The FGF23/Klotho axis in the regulation of mineral and metabolic homeostasis
-
Masanobu Kawai


Abstract
The function of fibroblast growth factor (FGF) 23 has been suggested to be multifaceted beyond its canonical function as a regulator of mineral metabolism. FGF23 was originally shown to play a central role in phosphate (Pi) and vitamin D metabolism, and a number of diseases associated with dysregulated Pi metabolism have been attributed to abnormal FGF23 signaling activities. The discovery of Klotho as a co-receptor for FGF23 signaling has also accelerated understanding on the molecular mechanisms underlying Pi and vitamin D metabolism. In addition to these canonical functions, FGF23 has recently been implicated in a number of metabolic diseases including chronic kidney disease-associated complications, cardiovascular diseases, and obesity-related disorders; however, the physiological significance and molecular mechanisms of these emerging roles of FGF23 remain largely unknown. Molecular and functional insights into the FGF23 pathway will be discussed in the present review, with an emphasis on its role in human disorders related to dysregulated Pi metabolism as well as metabolic disorders.
Introduction
Fibroblast growth factor (FGF) 23 is a secretory molecule that is mainly produced by osteoblastic cells, and was originally shown to function as a central regulator of phosphate (Pi) and vitamin D metabolism. Since its identification in 2000 [1], extensive studies have been conducted in an attempt to reveal the role of FGF23 in the pathogenesis of human diseases associated with dysregulated mineral metabolism. The findings of these studies led to the development of new strategies to combat these disorders, and clinical trials for the treatment of X-linked hypophosphatemic rickets using FGF23-neutralizing antibodies are ongoing [2], [3]. Besides these canonical functions, FGF23 has recently been implicated in metabolic homeostasis. For example, FGF23 has been associated with a higher risk of cardiovascular disease (CVD) [4], [5], [6], [7], [8]. Furthermore, relationships have been reported between FGF23 concentrations and clinical parameters involved in glucose metabolism as well as inflammation [9], [10], [11], [12], [13], [14]; however, the physiological significance and molecular mechanisms of these emerging roles of FGF23 remain largely unknown.
The discovery of α-Klotho (KL) as a co-receptor for FGF23 signaling has also accelerated understanding on the mechanisms responsible for the regulation of mineral metabolism [15]. Despite the critical roles of KL in FGF23 signaling, accumulating evidence has revealed a KL-independent pathway of FGF23, which may be involved in the non-canonical functions of FGF23 such as the development of cardiac hypertrophy. Thus, these novel findings have initiated a new era of research on the role of FGF23 signaling in human disorders. The canonical functions of FGF23/KL as a regulator of Pi and vitamin D metabolism will be discussed in the present review, with a focus on how its disturbance results in the development of human disorders associated with dysregulated mineral metabolism. The emerging roles of FGF23 beyond its involvement in mineral metabolism are also discussed.
Physiology of FGF23
The FGF23/KL axis
FGF23 was originally identified as a novel member of the FGF family [1] and is the gene responsible for hypophosphatemic rickets and osteomalacia [16], [17]. Although FGF23 was initially reported to be expressed in the brain, osteoblastic cells, particularly osteocytes, were subsequently identified as a physiological source of FGF23 [18], [19]. A number of other tissues including the thymus, small intestine, and heart have also been shown to express FGF23 [1], [16], but at lower levels and with an unknown physiological relevance. FGF23 contains a signal peptide in its N-terminus region and lacks the heparin-binding domain present in autocrine/paracrine FGFs [20]; therefore, FGF23 is able to escape from the extracellular matrix and function as an endocrine factor. Human FGF23 is a protein that comprises 251 amino acids (32 kDa), with rat and mouse FGF23 sharing 72% and 71% homologies to human FGF23, respectively. The N-terminal region of FGF23 is a FGF core homology domain and interacts with FGFRs, whereas the C-terminal domain is unique to FGF23 and binds KL [21], [22] (Figure 1).
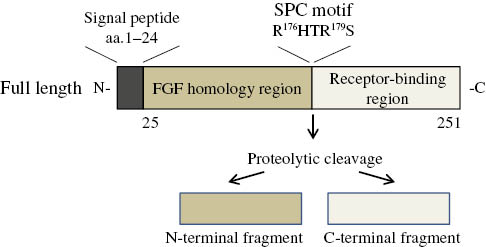
Structure of FGF23.
Full-length FGF23 is proteolytically cleaved at the SPC motif to produce inactive forms of FGF23.
Significant advances in understanding on the roles of FGF23 have been accomplished by the discovery of KL functioning as a co-receptor for FGF23 [15]. KL was originally identified as the gene responsible for premature aging-like symptoms such as a short lifespan, infertility, arteriosclerosis, skin atrophy, osteoporosis, and emphysema in mice [23]. KL is a 135 kDa single-pass transmembrane protein that consists of an extracellular domain, transmembrane domain, and intracellular carboxyl domain [24]. The extracellular domain contains KL1 and KL2 internal repeats and exhibits weak β-galactosidase activity [25], [26]. In addition to its membrane localization, KL is known to be secreted into the circulation (soluble KL: sKL) and functions as a humoral factor that plays multiple roles in anti-oxidation, anti-apoptosis, and ion transport [26], [27], [28]. Approximately 130 kDa sKL is produced through the ectodomain shedding of membrane-bound KL by disintegrin and metalloproteinase (ADAM)-10, ADAM-17, and β-site amyloid precursor protein-cleaving enzyme 1 (BACE1) [29], [30] and has been detected in the urine, serum, and cerebrospinal fluid of humans [31]. Previous studies reported that 65–70 kDa sKL is also produced by alternative splicing; however, its presence in humans has not yet been confirmed [32], [33] (Figure 2).
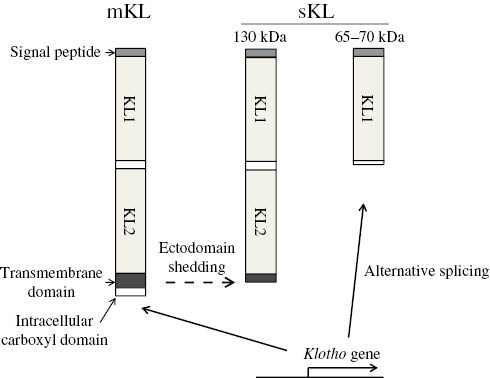
Structure of Klotho (KL).
Membrane-bound KL (mKL) is composed of two internal repeats (KL1 and KL2), a transmembrane domain, and short intracellular carboxyl domain. The proteolytic cleavage of mKL at the transmembrane domain results in the production of 130 kDa soluble KL (sKL), while 65–70 kDa sKL is produced by alternative splicing.
Post-translational regulation of FGF23
One of the important features of FGF23 regulation is that it is proteolytically cleaved at the site of a subtilisin-like proprotein convertase (SPC) motif (176RXXR179). Cleavage at this site by proteases such as furin results in the inactivation of FGF23 and production of N- and C-terminal fragments [22]. C-terminal fragments have been shown to interact with KL and, as a consequence, function as antagonists of the FGF23 signaling pathway; therefore, the cleavage of this site may add another level to the mechanism by which the proteolytic cleavage of FGF23 impairs the FGF23 signaling pathway [34].
The cleavage of FGF23 at the SPC motif is known to be regulated by post-translational modifications. The glycosylation of Thr178 in the SPC motif by GALNT3 (polypeptide N-acetylgalactosaminyltransferase 3) is crucial for the production and secretion of full-length biologically active FGF23 [35], [36]. The phosphorylation of FGF23 adds another level to its regulatory network such that phosphorylation at Ser180 within the SPC motif by FAM20C has recently been reported to inhibit O-glycosylation by GALNT3, resulting in accelerated cleavage at this site [37], [38] (Figure 1).
Regulation of FGF23 expression
The regulatory network of FGF23 expression in bone cells has been widely examined and several signaling cascades have been shown to regulate its expression. As activation of the FGF23 signaling pathway decreases circulating Pi and 1,25-dihydroxyvitamin D (1,25-(OH)2D) levels, it is important to determine whether any feedback loops are operative between FGF23 and Pi/1,25-(OH)2D. Pi and 1,25-(OH)2D have both been positively associated with FGF23 levels, indicating that FGF23 and Pi/1,25-(OH)2D mutually regulate each other and form a fine-tuned network in mineral metabolism. 1,25-(OH)2D is known to up-regulate the expression of FGF23 by activating the vitamin D receptor (VDR) [39], [40], [41]. Although VDR response elements have been reported in the mouse Fgf23 gene [41], the mode of in vivo regulation of human FGF23 by 1,25-(OH)2D has not yet been elucidated in detail.
In contrast to the regulation of FGF23 expression by 1,25-(OH)2D, it currently remains unclear whether Pi directly regulates the expression of FGF23. As accumulating evidence has indicated that extracellular Pi functions as a signaling molecule and transduces its signals in target cells, it is possible that Pi directly regulates the expression of FGF23 in bone cells. In support of this, in vivo animal models have revealed a relationship between dietary Pi loads and circulating FGF23 levels [42], [43], indicating that extracellular Pi induces the expression of FGF23; however, experimental evidence for the direct regulation of FGF23 by Pi is limited. Similar to the findings observed in mice, the amount of dietary Pi has been associated with serum FGF23 levels in humans [44], [45], although there is also evidence showing the lack of a relationship between these two parameters [46]. In order to determine the relationship between FGF23 and Pi, Scanni et al. analyzed the response of FGF23 to an acute Pi load in healthy humans, and found that an acute Pi load by an intravenous infusion or a duodenal Pi load increased FGF23 levels [47]. These increases were preceded by elevations in PTH, suggesting that the induction of FGF23 by an acute Pi load is partly mediated through an increase in PTH levels because PTH is known augment FGF23 levels. 1,25-(OH)2D concentrations were shown to be decreased after FGF23 levels increased [47].
