Development of heterocyclic-based anticancer agents: A comprehensive review
-
Mohammed Hadi Ali Al-Jumaili
 ,
Ekhlas Aziz Bakr
,
Ekhlas Aziz Bakr

Abstract
This review article summarizes the role of heterocyclic compounds as anticancer drugs used against various human cancers, including doxorubicin, cisplatin, paclitaxel, and resveratrol, which are among the most effective therapeutic agents. Chemotherapy, a treatment modality, exerts its effects on tumor cell DNA and often involves the use of low-molecular-weight medicines to selectively target and destroy cancer cells. However, systemic chemotherapy is associated with several side effects, such as nausea, vomiting, myelosuppression, and cardiotoxicity. Cancer remains one of the most prevalent and lethal diseases, characterized by uncontrolled cell division and abnormal cell growth driven by multiple genetic mutations. The etiopathogenesis of cancer is complex, but significant advancements have been made in treatment, particularly with the discovery of anticancer drugs, including cytotoxic chemotherapy, hormonal agents, and targeted therapies. Anticancer drugs are widely employed for the treatment of various cancers, such as breast, cervical, uterine, and kidney cancers. These drugs are classified into several categories, including alkylating agents, antimetabolites, antibiotics, and topoisomerase inhibitors. Among these, numerous heterocyclic compounds have shown promising anticancer properties. The goal of this review is to compile information on heterocyclic compounds used as anticancer drugs, highlighting their positive effects and therapeutic targets in cancer treatment and chemoprevention.
1 Introduction
Heterocyclic compounds have shown a significant potential as anticancer agents. These compounds are organic compounds that contain at least one heteroatom, such as nitrogen, oxygen, or sulfur, in their cyclic structure [1,2,3]. Several families of heterocyclic compounds have been extensively studied for their anticancer properties [4,5]. Cancer is the world’s second-deadliest disease, characterized by the loss of growth control in one or more cells, resulting in a solid mass of tumor-related cells or even liquid cancer such as blood or bone marrow-related cancer [6,7]. Many protein-coding genes, such as growth factors, growth factor receptors, antiapoptotic proteins, transcription factors, and tumor suppressors, are involved in most malignancies. In addition, there are some factors, such as tobacco, alcohol, and environmental toxins, that raise the risk of cancer [8,9]. According to the World Health Organization, the number of cases increased dramatically in 2018; there will be 18.1 million new cases of this disease and 9.6 million fatalities [10]. The most common types of cancer in men worldwide in terms of the number of new cases are cancers of the lung, prostate, colorectal, stomach, and liver cancer, while the most common types among women are breast, colorectal, lung, cervical, and thyroid cancers [11,12]. By 2030, the global death toll from this devastating disease is anticipated to rise to 21.6 billion [13]. Therefore, the advancement of research aimed at improving cancer therapy has increased, as a result of which new developments have occurred depending on the method of treatment. However, the primary treatments were surgery, chemotherapy, radiotherapy, hormonal therapy, and immune therapy. Chemotherapy uses medicines of lower molecular weight to kill tumor cells, or at least their proliferation is limited [14].
Anticancer drugs can be divided into different categories according to antimetabolites, antitubulin agents, DNA-interactive agents, and other mechanisms involving hormones, monoclonal antibodies, molecular targeting agents, and other biological agents [15,16]. Anticancer medications that stop cancer cells from dividing and proliferating have a low selectivity for cancer cells. Cytotoxic medicines with high potency work on both cells and organs in cancer cells and healthy living cells. That makes healthy living cells susceptible to the anticancer drug’s cytotoxic action affects DNA synthesis [17]. In addition, the use of large anticancer drug dosages through chemotherapy as well impacts patients’ immunity by influencing the body’s bimolecular protection system. However, the irrational use of unreasonable drugs is a significant health issue in medical practice today. This in turn leads to numerous consequences including, but not limited to, ineffective diagnosis, excessive prescription of especially antimicrobials and injections, creation of antibiotic resistance, adverse effects, and economic burden [18]. In this review, we will focus on the most common heterocyclic compounds that are discovered as antitumors until 2024.
2 Classification of anticancer drugs
Anticancer drugs are primarily classified into different groups based on their action mechanisms: chemotherapy, hormonal therapy, and targeted drugs (Figure 1).

Anticancer drug categories.
2.1 Chemotherapy drugs in cancer treatment
Chemotherapy, a critical tool in cancer treatment, has been in use since the 1940s, starting with nitrogen mustard-based drugs. It remains a central component of oncology, often combined with other therapeutic strategies. Chemotherapy works through monoclonal antibodies and small molecules that target proteins within tumor cells and disrupt DNA processes, effectively hindering cancer cell growth and survival.
Widely regarded as a frontline defense against cancer, chemotherapy utilizes a diverse range of antineoplastic drugs designed to address specific cancer types. These drugs are categorized based on their mechanisms of action and molecular targets, enabling more precise and effective treatment approaches (Figure 2).
Chemotherapy drugs.
2.1.1 Alkylating agents
Alkylating agents are the most widely used anticancer drugs, and they are essential components of combination chemotherapy [19]. These agents could cause direct DNA damage by introducing methyl or other alkyl groups into their nucleotide bases [20]. These are small molecules that can form adducts by covalently binding an alkyl group to electron-rich nucleophilic moieties and thus have a cytotoxic influence on cells engaged to DNA molecules [21]. In addition to deactivating cell respiration and intermediate metabolism by protein and enzyme alkylation, alkylating agents cause a reduction in DNA synthesis, genetic mutations, and chromosomal aberrations [22]. Melphalan, cyclophosphamide (CP), dacarbazine, procarbazine, temozolomide (TMZ), chlorambucil, and ifosfamide (IFO) are examples of anticancer drugs that contain alkylating agents (Table 1).
Alkylating agent drugs
| Alkylating agents | Drugs | Uses |
|---|---|---|
| Nitrogen mustards | CP | Lymphoma, leukemia Hodgkin’s disease, multiple myeloma (MM), sarcoma, lung, testicular, prostate, breast, lupus vasculopathy and ovarian carcinoma, rheumatoid arthritis |
| Chlorambucil | Non-Hodgkin’s lymphomas, Hodgkin’s, chronic lymphocytic leukemia (CLL) | |
| IFO | Breast cancer, lung cancer, cancer of the head and neck, cervical cancer and Ewing sarcoma | |
| Melphalan | Germ cells, breast cancer, lung cancer of the head and neck, cervical cancer, and Ewing sarcoma | |
| Mechlorethamine | Hodgkin’s disease, and in the topical treatment of mycosis fungoides and other lymphomas | |
| Other alkylating agents | TMZ | Metastatic melanoma, malignant melanoma, and primary brain tumor |
| Procarbazine | MM, multiform glioblastoma and Hodgkin lymphoma, and CNS malignancies | |
| Chlorozotocin | Neuroendocrine tumors, pneumonitis, hormone-sensitive cancers, such as ovarian, prostate, breast, and endometrium | |
| Alkyl sulfonates | Busulfa | Chronic myelocytic leukemia and polycythemia vera |
| Nitrosoureas | Carmustine | Pancreatic metastatic islets, cellular carcinoma |
| Lomustine | Hodgkin’s disease, lung and brain tumors, MM, melanoma, and brain tumors | |
| Ethylenimine derivative | Thiotepa | Adenocarcinoma of the breast and ovary, urinary bladder carcinoma, and urinary bladder carcinoma |
| Triazines | Dacarbazine | Metastatic malignant melanoma, Hodgkin’s disease, sarcoma, neuroblastoma, neuroendocrine brain tumors and primary tumors |
2.1.1.1 Nitrogen mustards
Nitrogen mustard-based substrates are cytotoxic organic compounds with chloroethylamine functional groups. It is considered a type of DNA bifunctional alkylating agent. During cell division, alkyl groups are transferred to DNA in the N-7 position of guanine, according to the chemical structure (Figure 3). DNA strand breaking and cross-linking of such two strands are all possibilities, as shown in Figure 4.

Alkylating agent drugs (nitrogen mustards).

Nitrogen Mustard mechanism of action.
2.1.1.1.1 CP
CP is a nitrogen mustard-containing compound, which could be inefficient, primarily in the liver, when metabolic activation is negatively affected. It is activated by hepatic transformation to form 4-hydroxycyclophosphamide, which breaks down into the ultimate alkylating agents [23,24]. The active metabolites of CP alkylate cellular macromolecules form covalent bonds that impede their separation. The most critical target is probably DNA, as alkylation limits cell division and disrupts gene expression. Following CP treatment, lymphocyte count and function are significantly reduced [23]. The mechanism of CP synthesis involves interfering with the replication of DNA, resulting in ribonucleic acid (RNA) formation. The effective metabolite of CP is phosphor amide mustard, which is produced by liver enzymes such as cytochrome P-450 during the metabolism of the drug in the body. This metabolite was created only in cells with low activity levels of aldehyde dehydrogenase. The phosphor amide metabolite at the guanine N-7 position forms cross-linkages within and between adjacent DNA strands, which results in the apoptosis of cells [25]. These types of drugs can be given either orally or parenterally. However, it is more common in rheumatology (intravenous administration). CP is an effective immunosuppressant and is, therefore, widely used for bone marrow transplantation and autoimmune disease treatment [26]. CP pharmacokinetics studies in kidney failure have generated contradictory findings. To date, scientists have not found any improvements in the presence of hepatic or renal sufficiency, which led to no dose adjustment in case of kidney failure, whereas others have reported substantially reduced drug clearance in the case of extreme renal insufficiency [27–29]. Myelosuppression is typically dose-limiting toxicity; escalation beyond this dosage range is restricted by cardiac toxicity. Synergistic hematopoietic toxicity may occur with concomitant use of allopurinol [30]. According to the Food and Drug Administration (FDA) agency in 1995, CP was approved to be a valuable drug for consumption [31,32]. From that time, CP started to be one of the most common medicines to treat tumor malignancies such as MM and breast cancer. Besides, it can be used in the treatment of various rheumatic diseases, including scar pemphigoid (also known as pemphigoid mucous membrane), juvenile dermatomyositis and soft tissue sarcoma, and thrombocytopenic purpura refractory therapy [33–35]. In contrast, the side effects of CP use are nausea, vomiting, amenorrhea, ovarian failure, sterility, cardiotoxicity, and immunosuppression, which is associated with an increased risk of secondary malignancy infection. Resistance mechanisms are similar to those of other mustards but also include increased expression of aldehyde dehydrogenase, which leads to enzymatic detoxification of the drug [36].
2.1.1.1.2 Chlorambucil
Chlorambucil belongs to the nitrogen mustard group of alkylating agents, which also form DNA cross-linking scaffolds. In 2013, the FDA approved chlorambucil in combination form as a therapy for patients with previously untreated CLL [37]. The mechanism of action for chlorambucil is similar to that of CP but relatively slow [17]. CLB’s mechanism of action has been explored in many tumor cell models. The agent hydrolyzes quickly and has a half-life of 30 min in water and 42 min in blood [23]. Passive diffusion was found to be the mechanism of drug uptake in rat Yoshida ascites sarcoma cells [24] and mouse models. CLB and other nitrogen mustards form DNA crosslinks and monoadducts in several cell types. DNA crosslinks may limit replication in growing cells, while DNA monoadducts may contribute to the agent’s cytotoxicity by inhibiting transcription [38]. This compound is used for patients with non-Hodgkin’s lymphomas, Hodgkin’s disease, non-Hodgkin’s lymphomas, and CLL [39–42]. The therapeutic use of this drug’s effectiveness is limited by toxic effects such as bone marrow suppression, anemia, and a weakened immune system. It also causes nausea, myotoxicity, and neurotoxicity, as well as secondary cancers [43].
2.1.1.1.3 IFO
IFO is a nitrogen mustard compound, which differs from CP in the location of chloroethyl moiety. It is commonly used as a racemic mixture intravenously to produce the active cytotoxic metabolites, which are activated by the liver cytochrome P450 microsomal system [44]. IFO was approved as an official drug in 1988 by the FDA agency [45]. The mechanism of action of IFO involves its metabolites by CYP450 enzymes in the liver. These effective metabolites (the derivatives of phosphor amide mustard and acrolein) bind to DNA and inhibit the synthesis of modified DNA. There are two types of mechanisms for IFO: first, it forms an intrastrand cross-linking reaction with DNA to generate the modified one, which causes cell damage; and second, it contributes to the apoptosis of the damaged cells. These active metabolites upregulate the reactive oxygen species, resulting in irreparable damage to DNA and ceasing the formation of proteins [46]. IFO has many major toxic side effects: hemorrhagic cystitis is responsible for the dose-limiting toxicity. However, the drug itself is not specifically harmful to kidneys, but its metabolite chloro-acetaldehyde in vitro and in vivo is harmful to renal tubular cells [47]. This type of drug is specifically recommended for patients with persistent cancers of the germ cells, breast cancer, lung cancer, cancer of the head and neck, and cervical cancer [48,49]. Cancer treatment with alkylating DNA agents, like IFO, can lead to gonadal toxicity, ovarian failure, and reduced fertility, leading to premature menopause due to numerous electrophilic aldehydes that result from chemotherapy [50,51].
2.1.1.1.4 Melphalan
Melphalan is an anti-cancer drug that prevents cancer cells in the body from growing and spreading. It has been an effective treatment for MM, also known as phenylalanine mustard and phenylalanine melphalan [52,53]. The mechanism of action of Melflufen, as a peptide–drug combination: the compound’s high lipophilicity allows for fast diffusion across the neoplastic membrane. The peptidase enzyme found in the cell acts on the peptide–drug combination, releasing the alkylating moiety from the carrier. The nitrogen mustard moiety enters the nucleus, which leads to DNA crosslinking.
Furthermore, Melphalan has been utilized in the treatment of MM since the 1960s [54]. In 2016, in the United States, this drug was not licensed for use as a high-dose conditioning therapy prior to hematopoietic progenitor (stem) cell transplantation in MM patients [55]. The mechanism of action is similar to bifunctional alkylating agents; its cytotoxicity seems to be linked to the degree of its DNA cross-linking. These covalent bonds bend DNA’s double helix, causing cytotoxicity and interfering with the polymerases’ ability to function, presumably by binding guanine at the N7 position. In addition, these adduct cell apoptosis can be caused by deletions, strand scissions, and the development of open rings. Thus, it is an efficient treatment against resting and rapidly dividing tumor cells [56,57]. In addition, Melphalan was utilized orally for MM, ovarian, and breast carcinomas and is used in treatment regimens in bone marrow transplantation at high dosages [58,59]. However, it has many side effects, such as nausea, diarrhea, vomiting, and ulcers in the mouth. Therefore, to decrease the microsites, the patients were recommended to consume ice chips to maximize circulation of blood to the oral mucosa and to improve drug distribution to the region, often referred to as cryotherapy [60].
2.1.1.1.5 Mechlorethamine
Mechlorethamine was the first nitrogen mustard drug used in clinical practice and the progenitor of antineoplastic alkylating agents, also known as chlormethine. It is readily and unimolecularly converted at physiological pH to a relatively stable aziridinium ion as a free base to turn the interaction of biomolecules into accessible nucleophilic centers. It was approved by the FDA in 1949 [61]. Mechlorethamine reacted with N-7 to create an interstrand cross-link of two separate guanines in DNA. The formation of the ammonium ion N-7 makes the guanine more acidic and thus tends to alter the balance in favor of the enol tautomer. In addition, N-7 alkylated guanosine is susceptible to hydrolysis and forms a basic guanosine [62]. Mechlorethamine is also utilized in the therapy of Hodgkin’s disease systemically and in the topical treatment of mycosis fungoides and other lymphomas [63]. Vomiting and nausea are the most common side effects of mechlorethamine; it typically begins within 30 min of injection and lasts for 8 h, as reported [64].
2.1.1.2 Other alkylating agents
2.1.1.2.1 TMZ
TMZ has been used as a standard anti-cancer drug for glioblastoma, often in conjunction with radiotherapy (Figure 5) [65]. It is a convenient oral drug adjunct that penetrates the blood–brain barrier [66]. In 1999, the FDA approved this drug for patient remediation with primary brain tumors [67]. The mechanism of TMZ metabolism includes DNA damage and repair. Under physiological conditions, TMZ is metabolized to MTIC, which is subsequently converted to its active form, a methyl diazonium ion. The electrophilic methyl diazonium ion functions as a methyl donor, donating its methyl group to negatively charged DNA to form DNA adducts. This alkylating activity occurs primarily at N7 of guanine, O3 of adenine, and O6 of guanine, and if left unrepaired, causes incorrect base pairing and causes breaking of the single and double-stranded DNA and the key endogenous DNA repair [68]. The cytotoxicity of TMZ is regulated by the addition of methyl groups to guanine at the N7 and O6 sites and adenine at the O3 site to genomic DNA. During subsequent DNA replication, alkylation of the O6 site on guanine leads to the addition of thymine instead of cytosine opposite methyl guanine, and this may result in cell death [69]. Adding TMZ to the standard treatment protocol was considered a significant advance in GB therapy [70]. TMZ is also utilized in the treatment of metastatic melanoma and is often utilized in the treatment of malignant melanoma. Few respiratory side effects were reported, mainly pharyngitis, infection of the upper respiratory tract sinusitis, and cough [71].

