Sortase-mediated backbone cyclization of proteins and peptides
-
Wim van ’t Hof


Abstract
Backbone cyclization has a profound impact on the biological activity and thermal and proteolytic stability of proteins and peptides. Chemical methods for cyclization are not always feasible, especially for large peptides or proteins. Recombinant Staphylococcus aureus sortase A shows potential as a new tool for the cyclization of both proteins and peptides. In this review, the scope and background of the sortase-mediated cyclization are discussed. High efficiency, versatility, and easy access make sortase A a promising cyclization tool, both for recombinant and chemo-enzymatic production methods.
Introduction: backbone-cyclized poteins and peptides
Cyclic antimicrobial peptides are not uncommon in nature. In fact, gramicidin S, one of the first large-scale used antibiotics, is a cyclodecapeptide produced by Bacillus brevis (Gause and Brazhnikova, 1944). In the years following the discovery of gramicidins, various cyclic peptide antibiotics have been isolated from bacterial sources, including daptomycin, champacyclin, ohmyungsamcin (all from Streptomyces spp.), and lactocyclin (from Lactobacillus spp.). Cyclic antimicrobial peptides and proteins have also been found in plants (cyclotides) and mammals (rhesus θ-defensin: RTD-1). The growing interest in small proteins and peptides in the last decades has led to the discovery of more and more cyclic molecules in natural sources. Also, outside the field of antimicrobial peptides, a number of cyclic peptides have been found with powerful biological activities, such as the immunosuppressant cyclosporin from fungal origin and Sunflower Trypsin Inhibitor-1, the most potent natural trypsin inhibitor known to date (Arnison et al., 2013).
The biosynthesis of cyclic proteins and peptides can proceed along different pathways. Some are directly synthesized from scratch, without involvement of an RNA template, by dedicated multi-enzyme complexes that stitch specific amino acids to each other. Examples are cyclosporine, daptomycin, and gramicidin S (Clark and Craik, 2010; Aboye et al., 2012). Others are DNA-encoded and ribosomally produced as linear precursors that are further post-translationally cyclized by intra-molecular head-to-tail ligation. The enzymes (endopeptidases or transpeptidases) involved in this ligation are mostly unknown (Craik et al., 1999; Clark and Craik, 2010; Katoh et al., 2011; Aboye et al., 2012). Only the endopeptidase responsible for the cyclization of plant cyclotides was recently identified (Nguyen et al., 2014). A special case constitutes the θ-defensins. These peptides are composed of two identical ribosomally produced linear peptide precursors, which, after translation, are ligated in an inter-molecular head-to-tail fashion into a cyclic dimer (Tang et al., 1999).
Backbone cyclization in principle endows proteins and peptides with several advantageous properties. Many proteins contain unstructured, highly flexible, N- and C-termini that are spatially close in the three-dimensional folded structure (Antos et al., 2009; Clark and Craik, 2010). Joining these termini by a peptide bond in theory will aid in stabilization of the native folded state of these proteins. In peptides, which lack a distinct three-dimensional structure, backbone cyclization may induce a constrained folded structure, with enhanced biological activity and stability. The constrained cyclized structure increases resistance against proteolytic degradation by exoproteases (Popp et al., 2011; Borra and Camarero, 2013). This has sparked the interest in backbone cyclization as a way to improve protein- and peptide-based therapeutics (Clark and Craik, 2010; Popp et al., 2011). Particularly in the mouth, infested with host and bacterial proteases, backbone cyclization may significantly help to improve the oral bioavailability of therapeutic peptides (Wong et al., 2012).
Chemical cyclization of peptides
Chemical cyclization of synthetic peptides has been a challenge to peptide chemists for many years (Tam and Wong, 2012). A relatively old and often used cyclization method uses the oxidative coupling of two additional cysteine residues, one at the N- and one at the C-terminus (Figure 1A). The drawback of this method is the formation of dimeric and multimeric side products due to the formation of inter-chain disulfide bridges. Furthermore, the disulfide bond is very susceptible to reducing agents.

Peptide cyclization by different chemical approaches.
(A) Cyclization by oxidative cystine-bridge formation. Peptides containing two cysteine residues are cyclized by cystine-bridge formation under mild oxidative conditions, e.g., air-oxidation, addition of Fe3+ salts, etc. (B) Cyclization using ‘click’ chemistry. Peptides are synthesized with an azido-lysine at one terminus (N-terminus in this scheme) and a propargyl-glycine at the other. Their side chains are fused by Cu1+-catalyzed Huisgen 1,3-dipolar cycloaddition to a 1,4-disubstituted 1,2,3-triazole that covalently links both termini to form a cyclized peptide. (C) Cyclization by formation of an N→C peptide bond during solid-phase peptide synthesis. Synthesis is started with a glutamic acid residue, attached to the resin by its side chain carboxyl group, with the α-carboxyl group protected as Dmab ester. After completion of the sequence, this orthogonal protecting group is removed by 2% hydrazine, and on-resin cyclization is achieved using the coupling reagent PyBOP. (D) Cyclization by NPL. Peptides and proteins with an N-terminal cysteine and a C-terminal thioester group spontaneously undergo a two-step N→C cyclization reaction. In the first transthioesterification step, the thiol group of the N-terminal cysteine places a nucleophilic attack on the C-terminal carbonyl moiety of the thioester bond, resulting in the formation of a new intramolecular thioester bond with concomitant release of the thiol RSH. This reversible reaction is followed by an irreversible intramolecular S→N acyl shift by a nucleophilic attack of the glycine amino group on the newly formed thioester group. This yields a backbone-cyclized peptide in which both termini are joined by a reduced (native) cysteine residue (figure adapted from Borra and Camarero, 2013).
More recently, the so-called click reaction is used for fast and easy cyclization of peptides. For this purpose, an ε-azidolysine is introduced at one (C- or N-) terminus and propargylglycine at the other. By addition of Cu+, azide alkyne 1,3-dipolar cycloaddition takes place, yielding a cyclic product in which the two side chains are covalently linked by the resulting 1,2,3-triazole linker, which is an order of magnitude stronger than a disulfide bond (Figure 1B).