PTH has also been identified as a positive regulator of skeletal FGF23 expression, as evidenced by clinical conditions in which PTH signaling is activated such as Jansen metaphyseal chondrodysplasia caused by a mutation in the PTH1R gene [48], [49]. In experimental animal models, intermittent injections of hPTH failed to induce FGF23 expression in mice lacking the PTH type 1 receptor in the limb mesenchyme, whereas it was induced in WT mice [50]. Chronic kidney disease (CKD) is an additional example of elevations in FGF23 levels being associated with increased PTH levels. The stimulatory role of PTH in FGF23 expression was experimentally proven in a rat CKD model in which parathyroidectomy reversed the elevated FGF23 levels in these rats [49]; however, evidence also exists that does not support this scenario [51]. In addition, clinical evidence for the relationship between PTH and FGF23 is limited. For example, FGF23 levels in patients with primary hyperparathyroidism were found to be similar to those in healthy controls [52]; therefore, the effects of PTH on FGF23 expression may be context-specific and further studies are clearly required to determine the role of PTH in the regulation of FGF23 expression.
We recently demonstrated that the expression of FGF23 was regulated by the sympathetic nervous system (SNS) [53]. As skeletal Fgf23 expression increases during the dark phase, in which food intake is stimulated in mice, we hypothesized that food intake may regulate FGF23 expression in the skeleton. SNS is activated by food intake; therefore, we determined whether the activation of SNS enhanced FGF23 expression. As expected, Fgf23 expression was induced by a β-adrenergic agonist in the skeleton and this effect was mediated through a cAMP-response element located in the promoter region in the Fgf23 gene. In order to better understand the relationships between food intake, the SNS, and FGF23 expression, mice were exclusively fed during the light phase. Under these conditions, a peak in SNS activity was shifted from the dark to light phase and this was associated with a peak expression of Fgf23 during the light phase [53]. Furthermore, when a β-blocker was concomitantly used, a peak in the expression of Fgf23 was not observed. These findings suggest that the timing of food intake determines the rhythmic expression profile of skeletal Fgf23, and also that food intake-driven sympathetic activation is critical in this regulation. This system may function as part of the systemic regulatory network maintaining Pi levels such that the enhanced influx of Pi from the diet is balanced by the food intake-associated induction of FGF23 to stimulate the excretion of Pi in the urine.
In addition to the factors described above, numerous factors have been implicated in the regulation of FGF23 expression. For example, activation of the FGF signaling pathway has been suggested to activate FGF23 expression, as demonstrated in patients with osteoglophonic dysplasia caused by the activation of mutations in the FGFR1 gene, which increases FGF23 levels [54]. In vivo and in vitro analyses also experimentally support this [55], [56], [57], [58]. The activation of FGFR by low molecular weight (LMW) FGF2 was shown to increase the expression of FGF23 through activation of NFAT pathway [57], [58]. In addition to the activation of membrane FGFR by FGF2, the intranuclear activation of FGFR1 by high molecular weight (HMW) FGF2 was also found to increase the expression of FGF23, possibly in a manner involving the activation of CREB pathway [57]. Hypoxia has been shown to induce FGF23 expression, and hypoxia inducible factor-1 (HIF1) is suggested to be involved in this regulation [59]. HIF1 has also been reported to be involved in the induction of FGF23 by inflammation and iron deficiency [60]. FGF23 levels were also reported to be positively associated with the biomarkers of inflammation, insulin resistance, myocardial infarction, and metabolic acidosis [9], [10], [11], [12], [13], [14]; however, the molecular mechanisms underlying the induction of FGF23 under these conditions have not yet been elucidated in detail (Figure 3).
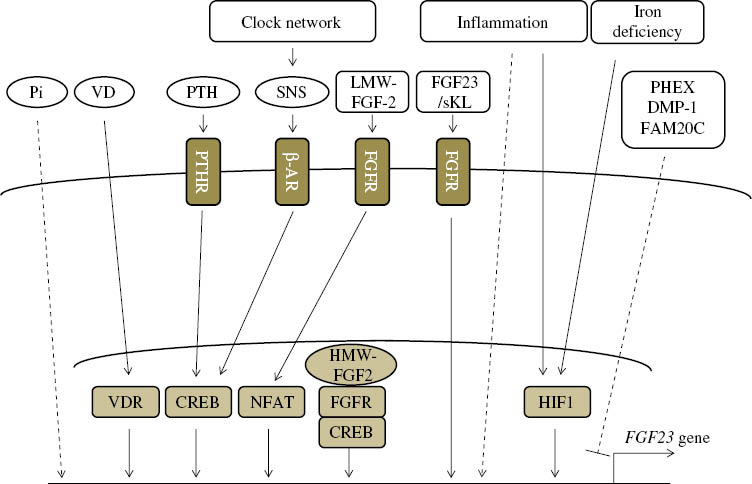
Regulation of FGF23 expression.
The numerous factors regulating FGF23 transcription have been identified, but the underlying molecular cascades remain largely unknown. VD, 1,25(OH)2 vitamin D; PTH, parathyroid hormone; PTHR, PTH receptor; SNS, sympathetic nervous system; β-AR, beta adrenergic receptor; LMW-FGF2, low molecular weight; FGFR, FGF receptor; sKL, soluble Klotho; VDR, vitamin D receptor; CREB, cAMP response element binding protein; NFAT, nuclear factor of activated T-cells, HMW-FGF2, high molecular weight FGF2; HIF1, hypoxia inducible factor-1.
The FGF23 signaling pathway
Since KL is required for FGF23 to exert its effects, KL-expressing tissues are considered to be the target tissues of the FGF23 signaling pathway. KL is expressed in various tissues including the parathyroid gland, placenta, pituitary, and choroid plexus of the brain [23], with the distal tubular epithelial cells of the kidney being the strongest expressers of KL [23]. In these tissues, FGF23 binds to FGF receptors and activates downstream pathways such as the extracellular signal-regulated kinases (ERK)/early growth response-1 (EGR-1) signal pathway. FGF23 has been shown to bind to FGFR1, 3c, and 4 in vitro [61], whereas the in vivo relevance of this complex may be restricted to FGFR1 [62], [63].
In contrast to the widely accepted tenet that membrane-bound KL is required for the FGF23 signaling pathway, accumulating evidence has revealed that FGF23 exerts its effects in cooperation with sKL, which also makes a complex with FGF23 and FGFR and activates downstream signaling cascades [61], [64], [65]. The in vivo implications of the FGF23/sKL complex were previously analyzed using a mouse model in which sKL was overexpressed [65]. The FGF23/sKL complex has been proposed to activate FGFRs and induce FGF23 expression in osteoblastic cells. It may also play a role in the regulation of chondrocyte biology such that FGF23/sKL binds to FGFR3 and suppresses chondrocyte proliferation [64]. These findings suggest that FGF23 transduces its signals in cells in which membrane-bound KL is not expressed; however, whether this scenario is operative under physiological conditions remains to be determined since these findings were based on the use of supraphysiological levels of sKL.
In addition to the KL-dependent effects of FGF23, accumulating evidence indicates the presence of a KL-independent function of FGF23. For example, FGF23 has been shown to induce left ventricular hypertrophy by activating the calcineurin-NFAT signaling pathway independent of KL, and the FGFR4/PLCγ pathway was subsequently shown to be activated by FGF23 in cardiac myocytes when KL was absent [66], [67]. Additionally, FGF23 has been reported to suppress the secretion of PTH in a KL-independent manner; however, KL-dependent signaling pathway also plays an important role in this regulation, [68], [69]. Although these findings emphasize the presence of the KL-independent signaling pathway of FGF23, the molecular mechanisms by which FGF23 binds and activates FGFR in the absence of KL remain unknown (Figure 4).
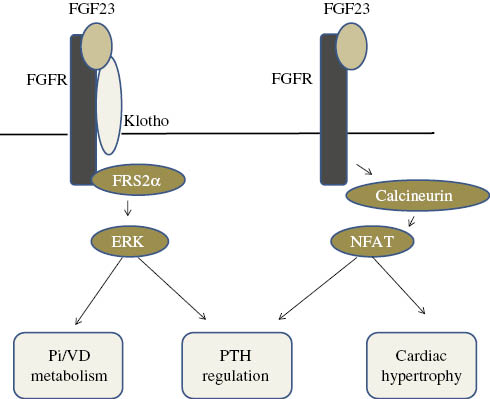
FGF23 signaling cascades.
FGF23 activates its downstream signaling molecules including FRS2α and ERK in co-operation with Klotho, and this is involved in the regulation of mineral metabolism and PTH production. In the absence of Klotho, FGF23 may activate the calcineurin/NFAT signaling pathway, regulate PTH production, and induce cardiac hypertrophy; however, the mode of binding between FGFR and FGF23 in this scenario has not yet been determined. Pi, Inorganic phosphate; VD, vitamin D.
Functions of the FGF23/KL signaling pathway
Regulation of Pi metabolism
Genetically engineered mouse models have revealed the central role of the FGF23/KL axis in the regulation of Pi homeostasis; Fgf23-deficient mice and Klotho-deficient mice displayed almost identical phenotypes in the context of Pi metabolism such that both animal models exhibit increases in Pi levels with reduced urinary Pi excretion [70]. In a similar manner, FGF23 transgenic mice display hypophosphatemia [71], [72]. The strict requirement of KL in this regulation is also evident in a mouse model in which the KL/FGF23 axis is disrupted. An injection of recombinant FGF23 (rFGF23) into WT or Fgf23-deficient mice caused a decrease in Pi levels, whereas this effect of rFGF23 was not observed in Klotho-deficient mice. The phosphaturic effects of FGF23 are mainly mediated through its actions in KL-expressing cells in the distal tubular epithelial cells of the kidney because the partial deletion of Klotho in these cells partly recapitulates the phenotypes of Fgf23-deficient and Klotho-deficient mice [73]. The administration of rFGF23 to WT mice was consistently shown to activate its downstream molecules in distal tubular epithelial cells and result in the development of hypophosphatemia [17], [74].
