeyer-Villiger monooxygenase facilitate the synthesis of chiral lactones
- Anticalins directed against vascular endothelial growth factor receptor 3 (VEGFR-3) with picomolar affinities show potential for medical therapy and in vivo imaging
- Development of a screening system for inteins active in protein splicing based on intein insertion into the LacZα-peptide
- A FACS-based screening strategy to assess sequence-specific RNA-binding of Pumilio protein variants in E. coli
- Review
- Alpha-synuclein at the intracellular and the extracellular side: functional and dysfunctional implications
- Research Articles/Short Communications
- Protein Structure and Function
- A novel plant enzyme with dual activity: an atypical Nudix hydrolase and a dipeptidyl peptidase III
- Comparison of the ability of mammalian eEF1A1 and its oncogenic variant eEF1A2 to interact with actin and calmodulin
- Structural analysis and interaction studies of acyl-carrier protein (acpP) of Staphylococcus aureus, an extraordinarily thermally stable protein
- Characterization of a new toxin from the entomopathogenic fungus Metarhizium anisopliae: the ribotoxin anisoplin