Another often-applied method for cyclization is direct introduction of a peptide bond between the N-terminal amino group and the C-terminal carboxyl group as the last coupling step during organic synthesis (Figure 1C). In solid-phase peptide synthesis, on-resin cyclization can be achieved by coupling glutamic acid to the synthesis resin via its side-chain (γ-) carboxyl group instead of its backbone (α-) carboxyl group, which is semi-permanently protected. After completion of the sequence by conventional coupling procedures, the α-carboxyl group of the resin-coupled glutamic acid will be available for reaction with the amino group of the last residue in the chain. By using an existing glutamate within the sequence of the peptide, a cyclic peptide without an additional Glu-insert between the termini is obtained (Bolscher et al., 2011). This method has an intrinsic shortcoming: conventional solid-phase synthesis reactions are kinetically driven to completion by generating excess of activated amino acid building blocks in the coupling steps. This results in rapid and efficient formation of the peptide bond. However, in on-resin backbone cyclization, the rate of coupling of the N- and C-termini in a one-to-one stoichiometry depends strongly on the length of the peptide. Longer peptides possess such a large degree of conformational freedom that cyclization requires much longer reaction times. This is not very compatible with the use of highly reactive, unstable, carboxylic intermediates. As a result, cyclization yields drop sharply for peptides longer than 10–15 residues (Tam and Wong, 2012; Borra and Camarero, 2013).
To overcome the problem of prolonged reaction times of activated intermediates, in the so-called native protein ligation (NPL) strategy, a stable C-terminal thioester is introduced, which reacts with an additional N-terminal cysteine (Tam and Lu, 1997). After an acyl shift from the thioester to the thiol side chain of cysteine, an S→N rearrangement takes place, driving the reaction to completion and resulting in an N→C backbone-cyclized peptide with a cysteine insert at the ligation site (Figure 1D). Cyclization of θ-defensins, containing two anti-parallel motifs of three cysteine residues separated by a single residue (CXCXC), is accelerated due to formation of an intermediate so-called cysteine zipper (Aboye et al., 2012; Tam and Wong, 2012; Borra and Camarero, 2013; Jia et al., 2014). The drawback of the NPL strategy is that it has been incompatible with standard Fmoc-protocols for many years, as C-terminal thioesters are not resistant to the repetitive alkaline conditions during Fmoc removal by piperidine (Aboye et al., 2012). Several strategies have been employed to overcome this incompatibility: (i) use of the non-nucleophilic bicyclic base 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) instead of piperidine during the Fmoc removal (Clippingdale et al., 2000); (ii) masking of the thioester group by introducing a stable group at the C-terminus which after completion of the whole sequence is converted into a thioester (Brask et al., 2003); and (iii) the use of alternative resin linkers that produce thioesters during acidic work-up after cleavage from the resin (Ingenito et al., 1999). The recent advances in masking strategies (Liu and Mayer, 2013; Zheng et al., 2013) and the use of dedicated resin-linkers (Hou et al., 2010; Ollivier et al., 2010, 2014; Hemu et al., 2013; Taichi et al., 2013) have made peptides with a C-terminal thioester group readily accessible for Fmoc-chemistry.
Application of sortase A, a transpeptidase from Gram-positive bacteria, for cyclization of peptides
In our research on histatins, a group of antimicrobial peptides present in human saliva, synthetic linear peptides have been in the focus for many years (Van’t Hof et al., 2013). We discovered that some histatins stimulate migration of epithelial cells through a stereospecific interaction, probably via a G protein-coupled receptor (Oudhoff et al., 2008, 2009). To explore the effect of conformational constraint, we became interested in the synthesis of a backbone-cyclized histatin variant. Although chemical synthesis of backbone-cyclized peptides has reached a high level of sophistication (Tam and Wong, 2012), in our hands, the chemical head-to-tail ligation of the 38-residue peptide histatin 1 resulted in disappointingly low yields. We therefore explored the feasibility of a semi-enzymatic cyclization method based on sortase A, a transpeptidase of S. aureus, which successfully had been used before for cyclization of proteins (Antos et al., 2009; Popp and Ploegh, 2011, Popp et al., 2011). This resulted in a virtually quantitative cyclization yield from the linear precursor peptide. The enzymatically and chemically cyclized histatins had comparable molecular activities, which were approximately 1000-fold higher than that of the natural linear parent peptide (Bolscher et al., 2011).
Physiological role of sortase A
Sortase A anchors proteins equipped with an LPXTG motif to the peptidoglycan layer in the cell wall of Gram-positive bacteria. Besides the LPXTG motif, many of these proteins contain a unique N-terminal sorting signal sequence and a C-terminal amphipathic leader sequence that facilitate secretion across the plasma membrane. The LPXTG motif, connecting the mature protein to the amphipathic leader sequence, is cleaved by sortase A. The processed mature protein is subsequently linked to the N-terminal penta-glycine motif of membrane-bound Lipid-II. In the following step, the trans-membrane phospholipid part of Lipid-II is cleaved off, and its protein-bearing glycopeptide part is incorporated into the developing peptidoglycan. Both the leader sequence and the phospholipid part of Lipid-II are internalized across the plasma membrane and recycled (Figure 2) (Schneewind and Missiakas, 2012). Sortase is a peptidyl-transferase harboring in its active site a well-conserved catalytic triad, consisting of Cys184, Arg197, and His120. The currently accepted mechanism of action of sortase is described in the so-called reverse protonation model (Figure 3). With the highly conserved proline and threonine of the LPXTG recognition motif held in position by hydrophobic interactions, the thiol side chain of Cys184, assisted by the imidazole side chain of His120, places a nucleophilic attack on the peptide bond between threonine and glycine. This results in a quaternary intermediate, stabilized by hydrogen bridges from Arg197, which is resolved by the donation of a proton from His120. This leads to the formation of a covalent thioester bond between sortase and its substrate with concomitant breaking of the threonine-glycine peptide bond. This thioester bond is very stable to hydrolysis by water molecules but is readily resolved by nucleophilic attack of a primary amine attached to a primary carbon, for instance the Cα atom of glycine (Proft, 2010). Isopeptide ligation, i.e., coupling to the ε-amino group of lysines, does not generally occur, because at physiological pH this group is protonated, resulting in decreased nucleophilicity. An exception is sortase-mediated isopeptide ligation in indolicidin-derived peptides (Dasgupta et al., 2011). It is assumed that in these peptides the pKa of the ε-amino group is decreased because it is surrounded by six tryptophans, creating a hydrophobic microenvironment. It is noteworthy that many Gram-positive bacteria produce specific sortases that are involved in the assembly of pili by isopeptide ligation. Pili, long protein fibers protruding from the cell wall that play a role in inter-bacterial contact, are made by stepwise polymerization of pilin subunits by ligation of their respective C-terminal LPXTG motifs and their highly conserved N-terminal lysines by isopeptide bonds (Spirig et al., 2011). Based on the results obtained with indolicidin-derived peptides, Dasgupta et al. (2011) speculated that this specificity is not an intrinsic property of the different sortases. Instead, the hydrophobic microenvironment of the lysine in the pilin motif may strongly reduce the pKa of the ε-amino group thereby increasing its nucleophilic activity.

Sortase-mediated assembly of the cell wall peptidoglycan in Staphylococcus aureus.