The phosphaturic effects of the FGF23/KL axis are predominantly dependent on its regulation of the renal expression and localization of type II Na+-dependent Pi co-transporters, particularly Npt2a and Npt2c. Activation of the FGF23 signaling pathway has been shown to suppress the expression and activities of Npt2a and Npt2c and reduce the renal reabsorption of Pi [74], [75]. In addition, as described later, activation of the FGF23 pathway is known to reduce 1,25-(OH)2D levels in the circulation, which, in turn, decreases the intestinal absorption of Pi and may contribute to reductions in circulating Pi levels in response to the activation of FGF23. It is important to note that renal Npt2a and 2c are predominantly expressed in the proximal tubular epithelial cells, which is distinct from the predominant expression of KL in the distal tubular epithelial cells [76], [77]; therefore, the mechanisms by which the activation of FGF23 signaling in distal tubular epithelial cells affects Npt2a and Npt2c expression in the proximal tubules remains unclear. Nevertheless, the existence of secretory molecules that mediate this connection has been suggested. The weak expression of KL in the proximal tubules may also explain the regulation of Npt2a and 2c by FGF23 [78]; however, further analyses are clearly needed in order to elucidate the underlying mechanisms.
Regulation of vitamin D metabolism
In addition to its critical roles in the regulation of Pi metabolism, the FGF23/KL axis also regulates the synthesis of 1,25-(OH)2D. Activation of the FGF23/KL signaling pathway decreases circulating levels of 1,25-(OH)2D by suppressing Cyp27b1 expression, which encodes 1-α-hydroxylase, an enzyme that converts 25-dihydroxyvitamin D to 1,25-(OH)2D and increases the expression of Cyp24a1, encoding for 24-hydroxylase, resulting in the inactivation of 1,25-(OH)2D [74]. The expression of KL in the parathyroid gland may also affect the synthesis of 1,25-(OH)2D. Animal studies have suggested that activation of the FGF23/KL pathway in the parathyroid gland suppresses the expression and secretion of PTH [69]; therefore, the effects of FGF23 on the suppression of 1,25-(OH)2D production may, at least partly, be mediated through the inhibited production of PTH because PTH is known to enhance the expression of Cyp27b1 in the kidney. Although increasing evidence from animal studies has demonstrated the suppressive effects of FGF23 on PTH production, it is still unclear whether this pathway is operative in humans because FGF23 fails to suppress PTH levels in some human disorders. For example, elevations in FGF23 levels in CKD patients are often associated with secondary hyperparathyroidism. Thus, the precise roles of FGF23 in the regulation of PTH expression under physiological and/or pathogenic conditions have yet to be determined.
Human diseases related to a dysregulated FGF23/KL axis
Enhanced FGF23/KL axis
X-linked hypophosphatemic rickets (XLH)
XLH is a disorder caused by a mutation in the PHEX gene (a phosphate-regulating gene with homologies to endopeptidases on the X chromosome). A loss-of-function mutation in this gene has been shown to enhance the production of FGF23 and cause hypophosphatemia [79]; however, the molecular mechanism by which the lack of PHEX increases FGF23 production currently remains unclear. A clinical trial on the use of neutralizing antibodies raised against FGF23 (KRN23) is currently underway, and has been reported to successfully reverse hypophosphatemia and increase 1,25-(OH)2D levels [2], [3]. However, the effectiveness of KRN23 in improving skeletal abnormalities has not yet been reported; therefore, future studies are needed in order to assess the effectiveness of this new therapy in XLH patients.
Autosomal-dominant hypophosphatemic rickets (ADHR)
A gain-of-function mutation in the FGF23 gene is responsible for ADHR, which is characterized by rickets, osteomalacia, a short stature, bone pain, and dental abscesses [16]. Mechanistically, the substitution of Arg to Gln at position 176 (R176Q) in the SPC motif has been shown to inhibit the proteolytic cleavage of FGF23, which results in activation of the FGF23/KL signaling pathway. The iron status has recently been linked to the development of ADHR phenotypes [59]. Using a mouse model in which the R176Q mutation was knocked-in, an iron deficiency was found to up-regulate the expression of Fgf23 in ADHR mice, but not in WT mice [59]. A similar scenario may occur in humans based on the clinical observation that the successful withdrawal of rickets medications was achieved in ADHR patients taking high doses of iron [80].
Autosomal-recessive hypophosphatemic rickets (ARHR) 1 and 2
Mutations in the DMP1 (dentin matrix protein 1) and ENPP1 (ectonucleotide pyrophosphatase/phosphodiesterase 1) genes are responsible for ARHR 1 and 2, respectively. Mutations in these genes up-regulate the expression of FGF23 and result in hypophosphatemic rickets; however, the mechanisms responsible are currently unknown [81], [82], [83].
Tumor-induced osteomalacia (TIO)
TIO is a rare paraneoplastic syndrome in which elevated FGF23 levels produced by endocrine tumors are associated with hypophosphatemia and inappropriately normal or low levels of 1,25-(OH)2D [84]. Abnormal mineral metabolism results in the development of osteomalacia and rickets, and clinical symptoms ares successfully treated by surgical removal of the tumors responsible.
Mutations in the FAM20C gene
Mutations in the FAM20C gene, known to be responsible for Raine syndrome, have recently been implicated in the autosomal-recessive form of the disorder characterized by elevations in FGF23 levels, hypophosphatemia, dental anomalies, and ectopic calcifications [85], [86]. The FAM20C protein was initially reported to be a kinase that phosphorylates SIBLING proteins (small integrin-binding ligand N-linked glycoproteins) such as DMP-1 and OPN [87]. FGF23 was subsequently shown to be phosphorylated at Ser 180 by FAM20C, and the phosphorylation of FGF23 at this site has been suggested to inhibit the O-glycosylation of FGF23 by GALNT3, which enhances the proteolytic cleavage of FGF23 [37], [38]; therefore, hypophosphatemia in these patients may be caused by decreases in the cleavage of FGF23 through the loss of functional mutations in the FAM20C gene.
Others
Elevated FGF23 levels have been detected in McCune-Albright syndrome, caused by an activating mutation in the GNAS gene [19], osteoglophonic dysplasia, caused by a heterozygous mutation in the FGFR1 gene [54], and Jansen metaphyseal chondrodysplasia, caused by a mutation in the PTH1R gene [48]. Chromosomal translocations with a breakpoint adjacent to the KLOTHO gene were previously reported to enhance KL expression and cause hypophosphatemic rickets due to enhanced stimulation of the FGF23/KL signaling pathway [88].
Reduced FGF23/KL axis
Familial tumoral calcinosis
Mutations in the FGF23 gene [89], [90] and GALNT3 gene [35], [91], [92], [93] cause hyperphosphatemic familial tumoral calcinosis. The loss of a functional mutation in the FGF23 gene enhances the susceptibility of the mutated protein to proteolytic cleavage, thereby decreasing intact FGF23 concentrations. As described above, GALNT3 is known to O-glycosylate FGF23 at Thr178 and protect FGF23 from proteolytic cleavage; therefore, the loss of a functional mutation in the GALNT3 gene results in the accelerated cleavage of FGF23, which causes hyperphosphatemia.
Mutation in the KLOTHO gene
The loss of a functional mutation in the KLOTHO gene was discovered in a patient with familial tumoral calcinosis [94]. This patient possesses a H193R mutation in the KLOTHO gene, and this mutated protein decreases FGF23 signaling and causes hyperphosphatemia.
Emerging roles of FGF23 in metabolic homeostasis
Adipose tissue biology
A growing body of evidence from clinical studies has highlighted the relationship between FGF23 and the metabolic status such as insulin resistance, dyslipidemia, and obesity [95]. Hanks et al. recently investigated the relationship between FGF23 and markers for insulin resistance, and reported a positive association between FGF23 and HOMA-IR [12]. Fernandez-Real et al. also described similar findings [96]. In the latter study, weight loss was associated with decreases in HOMA-IR and FGF23. Mirza et al. examined the relationship between FGF23 and obesity-related markers in elderly subjects, and found that FGF23 levels positively associated with fat mass and triglyceride levels, but not with HOMA-IR [97]. These findings suggest that the FGF23 signaling pathway plays important roles in the pathogenesis of obesity and obesity-associated complications; however, other studies reported a negative association between theses parameters [98], [99]. Therefore, further clinical investigations are clearly required in order to determine the role of FGF23 in the regulation of obesity in humans.
Animal models have also suggested the involvement of the FGF23/KL axis in the regulation of adipose tissue biology. Klotho-deficient mice exhibit a markedly smaller white adipose tissue (WAT) mass, which is associated with enhanced insulin sensitivity and low glucose levels. Intra-abdominal WAT is almost undetectable, whereas inguinal WAT (iWAT) is present, albeit at an extremely reduced amount [100]. Although a histological analysis of iWAT revealed the significantly reduced accumulation of lipid droplets, this was unlikely a consequence of lipodystrophy because of the absence of macrophage infiltration and fibrotic changes [100]. In vivo and in vitro analyses have been performed in order to investigate the mechanisms by which the lack of KL affects fat depots. KL has been proposed to have direct effects on adipocyte metabolism, and its overexpression in the preadipocytic cell line, 3T3-L1, has been shown to stimulate adipogenesis [101]. The expression levels of Kl were more than 1000-fold lower in white adipose tissue than in the kidney in C57BL/6 mice [100]; therefore, it is still unclear whether the expression of physiological levels of KL in adipose tissue has any impact on adipocyte biology. In addition to its direct effects on adipocytes, the lack of KL may affect adipocyte biology through alterations in mineral homeostasis. Consistent with this notion, decreases in WAT were found to be partially rescued by the correction of hyperphosphatemia in Klotho-deficient mice [102], suggesting that hyperphosphatemia is, at least partly, a cause of the decreased fat mass in these mice; however, the mechanisms by which increases in phosphate levels decrease the fat mass remain to be determined.
Regarding the role of FGF23 in BAT function, Klotho-deficient mice are hypothermic and not able to tolerate the cold [103]. The expression of Ucp1 is down-regulated in the BAT of Klotho-deficient mice [100], [103]. As the lack of Kl does not affect brown adipogenesis or the induction of Ucp1 by forskolin in mature brown adipocytes, BAT dysfunction in Klotho-deficient mice may not be a consequence of the lack of KL in BAT [100]. We recently demonstrated that hyperphosphatemia was responsible for impaired BAT function in Klotho-deficient mice [100]. Mechanistically, increases in extracellular Pi activate the AKT/mTORC pathway by suppressing PTEN, and mTORC1 activation causes a decrease in the mitochondrial membrane potential and enhances oxidative damage [100]. The blockade of mTORC1 by rapamycin was found to consistently improve BAT function in Klotho-deficient mice [100]. Taken together, these findings implicate FGF23 in the regulation of adipocyte biology; however, evidence to support this notion is still limited.