Other alkylating agents.
2.1.1.2.2 Procarbazine
Procarbazine is a moderate inhibitor of monoamine oxidase (MAOI), which was approved by the Food and Medications for therapy in 1969 (Figure 5) [72]. PCB is a prodrug, and its hepatic metabolism plays a significant role in the production of active metabolite species. Chemical breakdown and microsomal oxidation are two potential activation mechanisms. It is converted to azoprocarbazine by microsomal cytochrome P-450 oxidoreductase or mitochondrial MAOI enzymatic conversion. AzoPCB may be metabolized further through unknown mechanisms. [73]. To generate cytotoxic responses, procarbazine must first be metabolically activated. The mechanism of action of procarbazine is through trans methylation of the methionine group to t-RNA, which is blocked by the drug procarbazine and leads to triggers the cessation of protein synthesis. It can also directly target DNA by targeting the sulfahydrate protein groups found in the residual protein that are closely bound to DNA [74]. Procarbazine is utilized in Hodgkin’s lymphoma, non-lymphoma, Hodgkin’s brain tumors, melanoma, lung cancer, and MM [75]. Acute side effects of Procarbazine include nausea at large doses; nausea and vomiting can develop leukopenia, and thrombocytopenia develop hematologic toxicity within 3–4 weeks of administration [76].
2.1.1.2.3 Chlorozotocin
Urea is a type of alkylating agent used to treat neuroendocrine tumors (Figure 5). Several cases of moderate pneumonitis were associated with its use, which was managed by discontinuation of the drug and administration of corticosteroids [77].
2.1.1.2.4 Steroidal hormones
Steroids have been commonly used for drug delivery because they bind selectively to steroid hormone receptors located on the cancer cells’ surface [78]. Moreover, studies have shown that tumor growth and proliferation in certain forms of cancer, such as breast cancer, are directly linked to steroidal hormones. The chemical bonding of an alkylating chemical to steroidal nuclei as a carrier molecule was a promising strategy for increasing the absorption of nitrogen mustard into these hormone-sensitive tumors [79,80]. The significance of steroid hormones in the prevention and treatment of hormone-sensitive organisms’ cancers such as ovarian, prostate, and breast with endometrium. Steroids have shown that steroidal–mustard conjugates are more effective, selective, and less toxic [81].
2.1.1.3 Alkyl sulfonates
Alkyl sulfonates are a kind of alkylating agent, which are mainly used to treat neoplastic diseases. These modes of action all involve reactive amine alkylation on DNA. It is most efficient in rapidly proliferating tissues, which partially account for their immunosuppressive effects, as shown in Figure 6 [82].

Mechanism of action of alkyl sulfonates.
2.1.1.3.1 Busulfan
Busulfan is an alkylating agent used to treat myeloproliferative neoplasms (MPNs). It was utilized in a number of hematopoietic conditioning regimens, system cell transplantation hematopoietic cell transplantation (HCT), and in the control of chronic myelogenous leukemia in 1953 [83,84]. Furthermore, in 1999, Busulfan was approved by the FDA for the treatment of chronic myeloid leukemia (CML). It interacted with the protein during the hydrolysis process. The methane sulfonates are released, and carbonium ions are formed. DNA is alkylated by carbonium ions, resulting in the intervention of DNA replication and transcription of RNA. Its alkylation mechanism of action establishes an intrastrand crosslink between guanine and adenine [85]. It is also utilized as a conditioning agent in pediatrics and adults in conjunction with CP or clofarabine prior to bone marrow transplantation. However, it suppresses bone marrow in the same way as other antineoplastic alkylating medicines. Busulfan, on the other hand, can cause myelosuppression to last a long time [86].
2.1.1.4 Nitrosoureas
The nitrosourea drug is a class of nitrogen mustard-linked DNA and is considered the chemotherapeutic agent most widely used in the treatment of malignant PBT (Figure 7). It is an extremely lipid soluble, non-ionized, cell-cycle-non-specific agents that easily cross the blood–brain barrier. It spontaneously decomposes into two active intermediates, a group of isocyanates and chloroethyl diazohydroxide [87]. Their mechanism of action is established on the formation of diazohydroxide under basic conditions, which in turn generates a reactive for alkylation (Figure 8).
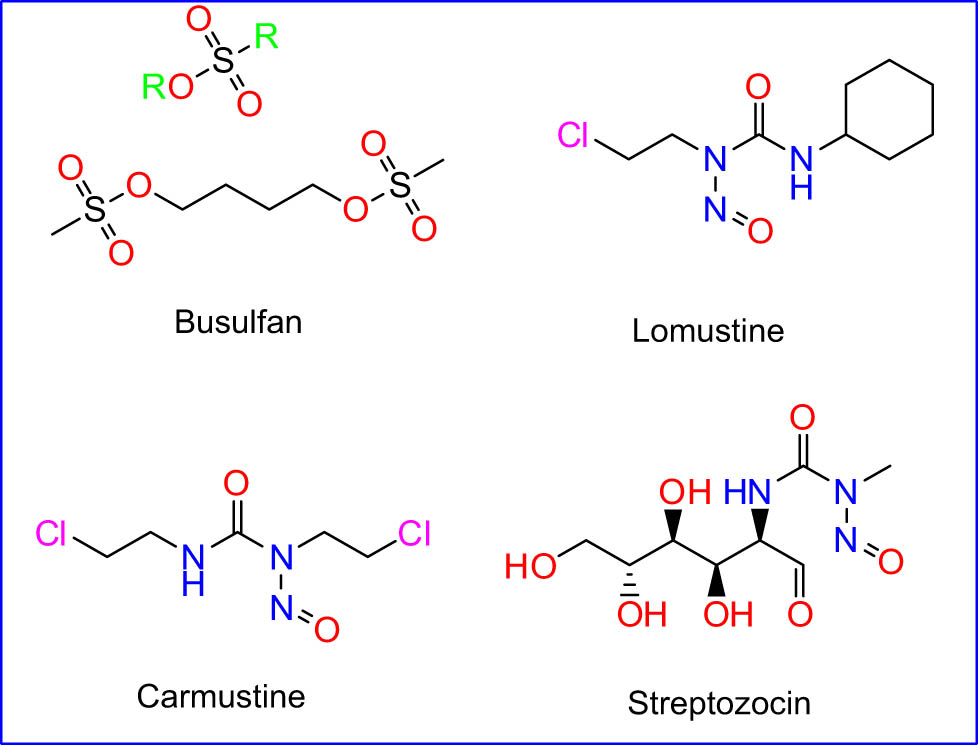
Alkyl sulfonates and nitrosoureas drug.

Mechanism of action of nitrosourea.
2.1.1.4.1 Lomustine
Lomustine is an oral chemotherapeutic efficient, highly lipid-soluble, and enters cells by passive diffusion. Lomustine spontaneously decomposes into a reactive center to cross-link DNA alkylation, DNA–DNA, and DNA–protein under aqueous conditions and at physiological pH [88]. In 1976, the FDA approved it for the treatment of certain diseases. Lomustine undergoes inclusive hepatic metabolism, mainly through cyclohexyl ring hydroxylation, to form metabolites of alkylating activity, which is at least equivalent and are believed to play a significant role in cytotoxic activity [89]. Nitrosourea subclass, 1-(2-chloroethyl)-3-cyclohexyl-1-nitrosourea (CCNU, lomustine) is most widely used against lymphoma and mast cell tumors due to its possibility to pass through the blood–brain barrier. CCNU is most often used alone or in multiagent protocols for brain tumors [24,90]. Myelosuppression with acute neutropenia followed by thrombocytopenia is the primary dose-limiting toxicity [91].
2.1.1.4.2 Carmustine
Alkylating nitrosourea is commonly utilized for treating malignancies. Since systemic administration of nitrosoureas was poorly successful in treating high-grade glioma, carmustine wafers were designed to deliver high local drug concentrations. In particular, carmustine was successful by alkylating DNA and RNA [92]. It has a low molecular weight, is extremely lipophilic, and penetrates deep into the central nervous system blood–brain barrier easily owing to its medicinal lipophilic nature [93]. The main action mechanism for this family drug is by binding to DNA and alkylation of DNA nitrogen bases in the duplex. However, several studies have confirmed that carmustine prevents cancer cell growth. The essential processes such as replication, transcription, and DNA translation are inhibited by the alkylation of DNA [94,95]. Carmustine is commonly used to treat various forms of human cancers, including glioblastoma, brainstem glioma, astrocytoma, metastatic brain tumor leukemia, and medulloblastoma [96,97]. Delayed myelosuppression, interstitial pneumonia, and platelet count are almost common, and severe disadvantages of carmustine might even be nausea and vomiting [98,99].
2.1.1.4.3 Streptozocin
Streptozocin (also known as streptozotocin) is an alkylating nitrosourea agent known to be absorbed into cells by the GLUT2 protein expressed by glucose to cause cell harm [100]. STZ is also a powerful alkylating agent used to methylate DNA directly and is extremely genotoxic [101]. It was approved by the FDA in 1982 and is used to treat metastatic cancers of pancreatic islets of Langerhans [102]. Streptozocin does not cross the blood–brain barrier quickly, nor is it highly myelosuppressive. Streptomyces achromogenes, which are especially toxic to the pancreatic beta cells producing insulin in mammals. It is used as a chemotherapeutic medication in the medical sector to treat some Langerhans islets cancers and a singular antineoplastic agent used to treat pancreatic metastatic islets cellular carcinoma [103]. Streptozocin is unique in its attraction to pancreatic islet cells. Because it is a vesicant, it should be given intravenously with caution. It is one of the most emetic agents, necessitating a large amount of antiemetic premedication. The side effect of this drug is the possibility to be toxic and thereby cause renal and hepatic failure [104,105].
2.1.1.5 Ethylenimine derivative
Ethylenimine is also called aziridine, which is made from ethylenimine. It is a three-pointed ring made up of two carbon atoms and one nitrogen atom, such as Thiotepa (Figure 9).

Ethylenimine derivative and Triazine drugs.
2.1.1.5.1 Thiotepa
Thiotepa is the first chemotherapeutic agent contra NMIBC FDA-approved. It is a nonspecific alkylating agent with low molecular weight (189 kDa) chemically used for nitrogen mustard [106]. According to routes, Thiotepa is a polyfunctional alkylating chemical, and it can generate crosslink reactions with DNA molecules. The aziridinyl groups’ mechanism draws a proton from the N7 guanine of DNA and performs a ring-opening reaction via nucleophilic attack [107]. The exact method by which they have anticancer properties is uncertain. However, they are categorized as alkylating agents, and it is assumed that they work by crosslinking DNA double-helix strands to halt tumor growth. This prevents the strands from uncoiling and separating, which is required for DNA replication. Cancer cells can no longer divide and die as a result [81]. Their toxicity appears to be dose-dependent, as there is a lesser likelihood of leucopenia. A leukocyte and platelet count should be acquired before each treatment because it is the system that is responsible for much toxicity. In 1994, thiotepa was approved by the FDA for the treatment of breast cancer, ovarian cancer, and bladder cancer. After that, it was used as a conditioning treatment prior to hematopoietic stem cell transplantation in 2007 [108]. Thiotepa has been utilized to treat a variety of neoplastic diseases, the most common of which are adenocarcinoma of the breast and ovary, urinary bladder carcinoma, and intracavitary effusions caused by diffuse or localized neoplastic diseases of various serosal cavities [109]. Acute side effects of thiotepa are similar to other alkylating agents like nausea, vomiting, diarrhea, alopecia, mucositis, bone marrow suppression, and rash, and uncommon but potentially serious adverse events involve severe myelosuppression, severe infections, sepsis, bleeding, and embryo-fetal toxicity [110].
2.1.1.6 Triazines
Triazine derivatives, particularly 1,2,4-triazine derivatives, have been identified as new antitumor agents. These compounds exhibit potent cytotoxic activities with fewer side effects [111].
2.1.1.6.1 Dacarbazine
Dacarbazine was used widely as a chemotherapeutic agent in the triazene family as an alkylating agent. A chemotherapeutic drug for the treatment of melanoma was approved by the FDA in 1975 [112,113]. Dacarbazine influences cells through three pathways as follows: carbon ion alkylation, metabolic inhibitory effects against DNA and RNA synthesis, and protein alkylating sulfhydryl groups [114]. DTIC was highly efficient and developed for clinical use. Therefore, it is used in the treatment of metastatic malignant melanoma and Hodgkin’s disease. Additionally, DTIC has moderate efficiency in sarcoma therapy, neuroblastoma, neuroendocrine brain tumors, and primary tumors [115]. The main side effects of dacarbazine include nausea and vomiting, which are common. This agent has been linked to mild bone marrow suppression. Patients taking moderate to soaring doses of DTIC also exhibit photosensitivity responses [116].
2.1.2 Antimetabolites
Antimetabolites are among the oldest and the first effective chemotherapeutic agents discovered. It is comparable to chemicals necessary with proper metabolic efficiency but is dissimilar enough to disrupt normal cell functions when antimetabolites are integrated into RNA and DNA during the S phase of cell growth; they cause cell death or block enzymes involved in nucleic acid creation. Because cells in the S phase are more sensitive to antimetabolites DNA synthesis, toxicity is prevalent in any tissue, for example, bone marrow and gastrointestinal tract tissues. This drug classification is described in Table 2 and contains antifolates and antipyrimidines “5-fluorouracil, capecitabine, eniluracile, hydroxyurea,” and antipurines “6-mercaptopurine,6-thioguanine” methotrexate, pemetrexed” [117].
Antimetabolite drugs
| Antimetabolites | Drugs | Use |
|---|---|---|
| 1. Folic acid analogues | Methotrexate | Leukemia: lymphocytic leukemia, acute lymphoid leukemia, and acute and CML |
| Pemetrexed | Non-small cell lung cancer, and treatment for lung, colon, pancreatic, breast, bladder, head and neck, and cervix and gastric cancers | |
| 2. Pyrimidine analogues | Fluorouracil | Breast, stomach, pancreatic, head and neck, esophageal, gastric, colorectal, and anal cancers |
| Capecitabine | Colon cancer and breast and gastric cancer | |
| Cytarabine | Lymphoma and leukemia | |
| Gemcitabine | Solid tumor pancreatic cancer | |
| 3. Purine analogues | Mercaptopurine and thioguanine | Hematological malignancies, chronic inflammatory diseases, and transplantation acute leukemia in oral chemotherapy |
| Fludarabine | CLL, hairy-cell leukemia, and non-Hodgkin’s lymphomas | |
| Pentostatin | Hairy cell leukemia and T cell lymphoma | |
| Cladribine | Hairy cell leukemia and acute myeloid for leukemia | |
| Clofarabine | Refractory acute myeloid and lymphoblastic leukemia | |
| Nelarabine | Relapsed and refractory T-ALL |
2.1.2.1 Folic acid analogues
The synthesis of folates plays a crucial role in RNA and DNA structures. Analogues of folate are also known as antimetabolites, which are considered the most used drug for cancer treatment. Since cancer cells have an aberrant proliferation compared to their normal counterparts, DNA and RNA synthesis inhibition is a very effective anticancer therapy strategy [118] (Figure 10).