The precursor proteins of cell-wall-anchored proteins contain several distinct domains: an N-terminal sorting sequence, the sortase motif LPXTG, an amphipathic prosegment, and a C-terminal positively charged leader peptide. In stage I of the incorporation into the cell wall, the N-terminal sorting motif facilitates recognition by trans-membrane transporter complexes that incorporate the precursors into the plasma membrane, with the amphipathic prosegment spanning the membrane with the C-terminal leader sequence sticking out into the cytoplasm, under proteolytic removal of the sorting motif. In stage II, sortase cleaves the LPXTG motif at threonine, forming a thioacyl complex between the mature protein and the cysteine residue in its active site. The amphipathic prosegment, flanked by a C-terminal glycine and the N-terminal leader sequence, is internalized over the cell membrane. In stage III, this complex is resolved by nucleophilic attack of the N-terminus of the penta-glycine branch of the isopeptide chain of lipid II. In stage IV, the glycopeptide moiety of lipid II, with the attached protein, is incorporated into the developing peptidoglycan moiety of the cell wall. The phospholipid part of lipid II is passively internalized over the cell membrane to be recycled into a new lipid II molecule that transverses the membrane by the action of a dedicated transporter protein (figure adapted from Maresso and Schneewind, 2008).

The reversed protonation model.
(i) The thiol side chain of Cys184 places a nucleophilic attack on the peptide bond between the threonine and glycine of the LPETG-recognition motif, facilitated by proton uptake by His120. (ii) This results in a quaternary transition state, stabilized by hydrogen bridges from Arg197, which is resolved by the donation of a proton by His120. (iii) This leads to the formation of a covalent thioester bond between sortase and its substrate with concomitant breaking of the threonine-glycine peptide bond and release of glycine. This thioester bond is very stable to hydrolysis by water molecules but is readily resolved by nucleophilic attack of a suitably exposed primary amine, e.g., the N-terminus of a glycine residue (figure adapted from Proft, 2010).
Sortase A as a tool for chemical ligation
In the last decades there has been increasing interest in the field of protein bioengineering in versatile tools that can conjugate all kinds of functional groups to existing proteins. Functionalization of proteins by bioconjugation provides access to many new, potentially interesting molecules (Heck et al., 2013). From the group of Gram-positive bacterial sortases, in particular sortase A from S. aureus has drawn much attention as a promising bio-conjugation tool. It has several properties that make it ideally suited for a wide range of protein ligation reactions. In the first place, it accepts a broad variety of substrates, as long as they contain the LPXTG recognition motif (in which X may be any amino acid). It is not very particular about the acyl-acceptor site either, a methylene-amino group, as in an N-terminal glycine residue suffices as resolving nucleophile, although the ligation reaction proceeds more efficiently with an N-terminal diglycine motif (Popp and Ploegh, 2011). As a result, only a small stretch of six amino acids, LPXTGG, ends up as ‘contamination’ between the two conjugation partners, reducing the risk on disturbance of the protein structure or unwanted biological side effects due to ligation. Together with the easy access to large quantities of bioactive recombinant sortase A, this versatility has made this enzyme a true workhorse in the fields of site-specific modification and functionalization of proteins (sortagging) and glycopeptides, site-specific ligation of peptides and proteins, and covalent immobilization of proteins, which contributed significantly to the scientific progress in these disciplines over the last decade (Proft, 2010; Popp and Ploegh, 2011; Wu and Guo, 2012).
Sortase A: cyclization vs. multimerization
The group of Ploegh at the Whitehead Institute was the first to describe in detail backbone-cyclization of proteins containing both a C-terminal LPETG cleavage motif and an N-terminal glycine acyl acceptor motif by sortase A. This provided an elegant new route to cyclize proteins and peptides (Antos et al., 2009; Bolscher et al., 2011; Wu et al., 2011).
Theoretically, proteins or peptides equipped with both the LPETG donor motif and an acceptor motif, e.g., -GG, may undergo cyclization (internal transpeptidation) as well as dimerization and trimerization (external transpeptidation). The ratio cyclic product: multimeric product depends both on the size of the substrate and on the concentration. Wu et al. (2011) found that for sortase A-catalyzed peptide cyclization the minimal size of substrate peptides is a 19-mer, including the sorting signal. With a 17-mer peptide a direct head-to tail cyclization product was obtained with 6% yield, whereas the major product was the cyclic dimer. In the same study, it was found that at increasing substrate concentrations, the yield of dimer and trimer products increased, whereas that of cyclic products decreased. The same effect was observed for butelase 1, the enzyme involved in the in vivo cyclization of plant cyclotides (Nguyen et al., 2014). This is in line with the reaction scheme depicted in Figure 4. Strikingly, 19-mer and longer peptides are virtually completely converted into a cyclic product, even at relatively high substrate/enzyme ratios, suggesting that with these substrates the cyclization reaction is preferred, presumably because the acceptor diglycine motif in these cases can come into close vicinity to the donor LPETG motif. Although under these conditions conversion virtually completely occurs, it must be kept in mind that the overall outcome is determined by the thermodynamic equilibrium. This is illustrated by the fact that in the presence of excess triglycine acting as a nucleophilic acceptor, cyclized G-Cre-LPETG-His6 (CG-Cre-LPET) is converted to the linear product G-Cre-LPETGGG (Antos et al., 2009).

Sortase-mediated reactions of peptides containing both an acceptor and a donor motif.
(A) After the LPETG recognition motif of the substrate (S) has bound to the sortase A (E) enzymatic site (ES), the resulting covalent intermediate LPET-sortase (*ES) can undergo two competing reactions: cyclization (intramolecular transpeptidation, pathway 1 → CS) or dimerization (intermolecular transpeptidation, pathway 2 → S2) and further oligomerization. The ratio of cyclic product/multimeric product depends both on the size of the substrate and on the enzyme/substrate ratio. The minimal size of substrate peptides is 19 residues, including the sorting signal and the diglycine acceptor motif. With a 17-mer peptide a direct head-to tail cyclization product was obtained with 6% yield, whereas the major product was the cyclic dimmer (pathway 3 → CS2). By increasing the substrate concentration, the yield of dimer and trimer products increased, whereas that of cyclic products decreased. The virtually complete conversion of 19-mer and longer peptides, even at relatively high substrate/enzyme ratios, suggests that for these substrates cyclization reaction is the preferred reaction (Wu et al., 2011). (B) Schematic representation of the molecular transpeptidation pathways. For short peptides cyclization is unfavorable, and cyclization becomes only possible after dimerization (pathway 3). The dimer may bind to sortase A in two fashions: by its central ligation LPETGG motif, leading to reversal of the dimerization, or by its C-terminal LPETG motif, allowing further cyclization and oligomerization.