CKD
In patients with CKD, FGF23 levels increase in the early stage of CKD and precede elevations in PTH and Pi levels [46], [104], [105], [106], [107]. As clinical studies have revealed a relationship between increases in FGF23 levels and a higher risk of CVD and mortality [5], [7], [8], [108], [109], [110], extensive studies have been conducted in order to test the hypothesis that increases in FGF23 levels may be pathogenically involved in the development of CKD-related complications, and the efficacy of the blockade of FGF23 signaling by FGF23-neutralizing antibodies has been examined in CKD animal models. However, the efficacy of this blockade has not been consistently reported. A previous study showed that the blockade of FGF23 signaling increased mortality, even though it ameliorated hyperparathyroidism [111]. It is also possible that FGF23 levels increase in order to reduce the retention of Pi as part of a compensatory mechanism; however, due to concomitant decreases in KL expression in the kidney [112], [113], this compensatory mechanism may not be effectively operative in CKD. Although the mechanisms responsible for increases in FGF23 levels in CKD are not fully understood, higher FGF23 mRNA levels in osteocytes and extra-skeletal tissues such as the heart, and the prolonged half-life of FGF23 in the circulation may contribute to the elevations in FGF23 levels observed in CKD [114], [115].
CVD
Increases in FGF23 levels have been associated with a higher risk of CVD regardless of the presence of renal dysfunction [4], [5], [6], [7], [8]. Despite increasing clinical evidence, it still remains unclear whether increases in FGF23 levels are pathogenic to these conditions. In order to elucidate the role of FGF23 in the development of CVD, an intravenous or intramyocardial injection of rFGF23 was administered to mice, and FGF23 was found to induce ventricular hypertrophy [66]. Cardiac hypertrophy was induced, even in mice lacking Klotho, suggesting the KL-independent effects of FGF23. The effects of FGF23 in the development of cardiac hypertrophy were subsequently shown to involve FGFR4 and its downstream activation of the PLCγ/calcineurin/NFAT pathway [67]. Additionally, FGF23 has been detected in cardiac myocytes under pathogenic conditions [116], further supporting the possibility of FGF23 as a cause of CVD. As Pi is also known to be associated with the development of CVD, the role of FGF23 in the development of CVD is, at least partly, mediated through alterations in Pi homeostasis. However, some clinical studies have identified FGF23 as a risk factor for CVD independent of Pi levels [116]. Regarding the role of KL in CVD, serum levels of sKL have been negatively associated with the risk of CVD in humans [117]. Animal models also demonstrated that the lack of KL in mice caused left ventricular hypertrophy. Since the lack of Klotho in mice causes an increase in FGF23 levels, the KL-independent effects of FGF23 may play a role in the development of cardiac hypertrophy even though sKL itself has been shown to have protective effects against cardiac hypertrophy by down-regulating TRPC6 channels [118]. Although accumulating evidence indicates the critical roles of FGF23 and KL in cardiovascular biology, the underlying mechanisms are complex and additional clinical and basic studies are clearly required in order to understand the precise roles of FGF23 and KL in this regulation.
Concluding remarks
A growing of body of evidence clearly demonstrates that the FGF23/KL signaling network plays central roles in the regulation of mineral metabolism, and multiple disorders associated with dysregulated Pi metabolism have been attributed to disturbances in this pathway. Our knowledge on FGF23/KL in the pathogenesis of these disorders has recently been expanded to the commencement of clinical trials for the treatment of XLH. Beyond concrete evidence for the role of FGF23 in Pi metabolism, clinical and basic studies have opened a new field in which FGF23 signaling is involved in the pathogenesis of metabolic diseases including CKD-related complications, obesity-related disorders, and CVD. These novel findings have clearly provided us with opportunities to target FGF23 in order to combat these disorders; however, clinical and basic evidence to support this notion is still limited. Notwithstanding, future studies will undoubtedly prove the roles of FGF23 in these disorders and provide us with opportunities to target FGF23 for the treatment of FGF23-related metabolic disorders.
Acknowledgments
This work was supported by a grant from JSPS KAKENHI (Grant Number 26461558).
References
1. Yamashita T, Yoshioka M, Itoh N. Identification of a novel fibroblast growth factor, FGF-23, preferentially expressed in the ventrolateral thalamic nucleus of the brain. Biochem Biophys Res Commun 2000;277:494–8.10.1006/bbrc.2000.3696Suche in Google Scholar PubMed
2. Imel EA, Zhang X, Ruppe MD, Weber TJ, Klausner MA, Ito T, Vergeire M, Humphrey JS, Glorieux FH, Portale AA, Insogna K, Peacock M, Carpenter T. Prolonged correction of serum phosphorus in adults with X-linked hypophosphatemia using monthly doses of KRN23. J Clin Endocrinol Metab 2015;100:2565–73.10.1210/jc.2015-1551Suche in Google Scholar PubMed PubMed Central
3. Carpenter TO, Imel EA, Ruppe MD, Weber TJ, Klausner MA, Wooddell MM, Kawakami T, Ito T, Zhang X, Humphrey J, Insogna KL, Peacock M. Randomized trial of the anti-FGF23 antibody KRN23 in X-linked hypophosphatemia. J Clin Invest 2014;124:1587–97.10.1172/JCI72829Suche in Google Scholar PubMed PubMed Central
4. Parker BD, Schurgers LJ, Brandenburg VM, Christenson RH, Vermeer C, Ketteler M, Shlipak MG, Whooley MA, Ix JH. The associations of fibroblast growth factor 23 and uncarboxylated matrix Gla protein with mortality in coronary artery disease: the Heart and Soul Study. Ann Intern Med 2010;152:640–8.10.7326/0003-4819-152-10-201005180-00004Suche in Google Scholar PubMed PubMed Central
5. Kendrick J, Cheung AK, Kaufman JS, Greene T, Roberts WL, Smits G, Chonchol M. FGF-23 associates with death, cardiovascular events, and initiation of chronic dialysis. J Am Soc Nephrol 2011;22:1913–22.10.1681/ASN.2010121224Suche in Google Scholar PubMed PubMed Central
6. Scialla JJ, Lau WL, Reilly MP, Isakova T, Yang HY, Crouthamel MH, Chavkin NW, Rahman M, Wahl P, Amaral AP, Hamano T, Master SR, Nessel L, Chai B, Xie D, Kallem RR, Chen J, Lash JP, Kusek JW, Budoff MJ, Giachelli CM, Wolf M. Fibroblast growth factor 23 is not associated with and does not induce arterial calcification. Kidney Int 2013;83:1159–68.10.1038/ki.2013.3Suche in Google Scholar PubMed PubMed Central
7. Isakova T, Xie H, Yang W, Xie D, Anderson AH, Scialla J, Wahl P, Gutierrez OM, Steigerwalt S, He J, Schwartz S, Lo J, Ojo A, Sondheimer J, Hsu CY, Lash J, Leonard M, Kusek JW, Feldman HI, Wolf M. Fibroblast growth factor 23 and risks of mortality and end-stage renal disease in patients with chronic kidney disease. J Am Med Assoc 2011;305:2432–9.10.1001/jama.2011.826Suche in Google Scholar PubMed PubMed Central
8. Gutierrez OM, Mannstadt M, Isakova T, Rauh-Hain JA, Tamez H, Shah A, Smith K, Lee H, Thadhani R, Juppner H, Wolf M. Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N Engl J Med 2008;359:584–92.10.1056/NEJMoa0706130Suche in Google Scholar PubMed PubMed Central
9. Krieger NS, Culbertson CD, Kyker-Snowman K, Bushinsky D. Metabolic acidosis increases fibroblast growth factor 23 in neonatal mouse bone. Am J Physiol Renal Physiol 2012;303:F431–6.10.1152/ajprenal.00199.2012Suche in Google Scholar PubMed PubMed Central
10. Munoz Mendoza J, Isakova T, Ricardo AC, Xie H, Navaneethan SD, Anderson AH, Bazzano LA, Xie D, Kretzler M, Nessel L, Hamm LL, Negrea L, Leonard MB, Raj D, Wolf M. Fibroblast growth factor 23 and Inflammation in CKD. Clin J Am Soc Nephrol 2012;7:1155–62.10.2215/CJN.13281211Suche in Google Scholar PubMed PubMed Central
11. Holecki M, Chudek J, Owczarek A, Olszanecka-Glinianowicz M, Bozentowicz-Wikarek M, Dulawa J, Mossakowska M, Zdrojewski T, Skalska A, Wiecek A. Inflammation but not obesity or insulin resistance is associated with increased plasma fibroblast growth factor 23 concentration in the elderly. Clin Endocrinol (Oxf) 2015;82:900–9.10.1111/cen.12759Suche in Google Scholar PubMed