Mechanism of action of folic acid analogues.
2.1.2.1.1 Methotrexate (MTX)
Antifolate compounds are potent and well-characterized compounds, which imitate folic acid as they have an additional methyl group at N10” (Figure 10). MTX is a folic acid antimetabolite that works by being competitively bound to the dihydrofolate reductase enzyme. MTX was approved in the United States in 1953 and in 1972 by the FDA to treat psoriasis [119,120]. It inhibits the formation of purines and pyrimidines in the cells and it could also inhibit the synthesis of DNA but may also interfere with protein synthesis that blocks the conversion of certain amino acids [121]. This inhibition results in diminished dihydrofolate conversion for tetrahydrofolate and impaired thymidic acid synthesis. Deficiencies of such acids hinder the synthesis of DNA and RNA [122]. MTX shows a toxic effect profile against normal proliferating tissues such as bone marrow and intestinal tissue to broaden the treatment efficacy to competitively inhibit intracellular MTX transport in tissues close to blood vessels; high doses of MTX are usually followed by low doses of leucovorin (5-formyl-THF) [123]. MTX is approved to treat all cases of leukemia, acute lymphocytic leukemia, acute lymphoid leukemia in childhood, and CML. Also, it is a common medication used to treat pain lymphoid leukemia in childhood [124,125]. In fact, the side effects of MTX appeared in bone marrow and gastrointestinal mucosa [126].
2.1.2.1.2 Pemetrexed
Pemetrexed was authorized as an FDA drug for the first time in 2004 for nonresectable pleural mesotheliomas in conjunction with cisplatin. It works as a cell cycle-specific and antimetabolic anticancer medication that blocks folic acid metabolism (Figure 11). Also, it works on the inhibition of enzymes, implicates purine and pyrimidine metabolism, and impairs the synthesis of RNA and DNA in tumors [127,128]. Reduced folate carrier (RFC) and folate receptor-a allow pemetrexed to enter cells (FR-a) and are intracellularly polyglutamated. Polyglutamation leads to a significant increase in pemetrexed’s affinity for enzymes involved in folate metabolism and a decrease in its affinity for RFC and FR. This process results in a prolonged intracellular lifespan of the drug [129]. Furthermore, pemetrexed is shown officially for the treatment of non-small cell lung cancer, whether locally advanced or metastatic, and for lung treatment, colon, pancreatic, breast, bladder, head and neck, and cervix and gastric cancers [130]. The most common side effects of this drug are diarrhea, mucositis, rash, tiredness, and myelosuppression. Therefore, recently published results confirmed to reduce myelosuppression and mucositis. Folic acid (1 mg daily) and vitamin B12 (1 mg injectable every 8 to 10 weeks) supplementation were prescribed [131].

Folic acid analogues.
2.1.2.2 Pyrimidine analogues
Pyrimidine antimetabolites that interfere with the synthesis of nucleic acids are known as analogues (Figure 12). They are hydrophilic molecules, in general, which require specialized membrane transporters for cell entry. Therefore, by the intracellular enzyme phosphorylation, such as deoxycytidine, the kinase will transform these drugs into active metabolites. Their antiproliferative effect is achieved through incorporation into DNA, resulting in chain termination and suppression of DNA composition (Figure 13) [132].

Pyrimidine analogues.

Mechanism of action of pyrimidine analogues.
2.1.2.2.1 Fluorouracil
The use of 5-fluorouracil (5-FU) as an anticancer agent in the United States was licensed for medical use in 1970 to treat breast cancer, colorectal cancer, stomach cancer, pancreatic cancer, head and neck, esophageal, gastric, colorectal, and anal cancers. Although the exact act mechanism of the drug is still to be fully known, it involves blocking the thymidylate synthase (TS) enzyme, resulting in a loss of phosphorylated deoxythymidine and a toxic accumulation of deoxyuridine [133,134]. It is an anti-cytotoxic but also an anti-neoplastic cell from the type of anti-metabolites, a natural pyrimidine base that is employed in the production of nucleic acids [135]. 5-FU is transformed into various active metabolites like fluorodeoxyuridine monophosphate (FdUMP), fluorouridine triphosphate (FUTP), and fluorodeoxyuridine triphosphate (FdUTP). It plays an important role in biological activities such as disrupting RNA synthesis and exerts anticancer influence through TS inhibition [136]. These formulations are all administered through the intravenous route. The main problem with this administration is that healthy tissues are exposed to harmful chemotherapeutic agents [137]. 5-FU was used in treatment protocols for breast, stomach, and pancreatic cancers [138]. Severe side effects of this drug in patients who receive it intravenously to treat malignant neoplasms include loss of appetite, nausea, fatigue, a metallic taste, and ulcers [139].
2.1.2.2.2 Capecitabine
Capecitabine is an antimetabolite 5-FU prodrug and was approved in 2001 for metastatic colon therapy [140]. Capecitabine is commonly used as a monotherapy for palliative chemotherapy in inoperable patients. Capecitabine is hydrolyzed in the liver and gradually transformed into the effective 5-FU drug. This drug is active as a single agent, but it can also be combined with other cytotoxic agents, including oxaliplatin, irinotecan, taxane, or cisplatin [141,142]. In its mechanism of action, thymidylate phosphorylase converts capecitabine to 5-FU. TS is inhibited by 5-FU, which leads to the inhibition of the development of thymidine monophosphate needed for de novo DNA synthesis. In many tumor cells and hepatocytes, high thymidylate phosphorylase levels are observed, making these cells more vulnerable to toxicity [143]. Capecitabine is currently being utilized in conjunction with preoperative radiotherapy, postoperative adjuvant therapy, or in conjunction with other chemotherapy agents that are effective against some cancer types [144]. However, capecitabine has been recorded by 5-FU to be used as a treatment for colon cancer and advanced breast and gastric cancers. Capecitabine’s side effects include stomatitis, extreme diarrhea, and minor nausea and vomiting. In addition, extreme “hand-foot syndrome, “palmar/plantar erythrodysesthesia,” and other dermatology benefits have been noted [145].
2.1.2.2.3 Cytarabine (CBN)
CBN is a structural analogue of pyrimidine and purine bases, and its functional role is to inhibit unchecked cancer cell growth by interfering with cell DNA synthesis [146]. It was approved by the FDA in 1969 as a potent drug of antileukemic substance [147]. The mechanism of action of this drug is that it is converted into a triphosphate inside the cell and competes to integrate itself into the DNA with cytidine. CBN’s sugar moiety hinders the molecule’s rotation within the DNA. Through the S steps of a cell cycle, DNA replication stops, giving it a particular medication for quickly proliferating cells like cancer cells. The replication and repair of DNA also stops because CBN inhibits DNA polymerase [148]. This drug is recommended for patients intravenously and it is quickly eliminated from the plasma with a half-life of 5–20 min, and the blood is delaminated in the liver [149]. CBN treats CNS disease with systemic because of the height-dose therapy; CBN can pass through the blood–brain barrier to reach cerebrospinal fluid concentrations between 40 and 50% of plasma concentrations [150]. CBN is also effective in treating acute lymphocytic leukemia, and it can be used in both children and adults to treat relapses of acute lymphocytic leukemia. In the treatment of non-Hodgkin lymphomas, Ara-Cis is used in conjunction with other agents. In terms of treatment of solid tumors, Ara-C is not especially useful [151]. CBN can also be used to treat other forms of lymphoma and leukemia [152]. Therefore, myelosuppression is the main side effect of this drug. In fact, high doses cause nausea and vomiting, mucositis, diarrhea, keratoconjunctivitis, and cerebellar toxicity [153].
2.1.2.2.4 Gemcitabine
Gemcitabine is a deoxycytidine analogue, which is helpful for treating several forms of solid tumors. It is an effective candidate for combination therapy and the combination of gemcitabine and DNA-damaging drugs such as alkylating agents and platinum complexes [154]. Gemcitabine was approved by the FDA in 1996 to treat advanced pancreatic cancer [155]. The mechanism of action of gemcitabine is that it mostly has anticancer properties and influences interfering with DNA synthesis. It possibly integrates its metabolites into DNA, thus abrogating successful DNA synthesis. In addition, it inhibits TS, the primary enzyme in the thymidine de novo production pathway. The alternative route to the thymidine salvage pathway represents the basis for tissue absorption of FLT [18F] [156]. Gemcitabine is an approved medicine for pancreatic cancer and lung cancer, with an objective response rate of 23.8% [157]. The commonly recorded side effects are bone marrow suppression, liver and renal issues, nausea, fever, rash, shortness of breath, and hair loss [158].
2.1.2.3 Purine analogues
Purine analogues are the most common class of heterocyclic compounds containing nitrogen, which play a crucial role in a wide range of living organisms’ functions, as shown in Figure 14.

Mechanism of action of purine analogues.
2.1.2.3.1 Mercaptopurine and thioguanine
6-Mercaptopurine and (thiopurines) are immunosuppressive drugs that have shown benefit in the maintenance of remission in both CD and UC [159]. 6-MP was first approved for medical use in the United States in 1953 [160]. Mercaptopurine’s key mechanism of action is that 6-MP thiopurines need intracellular activation to exert cytotoxicity, catalyzed by multiple enzymes. Thiopurine drugs’ cytotoxic effects are achieved by various mechanisms: incorporation of thio-deoxyguanosine triphosphate and thioguanine triphosphate into DNA and RNA, respectively, inhibition of de novo purine synthesis by methyl mercaptopurine nucleotides, inhibition of Rac1 inducing apoptosis, and inhibition of Rac1 inducing apoptosis. It is utilized mainly in relation to the remediation of leukemia, autoimmune disorders, and solid tumors. 6-MP is the most prescribed drug for acute lymphoblastic leukemia (ALL). Common side effects include rashes, flu-like symptoms, bone marrow suppression, hepatotoxicity, pancreatitis, nausea and vomiting, and bone marrow suppression. 6-MCP is commonly utilized to treat hematological malignancies, chronic inflammatory diseases, and transplantation acute leukemia in oral chemotherapy [161,162]. Some dose- and metabolism-dependent toxicity, such as leukopenia, thrombocytopenia, and malignancy, are linked to thiopurine methyltransferase (TPMT) deficiency. Other hypersensitivity reactions include rash, fever, arthralgia, pancreatitis, hepatitis, nausea, non-pancreatic stomach discomfort, and diarrhea [163].
2.1.2.3.2 Fludarabine
Fludarabine is a type of purine analogue that is the most widely used as a chemotherapeutic agent for B-cell CLL treatment dysfunction [164]. F-ara-ATP is the active metabolite in charge of the mechanism of action. This is due to competition with the naturally occurring metabolite deoxyadenosine triphosphate (dATP). The inclusion of a thymidylate in the template strand leads F-ara-ATP to compete with dATP for incorporation. The DNA polymerases are stopped or paused at the exact place in the DNA strand when a single F-ara-A molecule is integrated into it. Fludarabine’s inhibitory property makes it a chain terminator [164,165]. Hairy-cell leukemia, non-lymphomas, Hodgkin’s, and Waldeström’s macroglobulinemia are among the cancers that can be treated with this drug [166]. Fludarabine side effects include nausea, diarrhea, fever, rash, and dyspnea, as well as the development of myelosuppression. Fludarabine can cause cardiac and hematologic toxicity, as well as pain, paresthesia, visual disturbance, vomiting, anorexia, chills, and diaphoresis [167].
2.1.2.3.3 Pentostatin
Pentostatin is a purine analogue that induces T-cell apoptosis through adenosine deaminase inhibition. It is used as part of the conditioning regimen in HCT [168]. Pentostatin and related compounds, which are of particular interest in lymphoproliferative treatment, primarily target proliferative lymphocytes [169]. The key mechanism of pentostatin includes the inhibition of adenosine deaminase (ADA), causing dATP pools to accumulate, inhibiting ribonucleotide reductase, and changing the pools of other deoxynucleotides. High amounts of dATP, an inhibitor of RNR, the enzyme that removes the 2′-hydroxy group of the ribose ring during DNA modification, occur from accumulation. Hairy-cell leukemia, B-cell CLL, Waldenstrom’s macroglobulinemia, T-cell leukemias, cutaneous lymphomas, lymphomas, glucocorticoid-refractory acute, and chronic graft-versus-host disease have all been treated with pentostatin [170]. The main toxicities of pentostatin involve myelosuppression, nausea and vomiting, immunosuppression, acute renal failure, keratoconjunctivitis, fever, and elevations of liver function enzymes that have been recorded in higher doses. Neurologic toxicity, including somnolence, seizures, and coma, rarely appears in patients receiving standard-dose treatment [171].
2.1.2.3.4 Cladribine
Cladribine, or 2-CdA, is an analogue of purine that is resistant to ADA activity. Cladribine agent builds up in the lymphoid cells, likely because they are abundant in the deoxycytidine kinase enzyme [172]. Approved by the FDA in 1993 for medical use, this type of drug works primarily by incorporating itself into DNA as a fake nucleotide. This integration inhibits DNA strand elongation, DNA polymerase, and ribonucleotide reductase. Besides, when given as a single agent by continuous intravenous infusion, 40–90% of patients who underwent main therapy had significant responses (for 5–7 days, a 2-h daily infusion or subcutaneous bolus injections are used) [173]. Cladribine is most widely used for treating hairy cell leukemia and acute myeloid leukemia. Myelosuppression is the primary dose-limiting toxicity [174].
2.1.2.3.5 Clofarabine
Clofarabine is a purine analogue, which inhibits the enzyme ribonucleotide reductase and DNA synthesis [175]. It is analogous to cladribine as the hydrogen in the purine’s 2-position is substituted with chlorine. In addition, DNA strand elongation is inhibited by triphosphate metabolites and ribonucleotide reductase is inhibited, decreasing the deoxynucleotide pools required for DNA synthesis [176]. Clofarabine is a drug that is used to treat acute myeloid and lymphoblastic leukemia that has relapsed or become resistant to other treatments [143]. Clofarabine was approved as an FDA drug for medical use in 2004. Clofarabine is recommended in pediatric patients with recurrent refractory lymphoblastic. Severe myelosuppression has been described as a dose-limiting adverse effect, including neutropenia, anemia, and thrombocytopenia. Therefore, the risk of serious infection could be increased, and non-hematological adverse dose-limiting effects, including rashes, reversible hyperbilirubinemia, and increased aminotransferase activity, have been identified [177,178].
2.1.2.3.6 Nelarabine
Tetrahydrofuran-3,4-diol is a prodrug of the deoxyguanosine analogue 9-beta-d-arabinofuranosylguanine (Ara-G). It is considered a purine nucleoside analogue that is good for T cells. Ara-G triphosphate is then converted, accumulates preferentially in lymphoblasts, and inhibits DNA replication [179]. It reduces deoxynucleotide pools, which are required for DNA synthesis by inhibiting ribonucleotide reductase, and triphosphate metabolites are incorporated into DNA, where they inhibit strand elongation. In 2005, the FDA organization approved it for medical use. T-cell acute lymphoblastic leukemia (T-ALL) and T-cell lymphoblastic lymphoma are both treated with nelarabine (T-LBL) [180]. After infusion, nelarabine can be administered intravenously and is rapidly converted to Ara-G in the plasma by adenosine deaminase [179]. The active metabolite Ara-G has a half-life of 3 h, while nelarabine has a half-life of 30 min [171]. Some of the negative side effects include extreme sleepiness, seizures, peripheral neuropathies, cough, dyspnea, as well as paralysis, exhaustion, bone marrow suppression, and gastrointestinal and respiratory symptoms are the main side effects [181] (Figure 15).

Mercaptopurine derivatives.
2.1.3 Antibiotics
Antibiotics are generally used to inhibit cell division in chemotherapy and chemotherapeutic agents for microorganism-inserted cancer. Some of them introduce direct DNA damage and use antitumors to specifically inhibit topoisomerase II (Figure 16). They bind to DNA mainly through intercalation and obstruct DNA and RNA [182]. In 1940, Waksman and Woodruff discovered and published the first anticancer antibiotic (Actinomyces antibiotic “actinomycin D), which was extremely toxic to animals. Furthermore, Waksman and Woodruff subsequently proved the successful use of antibiotics in some childhood tumors in 1941. This was the beginning of the use of antibiotics in cancer chemotherapy, and since then, numerous natural or synthetic antibiotics have been used in cancer care. Some common antibiotic drugs are summarized in Table 3 [117,183,184].