Sortase-mediated backbone cyclization in living cells
An obvious limitation in the backbone cyclization of proteins and peptides is the insertion of additional sequences, i.e., the GG- and an LPXTG- motif, respectively, in the linear precursors. This excludes the sortase-mediated cyclizations of natural proteins and peptides lacking these motifs; the only suitable substrates comprise recombinant proteins and peptides or synthetic peptides elongated with these motifs in the proper fashion. The potential clinical use of cyclic proteins has prompted research to their direct synthesis in cells by recombinant co-expression of a suited linear precursor and sortase in the same cell. Since the Ca2+-dependent S. aureus sortase A is not functional intra-cellularly, the Ca2+-independent Streptocuccus pyogenes sortase A was used for this purpose (Strijbis et al., 2012; Zhang et al., 2013; Strijbis and Ploegh, 2014). In a proof-of-principle experiment, it was shown that co-expression of S. pyogenes sortase A and GG-GFP-LPETG-Myc (control epitope tag) resulted in efficient backbone cyclization in the cytosol and endoplasmic reticulum lumen of transfected Saccharomyces cerevisiae and human HEK293T cells (Strijbis et al., 2012).
Other recombinant methods to obtain cyclic polypeptides include expressed protein ligation (EPL) and intein-mediated protein trans-splicing (PTS). EPL is basically a recombinant extension of the native peptide ligation (Figure 1D). In EPL, the expressed target protein contains an additional glycine at its N-terminus and is C-terminally fused with a specially designed intein containing an additional cysteine near its N-terminus (Figure 5A). During intein-mediated cyclization, a thioester bond is formed between the side chain of the latter cysteine and the peptide bond linking the intein moiety with the target polypeptide. The resulting thioester bond is resolved by nucleophilic attack of the N-terminal amino group of the former cysteine (intramolecular aminolysis). Via an S→N acyl shift, this leads to the irreversible formation of a backbone-cyclized peptide (Austin et al., 2010; Gould et al., 2012). In PTS, inactive N- and C-intein fragments are recombinantly fused to the C- and N-terminus of the target polypeptide chain, which has been equipped with an extra N-terminal glycine (Figure 5B). Under the proper conditions, the two inactive intein fragments spontaneously associate into a single active intein domain. Similar to intron splicing, the intein fragment is excised from the chain, concomitantly linking the N- and C-terminus of the target polypeptide. It is claimed that PTS is a faster and more efficient cyclization method than EPL. In PTS the N- and C-terminus of the target polypeptide are actively pulled together by the incorporated intein moiety, whereas in EPL the reaction depends on random collisions between the N-terminal cysteine and the thioester bond.
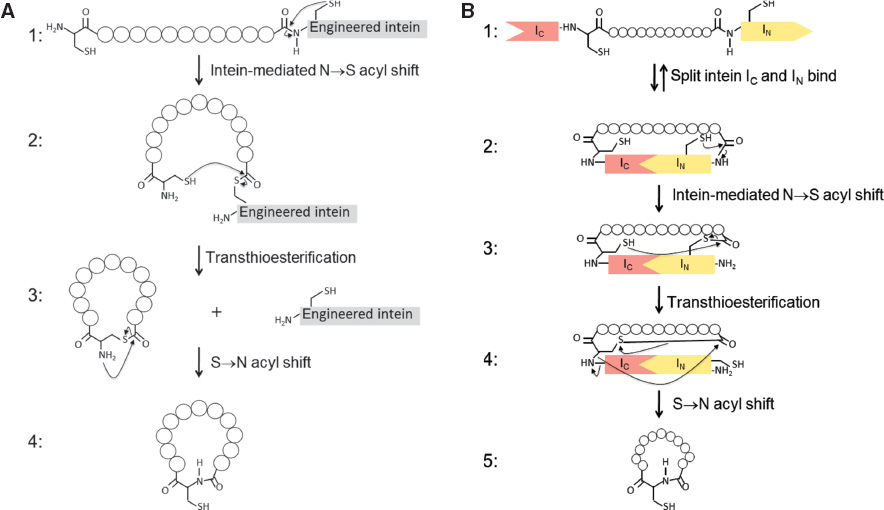
Intein-mediated recombinant production of cyclic peptides and proteins.
(A) Expressed protein ligation: (1) The target protein is expressed with an additional cysteine at its N-terminus and fused at its C-terminus with an engineered intein. After protein splicing, the intein is linked to the target protein by a thioester bond. (2) Nucleophilic attack of the N-terminal cysteine-SH group on this C-terminal thioester bond results in formation of a cyclic protein with concomitant release of the intein. (3) Intramolecular aminolysis of the thioester bond by the amino group of cysteine results in an S→N acyl shift and the formation of a backbone-cyclized peptide (4). (B) Intein-mediated protein trans-splicing (PTS). (1) Two inactive intein fragments are recombinantly fused to the N and C-terminus of the target protein. (2) Spontaneous association of the intein domains results in an active protein-splicing domain that after splicing is linked to the target protein by a thioester bond. (3) Nucleophilic attack of the N-terminal cysteine-SH group on the C-terminal thioester bond. (4) Aminolysis of the C-terminal thioester bond by the amino group of N-terminal cysteine results in an S→N acyl shift and the formation of a cyclic product with concomitant release of the intein (5). (figure adapted from Aboye and Camarero, 2012).
There is some debate about what should be the method of choice for recombinant production of backbone-cyclized polypeptides. EPL and PTS result in insertion of one additional cysteine residue, instead of the LPXTGG sequence after sortase-mediated cyclization. Furthermore, reaction rate of PTS-mediated cyclization is faster than that of sortase-mediated cyclization (reviewed in Katoh et al., 2011; Aboye and Camarero, 2012; Borra and Camarero, 2013). A point of discussion in favor of sortase A is that EPL and especially PTS require more extensive modification of the expression construct, which may reduce protein expression or affect protein solubility (Antos et al., 2009).
Biological (recombinant) and biochemical sortase-mediated backbone cyclization
The ability of sortase A to process a broad range of molecules, from very small to very large, makes it suited for a broad range of biotechnological cyclization reactions. The relative ease with which polypeptide-encoding genes can be fused with an N-terminal acyl-acceptor motif and a C-terminal acyl-donor motif allows the cost-effective recombinant production of many ‘linear’ precursors that can be efficiently cyclized by sortase A. In principle, by co-expression of S. pyogenes sortase A and the recombinant precursors, the road lies open to the direct recombinant production of cyclic proteins with potential high value for biology and medicine. However, this procedure is not yet fully developed. Although expression of a cyclic product in S. cerevisiae proceeded smoothly, expression in human cells led to a 50% yield (Strijbis et al., 2012). In the latter case, recombinant production of a cyclized product would make a subsequent purification step necessary. The standard procedure for recombinantly produced proteins, Ni2+-NTA-affinity purification, requires the incorporation of a hexa-histidine motif inside the cyclized backbone, either between the additional GG motif and the N-terminus of the protein or between its C-terminus and the LPXTG motif. This will result in a twice as long insert in the cyclic backbone, HHHHHHLPXTGG or LPXTGGHHHHHH instead of LPXTGG, which may have a more serious impact on the biological activity of the product. In recombinant production of ‘linear’ precursors, the hexa-histidine motif can be inserted C-terminally of the LPXTG motif. The hexa-histidine motif will then be released in the subsequent sortase-mediated cyclization in vitro (see examples in Table 1).