12. Hanks LJ, Casazza K, Judd SE, Jenny NS, Gutierrez O. Associations of fibroblast growth factor-23 with markers of inflammation, insulin resistance and obesity in adults. PLoS One 2015;10:e0122885.10.1371/journal.pone.0122885Suche in Google Scholar PubMed PubMed Central
13. Yamazaki M, Kawai M, Miyagawa K, Ohata Y, Tachikawa K, Kinoshita S, Nishino J, Ozono K, Michigami T. Interleukin-1-induced acute bone resorption facilitates the secretion of fibroblast growth factor 23 into the circulation. J Bone Miner Metab 2015;33:342–54.10.1007/s00774-014-0598-2Suche in Google Scholar PubMed
14. Andrukhova O, Slavic S, Odorfer KI, Erben RG. Experimental myocardial infarction upregulates circulating fibroblast growth factor-23. J Bone Miner Res 2015;30:1831–9.10.1002/jbmr.2527Suche in Google Scholar PubMed PubMed Central
15. Urakawa I, Yamazaki Y, Shimada T, Iijima K, Hasegawa H, Okawa K, Fujita T, Fukumoto S, Yamashita T. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature 2006;444:770–4.10.1038/nature05315Suche in Google Scholar PubMed
16. ADHR consortium. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat Genet 2000;26:345–8.10.1038/81664Suche in Google Scholar PubMed
17. Shimada T, Mizutani S, Muto T, Yoneya T, Hino R, Takeda S, Takeuchi Y, Fujita T, Fukumoto S, Yamashita T. Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc Natl Acad Sci USA 2001;98:6500–5.10.1073/pnas.101545198Suche in Google Scholar PubMed PubMed Central
18. Collins MT, Chebli C, Jones J, Kushner H, Consugar M, Rinaldo P, Wientroub S, Bianco P, Robey PG. Renal phosphate wasting in fibrous dysplasia of bone is part of a generalized renal tubular dysfunction similar to that seen in tumor-induced osteomalacia. J Bone Miner Res 2001;16:806–13.10.1359/jbmr.2001.16.5.806Suche in Google Scholar PubMed
19. Riminucci M, Collins MT, Fedarko NS, Cherman N, Corsi A, White KE, Waguespack S, Gupta A, Hannon T, Econs MJ, Bianco P, Gehron Robey, P. FGF-23 in fibrous dysplasia of bone and its relationship to renal phosphate wasting. J Clin Invest 2003;112:683–92.10.1172/JCI18399Suche in Google Scholar PubMed PubMed Central
20. Goetz R, Beenken A, Ibrahimi OA, Kalinina J, Olsen SK, Eliseenkova AV, Xu C, Neubert TA, Zhang F, Linhardt RJ, Yu X, White KE, Inagaki T, Kliewer SA, Yamamoto M, Kurosu H, Ogawa Y, Kuro-o M, Lanske B, Razzaque MS, Mohammadi M. Molecular insights into the klotho-dependent, endocrine mode of action of fibroblast growth factor 19 subfamily members. Mol Cell Biol 2007;27:3417–28.10.1128/MCB.02249-06Suche in Google Scholar PubMed PubMed Central
21. Yamazaki Y, Tamada T, Kasai N, Urakawa I, Aono Y, Hasegawa H, Fujita T, Kuroki R, Yamashita T, Fukumoto S, Shimada T. Anti-FGF23 neutralizing antibodies show the physiological role and structural features of FGF23. J Bone Miner Res 2008;23:1509–18.10.1359/jbmr.080417Suche in Google Scholar
22. Hu MC, Shiizaki K, Kuro-o M, Moe OW. Fibroblast growth factor 23 and Klotho: physiology and pathophysiology of an endocrine network of mineral metabolism. Annu Rev Physiol 2013;75:503–33.10.1146/annurev-physiol-030212-183727Suche in Google Scholar
23. Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, Ohyama Y, Kurabayashi M, Kaname T, Kume E, Iwasaki H, Iida A, Shiraki-Iida T, Nishikawa S, Nagai R, Nabeshima YI. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 1997;390:45–51.10.1038/36285Suche in Google Scholar
24. Ito S, Kinoshita S, Shiraishi N, Nakagawa S, Sekine S, Fujimori T, Nabeshima YI. Molecular cloning and expression analyses of mouse betaklotho, which encodes a novel Klotho family protein. Mech Dev 2000;98:115–9.10.1016/S0925-4773(00)00439-1Suche in Google Scholar
25. Imura A, Tsuji Y, Murata M, Maeda R, Kubota K, Iwano A, Obuse C, Togashi K, Tominaga M, Kita N, Tomiyama K, Iijima J, Nabeshima Y, Fujioka M, Asato R, Tanaka S, Kojima K, Ito J, Nozaki K, Hashimoto N, Ito T, Nishio T, Uchiyama T, Fujimori T. Alpha-Klotho as a regulator of calcium homeostasis. Science 2007;316:1615–8.10.1126/science.1135901Suche in Google Scholar PubMed
26. Xu Y, Sun Z. Molecular basis of Klotho: from gene to function in aging. Endocr Rev 2015;36:174–93.10.1210/er.2013-1079Suche in Google Scholar PubMed PubMed Central
27. Kurosu H, Yamamoto M, Clark JD, Pastor JV, Nandi A, Gurnani P, McGuinness OP, Chikuda H, Yamaguchi M, Kawaguchi H, Shimomura I, Takayama Y, Herz J, Kahn CR, Rosenblatt KP, Kuro-o M. Suppression of aging in mice by the hormone Klotho. Science 2005;309:1829–33.10.1126/science.1112766Suche in Google Scholar PubMed PubMed Central
28. Chang Q, Hoefs S, van der Kemp AW, Topala CN, Bindels RJ, Hoenderop JG. The beta-glucuronidase klotho hydrolyzes and activates the TRPV5 channel. Science 2005;310:490–3.10.1126/science.1114245Suche in Google Scholar PubMed
29. Chen CD, Podvin S, Gillespie E, Leeman SE, Abraham CR. Insulin stimulates the cleavage and release of the extracellular domain of Klotho by ADAM10 and ADAM17. Proc Natl Acad Sci USA 2007;104:19796–801.10.1073/pnas.0709805104Suche in Google Scholar PubMed PubMed Central
30. Bloch L, Sineshchekova O, Reichenbach D, Reiss K, Saftig P, Kuro-o M, Kaether C. Klotho is a substrate for alpha-, beta- and gamma-secretase. FEBS Lett 2009;583:3221–4.10.1016/j.febslet.2009.09.009Suche in Google Scholar PubMed PubMed Central
31. Imura A, Iwano A, Tohyama O, Tsuji Y, Nozaki K, Hashimoto N, Fujimori T, Nabeshima Y. Secreted Klotho protein in sera and CSF: implication for post-translational cleavage in release of Klotho protein from cell membrane. FEBS Lett 2004;565:143–7.10.1016/j.febslet.2004.03.090Suche in Google Scholar
32. Matsumura Y, Aizawa H, Shiraki-Iida T, Nagai R, Kuro-o M, Nabeshima Y. Identification of the human klotho gene and its two transcripts encoding membrane and secreted klotho protein. Biochem Biophys Res Commun 1998;242:626–30.10.1006/bbrc.1997.8019Suche in Google Scholar
33. Shiraki-Iida T, Aizawa H, Matsumura Y, Sekine S, Iida A, Anazawa H, Nagai R, Kuro-o M, Nabeshima Y. Structure of the mouse klotho gene and its two transcripts encoding membrane and secreted protein. FEBS Lett 1998;424:6–10.10.1016/S0014-5793(98)00127-6Suche in Google Scholar
34. Goetz R, Nakada Y, Hu MC, Kurosu H, Wang L, Nakatani T, Shi M, Eliseenkova AV, Razzaque MS, Moe OW, Kuro-o M, Mohammadi M. Isolated C-terminal tail of FGF23 alleviates hypophosphatemia by inhibiting FGF23-FGFR-Klotho complex formation. Proc Natl Acad Sci USA 2010;107:407–12.10.1073/pnas.0902006107Suche in Google Scholar PubMed PubMed Central
35. Frishberg Y, Ito N, Rinat C, Yamazaki Y, Feinstein S, Urakawa I, Navon-Elkan P, Becker-Cohen R, Yamashita T, Araya K, Igarashi T, Fujita T, Fukumoto S. Hyperostosis-hyperphosphatemia syndrome: a congenital disorder of O-glycosylation associated with augmented processing of fibroblast growth factor 23. J Bone Miner Res 2007;22:235–42.10.1359/jbmr.061105Suche in Google Scholar PubMed
36. Kato K, Jeanneau C, Tarp MA, Benet-Pages A, Lorenz-Depiereux B, Bennett EP, Mandel U, Strom TM, Clausen H. Polypeptide GalNAc-transferase T3 and familial tumoral calcinosis. Secretion of fibroblast growth factor 23 requires O-glycosylation. J Biol Chem 2006;281:18370–7.10.1074/jbc.M602469200Suche in Google Scholar PubMed
37. Tagliabracci VS, Engel JL, Wiley SE, Xiao J, Gonzalez DJ, Nidumanda Appaiah H, Koller A, Nizet V, White KE, Dixon JE. Dynamic regulation of FGF23 by Fam20C phosphorylation, GalNAc-T3 glycosylation, and furin proteolysis. Proc Natl Acad Sci USA 2014;111:5520–5.10.1073/pnas.1402218111Suche in Google Scholar PubMed PubMed Central
38. Lindberg I, Pang HW, Stains JP, Clark D, Yang AJ, Bonewald L, Li KZ. FGF23 is endogenously phosphorylated in bone cells. J Bone Miner Res 2015;30:449–54.10.1002/jbmr.2354Suche in Google Scholar PubMed PubMed Central
39. Collins MT, Lindsay JR, Jain A, Kelly MH, Cutler CM, Weinstein LS, Liu J, Fedarko NS, Winer KK. Fibroblast growth factor-23 is regulated by 1alpha,25-dihydroxyvitamin D. J Bone Miner Res 2005;20:1944–50.10.1359/JBMR.050718Suche in Google Scholar PubMed