Antibiotic drug structures.
Antibiotics drugs
| Antibiotics | Drugs | Use |
|---|---|---|
| Dactinomycin | — | Pediatric tumors, such as Wilms tumor, Ewing sarcoma, embryonal rhabdomyosarcoma, testicular tumors, and choriocarcinoma |
| Anthracenedione | — | Prostate cancer, which is reticent to hormonal therapies, lymphomas, and multiple sclerosis relapse-remitting, acute leukemias |
| Bleomycin | — | Hodgkin’s lymphoma, head and neck penile cancer, testicular cancer, cervical and vulvar squamous cell carcinomas |
| Mitomycin | — | Solid tumors |
| Anthracyclines | Mitoxantron | Myelogenous leukemia and acute lymphocytic leukemia |
| Doxorubicin | Acute leukemia, lymphoma, cancer of the liver, breast, and ovaries, Kaposi’s sarcoma, and cancer of the bone | |
| Epirubicin | Breast cancer, gastric and esophageal malignancies, and soft malignancies tissue sarcomas | |
| Idarubicin | Lymphoma |
2.1.3.1 Dactinomycin (actinomycin D)
Dactinomycin is a crystalline antibiotic, which is made up of two symmetric polypeptide chains attached to a central phenazone ring because of Streptomyces parvulus fermentation [185]. In early 1942, actinomycin was discovered and emanated from Selman Waksman’s pioneering work on soil microorganisms [186,187]. Dactinomycin was approved by the FDA in 1964 for medicinal use. The mechanism(s) of action of the drug is through the inhibition of RNA (RNA polymerases; transcription) and DNA-dependent synthesis by its non-covalent interaction with unique DNA motifs near transcriptional complexes. The minor grooves of DNA are bound to the DNA–RNA polymerase sites and have an “interfacial” (intercalator, noncovalent) action [188]. Dactinomycin is the most common treatment for pediatric tumors like Ewing sarcoma, Wilms tumor, and embryonic rhabdomyosarcoma. It has a significant role in the treatment of testicular tumors in choriocarcinoma, and its dose-limiting toxicity in mucositis and vein extravasation causes extreme necrosis of the tissue [189,190].
2.1.3.2 Bleomycin (BLM)
Bleomycin (BLM) is also known as Blenoxane. It is commonly used as an antibiotic glycopeptide antitumor generated by the Streptomyces bacterium. Similar to those obtained with radiotherapy, BLM induced breaks in DNA [191]. Bleomycin was first approved by FDA in 1973 [192]. The mechanism of action of bleomycin includes free radical induction. Bleomycin forms the Fe(ii) complex, which is oxidized to Fe(iii). This causes free radicals to reduce oxygen, which causes single- and double-strand breaks in cellular DNA, leading to damaged cells becoming gnomically unstable. It also prevents the angiogenesis of tumors [193]. Bleomycin removal correlates well with patients with renal impairment and should be given in lower doses based on their creatinine clearance. BLM is inactivated in the tissues by bleomycin hydrolase. In addition, tissues that lack this enzyme are more sensitive to the toxic influences of the drug, such as the lungs and skin [194]. It is recommended for palliative treatment of Hodgkin’s lymphoma, head and neck penile cancer, testicular cancer, and cervical and vulvar squamous cell carcinomas [195,196]. Bleomycin is easily cleaned out of the lungs, spleen, and kidney blood and concentrated in the liver and epithelial tissues. Approximately 80% is excreted within 24 h in the urine [197]. The major important toxic influences of bleomycin are included in the lungs and skin [198].
2.1.3.3 Mitomycin
Mitomycin is one of the common drugs derived from Streptomyces caespitosus. It was approved by the FDA in 2002. It inhibits DNA synthesis after intracellular activation by responding to DNA similar to alkylating agents. Mitomycin inhibits DNA synthesis and cross-links DNA at the O6 N2 positions of guanine and the N6 position of adenine [199]. This drug is utilized for the treatment of solid tumors; Mitomycin C has a cumulative and dose-dependent effect. When Mitomycin is used by itself, the occurrence of nephrotoxicity is less than 1%; however, in conjunction with 5-FU, nephrotoxicity occurs more frequently [200]. It has been used as a cytotoxic and is active against several tumors, esophagus, and bladder, including breast, stomach, and non-small cell lung cancer [108]. Side effects involve skin irritation (dermatitis) and ocular irritation because of contact or toxic contact conjunctivitis, headaches, nausea, dizziness, and hair and menstrual alterations [201].
2.1.3.4 Mitoxantrone
Mitoxantrone is a synthetic antibiotic commonly utilized as a chemotherapy medication for solid tumor therapy [202]. It is a medication accepted by the US FDA to treat adult acute myeloid leukemia in 1987. It was used for treatment of worsening relapsing-remitting multiple sclerosis in 2000 [203]. The mechanisms of action of this drug involve intercalation with the DNA molecules, which in turn induces by inhibiting topoisomerase II, and it disrupts single- and double-stranded DNA and decreases DNA repair. Mitoxantrone greatly inhibits the development of macrophages as well as B and T lymphocytes. Other cells are also killed, and migration of the activated leukocytes is suppressed, such as antigen-presenting cells [204]. Mitoxantrone greatly inhibits the development of macrophages in addition to B and T lymphocytes. Other cells are also killed, and migration of activated leukocytes is suppressed, such as antigen-presenting cells. Mitoxantrone is widely used to treat prostate cancer; it is resistant to hormonal therapies, lymphomas, and multiple sclerosis relapse-remitting acute leukemia [205]. There are three essential side effects of mitoxantrone, such as serious leukopenia, cardiac toxicity, and acute myelogenous leukemia. Furthermore, patients treated with mitoxantrone often run a high risk of opportunistic infections [204,206].
2.1.3.5 Anthracyclines
Anthracyclines are part of many procedures that are used to treat pediatric cancer. According to most groups’ oncology treatment classes, the hematologic toxicity of anthracycline agents is equivalent to their cardiotoxicity, and they have been utilized in different hematological and solid tumor malignancies (Figure 17) [207]. However, hematological toxicity, alopecia, and cardiotoxicity are the main side effects to be considered for these types of drugs [208].

Anthracycline drug structures.
2.1.3.5.1 Daunorubicin
Daunorubicin is just a cytotoxic antibiotic produced with anthracycline by Streptomyces peucetius subsp. It received approval from the FDA in the United States in 1998. The antitumor effects of daunorubicin are back to intercalation by multiple mechanisms affecting both anti-mitotic and anti-cytotoxic activities. It functions by intercalating the DNA base pairs to uncoil the DNA double helix and inhibit the topoisomerase II enzyme, resulting in single- and double-strand breaks, thereby inhibiting the synthesis of DNA and RNA. Daunorubicin also inhibits the function of the polymerase enzyme that dysregulates gene expression, leading to free radical DNA damage and eventually leading to apoptosis, mitochondrial injury, and programmed death of cells [209]. This drug is being used to treat solid tumors that develop in the breast bile ducts, endometrial tissue, the esophagus, and the liver, acute myelogenous leukemia, and acute lymphocytic leukemia. The main route of removal is primarily through the bile duct, with some urine excretion [209,210]. Daunorubicin has some side effects, such as various skin and hypersensitivity reactions in addition to severe myelosuppression, and other adverse events that occurred frequently were nausea, neutropenia, stomatitis, rash, mucositis, vomiting, anemia, and neutropenia [211].
2.1.3.5.2 Doxorubicin (Adriamycin)
Doxorubicin is the most commonly utilized anthracycline antibiotic, and it has one hydroxyl group that distinguishes doxorubicin from daunorubicin [212]. It was the first FDA-confirmed anticancer nanomedicine for the treatment of certain forms of cancer in 1995. Intercalation into DNA and the inhibition of topoisomerase II activity tend to be its mechanism of action, thereby blocking DNA synthesis and giving rise to cytotoxic and apoptotic cell death [213]. Dox is used to treat a wide variety of human cancers, including acute leukemia, lymphoma, cancer of the liver, breast, and ovaries, Kaposi’s sarcoma, and cancer of the bone [214]. Dose-dependent permanent side effects arise from Dox administration, some of which give rise to the development of cardiomyopathy, dyspnea, exercise intolerance, hepatotoxicity, and nephropathy [214].
2.1.3.5.3 Epirubicin (Ellence)
Epirubicin is a DNA-damaging anticancer drug. Consequently, epirubicin lowers mRNA due to better efficacy and fewer side effects, and it is favored over doxorubicin [106,215]. Epirubicin was confirmed by the FDA in 1999 to treat some types of cancer. The mechanism of action of Epirubicin is adoxorubicin drug resistance mechanisms P-gp and mutations in topoisomerase II are involved in drug resistance, analogues that cause DNA strand breaks due to damage caused by topoisomerase II or free radicals. Epirubicin is licensed for use in breast cancer patients and shows efficacy in both gastric and esophageal malignancies and soft malignancies. Tissue sarcomas are a primary target of treatment with epirubicin. Most of its metabolites are excreted via the biliary system [216,217]. However, severe side effects, including acute myelotoxicity and cumulative dose-related cardiotoxicity, have limited its clinical use [218].
2.1.3.5.4 Idarubicin
Idarubicin is a daunorubicin analogue that lacks the methoxy group on the C4 aglycone. It is an anthracycline that has a longer half-life than doxorubicin and is used to treat lymphoma. In preclinical studies, idarubicin demonstrated a better therapeutic index, particularly in terms of the rate of cardiotoxicity [219]. Idarubicin was FDA-approved for the treatment in 1990 [220]. This family of molecules’ antitumor mechanism involves interactions with DNA in a variety of ways, including intercalation, DNA strand breakage, and inhibition of the topoisomerase II enzyme. Some antineoplastic drugs that contain saccharides include etoposide, a semisynthetic-d-glucopyranoside analogue of podophyllotoxin. This medication inhibits DNA synthesis by forming a topoisomerase II and DNA complex [221]. IDA, which is used in acute myelogenous leukemia and ALL treatment, is extremely lipophilic compared to DOX and DNR and is imported into cells faster than the anthracyclines described above [222]. Idarubicin can have severe blood and bone marrow disorder side effects such as myelosuppression, and gastrointestinal, dermatologic, cardiologic, hepatic, or renal complications [223].
2.1.4 Topoisomerase inhibitors
Topoisomerases have been regarded as important targets for therapeutic intervention due to their crucial role in DNA replication and recombination, as well as transcription and repair, as summarized in Table 4. These enzymes can change DNA architecture, which is important in the cell cycle.
Topoisomerase inhibitor drugs of topoisomerase
| Topoisomerase inhibitors | Drugs | Uses |
|---|---|---|
| Topoisomerase I | CPT | Liver cancer, lung, and bladder |
| Irinotecan | Colorectal cancer patients and in patients of lung cancers | |
| Topotecan | Metastatic ovarian carcinoma, relapsed and refractory cervical cancer | |
| Topoisomerase II | Etoposide | Small cell lung cancer and testicular cancer Hodgkin’s and non-Hodgkin’s lymphomas, gastric cancer |
2.1.4.1 Topoisomerase inhibitors of topoisomerase
The inhibitors of topoisomerase I and topoisomerase II compounds inhibit topoisomerase enzymes in the S stage of the cell cycle by altering the phosphodiester backbone of DNA strands to cause changes in the DNA structure. Classical examples of topoisomerase I inhibitors are camptothecins (CPTs), topotecan, and irinotecan derivatives inhibiting topoisomerase I, and topoisomerase II inhibitors are antitumor antibiotics (doxorubicin) [183]. Pemetrexed is one of the cytotoxic agents used most often in non-squamous NSCLC conditions [224].
2.1.4.2 Topoisomerase I (TOP1)
Topoisomerase I (TOP1) is an enzyme that is present in both prokaryotes and eukaryotes. TOP1 is an important enzyme in the mammalian system for normal development [225]. These enzymes are mainly responsible for relaxing supercoiled DNA positively and/or negatively. To form a 5′-phosphotyrosine intermediate, Topia enzymes cleave a single strand of supercoiled DNA transiently. These enzymes contain E. coli Top A, which relaxes negatively supercoiled DNA preferentially [226].
2.1.4.2.1 CPT
CPT is a quinoline alkaloid inhibitor specific to the cytotoxic TOP1 utilized in traditional Chinese medicine to treat cancer [227]. CPT inhibits DNA and RNA (including ribosomal RNA) synthesis and causes DNA harm. These scientists noted that during the S-phase of the cell cycle, CPT is the most potent and predicted that the DNA replication fork could play a role in cell death induced by CPT. Later studies suggested that at both S and G2 levels, CPT stopped the cell cycle, which was needed for CPT cytotoxicity [228]. During the treatment of many cancers, including colorectal cancer, liver cancer, lung cancer, and bladder cancer, CPT is commonly used [229]. The use of CPT is restricted by disadvantages such as water insolubility, low target ability, and natural tissue toxicity. To avoid these drawbacks, lower molecular weight drugs are conjugated to create higher molecular weight polymers [230].
2.1.4.2.2 Irinotecan (Camptosar)
Irinotecan is one of the considered derivatives of water-soluble CPT, which also inhibits TOP1. It is transformed into an effective metabolite, SN-38, by carboxylesterases after administration [231]. Irinotecan was approved to treat cervical, lung, and ovarian cancers in Japan in 1994 as the first member of this drug community. Its use was authorized in Europe in 1995 and the USA in 1996 [232]. The ternary irinotecan-TOP1-nicked DNA complex turns off the nicked strand’s relegation and prevents the release of topoisomerase. Collision with advancing replication forks of the shaped complex results in the formation of a lethal double-strand break (DSB). This leads to the fact that checkpoint signaling for DNA damage results in DNA replication failure, breaks of the DNA chain, and cell death [233,234]. This type of drug is recommended to be used in colorectal cancer patients, and in patients with lung cancers [235,236]. The much more common adverse effects of irinotecan use are diarrhea, vomiting, bone marrow suppression, and fever. Colitis, anaphylactic reaction, and thrombosis are other side effects that have been reported [237].
2.1.4.2.3 Topotecan (Hycamtin)
Topotecan is a semi-synthetic derivative that is structurally similar to CPT. It is an inhibitor of TOP1 and has demonstrated repeated disease as a single agent’s activities and remains the only second-line agent that has been recommended by the FDA and can be used in patients with SCLC [235,238]. In 1996, the US FDA approved intravenous topotecan (IV) infusion for many forms of cancer, and its oral capsule formulation was approved in 2007 [239]. Topotecan’s mechanism of action precludes topoisomerase from relegating split DNA strands caused by the collision of DNA replication forks with a ternary complex of TOP1, DNA, and topotecan, resulting in DNA damage. Topoisomerase inactivation causes apoptosis and cell death, as shown in Figure 18 [240,241]. Topotecan is approved as a second-line treatment in case of cell lung cancer, as well as for metastatic ovarian carcinoma, relapsed, and refractor cervical cancer [238]. Toxicities regarding topotecan involve bone marrow suppression, diarrhea, nausea, constipation) stomatitis, and fatigue [143].

Mechanism of action and resistance of irinotecan to SN-38.
2.1.4.3 Topoisomerase II
Type II DNA topoisomerases (TOP2), Class II topoisomerase enzymes, are responsible for resolving DNA topological problems (Figure 19). Enzymes tackle DNA topological challenges by causing transitory double-stranded breaks in critical biological processes, including transcription, replication, recombination, DNA repair, chromatin remodeling, chromosome condensation, and segregation [242].

Topoisomerase drug structure.
2.1.4.3.1 Etoposide (Etopophos)
Etoposide is a variety of alkaloids produced from the plant etoposide Podophyllum palatium mandrake plant in the late S and G2 stages of the cell cycle; it has cell cycle-specific action [213]. It was approved by the FDA and consumed as a cancer therapy in 1983 [243]. The action mechanisms of the etoposide cell cycle are predominantly in the later S and G2 phases. DNA unwinding replication is aided by topoisomerase II. It produces and repairs double-stranded DNA breaks during the replication process. Etoposide binds to topoisomerase II in a reversible manner, preventing it from repairing double-stranded DNA breaks. The etoposide–topoisomerase II complex initiates a mutagenesis and cell-death pathway in tumor cells with higher levels of topoisomerase II enzymes, which is more efficient in tumor cells, as shown in Figure 20 [244]. Etoposide is used to treat various cancers, which include small cell lung cancer and testicular cancer, and often a bone marrow conditioning agent in combination with other chemotherapeutic agents and Hodgkin’s and non-lymphomas, Hodgkin’s gastric cancer, and ovarian cancer [245,246]. The side effects of this drug include myelosuppression, anorexia, nausea/vomiting, alopecia, mucositis, diarrhea, fever, bronchospasm, hypersensitivity reactions, skin alterations in dyspnea, hypotension, and radiation recall [90].