Recombinant proteins that have been cyclized with recombinant S. aureus sortase A.
| Recombinant protein precursor | Remarks |
|---|---|
| G-Cre-LPETG-His6 human recombinase | Reversibility of backbone cyclization by incubation with sortase in the presence of the acyl-receptor triglycine (Antos et al., 2009) |
| G5-eGFP-LPETG-His6 enhanced green fluorescent protein | Resistance to thermal and chemical denaturation (monitored by loss of fluorescence) (Antos et al., 2009) |
| G-His6-p97-LPETG-XX human hexameric AAA-ATPase | Formation of higher order aggregates, incubation of cyclic p97 with sortase in the presence of acyl-receptor diglycine yields the linear product (Antos et al., 2009) |
| G-UCHL3 human ubiquitin C-terminal hydrolase | Retention of activity (Antos et al., 2009) |
| GG-GCSF-3-LPETG-His6 human granulocyte colony-stimulating factor | Retention of activity in NFS-60 cell proliferation assay Enhanced resistance to thermal denaturation (Popp et al., 2011) |
| GG-IFNα2-LPETG-His6 human interferon-α | Retention of activity in Daudi cell proliferation inhibition assay Enhanced resistance to thermal denaturation (Popp et al., 2011) |
| GG-EPO-LPETG-His6 human erythropoietin | Retention of activity in Ba/F3 cell proliferation assay (Popp et al., 2011) |
The ability of sortase A to cyclize relatively small polypeptides makes it an ideal tool for use in combination with organic peptide synthesis, which allows incorporation of a myriad of non-coded ‘unnatural’ amino acids, D-amino acids, or even non-peptidic building blocks, as in linear azol(in)e-containing peptides (Arnison et al., 2013). Chemical synthesis followed by biochemical cyclization provides a powerful method that gives easy access to a broad range of backbone-cyclized products that are not accessible to recombinant production.
Another feature unique for chemical peptide synthesis is the ease to start the synthesis of linear precursors at any given residue within the sequence. This allows incorporation of the cyclization insert in any given part of the cyclized product, thus affording rational design of the linear peptide precursors. This has several benefits. In the first place, it offers the possibility to position the LPXTGG-linker at a safe distance from the known active domain or recognition site of the peptide (Jia et al., 2014). If present, existing motifs within the sequence can be used, so that the size of the inserts is diminished, which minimizes even more the risk on deleterious biological side effects. For example, in the synthesis of the linear kalata B1 precursor, the starting residue was assigned in such a manner that the LPV motif within the sequence became part of the C-terminal LPXTGG motif, thus reducing the length of the cyclization insert after sortase-mediated cyclization by three amino acids (Jia et al., 2014). Precise control of the chain length and corresponding ring size after cyclization may be very important in some cases. For instance, the antimicrobial activity of cyclized histatin-derived peptides strongly depended on the ring size, suggesting that the cyclization facilitates adoption of a helical structure, which is supposed to be essential for the membrane-disturbing properties of these peptides (Brewer and Lajoie, 2002).
The versatility and flexibility of recombinant and semi-synthetic production allow high-throughput synthesis of cyclized peptides, making sortase A not only suited for both large-scale production but also for exploratory research purposes.
Importance of sortase A for the chemo-enzymatic production of backbone-cyclized peptides
From the point of view of the peptide chemist, sortase A from S. aureus presents a welcome new tool for the backbone cyclization of synthetic peptides. Conveniently, the upper limit for conventional chemical backbone cyclization more or less coincides with the lower limit for sortase-mediated cyclization. The minimum length for linear sortase substrates is 12 amino acids, LPXTG and GG motifs excluded; shorter substrates result in cyclic dimers (Wu et al., 2011). Sortase-mediated cyclization is an easy and very efficient, almost quantitative, reaction. Recombinant, hexa-histidine-tagged sortase is readily removed by Ni2+-NTA purification from the reaction mixture, essentially containing only sortase-cyclized product and a small diglycine leaving group. If Ni2+-NTA-affinity purification is not feasible, e.g., when the peptide product is rich in histidines as in histatins, HPLC-purification can be employed, without substantial loss of activity of the recovered, or as we may say in this context ‘recycled’, sortase (Bolscher et al., 2011). Although the number of reported cases of enzymatic sortase-mediated cyclization of synthetic peptide precursors is still limited, the scope and applicability of this method appears promising (Table 2).
Synthetic peptides that have been cyclized with recombinant S. aureus sortase A.
| Synthetic peptide precursor | Remarks |
|---|---|
| GG-histatin 1-LPETGG human salivary peptide | Increased molar activity in migration assay |
| (Bolscher et al., 2011) | |
| GG-VTGalNacSGalNac APDTGalNac-LPKTGGS human MUC1 glycopeptide | Minimum length for sortase-mediated cyclization is 12 residues (excluding the LPXTG motif), MUC1 glycopeptide yielded mainly cyclic homodimer |
| (Wu et al., 2011) | |
| GGG-kalata B1-TGG plant cyclotide | Strongly reduced hemolytic activity, slightly reduced proteolytic stability |
| Contains three disulfide bridges | |
| (Jia et al., 2014) | |
| G-α-conotoxin Vc 1.1-GLPETGGS marine cone shell neurotoxin | Improved pharmacokinetics (orally active) |
| Contains two disulfide bridges | |
| (Jia et al., 2014) | |
| GG-SFTI-1-LPETGG sunflower trypsin inhibitor | Minor effect on conformational constraint |
| Contains one disulfide bridge | |
| (Jia et al., 2014) | |
| GG-LFcinB17-41-LPETGG bovine lactoferricin | Improved antimicrobial activity at physiological ionic strength |
| (Arias et al., 2014) | |
| GG-LFcinB20-35-LPETGG bovine lactoferricin loop | Reduced antimicrobial activity |
| (Arias et al., 2014) | |
| GGG-MCoTI-II-LPETGG plant cyclotide | Engineered, more efficient, SrtA (Stanger et al., 2014) |
Conclusion: sortase as a promising tool for backbone cyclization of proteins and peptides
As the above examples illustrate, recombinant sortase A from S. aureus presents a promising new tool for the backbone cyclization of both proteins and peptides. The enzyme can be produced on a large scale by recombinant expression followed by Ni2+-NTA-affinity chromatography. It recognizes a broad range of substrates, provided they contain an LPXTG acyl-donor site at the C-terminus and a GG acyl-acceptor site at the N-terminus, and it cyclizes linear precursors longer than 19 amino acids readily and with high yields. The incorporated LPXTGG motif is relatively small, which minimizes the risk on adverse biological or immunological side reactions upon use of the cyclized product in vivo. The requirement of LPXTG and GG motifs in the linear precursor restricts the origin of the sortase substrates that can be cyclized to recombinant or synthetic sources. Preliminary data show that sortase is still active when immobilized to a solid support (Steinhagen et al., 2013), suggesting the feasibility of industrial-scale production of sortase-cyclized proteins and peptides in the near future. Furthermore, a more efficient SrtA variant has recently been produced using a directed evolution strategy (Table 2) (Stanger et al., 2014). Four mutations (P94S, D160N, D165A, and K196T) within the active domain were identified, which, when combined (m4SrtA), resulted in increased in vitro cyclization efficiency for rMCoI-II, a recombinantly produced cyclotide. With m4SrtA the maximum cyclization yield of 80% was obtained within 48 h, whereas with wild type SrtA only 20% yield was obtained after 96 h.