40. Kolek OI, Hines ER, Jones MD, LeSueur LK, Lipko MA, Kiela PR, Collins JF, Haussler MR, and Ghishan FK. 1alpha,25-Dihydroxyvitamin D3 upregulates FGF23 gene expression in bone: the final link in a renal-gastrointestinal-skeletal axis that controls phosphate transport. Am J Physiol Gastrointest Liver Physiol 2005;289:G1036–42.10.1152/ajpgi.00243.2005Suche in Google Scholar PubMed
41. Liu S, Tang W, Zhou J, Stubbs JR, Luo Q, Pi M, and Quarles LD. Fibroblast growth factor 23 is a counter-regulatory phosphaturic hormone for vitamin D. J Am Soc Nephrol 2006;17:1305–15.10.1681/ASN.2005111185Suche in Google Scholar PubMed
42. Perwad F, Azam N, Zhang MY, Yamashita T, Tenenhouse HS, and Portale AA. Dietary and serum phosphorus regulate fibroblast growth factor 23 expression and 1,25-dihydroxyvitamin D metabolism in mice. Endocrinology 2005;146:5358–64.10.1210/en.2005-0777Suche in Google Scholar PubMed
43. Saito H, Maeda A, Ohtomo S, Hirata M, Kusano K, Kato S, Ogata E, Segawa H, Miyamoto K, Fukushima N. Circulating FGF-23 is regulated by 1alpha,25-dihydroxyvitamin D3 and phosphorus in vivo. J Biol Chem 2005;280:2543–9.10.1074/jbc.M408903200Suche in Google Scholar PubMed
44. Ferrari SL, Bonjour JP, Rizzoli R. Fibroblast growth factor-23 relationship to dietary phosphate and renal phosphate handling in healthy young men. J Clin Endocrinol Metab 2005;90:1519–24.10.1210/jc.2004-1039Suche in Google Scholar PubMed
45. Burnett SM, Gunawardene SC, Bringhurst FR, Juppner H, Lee H, Finkelstein JS. Regulation of C-terminal and intact FGF-23 by dietary phosphate in men and women. J Bone Miner Res 2006;21:1187–96.10.1359/jbmr.060507Suche in Google Scholar PubMed
46. Larsson T, Nisbeth U, Ljunggren O, Juppner H, Jonsson KB. Circulating concentration of FGF-23 increases as renal function declines in patients with chronic kidney disease, but does not change in response to variation in phosphate intake in healthy volunteers. Kidney Int 2003;64:2272–9.10.1046/j.1523-1755.2003.00328.xSuche in Google Scholar PubMed
47. Scanni R, vonRotz M, Jehle S, Hulter HN, Krapf R. The human response to acute enteral and parenteral phosphate loads. J Am Soc Nephrol 2014;25:2730–9.10.1681/ASN.2013101076Suche in Google Scholar PubMed PubMed Central
48. Brown WW, Juppner H, Langman CB, Price H, Farrow EG, White KE, McCormick KL. Hypophosphatemia with elevations in serum fibroblast growth factor 23 in a child with Jansen’s metaphyseal chondrodysplasia. J Clin Endocrinol Metab 2009;94:17–20.10.1210/jc.2008-0220Suche in Google Scholar
49. Lavi-Moshayoff V, Wasserman G, Meir T, Silver J, Naveh-Many T. PTH increases FGF23 gene expression and mediates the high-FGF23 levels of experimental kidney failure: a bone parathyroid feedback loop. Am J Physiol Renal Physiol 2010;299:F882–9.10.1152/ajprenal.00360.2010Suche in Google Scholar PubMed
50. Fan Y, Bi R, Densmore MJ, Sato T, Kobayashi T, Yuan Q, Zhou X, Erben RG, Lanske B. Parathyroid hormone 1 receptor is essential to induce FGF23 production and maintain systemic mineral ion homeostasis. FASEB J 2016;30:428–40.10.1096/fj.15-278184Suche in Google Scholar PubMed PubMed Central
51. Samadfam R, Richard C, Nguyen-Yamamoto L, Bolivar I, Goltzman D. Bone formation regulates circulating concentrations of fibroblast growth factor 23. Endocrinology 2009;150:4835–45.10.1210/en.2009-0472Suche in Google Scholar PubMed
52. Tebben PJ, Singh RJ, Clarke BL, Kumar R. Fibroblast growth factor 23, parathyroid hormone, and 1alpha,25-dihydroxyvitamin D in surgically treated primary hyperparathyroidism. Mayo Clin Proc 2004;79:1508–13.10.4065/79.12.1508Suche in Google Scholar PubMed
53. Kawai M, Kinoshita S, Shimba S, Ozono K, Michigami T. Sympathetic activation induces skeletal Fgf23 expression in a circadian rhythm-dependent manner. J Biol Chem 2014;289:1457–66.10.1074/jbc.M113.500850Suche in Google Scholar PubMed PubMed Central
54. Farrow EG, Davis SI, Mooney SD, Beighton P, Mascarenhas L, Gutierrez YR, Pitukcheewanont P, White KE. Extended mutational analyses of FGFR1 in osteoglophonic dysplasia. Am J Med Genet A 2006;140:537–9.10.1002/ajmg.a.31106Suche in Google Scholar PubMed
55. Xiao Z, Huang J, Cao L, Liang Y, Han X, Quarles LD. Osteocyte-specific deletion of Fgfr1 suppresses FGF23. PLoS One 2014;9:e104154.10.1371/journal.pone.0104154Suche in Google Scholar PubMed PubMed Central
56. Miyagawa K, Yamazaki M, Kawai M, Nishino J, Koshimizu T, Ohata Y, Tachikawa K, Mikuni-Takagaki Y, Kogo M, Ozono K, Michigami T. Dysregulated gene expression in the primary osteoblasts and osteocytes isolated from hypophosphatemic Hyp mice. PLoS One 2014;9:e93840.10.1371/journal.pone.0093840Suche in Google Scholar PubMed PubMed Central
57. Han X, Xiao Z, Quarles LD. Membrane and integrative nuclear fibroblastic growth factor receptor (FGFR) regulation of FGF-23. J Biol Chem 2015;290:10447–59.10.1074/jbc.M114.609230Suche in Google Scholar PubMed PubMed Central
58. Xiao L, Naganawa T, Lorenzo J, Carpenter TO, Coffin JD, Hurley MM. Nuclear isoforms of fibroblast growth factor 2 are novel inducers of hypophosphatemia via modulation of FGF23 and KLOTHO. J Biol Chem 2010;285:2834–46.10.1074/jbc.M109.030577Suche in Google Scholar PubMed PubMed Central
59. Farrow EG, Yu X, Summers LJ, Davis SI, Fleet JC, Allen MR, Robling AG, Stayrook KR, Jideonwo V, Magers MJ, Garringer HJ, Vidal R, Chan RJ, Goodwin CB, Hui SL, Peacock M, White KE. Iron deficiency drives an autosomal dominant hypophosphatemic rickets (ADHR) phenotype in fibroblast growth factor-23 (Fgf23) knock-in mice. Proc Natl Acad Sci USA 2011;108:E1146–55.10.1073/pnas.1110905108Suche in Google Scholar PubMed PubMed Central
60. David V, Martin A, Isakova T, Spaulding C, Qi L, Ramirez V, Zumbrennen-Bullough KB, Sun CC, Lin HY, Babitt JL, Wolf M. Inflammation and functional iron deficiency regulate fibroblast growth factor 23 production. Kidney Int. 2015 Nov 4. doi: 10.1038/ki.2015.290. [Epub ahead of print].Suche in Google Scholar PubMed PubMed Central
61. Kurosu H, Ogawa Y, Miyoshi M, Yamamoto M, Nandi A, Rosenblatt KP, Baum MG, Schiavi S, Hu MC, Moe OW, Kuro-o M. Regulation of fibroblast growth factor-23 signaling by klotho. J Biol Chem 2006;281:6120–3.10.1074/jbc.C500457200Suche in Google Scholar PubMed PubMed Central
62. Liu S, Vierthaler L, Tang W, Zhou J, Quarles LD. FGFR3 and FGFR4 do not mediate renal effects of FGF23. J Am Soc Nephrol 2008;19:2342–50.10.1681/ASN.2007121301Suche in Google Scholar PubMed PubMed Central
63. Gattineni J, Bates C, Twombley K, Dwarakanath V, Robinson ML, Goetz R, Mohammadi M, Baum M. FGF23 decreases renal NaPi-2a and NaPi-2c expression and induces hypophosphatemia in vivo predominantly via FGF receptor 1. Am J Physiol Renal Physiol 2009;297:F282–91.10.1152/ajprenal.90742.2008Suche in Google Scholar PubMed PubMed Central
64. Kawai M, Kinoshita S, Kimoto A, Hasegawa Y, Miyagawa K, Yamazaki M, Ohata Y, Ozono K, Michigami T. FGF23 suppresses chondrocyte proliferation in the presence of soluble alpha-Klotho both in vitro and in vivo. J Biol Chem 2013;288:2414–27.10.1074/jbc.M112.410043Suche in Google Scholar PubMed PubMed Central
65. Smith RC, O’Bryan LM, Farrow EG, Summers LJ, Clinkenbeard EL, Roberts JL, Cass TA, Saha J, Broderick C, Ma YL, Zeng QQ, Kharitonenkov A, Wilson JM, Guo Q, Sun H, Allen MR, Burr DB, Breyer MD, White KE. Circulating alphaKlotho influences phosphate handling by controlling FGF23 production. J Clin Invest 2012;122:4710–5.10.1172/JCI64986Suche in Google Scholar PubMed PubMed Central
66. Faul C, Amaral AP, Oskouei B, Hu MC, Sloan A, Isakova T, Gutierrez OM, Aguillon-Prada R, Lincoln J, Hare JM, Mundel P, Morales A, Scialla J, Fischer M, Soliman EZ, Chen J, Go AS, Rosas SE, Nessel L, Townsend RR, Feldman HI, St John Sutton M, Ojo A, Gadegbeku C, Di Marco GS, Reuter S, Kentrup D, Tiemann K, Brand M, Hill JA, Moe OW, Kuro OM, Kusek JW, Keane MG, Wolf M. FGF23 induces left ventricular hypertrophy. J Clin Invest 2011;121:4393–408.10.1172/JCI46122Suche in Google Scholar PubMed PubMed Central