Mechanism of action and resistance of Etoposide.
2.1.5 Recent heterocyclic compound-based anticancer reactivity
Spiro heterocyclic compounds have crucial pharmacological biological and chemical properties such as antimicrobial, antiviral, psychotic, anti-inflammatory, and anticancer [247]. Sustainable protocols for the synthesis of novel spiro quinoxaline-pyrimidone-based heterocyclic compounds were successfully developed. Due to the high demand for Spiro heterocyclic compounds, currently, they are used as potent inhibitors of the proliferation of cancer cell antibacterial applications [248–250]. Therefore, it is tremendously important to develop synthetic methodologies to prepare Spiro heterocyclic compounds. Bhupesh and co-workers confirm that the spiro quinoxaline-pyrimidone-based heterocyclic compounds (RD 1–RD 12) were synthesized by using mild conditions and characterized via NMR, IR, and mass analysis techniques [251]. It is important at this stage to discuss the preparation strategy of these valuable heterocyclic compounds. A multi-component reaction methodology was used; the crude product obtained was collected, followed by simple purification to get the desired product in powder form. A wide range of pharmaceutical applications are used to demonstrate the potent activity of these compounds. A minimum inhibitory concentration was observed for all prepared spiro compounds toward bacteria by observing turbidity in the vial. In addition, various activities, such as the binding strength with DNA, molecular docking study, and cytotoxicity assay, were carried out to evaluate the biological activity. These studies demonstrated that all the spiro compounds bind efficiently with certain receptors and may work as a potent anticancer agent (Figure 21).
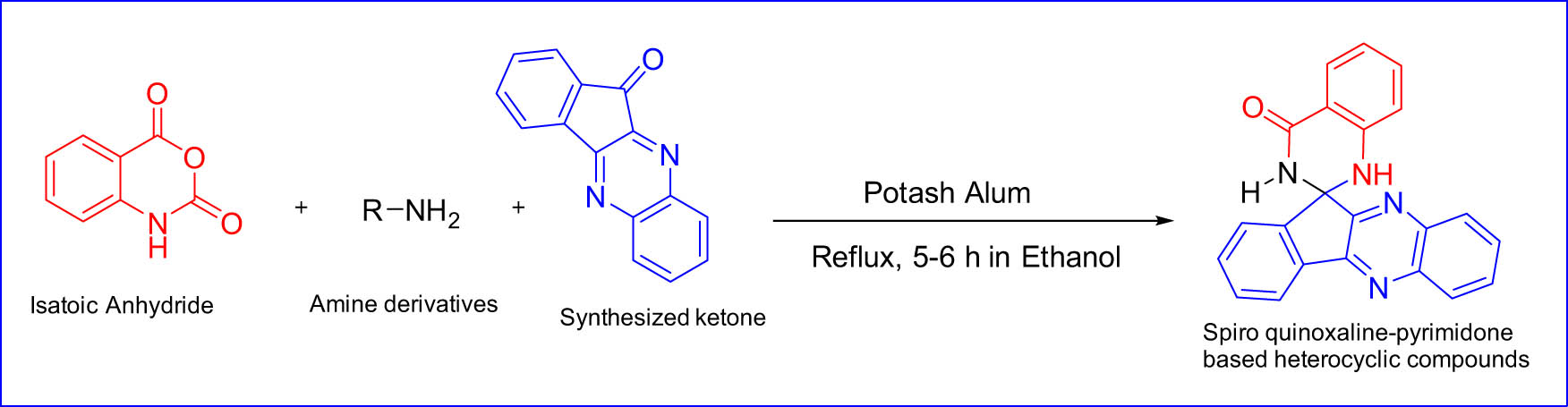
Spiro compounds as a potent anticancer agent.
In addition to this work, another research paper was published in 2022 by Svetlana and co-workers in which novel peroxide-based hybrid compounds were synthesized and characterized, which proves their applications as inhibitory activity against SARS-CoV-2 and leukemia cell lines. Notably, the authors found that some of these hybrid compounds, such as arsenic and peroxide-based hybrid compounds, are more potent against K562 leukemia and the reference anti-leukemia drug doxorubicin with high inhibition values (81–83%). A new class of peroxide-based hybrid compounds was demonstrated by performing a click reaction on the alkyne group of the linker and the aromatic azide group (Figure 22). The resulting compounds were tested against SARS-CoV-2 and cancer to prove their applications as an excellent candidate drug to treat cancer and SARS-CoV-2 infections.

Synthesis route of peroxide-based hybrid compounds.
Auhmani and co-workers published a new research paper in which a novel heterocyclic scaffold as an anticancer agent based on thymol [252]. The target compounds were synthesized via cyclization of the corresponding thymol compounds followed by characterization of the structure by IR, 1HNMR, 13CNMR, HRMS, and single-crystal X-ray diffraction. Interestingly, some target compounds, such as limonene-based thiosemicarbazone derivatives, show significant inhibitory effects on the growth of cancer cell lines and prostate cells. In contrast, due to the high biological activity of some thiosemicarbazones such as thiadiazoles, thiazoline, and thiazolidin-4-ones as anti-inflammatory, antidiabetic, and anticancer, some compounds containing 1,3,4-thiadiazole scaffold in their structure exhibits significant antitumor activity against the tested cell lines. Figure 23 shows the synthetic procedures for the target thymol compounds.

The synthetic procedures for the target thiosemicarbazones and their cyclization. Reagents and conditions: (i) CH3I, K2CO3, acetone, 12 h; (ii) AcCl, AlCl3, CH2 Cl2, 0°C, 2 h; (iii) Thiosemicarbazides, AcOH (few drops), EtOH, reflux, 12 h; (iv) Ac2O, N(Et)3, CH2Cl2, 5 h; (v) BrCH2COOEt, sodium acetate, EtOH, reflux, 8 h; (vi) DMAD, sodium acetate, EtOH, reflux, 4 h; (vii) Phenacyl bromide, sodium acetate, EtOH, reflux, 4 h.
In 2023, Xu and co-workers published a new research paper in which various Allosecurinine derivatives were synthesized along with the study mechanism of action of these compounds as anticancer agents [253]. The authors also tested the structure–activity of these anticancer families against nine human cancer cell lines and the possibility of working. A library of 23 novel Allosecurinine derivatives was designed and synthesized using the Baylis–Hillman reaction. The interaction between Allosecurinine and various types of aldehydes was facilitated by TiCl4, as shown in Scheme 1 and Figure 24.

Synthetic route of Allosecurinine derivatives.
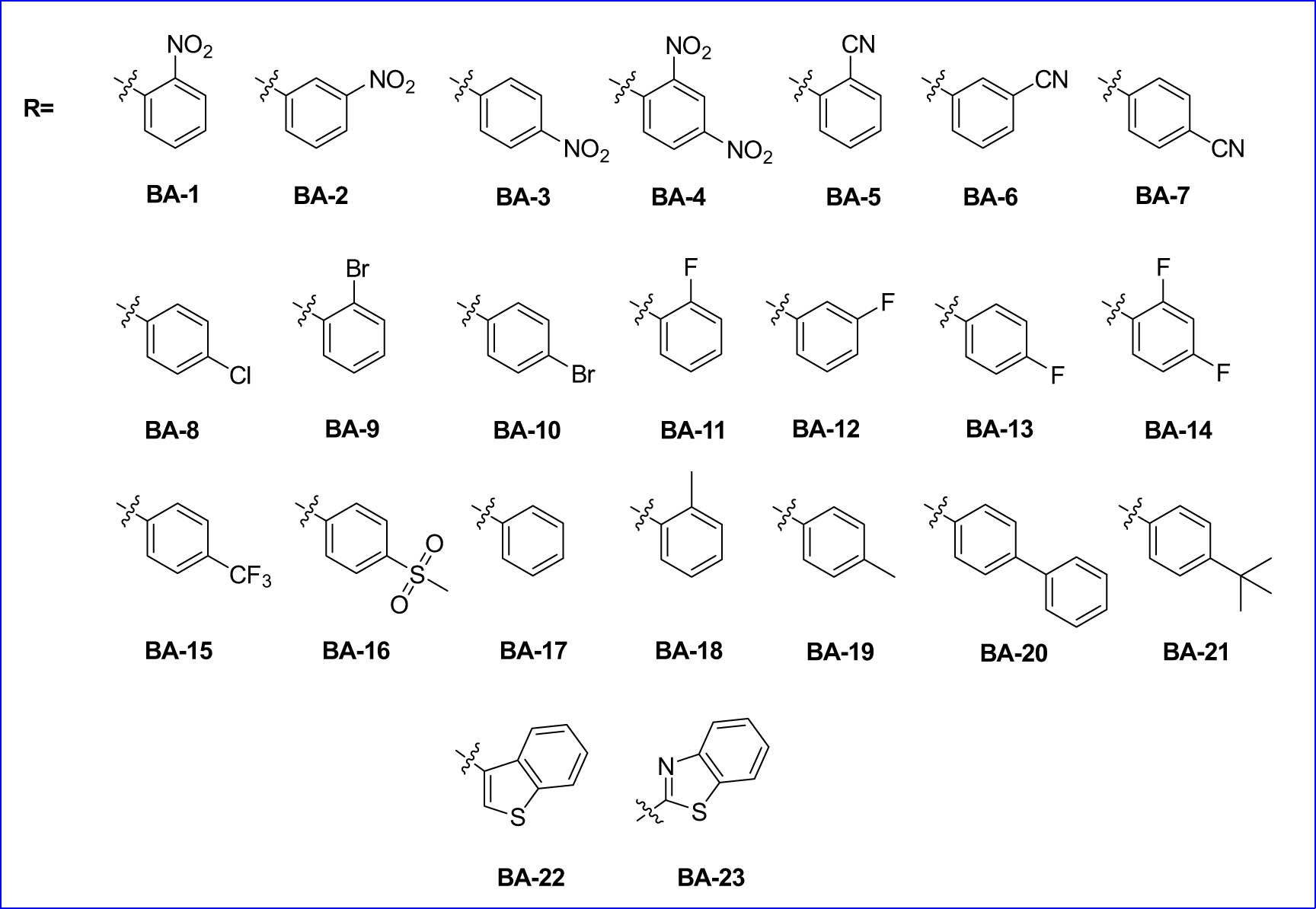
Structures of substrate scope for target compounds.
After the synthesis of Allosecurinine derivatives, antitumor activities in vitro and mechanism studies exhibited that these compounds could induce mitochondrial membrane potential within cancer cells as well as decrease the levels of antiapoptotic protein and induce apoptosis in leukemia cell lines. Interestingly, compound BA-3 proved to be the best among Allosecurinine derivatives to induce cell death through apoptosis by analysis using FCM technology as well as the high activity against tumor activities. To the best of the author's knowledge, this is the first example of the synthesis and development of Allosecurinine derivatives in the discovery of anticancer drugs, especially for leukemia.
Recently, a new study has been published, which focused on design, synthesis, and antiproliferative activities for a series of thiazolyl pyrazole hybrid derivatives as novel heterocyclic compound-based antitumor agents [254]. The antiproliferative effects of the target compounds were biologically evaluated, and the structure–activity relationship (SAR) was tested against cancer and normal cells by using an MTT assay. Initially, the synthetic approach of the target compounds was conducted via conversion of substituted acetophenone under Vilsmeier Haack conditions (POCl3, DMF at 80°C for 1 h) to generate the target aldehyde compounds in high yield by the neutralization reaction (Scheme 2).