The effect of sortase-mediated backbone cyclization on the biological activity and thermal- and proteolytic stability of several proteins and peptides is impressing (Antos et al., 2009; Popp and Ploegh, 2011, Popp et al., 2011; Bolscher et al., 2011). The variety of substrates reviewed here illustrates the versatility of sortase and promises a broad application in the search for backbone-cyclized protein- and peptide-based therapeutics in the years to come.
Only time will tell if all sortase A will last long enough to fulfil all these promises. During the preparation of this review, Nguyen et al. (2014) reported the identification of butelase 1, a ligase from the tropical plant Clitoria ternatea involved in the cyclization of cyclotides, which possesses several promising features. Like sortase, it cyclizes various peptides of plant and animal origin, such as kalata B1, Sunflower Trypsin Inhibitor-1, conotoxins, and histatins, very efficiently, with yields over 95%. Compared to sortase A, cyclization was achieved at a much lower enzyme-to-peptide ratio (1:400 vs. 1:3) and with much shorter reaction times (0.2 h for histatin vs. 16 h). Its structural demands from substrate peptides are even less strict than those of sortase. The C-terminal motif consists of the sequence NHV, or to some extent DHV, which is cleaved at the asparagine- (or aspartate)-histidine peptide bond. The N-terminal acceptor site contains two amino acids, of which the N-terminal residue is rather promiscuous (anything but D, E, or P, lower yields for S, T, and Y) and the penultimate N-terminal residue rather critical (strictly hydrophobic: C, I, L, or V). This results in smaller residual inserts after cyclization, three amino acids instead of six for sortase, minimizing the chance of adversary side reactions. This promiscuity furthermore increases the opportunities to prepare cyclic peptides without a residual insert by searching for suited motifs within the sequence when designing the linear substrate peptide, for instance the motif NYL in histatins, minimizing the chances of adversary side reactions even further.
As soon as sufficient amounts of butelase 1 will be available, it may become an interesting alternative cyclization tool to sortase A.
Funding: ZonMW (Grant/Award Number: ‘50-51700-98-055’), University of Amsterdam, (Grant/Award Number: ‘for research into the focal point Oral infections’).
References
Aboye, R.L. and Camarero, J.A. (2012). Biological synthesis of circular polypeptides. J. Biol. Chem. 287, 27026–27032.10.1074/jbc.R111.305508Search in Google Scholar PubMed PubMed Central
Aboye, T.L., Li, Y., Majumder, S., Hao, J., Shekhtman, A., and Camarero, J.A. (2012). Efficient one-pot cyclization/folding of rhesus θ-defensin-1 (RTD-1). Bioorg. Med. Chem. Lett. 22, 2823–2826.10.1016/j.bmcl.2012.02.080Search in Google Scholar PubMed PubMed Central
Antos, J.M., Popp, M.W., Ernst, R., Chew, G.L., Spooner, E., and Ploegh, H.L. (2009). A straight path to circular proteins. J. Biol. Chem. 284, 16028–16036.10.1074/jbc.M901752200Search in Google Scholar PubMed PubMed Central
Arias, M., McDonald, L.J., Haney, E.F., Nazmi, K., Bolscher, J.G.M., and Vogel, H.J. (2014). Bovine and human lactoferricin peptides: chimeras and new cyclic analogs. Biometals 27, 935–948.10.1007/s10534-014-9753-4Search in Google Scholar PubMed
Arnison, P.G., Bibb, M.J., Bierbaum, G., Bowers, A.A., Bugni, T.S., Bulaj, G., Camarero, J.A., Campopiano, D.J., Challis, G.L., Clardy, J., et al. (2013). Ribosomally synthesized and post-translationally modified peptide natural products: overview and recommendations for a universal nomenclature. Nat. Prod. Rep. 30, 108–160.10.1039/C2NP20085FSearch in Google Scholar PubMed PubMed Central
Austin, J., Kimura, R.H., Woo, Y.H., and Camarero, J.A. (2010). In vivo biosynthesis of an Ala-scan library based on the cyclic peptide SFTI-1. Amino Acids 38, 1313–1322.10.1007/s00726-009-0338-4Search in Google Scholar PubMed PubMed Central
Bolscher, J.G.M., Oudhoff, M.J., Nazmi, K., Antos, J.M., Guimaraes, C.P., Spooner, E., Haney, E.F., Garcia Vallejo, J.J., Vogel, H.J., van’t Hof, W., et al. (2011). Sortase A as a tool for high-yield histatin cyclization. FASEB J. 25, 2650–2658.10.1096/fj.11-182212Search in Google Scholar PubMed
Borra, R. and Camarero J.A. (2013). Recombinant expression of backbone-cyclized polypeptides. Biopolymers 100, 502–509.10.1002/bip.22306Search in Google Scholar PubMed PubMed Central
Brask, J., Albericio, F., and Jensen, K.J. (2003). Fmoc solid-phase synthesis of peptide thioesters by masking as trithioortho esters. Org. Lett. 5, 2951–2953.10.1021/ol0351044Search in Google Scholar PubMed
Brewer, D. and Lajoie, G. (2002). Structure-based design of potent histatin analogues. Biochemistry 41, 5526–5536.10.1021/bi015926dSearch in Google Scholar PubMed
Clark, R.J. and Craik, D.J. (2010). Native chemical ligation applied to the synthesis and bioengineering of circular peptides and proteins. Biopolymers 94, 414–422.10.1002/bip.21372Search in Google Scholar
Clippingdale, A.B., Barrow, C.J., and Wade, J.D. (2000). Peptide thioester preparation by Fmoc solid phase peptide synthesis for use in native chemical ligation. J. Pept. Sci. 6, 225–234.10.1002/(SICI)1099-1387(200005)6:5<225::AID-PSC244>3.0.CO;2-TSearch in Google Scholar