67. Grabner A, Amaral AP, Schramm K, Singh S, Sloan A, Yanucil C, Li J, Shehadeh LA, Hare JM, David V, Martin A, Fornoni A, Di Marco GS, Kentrup D, Reuter S, Mayer AB, Pavenstadt H, Stypmann J, Kuhn C, Hille S, Frey N, Leifheit-Nestler M, Richter B, Haffner D, Abraham R, Bange J, Sperl B, Ullrich A, Brand M, Wolf M, Faul C. Activation of cardiac fibroblast growth factor receptor 4 causes left ventricular hypertrophy. Cell Metab 2015;22:1020–32.10.1016/j.cmet.2015.09.002Suche in Google Scholar PubMed PubMed Central
68. Olauson H, Lindberg K, Amin R, Sato T, Jia T, Goetz R, Mohammadi M, Andersson G, Lanske B, Larsson TE. Parathyroid-specific deletion of Klotho unravels a novel calcineurin-dependent FGF23 signaling pathway that regulates PTH secretion. PLoS Genet 2013;9:e1003975.10.1371/journal.pgen.1003975Suche in Google Scholar PubMed PubMed Central
69. Ben-Dov IZ, Galitzer H, Lavi-Moshayoff V, Goetz R, Kuro-o M, Mohammadi M, Sirkis R, Naveh-Many T, Silver J. The parathyroid is a target organ for FGF23 in rats. J Clin Invest 2007;117:4003–8.10.1172/JCI32409Suche in Google Scholar PubMed PubMed Central
70. Shimada T, Kakitani M, Yamazaki Y, Hasegawa H, Takeuchi Y, Fujita T, Fukumoto S, Tomizuka K, Yamashita T. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J Clin Invest 2004;113:561–8.10.1172/JCI200419081Suche in Google Scholar
71. Shimada T, Urakawa I, Yamazaki Y, Hasegawa H, Hino R, Yoneya T, Takeuchi Y, Fujita T, Fukumoto S, Yamashita T. FGF-23 transgenic mice demonstrate hypophosphatemic rickets with reduced expression of sodium phosphate cotransporter type IIa. Biochem Biophys Res Commun 2004;314:409–14.10.1016/j.bbrc.2003.12.102Suche in Google Scholar PubMed
72. Larsson T, Marsell R, Schipani E, Ohlsson C, Ljunggren O, Tenenhouse HS, Juppner H, Jonsson KB. Transgenic mice expressing fibroblast growth factor 23 under the control of the alpha1(I) collagen promoter exhibit growth retardation, osteomalacia, and disturbed phosphate homeostasis. Endocrinology 2004;145:3087–94.10.1210/en.2003-1768Suche in Google Scholar PubMed
73. Olauson H, Lindberg K, Amin R, Jia T, Wernerson A, Andersson G, Larsson TE. Targeted deletion of Klotho in kidney distal tubule disrupts mineral metabolism. J Am Soc Nephrol 2012;23:1641–51.10.1681/ASN.2012010048Suche in Google Scholar PubMed PubMed Central
74. Shimada T, Hasegawa H, Yamazaki Y, Muto T, Hino R, Takeuchi Y, Fujita T, Nakahara K, Fukumoto S, Yamashita T. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res 2004;19:429–35.10.1359/JBMR.0301264Suche in Google Scholar PubMed
75. Segawa H, Kawakami E, Kaneko I, Kuwahata M, Ito M, Kusano K, Saito H, Fukushima N, Miyamoto K. Effect of hydrolysis-resistant FGF23-R179Q on dietary phosphate regulation of the renal type-II Na/Pi transporter. Pflugers Arch 2003;446:585–92.10.1007/s00424-003-1084-1Suche in Google Scholar PubMed
76. Farrow EG, Summers LJ, Schiavi SC, McCormick JA, Ellison DH, White KE. Altered renal FGF23-mediated activity involving MAPK and Wnt: effects of the Hyp mutation. J Endocrinol 2010;207:67–75.10.1677/JOE-10-0181Suche in Google Scholar PubMed PubMed Central
77. Farrow EG, Davis SI, Summers LJ, White KE. Initial FGF23-mediated signaling occurs in the distal convoluted tubule. J Am Soc Nephrol 2009;20:955–60.10.1681/ASN.2008070783Suche in Google Scholar PubMed PubMed Central
78. Hu MC, Shi M, Zhang J, Pastor J, Nakatani T, Lanske B, Razzaque MS, Rosenblatt KP, Baum MG, Kuro-o M, Moe OW. Klotho: a novel phosphaturic substance acting as an autocrine enzyme in the renal proximal tubule. FASEB J 2010;24:3438–50.10.1096/fj.10-154765Suche in Google Scholar PubMed PubMed Central
79. Jonsson KB, Zahradnik R, Larsson T, White KE, Sugimoto T, Imanishi Y, Yamamoto T, Hampson G, Koshiyama H, Ljunggren O, Oba K, Yang IM, Miyauchi A, Econs MJ, Lavigne J, Juppner H. Fibroblast growth factor 23 in oncogenic osteomalacia and X-linked hypophosphatemia. N Engl J Med 2003;348:1656–63.10.1056/NEJMoa020881Suche in Google Scholar PubMed
80. Kapelari K, Kohle J, Kotzot D, Hogler W. Iron supplementation associated with loss of phenotype in autosomal dominant hypophosphatemic rickets. J Clin Endocrinol Metab 2015;100:3388–92.10.1210/jc.2015-2391Suche in Google Scholar PubMed
81. Lorenz-Depiereux B, Bastepe M, Benet-Pages A, Amyere M, Wagenstaller J, Muller-Barth U, Badenhoop K, Kaiser SM, Rittmaster RS, Shlossberg AH, Olivares JL, Loris C, Ramos FJ, Glorieux F, Vikkula M, Juppner H, Strom TM. DMP1 mutations in autosomal recessive hypophosphatemia implicate a bone matrix protein in the regulation of phosphate homeostasis. Nat Genet 2006;38:1248–50.10.1038/ng1868Suche in Google Scholar PubMed PubMed Central
82. Levy-Litan V, Hershkovitz E, Avizov L, Leventhal N, Bercovich D, Chalifa-Caspi V, Manor E, Buriakovsky S, Hadad Y, Goding J, Parvari R. Autosomal-recessive hypophosphatemic rickets is associated with an inactivation mutation in the ENPP1 gene. Am J Hum Genet 2010;86:273–8.10.1016/j.ajhg.2010.01.010Suche in Google Scholar PubMed PubMed Central
83. Lorenz-Depiereux B, Schnabel D, Tiosano D, Hausler G, Strom TM. Loss-of-function ENPP1 mutations cause both generalized arterial calcification of infancy and autosomal-recessive hypophosphatemic rickets. Am J Hum Genet 2010;86:267–72.10.1016/j.ajhg.2010.01.006Suche in Google Scholar PubMed PubMed Central
84. Chong WH, Molinolo AA, Chen CC, Collins MT. Tumor-induced osteomalacia. Endocr Relat Cancer 2011;18:R53–77.10.1530/ERC-11-0006Suche in Google Scholar PubMed PubMed Central
85. Rafaelsen SH, Raeder H, Fagerheim AK, Knappskog P, Carpenter TO, Johansson S, Bjerknes R. Exome sequencing reveals FAM20c mutations associated with fibroblast growth factor 23-related hypophosphatemia, dental anomalies, and ectopic calcification. J Bone Miner Res 2013;28:1378–85.10.1002/jbmr.1850Suche in Google Scholar PubMed
86. Takeyari S, Yamamoto T, Kinoshita Y, Fukumoto S, Glorieux FH, Michigami T, Hasegawa K, Kitaoka T, Kubota T, Imanishi Y, Shimotsuji T, Ozono K. Hypophosphatemic osteomalacia and bone sclerosis caused by a novel homozygous mutation of the FAM20C gene in an elderly man with a mild variant of Raine syndrome. Bone 2014;67:56–62.10.1016/j.bone.2014.06.026Suche in Google Scholar PubMed
87. Tagliabracci VS, Engel JL, Wen J, Wiley SE, Worby CA, Kinch LN, Xiao J, Grishin NV, Dixon JE. Secreted kinase phosphorylates extracellular proteins that regulate biomineralization. Science 2012;336:1150–3.10.1126/science.1217817Suche in Google Scholar PubMed PubMed Central
88. Brownstein CA, Adler F, Nelson-Williams C, Iijima J, Li P, Imura A, Nabeshima Y, Reyes-Mugica M, Carpenter TO, Lifton RP. A translocation causing increased alpha-klotho level results in hypophosphatemic rickets and hyperparathyroidism. Proc Natl Acad Sci USA 2008;105:3455–60.10.1073/pnas.0712361105Suche in Google Scholar PubMed PubMed Central
89. Benet-Pages A, Orlik P, Strom TM, Lorenz-Depiereux B. An FGF23 missense mutation causes familial tumoral calcinosis with hyperphosphatemia. Hum Mol Genet 2005;14:385–90.10.1093/hmg/ddi034Suche in Google Scholar PubMed
90. Araya K, Fukumoto S, Backenroth R, Takeuchi Y, Nakayama K, Ito N, Yoshii N, Yamazaki Y, Yamashita T, Silver J, Igarashi T, Fujita T. A novel mutation in fibroblast growth factor 23 gene as a cause of tumoral calcinosis. J Clin Endocrinol Metab 2005;90:5523–7.10.1210/jc.2005-0301Suche in Google Scholar PubMed
91. Frishberg Y, Topaz O, Bergman R, Behar D, Fisher D, Gordon D, Richard G, Sprecher E. Identification of a recurrent mutation in GALNT3 demonstrates that hyperostosis-hyperphosphatemia syndrome and familial tumoral calcinosis are allelic disorders. J Mol Med (Berl) 2005;83:33–8.10.1007/s00109-004-0610-8Suche in Google Scholar PubMed
92. Garringer HJ, Fisher C, Larsson TE, Davis SI, Koller DL, Cullen MJ, Draman MS, Conlon N, Jain A, Fedarko NS, Dasgupta B, White KE. The role of mutant UDP-N-acetyl-alpha-D-galactosamine-polypeptide N-acetylgalactosaminyltransferase 3 in regulating serum intact fibroblast growth factor 23 and matrix extracellular phosphoglycoprotein in heritable tumoral calcinosis. J Clin Endocrinol Metab 2006;91:4037–42.10.1210/jc.2006-0305Suche in Google Scholar PubMed
93. Ichikawa S, Baujat G, Seyahi A, Garoufali AG, Imel EA, Padgett LR, Austin AM, Sorenson AH, Pejin Z, Topouchian V, Quartier P, Cormier-Daire V, Dechaux M, Malandrinou F, Singhellakis PN, Le Merrer M, Econs MJ. Clinical variability of familial tumoral calcinosis caused by novel GALNT3 mutations. Am J Med Genet A 2010;152A:896–903.10.1002/ajmg.a.33337Suche in Google Scholar PubMed PubMed Central