Total synthesis scheme of the target compounds.
On the other hand, the final product was studied further by converting the aldehyde functional group in the target compounds into primary alcohol to examine the antitumor reactivity of these compounds by using NaBH4 as a reducing agent (Scheme 3).
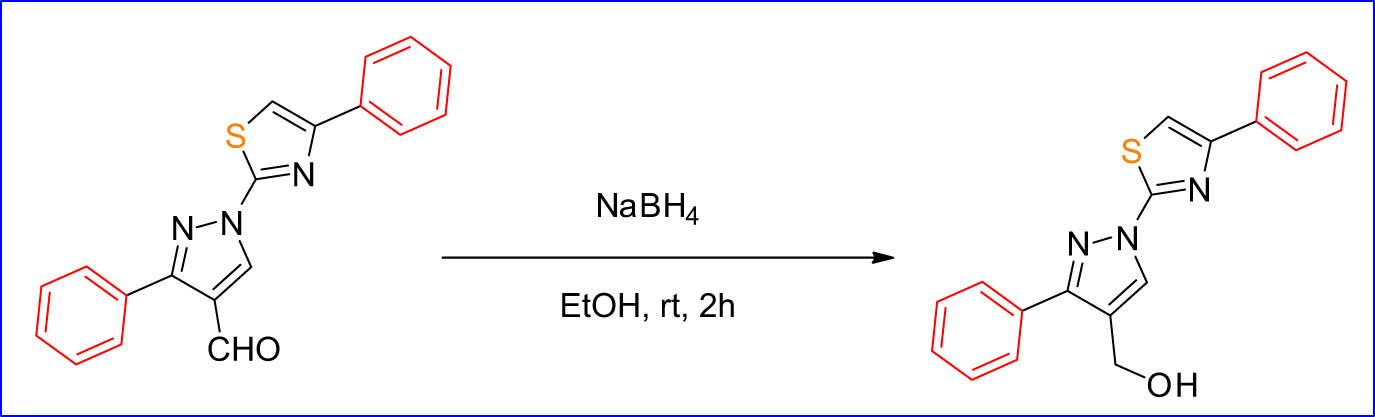
NaBH4 as a reducing agent.
The target compounds were investigated by 1H-NMR and 13C-NMR spectroscopy. The functional group of aldehydes clearly disappeared and reduced to –OH proton. Furthermore, the compounds under study had a higher antiproliferative effect in the cancer cells. Remarkably, the targeted compounds appear to have a pharmacokinetic property, which can make them a potent drug candidate after successful molecular modifications.
3 Conclusion
The chemistry of nitrogen-, oxygen-, and sulfur-containing monocyclic, polynuclear, and heterocyclic compounds have a significant role in biological activities and prospects for medicinal chemistry. Cancer is considered the most common cause of death across the world. The time from the past up to the present has witnessed a significant development in the field of cancer therapy by the development of innovative delivery technologies. Many cellular and molecular pathways growth in the sector of the treatment of cancer have been identified. Regrettably, all recently obtainable anticancer medications had flaws like undesirable effects and refusal to accept. In the current research, the most potent and privileged-based drugs are highlighted, which we believe are the most successful treatment tools in the fight of current years. These compounds have been divided into ten groups based on similarities in their chemical structures. The use of chemicals has been the most effective treatment approach in recent years. They show promise as candidates for the discovery and development of cancer drugs. This work hopefully presents assistance for chemists in medicinal areas to obtain a powerful design and development of new, strong anticancer drugs.
Acknowledgments
The author Dr. Mohammed Hadi Ali Al-Jumaili would like to express his special appreciation to University of Fallujah, College of Applied Science. Mohammed Hadi Ali Al-Jumaili, PhD. Department of Applied Chemistry, University of Fallujah, Al-Anbar, Iraq.
-
Funding information: Authors state no funding involved.
-
Author contributions: The authors contributed to the conceptualization and writing of the original draft. Mohammed Hadi Ali Al-Jumaili, reviewed, edited, and approved the final version. The authors have read and agreed to the published version of the article.
-
Conflict of interest: Authors state no conflict of interest.
-
Data availability statement: Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
References
[1] Mermer A, Keles T, Sirin Y. Recent studies of nitrogen containing heterocyclic compounds as novel antiviral agents: A review. Bioorg Chem. 2021;114:105076.10.1016/j.bioorg.2021.105076Search in Google Scholar PubMed
[2] Dawood RS, Hamed AS. Synthesis and characterization of new 1, 3-benzodioxole derivatives based on Suzuki-Miyaura coupling reaction. Res J Chem Env. 2019;23:14–21.Search in Google Scholar
[3] Hussein MA, Al‐Jumaili MHA, Sabi AA, Abdulhussein HA. Indium‐catalyzed direct amidation reaction of carboxylic acids and in silico study for screening the activity of potential therapeutics of the synthesized products. ChemistrySelect. 2024;9(37):e202403097.10.1002/slct.202403097Search in Google Scholar
[4] Al-Jumaili MHA, Hamad AA, Hashem HE, Hussein AD, Muhaidi MJ, Ahmed MA, et al. Comprehensive review on the Bis–heterocyclic compounds and their anticancer efficacy. J Mol Struct. 2023;1271:133970.10.1016/j.molstruc.2022.133970Search in Google Scholar
[5] Devi MM, Devi KS, Singh OM, Singh TP. Synthesis of imidazole derivatives in the last 5 years: An update. Heterocycl Commun. 2024;30(1):20220173.10.1515/hc-2022-0173Search in Google Scholar
[6] Nussbaumer S, Bonnabry P, Veuthey JL, Fleury-Souverain S. Analysis of anticancer drugs: a review. Talanta. 2011;85(5):2265–89. 10.1016/j.talanta.2011.08.034.Search in Google Scholar PubMed
[7] Al-Jumaili M, Siddique F, Abul Qais F, Hashem HE, Chtita S, Rani A, et al. Analysis and prediction pathways of natural products and their cytotoxicity against HeLa cell line protein using docking, molecular dynamics and ADMET. J Biomol Struct Dyn. 2023;41:765–77.10.1080/07391102.2021.2011785Search in Google Scholar PubMed
[8] Li J, Li L, Liu Y, Zhang J, Shi C, Zhou S, et al. Design, synthesis, and anticancer activity of novel 4, 6-dimorpholinyl-1, 3, 5-triazine compounds. Heterocycl Commun. 2023;29(1):20220152.10.1515/hc-2022-0152Search in Google Scholar
[9] Millimouno FM, Dong J, Yang L, Li J, Li X. Targeting apoptosis pathways in cancer and perspectives with natural compounds from mother nature. Cancer Prev Res. 2014;7(11):1081–107. 10.1158/1940-6207.CAPR-14-0136.Search in Google Scholar PubMed
[10] Kruk J, Aboul-Enein BH, Bernstein J, Gronostaj M. Psychological stress and cellular aging in cancer: a meta-analysis. Oxid Med Cell Longev. 2019;2019:1270397.10.1155/2019/1270397Search in Google Scholar PubMed PubMed Central
[11] The International Agency for Research on Cancer. Available online: https://www.who.int/cancer/PRGlobocanFinal.pdf (accessed on 11 September 2019.Search in Google Scholar
[12] Hassan AS, Hame AS. Study of antimicrobial activity of new prepared seven membered rings (Oxazepine). Res J Biotechnol. 2019;14(1):109–18.Search in Google Scholar
[13] Sophy MA, Reheim MA. Bis (2-cyanoacetohydrazide) as precursors for synthesis of novel azoles/azines and their biological evaluation. Heterocycl Commun. 2023;29(1):20220167.10.1515/hc-2022-0167Search in Google Scholar
[14] Espinosa E, Zamora P, Feliu J, Barón MG. Classification of anticancer drugs – a new system based on therapeutic targets. Cancer Treat Rev. 2003;29(6):515–23. 10.1016/S0305-7372(03)00116-6 Search in Google Scholar
[15] Thurston DE. Chemistry and pharmacology of anticancer drugs. Boca Raton, Florida, USA: CRC Press; 2006.10.1201/9781420008906Search in Google Scholar
[16] Al-Jumaili MH, Al Hdeethi MK. Study of selected flavonoid structures and their potential activity as breast anticancer agents. Cancer Inform. 2021;20:11769351211055160.10.1177/11769351211055160Search in Google Scholar PubMed PubMed Central
[17] Taşkın-Tok T, Gowder S. Anticancer drug – friend or foe. Pharmacol Ther. 2014;253.10.5772/58552Search in Google Scholar
[18] Mugada V, Paruchuri A, Munagala M. Drug utilization evaluation of anticancer drugs in a tertiary care teaching hospital: A descriptive observational study. J App Pharm Sci. 2016;6:98–101. 10.7324/JAPS.2016.601013.Search in Google Scholar
[19] DeVita VT, Lawrence TS, Rosenberg SA, DePinho RA, Weinberg RA. Principles and practice of oncology. 8th edn. Philadelphia, PA: Lippincott-Raven; 2008.Search in Google Scholar
[20] Izbicka E, Tolcher AW. Development of novel alkylating drugs as anticancer agents. Curr Opin Invest Drugs (London, England: 2000). 2004;5(6):587.Search in Google Scholar
[21] Bharti AC, Vishnoi K, Singh SM, Aggarwal BB. Pathways linked to cancer chemoresistance and their targeting by nutraceuticals. Role of nutraceuticals in cancer chemosensitization. Cambridge, Massachusetts, USA: Academic Press; 2018. p. 1–30. 10.1016/B978-0-12-812373-7.00001-2.Search in Google Scholar
[22] Hall AG, Tilby MJ. Mechanisms of action of, and modes of resistance to, alkylating agents used in the treatment of haematological malignancies. Blood Rev. 1992;6(3):163–73. 10.1016/0268-960X(92)90028-O.Search in Google Scholar PubMed
[23] Flombaum CD. Nephrotoxicity of chemotherapy agents and chemotherapy administration in patients with renal disease. Cancer and the kidney: the frontier of nephrology and oncology. Oxford (United Kingdom): Oxford University Press; 2011. p. 115–76.10.1093/med/9780199580194.003.0005Search in Google Scholar
[24] Gustafson DL, Page RL. Cancer chemotherapy. Withrow and MacEwen’s small animal clinical oncology. St. Louis, Missouri, USA: Elsevier Saunders; 2013. p. 157–79.Search in Google Scholar
[25] Dixit P, Jeyaseelan C, Gupta D. Nitrogen mustard: a promising class of anti-cancer chemotherapeutics–a review. Biointerface Res Appl Chem. 2022;13:135–61.10.33263/BRIAC132.161Search in Google Scholar
[26] Ogino MH, Tadi P. Cyclophosphamide. StatPearls [Internet]. Treasure Island, Florida, USA: StatPearls Publishing; 2020.Search in Google Scholar
[27] Bagley Jr CM, Bostick FW, DeVita Jr VT. Clinical pharmacology of cyclophosphamide. Cancer Res. 1973;33(2):226–33.Search in Google Scholar
[28] Moore MJ. Clinical pharmacokinetics of cyclophosphamide. Clin Pharmacokinet. 1991;20(3):194–208.10.2165/00003088-199120030-00002Search in Google Scholar PubMed
[29] Bramwell V, Calvert RT, Edwards G, Scarffe H, Crowther D. The disposition of cyclophosphamide in a group of myeloma patients. Cancer Chemother Pharmacol. 1979;3(4):253–9.10.1007/BF00254741Search in Google Scholar PubMed
[30] Bassan R, Fumagalli M, Chiaretti S, Audisio E, Cascavilla N, Paolini S, et al. Phase II trial with sequential clofarabine and cyclophosphamide for refractory and relapsed philadelphia-negative adult acute lymphoblastic leukemia. Results of the GIMEMA LAL 1610 protocol. Leukemia Lymphoma. 2019;60(14):3482–92.10.1080/10428194.2019.1639170Search in Google Scholar PubMed
[31] Burkard ME, Wisinski KB, Njiaju UO, Donohue S, Hegeman R, Stella A, et al. Feasibility of 4 cycles of docetaxel and cyclophosphamide every 14 days as an adjuvant regimen for breast cancer: a Wisconsin Oncology Network study. Clin Breast Cancer. 2014;14(3):205–11.10.1016/j.clbc.2013.10.018Search in Google Scholar PubMed PubMed Central
[32] Rivera DR, Grothen A, Ohm B, McNeel TS, Brennan S, Lam C, et al. Utilization of the cancer medications enquiry database (CanMED)-national drug codes (NDC): Assessment of systemic breast cancer treatment patterns. JNCI Monogr. 2020;55(2020):46–52.10.1093/jncimonographs/lgaa002Search in Google Scholar PubMed PubMed Central
[33] Clowse MB, McCune WJ. General toxicity of cyclophosphamide in rheumatic diseases. Waltham, Massachusetts, USA: Wolters Kluwer; 2020.Search in Google Scholar
[34] Munyangango EM, Le Roux-Villet C, Doan S, Pascal F, Soued I, Alexandre M, et al. Oral cyclophosphamide without corticosteroids to treat mucous membrane pemphigoid. Br J Dermatol. 2013;168(2):381–90.10.1111/bjd.12041Search in Google Scholar PubMed
[35] Rosa Neto NS, Goldenstein-Schainberg C. Dermatomiositejuvenil: revisão e atualizaçãoempatogênese e tratamento. RevistaBrasileira de Reumatologia. 2010;50(3):299–312.10.1590/S0482-50042010000300010Search in Google Scholar
[36] Prendergast GC, Jaffee EM, eds. Cancer immunotherapy: immune suppression and tumor growth. Cambridge, Massachusetts, USA: Academic Press; 2013.Search in Google Scholar
[37] Lee HZ, Miller BW, Kwitkowski VE, Ricci S, DelValle P, Saber H, et al. US Food and drug administration approval: obinutuzumab in combination with chlorambucil for the treatment of previously untreated chronic lymphocytic leukemia. Clin Cancer Res. 2014;20(15):3902–7.10.1158/1078-0432.CCR-14-0516Search in Google Scholar PubMed
[38] Begleiter A, Mowat M, Israels LG, Johnston JB. Chlorambucil in chronic lymphocytic leukemia: mechanism of action. Leukemia Lymphoma. 1996;23(3–4):187–201.10.3109/10428199609054821Search in Google Scholar PubMed
[39] Winkfield KM, Tsang RW, Gospodarowicz MK. Non-Hodgkin’s Lymphoma. Clinical radiation oncology. Philadelphia, Pennsylvania, USA: Elsevier; 2016. p. 1524–46.10.1016/B978-0-323-24098-7.00077-0Search in Google Scholar
[40] Delgado J, Villamor N, Nadeu F, Campo E. Chronic Lymphocytic Leukemia: Pathology and Genetics. Barcelona: Haematologica Young Authors’ Award; 2019. p. 405–17.Search in Google Scholar
[41] Rogers KA, Huang Y, Ruppert AS, Awan FT, Heerema NA, Hoffman C, et al. Phase 1b study of obinutuzumab, ibrutinib, and venetoclax in relapsed and refractory chronic lymphocytic leukemia. Blood. 2018;132(15):1568–72.10.1182/blood-2018-05-853564Search in Google Scholar PubMed PubMed Central
[42] Awan FT, Byrd JC. Chronic lymphocytic leukemia. Abeloff’s Clinical Oncology. Content Repository Only. 2020;6:1850–71.10.1016/B978-0-323-47674-4.00099-2Search in Google Scholar
[43] Ponticelli C, Glassock RJ. Prevention of complications from use of conventional immunosuppressants: a critical review. J Nephrol. 2019;32:1–20.10.1007/s40620-019-00602-5Search in Google Scholar PubMed
[44] Furlanut M, Franceschi L. Pharmacology of ifosfamide. Oncology. 2003;65(Suppl. 2):2–6.10.1159/000073350Search in Google Scholar PubMed
[45] Kuhar MB, Kuhar BM. Evaluation of selected new drugs: 1988. AAOHN J. 1989;37(10):428–33.10.1177/216507998903701005Search in Google Scholar
[46] Gangireddy M, Nookala. V. Ifosfamide. StatPearls [Internet]. Treasure Island, Florida, USA: StatPearls Publishing; 2019.Search in Google Scholar
[47] Najafi T. Chemotherapy-induced oxidative stress and infertility. Int J Womens Health Reprod Sci. 2017;5(2):80–1.10.15296/ijwhr.2017.15Search in Google Scholar
[48] Hatamluyi B, Lorestani F, Es’haghi Z. Au/Pd@ rGO nanocomposite decorated with poly (L-Cysteine) as a probe for simultaneous sensitive electrochemical determination of anticancer drugs, Ifosfamide and Etoposide. Biosens Bioelectron. 2018;120:22–9.10.1016/j.bios.2018.08.008Search in Google Scholar PubMed
[49] Oprea AD. Chemotherapy agents with known pulmonary side effects and their anesthetic and critical care implications. J Cardiothorac Vasc Anesth. 2017;31(6):2227–35.10.1053/j.jvca.2015.06.019Search in Google Scholar PubMed
[50] Williams D, Crofton PM, Levitt G. Does ifosfamide affect gonadal function? Pediatr Blood Cancer. 2008;50(2):347–51.10.1002/pbc.21323Search in Google Scholar PubMed
[51] Conklin KA. Chemotherapy-associated oxidative stress: impact on chemotherapeutic effectiveness. Integr Cancer Ther. 2004;3(4):294–300.10.1177/1534735404270335Search in Google Scholar PubMed
[52] Mirkamali ES, Ahmadi R, Kalateh K, Zarei G. Adsorption of melphalan anticancer drug on the surface of fullerene (C24): a comprehensive DFT study. Nanomed J. 2019;6(2):112–9.Search in Google Scholar
[53] Krasnov VP, Korolyova MA, Vodovozova E. Nano-sized melphalan and sarcolysine drug delivery systems: synthesis and prospects of application. Russian Chem Rev. 2013;82(8):783.10.1070/RC2013v082n08ABEH004358Search in Google Scholar
[54] Cook G, Morris CT. Evolution or revolution in multiple myeloma therapy and the role of the UK. Br J Haematol. 2020;191(4):542–51.10.1111/bjh.17148Search in Google Scholar PubMed
[55] Aljitawi OS, Ganguly S, Abhyankar SH, Ferree M, Marks R, Pipkin JD, et al. Phase IIa cross-over study of propylene glycol-free melphalan (LGD-353) and alkeran in multiple myeloma autologous transplantation. Bone Marrow Transplant. 2014;49(8):1042–5.10.1038/bmt.2014.120Search in Google Scholar PubMed
[56] Esma F, Salvini M, Troia R, Boccadoro M, Larocca A, Pautasso C. Melphalan hydrochloride for the treatment of multiple myeloma. Expert Opin Pharmacother. 2017;18(11):1127–36.10.1080/14656566.2017.1349102Search in Google Scholar PubMed