Craik, D.J., Daly, N.L, Bond, T., and Waine, C. (1999). Plant cyclotides: a unique family of cyclic and knotted proteins that defines the cyclic cystine knot structural motif. J. Mol. Biol. 294, 1327–1336.10.1006/jmbi.1999.3383Search in Google Scholar
Dasgupta, S., Samantaray, S., Sahal, D., and Roy, R.P. (2011). Isopeptide ligation catalyzed by quintessential sortase A: mechanistic cues from cyclic and branched oligomers of indolicidin. J. Biol. Chem. 286, 23996–24006.10.1074/jbc.M111.247650Search in Google Scholar
Gause, G.F. and Brazhnikova, M.G. (1944). Gramicidin S and its use in the treatment of infected wounds. Nature 154, 703–703.10.1038/154703a0Search in Google Scholar
Gould, A., Majumder, S., Garcia, A.E., Carlsson, P., Shekhtman, A., and Camarero J.A. (2012). Recombinant production of rhesus θ-defensin-1 (RTD-1) using a bacterial expression system. Mol. Biosyst. 8, 1359–1365.10.1039/c2mb05451eSearch in Google Scholar
Heck, T., Faccio, G., Richter, M., and Thöny-Meyer, L. (2013). Enzyme-catalized protein crosslinking. Appl. Microbiol. Biotechnol. 9, 461–475.10.1007/s00253-012-4569-zSearch in Google Scholar
Hemu, X., Taichi, M., Qiu, Y., Liu, D.X., and Tam, J.P. (2013). Biomimetic synthesis of cyclic peptides using novel thioester surrogates. Biopolymers 100, 492–501.10.1002/bip.22308Search in Google Scholar
Hou, W., Zhang, X., Li, F., and Liu, C.F. (2010). PeptidylN,N-bis(2-mercaptoethyl)-amides as thioester precursors for native chemical ligation. Org. Lett. 13, 386–389.10.1021/ol102735kSearch in Google Scholar
Ingenito, R., Bianchi, E., Fattori, D., and Pessi, A. (1999). Solid phase synthesis of peptide C-terminal thioesters by Fmoc/t-Bu chemistry. J. Am. Chem. Soc. 121, 11369–11374.10.1021/ja992668nSearch in Google Scholar
Jia, X., Kwon, S., Wang, C.I., Huang, Y.H., Chan L.Y., Tan, C.C., Rosengren, K.J., Mulvenna, J.P., Schroeder, C.I. and Craik, D.J. (2014). Semienzymetic cyclization of disulfide-rich peptides using sortase A. J. Biol. Chem. 289, 6627–6638.10.1074/jbc.M113.539262Search in Google Scholar PubMed PubMed Central
Katoh, T., Goto, Y., Reza, M.S., and Suga, H. (2011). Ribosomal synthesis of backbone macrocyclic peptides. Chem. Commun. 47, 9946–9958.10.1039/c1cc12647dSearch in Google Scholar PubMed
Liu, F. and Mayer, J.P. (2013). An Fmoc compatible, O to S shift-mediated procedure for the preparation of C-terminal thioester peptides. J. Org. Chem. 78, 9848–9856.10.1021/jo4015112Search in Google Scholar PubMed
Maresso, A.W. and Schneewind, O. (2008) Sortase as a target of anti-infective therapy. Pharmacol. Rev. 60, 128–141.Search in Google Scholar
Nguyen, G.K., Wang, S., Qiu, Y., Hemu, X., Lian Y., and Tam J.P. (2014). Butelase 1 is an Asx-specific ligase enabling peptide macrocyclization and synthesis. Nat. Chem. Biol. 10, 732–738.10.1038/nchembio.1586Search in Google Scholar PubMed
Ollivier, N., Dheur, J., Mhidia, R., Blanpain, A., and Melnyk, O. (2010). Bis(2-sulfanylethyl)amino native peptide ligation. Org. Lett. 12, 5238–5241.10.1021/ol102273uSearch in Google Scholar PubMed
Ollivier, N., Raibaut, L., Blanpain, A., Desmet, R., Dheur, J., Mhidia, R., Boll, E., Drobecq, H., Pira, S.L., and Melnyk, O. (2014). Tidbits for the synthesis of bis(2-sulfanylethyl)amido (SEA) polystyrene resin, SEA peptides and peptide thioesters. J Pept. Sci. 20, 92–97.10.1002/psc.2580Search in Google Scholar PubMed
Oudhoff, M.J., Bolscher, J.G.M., Nazmi, K., Kalay, H., van’t Hof, W., Nieuw-Amerongen, A.V., and Veerman, E.C.I. (2008). Histatins are the major wound-closure stimulating factors in human saliva as identified in a cell culture assay. FASEB J. 22, 3805–3812.10.1096/fj.08-112003Search in Google Scholar PubMed
Oudhoff, M.J., Kroeze, K.L., Nazmi, K., van den Keijbus, P.A.M., van’t Hof, W., Fernandez-Boria, M., Hordijk, P.L., Gibbs, S., Bolscher, J.G.M., and Veerman, E.C.I. (2009). Structure-activity analysis of histatin, a potent wound healing peptide from human saliva: cyclization of histatin potentiates molecular activity 1000-fold. FASEB J. 23, 3928–3935.10.1096/fj.09-137588Search in Google Scholar PubMed
Popp, M.W. and Ploegh, H.L. (2011). Making and breaking peptide bonds: protein engineering using sortase A. Angew. Chem. Int. Ed. Engl. 50, 5024–5032.10.1002/anie.201008267Search in Google Scholar PubMed
Popp, M.W., Dougan, S.K., Chuang, T.Y., Spooner, E., and Ploegh, H.L. (2011). Sortase-catalyzed transformations that improve the properties of cytokines. Proc. Natl. Acad. Sci. USA 22, 3169–3174.10.1073/pnas.1016863108Search in Google Scholar PubMed PubMed Central
Proft, T. (2010). Sortase-mediated protein ligation: an emerging biotechnology tool for protein modification and immobilisation. Biotechnol. Lett. 32, 1–10.10.1007/s10529-009-0116-0Search in Google Scholar
Schneewind, O. and Missiakas, D.M. (2012). Protein secretion and surface display in Gram-positive bacteria. Phil. Trans. R. Soc. B. Biol. Sci. 367, 1123–1139.10.1098/rstb.2011.0210Search in Google Scholar
Spirig, T., Weiner, E.M., and Clubb, R.T. (2011). Sortase enzymes in Gram-positive bacteria. Mol. Microbiol. 82, 1044–1059.10.1111/j.1365-2958.2011.07887.xSearch in Google Scholar
Stanger, K., Maurer, T., Kaluarachchi, H., Coons, M., Franke, Y., and Hannoush, R.N. (2014). Backbone cyclization of a recombinant cystine-knot peptide. FEBS Lett. 588, 4487–4496.10.1016/j.febslet.2014.10.020Search in Google Scholar