94. Ichikawa S, Imel EA, Kreiter ML, Yu X, Mackenzie DS, Sorenson AH, Goetz R, Mohammadi M, White KE, Econs MJ. A homozygous missense mutation in human KLOTHO causes severe tumoral calcinosis. J Clin Invest 2007;117:2684–91.10.1172/JCI31330Suche in Google Scholar PubMed PubMed Central
95. Razzaque MS. The role of Klotho in energy metabolism. Nat Rev Endocrinol 2012;8:579–87.10.1038/nrendo.2012.75Suche in Google Scholar PubMed PubMed Central
96. Fernandez-Real JM, Puig J, Serrano M, Sabater M, Rubio A, Moreno-Navarrete JM, Fontan M, Casamitjana R, Xifra G, Ortega FJ, Salvador J, Fruhbeck G, Ricart W. Iron and obesity status-associated insulin resistance influence circulating fibroblast-growth factor-23 concentrations. PLoS One 2013;8:e58961.10.1371/journal.pone.0058961Suche in Google Scholar PubMed PubMed Central
97. Mirza MA, Alsio J, Hammarstedt A, Erben RG, Michaelsson K, Tivesten A, Marsell R, Orwoll E, Karlsson MK, Ljunggren O, Mellstrom D, Lind L, Ohlsson C, Larsson TE. Circulating fibroblast growth factor-23 is associated with fat mass and dyslipidemia in two independent cohorts of elderly individuals. Arterioscler Thromb Vasc Biol 2011;31:219–27.10.1161/ATVBAHA.110.214619Suche in Google Scholar PubMed
98. Wojcik M, Dolezal-Oltarzewska K, Janus D, Drozdz D, Sztefko K, Starzyk JB. FGF23 contributes to insulin sensitivity in obese adolescents – preliminary results. Clin Endocrinol (Oxf) 2012;77:537–40.10.1111/j.1365-2265.2011.04299.xSuche in Google Scholar PubMed
99. Wojcik M, Janus D, Dolezal-Oltarzewska K, Drozdz D, Sztefko K, Starzyk JB. The association of FGF23 levels in obese adolescents with insulin sensitivity. J Pediatr Endocrinol Metab 2012; 25:687–90.10.1515/jpem-2012-0064Suche in Google Scholar PubMed
100. Kawai M, Kinoshita S, Ozono K, Michigami T. Inorganic phosphate activates the AKT/mTORC1 pathway and shortens the life span of an α-Klotho-deficient model. J Am Soc Nephrol 2016. In press.10.1681/ASN.2015040446Suche in Google Scholar PubMed PubMed Central
101. Chihara Y, Rakugi H, Ishikawa K, Ikushima M, Maekawa Y, Ohta J, Kida I, Ogihara T. Klotho protein promotes adipocyte differentiation. Endocrinology 2006;147:3835–42.10.1210/en.2005-1529Suche in Google Scholar
102. Ohnishi M, Razzaque MS. Dietary and genetic evidence for phosphate toxicity accelerating mammalian aging. FASEB J 2010;24:3562–71.10.1096/fj.09-152488Suche in Google Scholar
103. Mori K, Yahata K, Mukoyama M, Suganami T, Makino H, Nagae T, Masuzaki H, Ogawa Y, Sugawara A, Nabeshima Y, Nakao K. Disruption of klotho gene causes an abnormal energy homeostasis in mice. Biochem Biophys Res Commun 2000;278:665–70.10.1006/bbrc.2000.3864Suche in Google Scholar
104. Isakova T, Wahl P, Vargas GS, Gutierrez OM, Scialla J, Xie H, Appleby D, Nessel L, Bellovich K, Chen J, Hamm L, Gadegbeku C, Horwitz E, Townsend RR, Anderson CA, Lash JP, Hsu CY, Leonard MB, Wolf M. Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney Int 2011;79:1370–8.10.1038/ki.2011.47Suche in Google Scholar
105. Levin A, Bakris GL, Molitch M, Smulders M, Tian J, Williams LA, Andress DL. Prevalence of abnormal serum vitamin D, PTH, calcium, and phosphorus in patients with chronic kidney disease: results of the study to evaluate early kidney disease. Kidney Int 2007;71:31–8.10.1038/sj.ki.5002009Suche in Google Scholar
106. Nakano C, Hamano T, Fujii N, Matsui I, Tomida K, Mikami S, Inoue K, Obi Y, Okada N, Tsubakihara Y, Isaka Y, Rakugi H. Combined use of vitamin D status and FGF23 for risk stratification of renal outcome. Clin J Am Soc Nephrol 2012;7:810–9.10.2215/CJN.08680811Suche in Google Scholar
107. Portale AA, Wolf M, Juppner H, Messinger S, Kumar J, Wesseling-Perry K, Schwartz GJ, Furth SL, Warady BA, Salusky IB. Disordered FGF23 and mineral metabolism in children with CKD. Clin J Am Soc Nephrol 2014;9:344–53.10.2215/CJN.05840513Suche in Google Scholar
108. Block GA, Klassen PS, Lazarus JM, Ofsthun N, Lowrie EG, Chertow GM. Mineral metabolism, mortality, and morbidity in maintenance hemodialysis. J Am Soc Nephrol 2004;15:2208–18.10.1097/01.ASN.0000133041.27682.A2Suche in Google Scholar
109. Baia LC, Humalda JK, Vervloet MG, Navis G, Bakker SJ, de Borst MH. Fibroblast growth factor 23 and cardiovascular mortality after kidney transplantation. Clin J Am Soc Nephrol 2013;8:1968–78.10.2215/CJN.01880213Suche in Google Scholar
110. Wolf M, Molnar MZ, Amaral AP, Czira ME, Rudas A, Ujszaszi A, Kiss I, Rosivall L, Kosa J, Lakatos P, Kovesdy CP, Mucsi I. Elevated fibroblast growth factor 23 is a risk factor for kidney transplant loss and mortality. J Am Soc Nephrol 2011;22:956–66.10.1681/ASN.2010080894Suche in Google Scholar
111. Shalhoub V, Shatzen EM, Ward SC, Davis J, Stevens J, Bi V, Renshaw L, Hawkins N, Wang W, Chen C, Tsai MM, Cattley RC, Wronski TJ, Xia X, Li X, Henley C, Eschenberg M, Richards WG. FGF23 neutralization improves chronic kidney disease-associated hyperparathyroidism yet increases mortality. J Clin Invest 2012;122:2543–53.10.1172/JCI61405Suche in Google Scholar PubMed PubMed Central
112. Koh N, Fujimori T, Nishiguchi S, Tamori A, Shiomi S, Nakatani T, Sugimura K, Kishimoto T, Kinoshita S, Kuroki T, Nabeshima Y. Severely reduced production of klotho in human chronic renal failure kidney. Biochem Biophys Res Commun 2001;280:1015–20.10.1006/bbrc.2000.4226Suche in Google Scholar PubMed
113. Asai O, Nakatani K, Tanaka T, Sakan H, Imura A, Yoshimoto S, Samejima K, Yamaguchi Y, Matsui M, Akai Y, Konishi N, Iwano M, Nabeshima Y, Saito Y. Decreased renal alpha-Klotho expression in early diabetic nephropathy in humans and mice and its possible role in urinary calcium excretion. Kidney Int 2012;81:539–47.10.1038/ki.2011.423Suche in Google Scholar PubMed
114. Christov M, Waikar SS, Pereira RC, Havasi A, Leaf DE, Goltzman D, Pajevic PD, Wolf M, Juppner H. Plasma FGF23 levels increase rapidly after acute kidney injury. Kidney Int 2013;84:776–85.10.1038/ki.2013.150Suche in Google Scholar PubMed PubMed Central
115. Olauson H, Vervloet MG, Cozzolino M, Massy ZA, Urena Torres P, Larsson TE. New insights into the FGF23-Klotho axis. Semin Nephrol 2014;34:586–97.10.1016/j.semnephrol.2014.09.005Suche in Google Scholar PubMed
116. Scialla JJ, Wolf M. Roles of phosphate and fibroblast growth factor 23 in cardiovascular disease. Nat Rev Nephrol 2014;10:268–78.10.1038/nrneph.2014.49Suche in Google Scholar PubMed
117. Semba RD, Cappola AR, Sun K, Bandinelli S, Dalal M, Crasto C, Guralnik JM, Ferrucci L. Plasma klotho and cardiovascular disease in adults. J Am Geriatr Soc 2011;59:1596–601.10.1111/j.1532-5415.2011.03558.xSuche in Google Scholar PubMed PubMed Central
118. Xie J, Cha SK, An SW, Kuro OM, Birnbaumer L, Huang CL. Cardioprotection by Klotho through downregulation of TRPC6 channels in the mouse heart. Nat Commun 2012;3:1238.10.1038/ncomms2240Suche in Google Scholar PubMed PubMed Central
©2016 Walter de Gruyter GmbH, Berlin/Boston
Artikel in diesem Heft
- Frontmatter
- Editorial Preface
- Preface to special issue on ‘Endocrine functions of bone: new (patho)-physiological and clinical insights’
- Topic A: Endocrine Functions of Bone and Bone Marrow Adipose Tissue: Hormonal, Molecular, Nutritional Mechanisms
- Review Articles
- The “soft” side of the bone: unveiling its endocrine functions
- Bone marrow adipose tissue as an endocrine organ: close to the bone?
- The dietary protein, IGF-I, skeletal health axis
- The FGF23/Klotho axis in the regulation of mineral and metabolic homeostasis
Artikel in diesem Heft
- Frontmatter
- Editorial Preface
- Preface to special issue on ‘Endocrine functions of bone: new (patho)-physiological and clinical insights’
- Topic A: Endocrine Functions of Bone and Bone Marrow Adipose Tissue: Hormonal, Molecular, Nutritional Mechanisms
- Review Articles
- The “soft” side of the bone: unveiling its endocrine functions
- Bone marrow adipose tissue as an endocrine organ: close to the bone?
- The dietary protein, IGF-I, skeletal health axis
- The FGF23/Klotho axis in the regulation of mineral and metabolic homeostasis