[57] Weber GF. DNA damaging drugs. Molecular therapies of cancer. Cham: Springer; 2015. p. 9–112.10.1007/978-3-319-13278-5_2Search in Google Scholar
[58] Mateos MV, Cavo M, Blade J, Dimopoulos MA, Suzuki K, Jakubowiak A, et al. Overall survival with daratumumab, bortezomib, melphalan, and prednisone in newly diagnosed multiple myeloma (ALCYONE): a randomised, open-label, phase 3 trial. Lancet. 2020;395(10218):132–41.10.1016/S0140-6736(19)32956-3Search in Google Scholar PubMed
[59] Duan J, Chen L, Tu J, Cao L, Xiao X. Folate-grafted glycyl-glycine-melphalan conjugate self-assembled amphilphilcnanomicelles augmented drug delivery, cytotoxicity and cellular uptake in human ovarian cancer cells. J Microencapsul. 2020;3:1–30.Search in Google Scholar
[60] de Paula Eduardo F, Bezinelli LM, da Graça Lopes RM, Nascimento Sobrinho JJ, Hamerschlak N, Correa L. Efficacy of cryotherapy associated with laser therapy for decreasing severity of melphalan‐induced oral mucositis during hematological stem‐cell transplantation: a prospective clinical study. Hematological Oncol. 2015;33(3):152–8.10.1002/hon.2133Search in Google Scholar PubMed
[61] Magalhaes LG, Ferreira LL, Andricopulo AD. Recent advances and perspectives in cancer drug design. An da Acad Brasileira de Ciências. 2018;90(1):1233–50.10.1590/0001-3765201820170823Search in Google Scholar PubMed
[62] Silverman RB, Holladay MW. The organic chemistry of drug design and drug action. Cambridge, Massachusetts, USA: Academic Press; 2014.Search in Google Scholar
[63] Collins JM. Cancer pharmacology. Abeloff's Clinical Oncology. Elsevier; 2020. p. 411–9.10.1016/B978-0-323-47674-4.00025-6Search in Google Scholar
[64] Chu E, DeVita Jr VT. Physicians’ cancer chemotherapy drug manual 2020. Burlington, Massachusetts, USA: Jones & Bartlett Learning; 2019.Search in Google Scholar
[65] Wei L, Lei J, Luo T, Wu L. Influence of decoration with Si and Ge atoms on the sensitivity of boron nitride nanocone towards temozolomide anticancer drugs. Struct Chem. 2020;31(6):2321–31.10.1007/s11224-020-01576-ySearch in Google Scholar
[66] Arevalo OJ, Valenzuela R, Esquenazi Y, Rao M, Tran B, Zhu J, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a practical approach for gliomas, part 1. Basic tumor genetics. Neurographics. 2017;7(5):334–43.10.3174/ng.9170230Search in Google Scholar
[67] Hu C, Lin Q, Liu C, Liu J, Chen X, Li X, et al. Bioequivalence study of 20-mg and 100-mg temozolomide capsules (TOZ309 and Temodal®) in glioma patients in China. Cancer Chemother Pharmacol. 2020;86(6):793–801.10.1007/s00280-020-04175-0Search in Google Scholar PubMed
[68] Teraiya M, Perreault H, Chen VC. An overview of glioblastoma multiforme and temozolomide resistance: can LC-MS-based proteomics reveal the fundamental mechanism of temozolomide resistance? Front Oncol. 2023;13:1166207.10.3389/fonc.2023.1166207Search in Google Scholar PubMed PubMed Central
[69] Lee SY. Temozolomide resistance in glioblastoma multiforme. Genes & Dis. 2016;3(3):198–210.10.1016/j.gendis.2016.04.007Search in Google Scholar PubMed PubMed Central
[70] Strobel H, Baisch T, Fitzel R, Schilberg K, Siegelin MD, Karpel-Massler G, et al. Temozolomide and other alkylating agents in glioblastoma therapy. Biomedicines. 2019;7(3):69.10.3390/biomedicines7030069Search in Google Scholar PubMed PubMed Central
[71] Campana D, Walter T, Pusceddu S, Gelsomino F, Graillot E, Prinzi N, et al. Correlation between MGMT promoter methylation and response to temozolomide-based therapy in neuroendocrine neoplasms: an observational retrospective multicenter study. Endocrine. 2018;60(3):490–8.10.1007/s12020-017-1474-3Search in Google Scholar PubMed
[72] Massoud M, Armand JP, Ribrag V. Procarbazine in haematology: an old drug with a new life? Eur J Cancer. 2004;40(13): 1924–7.10.1016/j.ejca.2004.05.015Search in Google Scholar PubMed
[73] Bassem T, Salem P, Petruska PJ. Procarbazine for non-Hodgkin’s lymphoma. Leukemia Lymphoma. 2006;47(4):637–40.10.1080/10428190600591517Search in Google Scholar PubMed
[74] ProcarbazineIn Meyler’s Side Effects of Drugs (Sixteenth Edition); 2016.Search in Google Scholar
[75] Respiratory Toxicology K.Y. Yoneda, C.E. Cross, in Comprehensive Toxicology; 2010.Search in Google Scholar
[76] Kwok KK, Vincent EC, Gibson JN. Antineoplastic drugs. Pharmacology and therapeutics for dentistry. Chapter. 2017;2017:530–62.Search in Google Scholar
[77] Macdonald JS, Hoth D, Schein PS. Preclinical and clinical studies on chlorozotocin, a new nitrosourea with decreased bone marrow toxicity. New anticancer drugs. Berlin, Heidelberg: Springer; 1980. p. 83–9.10.1007/978-3-642-81392-4_9Search in Google Scholar PubMed
[78] Birudukota N, Mudgal MM, Shanbhag V. Discovery and development of azasteroids as anticancer agents. Steroids. 2019;152:108505.10.1016/j.steroids.2019.108505Search in Google Scholar PubMed
[79] Trafalis D, Dalezis P, Geromichalou E, Sagredou S, Sflakidou E, Voura M, et al. Discovery of steroidal lactam conjugates of POPAM-NH2 with potent anticancer activity. Future Med Chem. 2020;12(1):19–35.10.4155/fmc-2019-0255Search in Google Scholar PubMed
[80] Toale KM, Johnson TN, Ma MQ. Chemotherapy-induced toxicities. Oncologic emergency medicine. Cham: Springer; 2016. p. 381–406.10.1007/978-3-319-26387-8_33Search in Google Scholar
[81] Vardanyan R, Hruby V. Synthesis of best-seller drugs. Cambridge, Massachusetts, USA: Academic press; 2016.Search in Google Scholar
[82] McCulloh RJ. Infections associated with biologics. Infectious diseases. Philadelphia, Pennsylvania, USA: Elsevier; 2017. p. 1377–81.10.1016/B978-0-7020-6285-8.00159-3Search in Google Scholar
[83] Schofield RC, Landau HJ, Giralt SA, Shah GL, Scordo M, Lin A, et al. Measurement of the DNA alkylating agents busulfan and melphalan in human plasma by mass spectrometry. J Chromatogr B. 2019;1125:121711.10.1016/j.jchromb.2019.121711Search in Google Scholar PubMed PubMed Central
[84] Patel R, Tadi P. Busulfan. StatPearls [Internet]. Treasure Island, Florida, USA: StatPearls Publishing; 2020.Search in Google Scholar
[85] Karthick T, Tandon P. Computational approaches to find the active binding sites of biological targets against busulfan. J Mol Model. 2016;22(6):142.10.1007/s00894-016-3015-zSearch in Google Scholar PubMed
[86] Ruutu T, van der Werf S, van Biezen A, Backman JT, Peczynski C, Kröger N, et al. Use of busulfan in conditioning for allogeneic hematopoietic stem cell transplantation in adults: a survey by the Transplant Complications Working Party of the EBMT. Bone Marrow Transplant. 2019;54(12):2013–9.10.1038/s41409-019-0579-0Search in Google Scholar PubMed
[87] Avgeropoulos NG, Newton HB. Clinical pharmacology of brain tumor chemotherapy. Handbook of brain tumor chemotherapy, molecular therapeutics, and immunotherapy. Cambridge, Massachusetts, USA: Academic Press; 2018. p. 21–44.10.1016/B978-0-12-812100-9.00002-4Search in Google Scholar
[88] Dedeaux AM, Flesner BK, Reinhart JM, Langohr IM, Husnik R, Geraci SN, et al. Biochemical, functional, and histopathologic characterization of lomustine-induced liver injury in dogs. Am J Vet Res. 2020;81(10):810–20.10.2460/ajvr.81.10.810Search in Google Scholar PubMed
[89] Green synthesis and biological evaluation of anticancer drugsBimal Krishna Banik, Biswa Mohan Sahoo, in Green Approaches in Medicinal Chemistry for Sustainable Drug Design; 2020.Search in Google Scholar
[90] Marosi C. Complications of chemotherapy in neuro-oncology. Handbook of clinical neurology. Vol. 105, Philadelphia, Pennsylvania, USA: Elsevier; 2012. p. 873–85.10.1016/B978-0-444-53502-3.00029-XSearch in Google Scholar PubMed
[91] Hay JK, Larson VS. Lomustine (CCNU) and prednisone chemotherapy for high-grade completely excised canine mast cell tumors. Can Vet J. 2019;60(12):1326.Search in Google Scholar
[92] Jilani K, Lang F. Carmustine-induced phosphatidylserine translocation in the erythrocyte membrane. Toxins. 2013;5(4):703–16.10.3390/toxins5040703Search in Google Scholar PubMed PubMed Central
[93] Hadidi S, Shiri F, Norouzibazaz M. A DFT study of the degradation mechanism of anticancer drug carmustine in an aqueous medium. Struct Chem. 2019;30(4):1315–21.10.1007/s11224-019-1285-7Search in Google Scholar
[94] Zhuang L, Gao J, Zeng Y, Yu F, Zhang B, Li M, et al. HPLC method validation for the quantification of lomustine to study pharmacokinetics of thermosensitive liposome-encapsulated lomustine containing iohexol for CT imaging in C6 glioma rats. Eur J Drug Metab Pharmacokinet. 2011;36(2):61–9.10.1007/s13318-011-0030-4Search in Google Scholar PubMed
[95] Sukumari-Ramesh S, Prasad N, Alleyne CH, Vender JR, Dhandapani KM. Overexpression of Nrf2 attenuates Carmustine-induced cytotoxicity in U87MG human glioma cells. BMC Cancer. 2015;15(1):118.10.1186/s12885-015-1134-zSearch in Google Scholar PubMed PubMed Central
[96] Hadidi S, Shiri F, Norouzibazaz M. Conversion mechanism and isomeric preferences of the cis and trans isomers of anti-cancer medicine carmustine; a double hybrid dft calculation. Chem Phys. 2019;522:39–43.10.1016/j.chemphys.2019.02.013Search in Google Scholar
[97] Puyo S, Montaudon D, Pourquier P. From old alkylating agents to new minor groove binders. Crit Rev Oncol/Hematol. 2014;89(1):43–61.10.1016/j.critrevonc.2013.07.006Search in Google Scholar PubMed
[98] Türker L. Interaction of carmustine tautomers with adenine-DFT study. Earthline J Chem Sci. 2020;5(1):63–76.10.34198/ejcs.5121.6376Search in Google Scholar
[99] Baldo BA, Pham NH, Baldo BA, Pham NH. Non-targeted drugs for cancer therapy. Drug allergy. Cham: Springer; p. 645–82.10.1007/978-3-030-51740-3_15Search in Google Scholar
[100] Okusaka T, Ueno H, Morizane C, Kondo S, Sakamoto Y, Takahashi H, et al. Cytotoxic chemotherapy for pancreatic neuroendocrine tumors. J Hepato‐Biliary‐Pancreatic Sci. 2015;22(8):628–33.10.1002/jhbp.257Search in Google Scholar PubMed
[101] Deshpande AS, Ramireddy S, Sudandiradoss C, Noor A, Sen P. Streptozocin; a GLUT2 binding drug, interacts with human serum albumin at loci h6DOM3-h7DOM3. Int J Biol Macromol. 2019;128:923–33.10.1016/j.ijbiomac.2019.01.217Search in Google Scholar PubMed
[102] Thakare R, Kaul G, Shukla M, Kesharwani P, Srinivas N, Dasgupta A, et al. Repurposing nonantibiotic drugs as antibacterials. Drug discovery targeting drug-resistant bacteria. Cambridge, Massachusetts, USA: Academic Press; 2020. p. 105–38.10.1016/B978-0-12-818480-6.00005-9Search in Google Scholar
[103] Abdollahi M, Hosseini A. Streptozotocin. Cambridge, Massachusetts, USA: Academic Press; 2014. p. 402–4.10.1016/B978-0-12-386454-3.01170-2Search in Google Scholar
[104] Rubach M. Management of extravasation of antineoplastic agents. Oncol Clin Pract. 2018;14(1):15–22.Search in Google Scholar
[105] Apich MG. Saunders handbook of veterinary drugs-e-book: small and large animal. Philadelphia, Pennsylvania, USA: Elsevier Health Sciences; 2015.Search in Google Scholar
[106] Kim SI, Choo SH. Intravesical chemotherapy. Bladder Cancer. 2018;19:263–76. 10.1016/B978-0-12-809939-1.00019-9.Search in Google Scholar
[107] Torabifard H, Fattahi A. DFT study on Thiotepa and Tepa interactions with their DNA receptor. Struct Chem. 2013;24(1):1–11.10.1007/s11224-012-0020-4Search in Google Scholar
[108] Avendaño C, Menendez JC. Medicinal chemistry of anticancer drugs. Amsterdam, Netherlands: Elsevier; 2015.Search in Google Scholar
[109] Beilke LD. Thiotepa. Encyclopedia of toxicology. Cambridge, Massachusetts, USA: Academic Press; 2014. p. 551–2. 10.1016/B978-0-12-386454-3.00200-1.Search in Google Scholar
[110] Eder S, Labopin M, Finke J, Bunjes D, Olivieri A, Santarone S, et al. Safety and efficacy of thiotepa-based conditioning for allogeneic transplantation in AML: a survey from the ALWP of the EBMT. Bone Marrow Transplant. 2017;52(2):238–44.10.1038/bmt.2016.239Search in Google Scholar PubMed
[111] Modh RP, Patel AC, Chikhalia KH. Design, synthesis, antibacterial, and antifungal studies of novel 3-substituted coumarinyl-triazine derivatives. Heterocycl Commun. 2013;19(5):343–9.10.1515/hc-2013-0104Search in Google Scholar
[112] Cun B, Song X, Jia R, Zhao X, Wang H, Ge S, et al. Combination of oncolytic adenovirus and dacarbazine enhances antitumor ability against uveal melanoma cells via cell cycle block. Cancer Biol Ther. 2012;13(2):77–84.10.4161/cbt.13.2.18436Search in Google Scholar PubMed
[113] Luke JJ, Schwartz GK. Chemotherapy in the management of advanced cutaneous malignant melanoma. ClDermatology. 2013;31(3):290–7.10.1016/j.clindermatol.2012.08.016Search in Google Scholar PubMed PubMed Central
[114] Nabiuni M, Yazadanian Z, Amirpour Asl S, Dorazehi F, Karimzadeh-Bardei L. Anti-cancereffects of 1, 3-bis (2-ethoxyphenyl) on 4T1 breast cancer cell line, an in-vivostudy. Stud Med Sci. 2020;30(12):1016–24.Search in Google Scholar
[115] Chabner BA, Longo DL, eds. Cancer chemotherapy, immunotherapy, and biotherapy: principles and practice. Philadelphia, Pennsylvania, USA: Wolters Kluwer; 2019.Search in Google Scholar
[116] Milijašević B, Stefanović D, Lalić-Popović M, Tomić Z, Kolarović J, Lalošević D, et al. Acute toxic effects of single dose dacarbazine: hematological and histological changes in an animal model. Biotech Histochem. 2014;89(8):583–90.10.3109/10520295.2014.918653Search in Google Scholar PubMed
[117] Thirumaran R, Prendergast GC, Gilman PB. Cytotoxic chemotherapy in clinical treatment of cancer. Cancer immunotherapy. Cambridge, Massachusetts, USA: Academic Press; 2007. p. 101–16. 10.1016/B978-012372551-6/50071-7.Search in Google Scholar
[118] Peters GJ. Novel developments in the use of antimetabolites. Nucleosides Nucleotides Nucleic Acids. 2014;33(4–6):358–74.10.1080/15257770.2014.894197Search in Google Scholar PubMed
[119] Bezabeh S, Mackey AC, Kluetz P, Jappar D, Korvick J. Accumulating evidence for a drug–drug interaction between methotrexate and proton pump inhibitors. Oncologist. 2012;17(4):550.10.1634/theoncologist.2011-0431Search in Google Scholar PubMed PubMed Central
[120] Czarnecka-Operacz M, Sadowska-Przytocka A. The possibilities and principles of methotrexate treatment of psoriasis–the updated knowledge. Adv Dermatology Allergology/Postȩpy Dermatologii i Alergologii. 2014;31(6):392.10.5114/pdia.2014.47121Search in Google Scholar PubMed PubMed Central
[121] Pontinha ADR, Jorge SMA, Diculescu VC, Vivan M, Oliveira‐Brett AM. Antineoplasic drug methotrexate redox mechanism using a glassy carbon electrode. Electroanalysis. 2012;24(4):917–23. 10.1002/elan.201100558.Search in Google Scholar
[122] Nedelcu RI, Balaban M, Turcu G, Brinzea A, Ion DA, Antohe M, et al. Efficacy of methotrexate as anti‑inflammatory and anti‑proliferative drug in dermatology: Three case reports. Exp Ther Med. 2019;18(2):905–10.10.3892/etm.2019.7511Search in Google Scholar PubMed PubMed Central
[123] Reid MA, Sanderson SM, Locasale JW. Cancer metabolism. In Abeloff’s clinical oncology. Philadelphia, Pennsylvania, USA: Content Repository Only; 2020. p. 127–38.10.1016/B978-0-323-47674-4.00009-8Search in Google Scholar
[124] Mahbub A, Le Maitre C, Haywood-Small S, Cross N, Jordan-Mahy N. Dietary polyphenols influence antimetabolite agents: methotrexate, 6-mercaptopurine and 5-fluorouracil in leukemia cell lines. Oncotarget. 2017;8(62):104877–93. 10.18632/oncotarget.20501.Search in Google Scholar PubMed PubMed Central