Steinhagen, M., Zunker, K., Nordsieck, K., and Beck-Sickinger, A.G. (2013). Large scale modification of biomolecules using immobilized sortase A from Staphylococcus aureus. Bioorg. Med. Chem. 21, 3504–3510.10.1016/j.bmc.2013.03.039Search in Google Scholar
Strijbis, K. and Ploegh, H.L. (2014). Secretion of circular proteins using sortase. Methods Mol. Biol. 1174, 73–83.10.1007/978-1-4939-0944-5_5Search in Google Scholar
Strijbis, K., Spooner, E., and Ploegh, H.L. (2012). Protein ligation in living cells using sortase. Traffic 13, 780–789.10.1111/j.1600-0854.2012.01345.xSearch in Google Scholar
Taichi, M., Hemu, X., Qiu Y., and Tam, J.P. (2013). A thioethylalkylamido (TEA) thioester surrogate in the synthesis of a cyclic peptide via a tandem acyl shift. Org. Lett. 15, 2620–2623.10.1021/ol400801kSearch in Google Scholar
Tam, J.P. and Lu, Y.-A. (1997). Synthesis of large cyclic cystine-knot peptide by orthogonal coupling strategy using unprotected peptide precursor. Tetrahedron Lett. 38, 5599–5602.10.1016/S0040-4039(97)01271-9Search in Google Scholar
Tam, J.P. and Wong, C.T. (2012). Chemical synthesis of circular proteins. J. Biol. Chem. 287, 27020–27025.10.1074/jbc.R111.323568Search in Google Scholar PubMed PubMed Central
Tang, Y.Q., Yuan, J., Osapay, K., Tran, D., Miller, C.J., Ouellette, A.J., and Selsted, M.E. (1999). A cyclic antimicrobial peptide produced in primate leukocytes by the ligation of two truncated α-defensins. Science 286, 498–502.10.1126/science.286.5439.498Search in Google Scholar PubMed
Van’t Hof, W., Oudhoff, M.J., and Veerman, E.C.I. (2013). Histatins: multifunctional salivary antimicrobial peptides. In: Antimicrobial Peptides and Innate Immunity, P.S. Hiemstra and S.A.J. Zaat, eds. Progress in Inflammation Research (Basel, Switzerland: Springer), pp. 167–181.Search in Google Scholar
Wong, C.T., Rowlands, D.K., Wong, C.H., LO, T.W., Nguyen, G.K., and Tam, J.P. (2012). Orally active peptide bradykinin B1 receptor antagonists engineered from a cyclotide scaffold for inflammatory pain treatment. Angew. Chem. Int. Ed. Engl. 51, 5620–5624.10.1002/anie.201200984Search in Google Scholar PubMed
Wu, Z. and Guo, Z. (2012). Sortase-mediated transpeptidation for site-specific modification of peptides, glycopeptides, and proteins, J. Carbohydr. Chem. 31, 48–66.10.1080/07328303.2011.635251Search in Google Scholar PubMed PubMed Central
Wu, Z., Guo, X., and Guo, Z. (2011). Sortase A-catalyzed peptide cyclization for the synthesis of macrocyclic peptides and glycopeptides. Chem. Commun. 47, 9218–9220.10.1039/c1cc13322eSearch in Google Scholar PubMed PubMed Central
Zhang, J., Yamaguchi, S., Hirakawa, H., and Nagamune, T. (2013). Intracellular protein cyclization catalyzed by exogenously transduced Streptococcus pyogenes sortase A. J. Biosci. Bioeng. 116, 298–301.10.1016/j.jbiosc.2013.03.006Search in Google Scholar PubMed
Zheng, J.S., Tang, S., Qi, Y.K., Wang, Z.P., and Liu, L. (2013). Chemical synthesis of proteins using peptide hydrazides as thioester surrogates. Nat. Protoc. 8, 2483–2495.10.1038/nprot.2013.152Search in Google Scholar PubMed
©2015 by De Gruyter
Articles in the same Issue
- Frontmatter
- Reviews
- Sortase-mediated backbone cyclization of proteins and peptides
- Making the LINC: SUN and KASH protein interactions
- Enhancers, enhancers – from their discovery to today’s universe of transcription enhancers
- Minireview
- Recent advances and concepts in substrate specificity determination of proteases using tailored libraries of fluorogenic substrates with unnatural amino acids
- Research Articles/Short Communications
- Genes and Nucleic Acids
- microRNA-210 is involved in the regulation of postmenopausal osteoporosis through promotion of VEGF expression and osteoblast differentiation
- Protein Structure and Function
- C-terminal truncation of a Tat passenger protein affects its membrane translocation by interfering with receptor binding
- Aspartate 496 from the subsite S2 drives specificity of human dipeptidyl peptidase III
- Proteolysis
- Evolutionary divergence of Threonine Aspartase1 leads to species-specific substrate recognition
- Purification and characterisation of recombinant His-tagged RgpB gingipain from Porphymonas gingivalis
Articles in the same Issue
- Frontmatter
- Reviews
- Sortase-mediated backbone cyclization of proteins and peptides
- Making the LINC: SUN and KASH protein interactions
- Enhancers, enhancers – from their discovery to today’s universe of transcription enhancers
- Minireview
- Recent advances and concepts in substrate specificity determination of proteases using tailored libraries of fluorogenic substrates with unnatural amino acids
- Research Articles/Short Communications
- Genes and Nucleic Acids
- microRNA-210 is involved in the regulation of postmenopausal osteoporosis through promotion of VEGF expression and osteoblast differentiation
- Protein Structure and Function
- C-terminal truncation of a Tat passenger protein affects its membrane translocation by interfering with receptor binding
- Aspartate 496 from the subsite S2 drives specificity of human dipeptidyl peptidase III
- Proteolysis
- Evolutionary divergence of Threonine Aspartase1 leads to species-specific substrate recognition
- Purification and characterisation of recombinant His-tagged RgpB gingipain from Porphymonas gingivalis