[125] Hagner N, Joerger M. Cancer chemotherapy: targeting folic acid synthesis. Cancer Manag Res. 2010;2:293–301.10.2147/CMAR.S10043Search in Google Scholar
[126] Gaies E, Jebabli N, Trabelsi S, Salouage I, Charfi R, Lakhal M, et al. Methotrexate side effects: review article. J Drug Metab Toxicol. 2012;3(4):1–5.10.4172/2157-7609.1000125Search in Google Scholar
[127] Pan Z, Yang G, Cui J, Li W, Li Y, Gao P, et al. A pilot phase 1 study of intrathecal pemetrexed for refractory leptomeningeal metastases from non-small-cell lung cancer. Front Oncol. 2019;9:838. 10.3389/fonc.2019.00838.Search in Google Scholar PubMed PubMed Central
[128] Perazella MA, Shirali A. Kidney disease caused by therapeutic agents. National kidney foundation primer on kidney diseases. Amsterdam, Netherlands: Elsevier; 2014. p. 326–36. 10.1016/B978-1-4557-4617-0.00037-6.Search in Google Scholar
[129] Glezerman IG. Drug nephropathies. Renal disease in cancer patients; Amsterdam, Netherlands: Elsevier; 2014. p. 195–208. 10.1016/B978-0-12-415948-8.00013-1.Search in Google Scholar
[130] Wilson PM, Danenberg PV, Johnston PG, Lenz HJ, Ladner RD. Standing the test of time: targeting thymidylate biosynthesis in cancer therapy. Nat Rev Clin Oncol. 2014;11(5):282.10.1038/nrclinonc.2014.51Search in Google Scholar PubMed
[131] Azzoli CG, Patel JD, Krug LM, Miller V, James L, Kris MG, et al. Pralatrexate with vitamin supplementation in patients with previously treated, advanced non-small cell lung cancer: safety and efficacy in a phase 1 trial. J Thorac Oncol. 2011;6(11):1915–22.10.1097/JTO.0b013e31822adb19Search in Google Scholar PubMed
[132] Aronson JK. A worldwide yearly survey of new data in adverse drug reactions. Vol. 33. Amsterdam, Netherlands: Elsevier; 2011. p. 669–90.Search in Google Scholar
[133] Ryan ET, Hill DR, Solomon T, Aronson N, Endy TP. Hunter’s tropical medicine and emerging infectious diseases E-Book. Philadelphia, Pennsylvania, USA: Elsevier Health Sciences; 2019.Search in Google Scholar
[134] Perez RL, Münz F, Vidoni D, Rühle A, Trinh T, Sisombath S, et al. Mesenchymal stem cells preserve their stem cell traits after exposure to antimetabolite chemotherapy. Stem Cell Res. 2019;40:101536.10.1016/j.scr.2019.101536Search in Google Scholar PubMed
[135] Hietanen KE, Järvinen TA, Huhtala H, Tolonen TT, Kuokkanen HO, Kaartinen IS. Treatment of keloid scars with intralesional triamcinolone and 5-fluorouracil injections–a randomized controlled trial. J Plast Reconstructive Aesthetic Surg. 2019;72(1):4–11.10.1016/j.bjps.2018.05.052Search in Google Scholar PubMed
[136] Dey DK, Chang SN, Vadlamudi Y, Park JG, Kang SC. Synergistic therapy with tangeretin and 5-fluorouracil accelerates the ROS/JNK mediated apoptotic pathway in human colorectal cancer cell. Food Chem Toxicol. 2020;143:111529.10.1016/j.fct.2020.111529Search in Google Scholar PubMed
[137] Janardhanam LS, Indukuri VV, Verma P, Dusane AC, Venuganti VV. Functionalized layer-by-layer assembled film with directional 5-fluorouracil release to target colon cancer. Mater Sci Eng: C. 2020;115:111118.10.1016/j.msec.2020.111118Search in Google Scholar PubMed
[138] Beguin E, Gray MD, Logan KA, Nesbitt H, Sheng Y, Kamila S, et al. Magnetic microbubble mediated chemo-sonodynamic therapy using a combined magnetic-acoustic device. J Controlled Rel. 2020;317:23–33.10.1016/j.jconrel.2019.11.013Search in Google Scholar PubMed
[139] Gillis A, Eminger L. Hypogeusia and hyposmia with topical 5-fluorouracil treatment. JAAD Case Rep. 2020;6(7):650.10.1016/j.jdcr.2020.04.023Search in Google Scholar PubMed PubMed Central
[140] Popa EC, Shah. MA. Capecitabine in the treatment of esophageal and gastric cancers. Expert Opin Invest Drugs. 2013;22(12):1645–57.10.1517/13543784.2013.842974Search in Google Scholar PubMed
[141] Castellanos MI, Childress KJ, Ramirez M, Venkatramani R. Fetal exposure to capecitabine and temozolomide during the first trimester: A case report. J Gynecol Obstet Hum Reprod. 2020;49:101881.10.1016/j.jogoh.2020.101881Search in Google Scholar PubMed
[142] Lam SW, Guchelaar HJ, Boven E. The role of pharmacogenetics in capecitabine efficacy and toxicity. Cancer Treat Rev. 2016;50:9–22. 10.1016/j.ctrv.2016.08.001.Search in Google Scholar PubMed
[143] Sprangers B, Cosmai L, Porta C. 16 conventional chemotherapy. Onco-Nephrology E-Book. Amsterdam, Netherlands: Elsevier; 2019. p. 128.Search in Google Scholar
[144] Park JY. Analysis of data on capecitabine-related adverse drug reactions from the Korean adverse event reporting system database. Eur J Oncol Nurs. 2018;34:55–60. 10.1016/j.ejon.2018.03.004.Search in Google Scholar PubMed
[145] Wu J. Methods and compositions for the treatment of cancer combining an anti-smic antibody and immune checkpoint inhibitors. U.S. Patent Application No. 16/318, 928; 2016.Search in Google Scholar
[146] Sivodia C, Sinha. A. Assessment of graphite electrode on the removal of anticancer drug cytarabine via indirect electrochemical oxidation process: Kinetics & pathway study. Chemosphere. 2020;243:125456.10.1016/j.chemosphere.2019.125456Search in Google Scholar PubMed
[147] Prabhu RH, Patravale VB. Marine‐derived pharmaceuticals for oncotherapy: Clinical trial and FDA‐approved compounds. Encycl Mar Biotechnol. 2020;4:2607–18.10.1002/9781119143802.ch117Search in Google Scholar
[148] Faruqi A, Tadi P. Cytarabine. StatPearls [Internet]. Florida, USA: StatPearls Publishing; 2020.Search in Google Scholar
[149] Zuckerman T, Ram R, Akria L, Koren-Michowitz M, Hoffman R, Henig I, et al. BST-236, a novel cytarabine prodrug for patients with acute leukemia unfit for standard induction: a phase 1/2a study. Blood Adv. 2019;3(22):3740–9.10.1182/bloodadvances.2019000468Search in Google Scholar PubMed PubMed Central
[150] Lenk L, Vogiatzi F, Schewe DM. When the bond breaks–targeting adhesion of leukemia cells to the meninges. Haematologica. 2020;105(8):1991.10.3324/haematol.2020.253609Search in Google Scholar PubMed PubMed Central
[151] Freeman III BB, Pizzorno G. Pyrimidine antimetabolites. Cambridge, Massachusetts, USA: Academic Press; 2014.10.1016/B978-0-12-801238-3.04429-9Search in Google Scholar
[152] Anstee NS, Bilardi RA, Ng AP, Xu Z, Robati M, Vandenberg CJ, et al. Impact of elevated anti-apoptotic MCL-1 and BCL-2 on the development and treatment of MLL-AF9 AML in mice. Cell Death Differ. 2019;26(7):1316–13.10.1038/s41418-018-0209-1Search in Google Scholar PubMed PubMed Central
[153] Pandya BJ, Chen CC, Medeiros BC, McGuiness CB, Wilson SD, Walsh EH, et al. Economic and clinical burden of acute myeloid leukemia episodes of care in the United States: A retrospective analysis of a commercial payer database. J Managed Care Specialty Factors Impacting Pharm Prices Affordability: Narrative Review, Pharm. 2020;9:1–11.10.18553/jmcp.2020.19220Search in Google Scholar PubMed PubMed Central
[154] Biersack B. Interplay of non-coding RNAs and approved antimetabolites such as gemcitabine and pemetrexed in mesothelioma. Non-coding RNA Res. 2018;3(4):213–25.10.1016/j.ncrna.2018.11.001Search in Google Scholar PubMed PubMed Central
[155] Moysan E, Bastiat G, Benoit JP. Gemcitabine versus modified gemcitabine: a review of several promising chemical modifications. Mol Pharmaceutics. 2013;10(2):430–44.10.1021/mp300370tSearch in Google Scholar PubMed
[156] Schelhaas S, Held A, Wachsmuth L, Hermann S, Honess DJ, Heinzmann K, et al. Gemcitabine mechanism of action confounds early assessment of treatment response by 3′-deoxy-3′-[18F] fluorothymidine in preclinical models of lung cancer. Cancer Res. 2016;76(24):7096–105.10.1158/0008-5472.CAN-16-1479Search in Google Scholar PubMed
[157] Shi Y, Wang Y, Qian J, Yan X, Han Y, Yao N, et al. MGMT expression affects the gemcitabine resistance of pancreatic cancer cells. Life Sci. 2020;259:118148.10.1016/j.lfs.2020.118148Search in Google Scholar PubMed
[158] Alam S, Illo C, Ma YT, Punia P. Gemcitabine-induced cardiotoxicity in patients receiving adjuvant chemotherapy for pancreatic cancer: a case series. Case Rep Oncol. 2018;11(1):221–7.10.1159/000488139Search in Google Scholar PubMed PubMed Central
[159] Kwok KK, Vincent EC, Gibson JN. Antineoplastic drugs. Pharmacology and therapeutics for dentistry. 7th ed. 2017. p. 530–62. 10.1016/B978-0-323-39307-2.00036-9.Search in Google Scholar
[160] Nadhum SA, Kamoon RA, Mohammed MH. 6-Mercaptopurine derivatives: maintenance therapy of acute lymphoblastic Leukemia: a review. Biochem Cell Arch. 1987;8:65–71.Search in Google Scholar
[161] Daldal YD, Demiralay EÇ. Chromatographic and UV–visible spectrophotometric pKa determination of some purine antimetabolites. J Mol Liq. 2020;317:113930. 10.1016/j.molliq.2020.113930.Search in Google Scholar
[162] Tuncbilek M, Kucukdumlu A, Guven EB, Altiparmak D, Cetin-Atalay R. Synthesis of novel 6-substituted amino-9-(β-d-ribofuranosyl) purine analogs and their bioactivities on human epithelial cancer cells. Bioorg Med Chem Lett. 2018;28(3):235–9.10.1016/j.bmcl.2017.12.070Search in Google Scholar PubMed
[163] Chande N, Patton PH, Tsoulis DJ, Thomas BS, MacDonald JK. Azathioprine or 6‐mercaptopurine for maintenance of remission in Crohn’s disease. Cochrane Database Syst Rev. 2015;10:CD000067.10.1002/14651858.CD000067.pub3Search in Google Scholar PubMed PubMed Central
[164] Han X, Hao H, Li Q, Liu C, Lei J, Yu F, et al. The interaction mechanism between fludarabine and human serum albumin researched by comprehensive spectroscopic methods and molecular docking technique. Spectrochim Acta Part A, Mol Biomol Spectrosc. 2020;233:118170.10.1016/j.saa.2020.118170Search in Google Scholar PubMed
[165] Şenel P, Agar S, Sayin VO, Altay F, Yurtsever M, Gölcü A. Elucidation of binding interactions and mechanism of Fludarabine with dsDNA via multispectroscopic and molecular docking studies. J Pharm Biomed Anal. 2020;179:112994.10.1016/j.jpba.2019.112994Search in Google Scholar PubMed
[166] Cividini F, Cros-Perrial E, Pesi R, Machon C, Allegrini S, Camici M, et al. Cell proliferation and drug sensitivity of human glioblastoma cells are altered by the stable modulation of cytosolic 5′-nucleotidase II. Int J Biochem Cell Biol. 2015;65:222–9.10.1016/j.biocel.2015.06.011Search in Google Scholar PubMed
[167] Qiu L, Liu J, Wang Z, Hu W, Huang Q, Zhou Y. ZGDHu-1 and fludarabine have a synergistic effect on apoptosis of chronic lymphocytic leukemia cells. Oncol Rep. 2015;34(3):1239–48.10.3892/or.2015.4115Search in Google Scholar PubMed
[168] Bethge WA, Kerbauy FR, Santos EB, Gooley T, Storb R, Sandmaier BM. Extracorporeal photopheresis combined with pentostatin in the conditioning regimen for canine hematopoietic cell transplantation does not prevent GVHD. Bone Marrow Transplant. 2014;49(9):1198–204.10.1038/bmt.2014.137Search in Google Scholar PubMed PubMed Central
[169] Aronson JK, ed. Meyler’s side effects of drugs: the international encyclopedia of adverse drug reactions and interactions. Amsterdam, Netherlands: Elsevier; 2015.Search in Google Scholar
[170] Desai C. Meyler’s side effects of drugs: The international encyclopedia of adverse drug reactions and interactions. Indian J Pharmacol. 2016;48(2):224.10.4103/0253-7613.178821Search in Google Scholar
[171] Gerson SL, Caimi PF, William BM, Creger RJ. Pharmacology and molecular mechanisms of antineoplastic agents for hematologic malignancies. Hematology. Philadelphia, Pennsylvania, USA: Elsevier; 2018. p. 849–912.Search in Google Scholar
[172] Getta BM, Park JH, Tallman. MS. Hairy cell leukemia: past, present and future. Best Pract Res Clin Haematol. 2015;28(4):269–72.10.1016/j.beha.2015.10.015Search in Google Scholar PubMed PubMed Central
[173] Treon SP, Castillo JJ, Hunter ZR, Merlini G. Waldenström macroglobulinemia/lymphoplasmacytic lymphoma. Hematology. Philadelphia, Pennsylvania, USA: Elsevier; 2018. p. 1419–31.10.1016/B978-0-323-35762-3.00087-1Search in Google Scholar
[174] Abou Dalle I, Ravandi F. Moxetumomabpasudotox for the treatment of relapsed and/or refractory hairy cell leukemia. Expert Rev Hematol. 2019;12(9):707–14.10.1080/17474086.2019.1643231Search in Google Scholar PubMed
[175] Dasari A, Choi J-S, Berdis AJ. Chemotherapeutic intervention by inhibiting DNA polymerases. DNA repair in cancer therapy. Cambridge, Massachusetts, USA: Academic Press; 2016. p. 179–224.10.1016/B978-0-12-803582-5.00007-3Search in Google Scholar
[176] Durmaz O, Demirkaya M, Sevinir B. Recombinant human erythropoietin β: the effect of weekly dosing on anemia, quality of life, and long-term outcomes in pediatric cancer patients. Pediatric Hematol Oncol. 2011;28:461–8.10.3109/08880018.2011.570857Search in Google Scholar PubMed
[177] Podoltsev NA, Stahl M, Zeidan AM, Gore SD. Selecting initial treatment of acute myeloid leukaemia in older adults. Blood Rev. 2017;31(2):43–62.10.1016/j.blre.2016.09.005Search in Google Scholar PubMed
[178] Podolski AJ, Gucalp R. GI toxicities from cancer therapy. Geriatric Gastroenterology. Cham: Springer International Publishing, 2021. pp. 341–79.10.1007/978-3-030-30192-7_93Search in Google Scholar
[179] Abaza Y, Kantarjian HM, Faderl S, Jabbour E, Jain N, Thomas D, et al. Hyper‐CVAD plus nelarabine in newly diagnosed adult T‐cell acute lymphoblastic leukemia and T‐lymphoblastic lymphoma. Am J Hematol. 2018;93(1):91–9.10.1002/ajh.24947Search in Google Scholar PubMed
[180] Kadia TM, Gandhi V. Nelarabine in the treatment of pediatric and adult patients with T-cell acute lymphoblastic leukemia and lymphoma. Expert Rev Hematol. 2017;10(1):1–8.10.1080/17474086.2017.1262757Search in Google Scholar PubMed PubMed Central
[181] Pandey RK, DiPippo A, Kadia T, Pemmaraju N, Workeneh BT, Jabbour E, et al. Nelarabine-related rhabdomyolysis in a patient with T-cell acute lymphoblastic leukemia. Leukemia Lymphoma. 2020;11:1–3.10.1080/10428194.2020.1791852Search in Google Scholar PubMed
[182] Saeidnia S. New approaches to natural anticancer drugs. Springer International Publishing; 2015; Prendergast GC, Jaffee EM, eds. Cancer immunotherapy: immune suppression and tumor growth. Cambridge, Massachusetts, USA: Academic Press; 2013.Search in Google Scholar
[183] Jeswani G, Paul SD. Recent advances in the delivery of chemotherapeutic agents. Nano-and microscale drug delivery systems. Elsevier; 2017. p. 281–98. 10.1016/B978-0-323-52727-9.00015-7; Saeidnia S. New approaches to natural anticancer drugs. Cham, Switzerland: Springer International Publishing; 2015.Search in Google Scholar
[184] de Felício R, de Oliveira AL, Scopel M, Debonsi HM. 13 Microbial Natural Products. Mol Diversity Environ Prokaryotes. 2016;295–326.10.1201/9781315381909-14Search in Google Scholar
[185] Gustafson DL, Page RL. Cancer chemotherapy. Withrow and MacEwen s small animal clinical oncology. Elsevier; 2013. pp. 157–79.10.1016/B978-1-4377-2362-5.00011-6Search in Google Scholar
[186] Podolsky SH. From global recognition to global health: Antimicrobials and the Nobel Prize, 1901–2015. Attributing excellence in medicine. Leiden, Netherlands: Brill Rodopi; 2019. p. 81–96.10.1163/9789004406421_006Search in Google Scholar
[187] Woodruff HB. Selman A. Waksman, winner of the 1952 Nobel Prize for physiology or medicine. Appl Environ Microbiol. 2014;80(1):2–8.10.1128/AEM.01143-13Search in Google Scholar PubMed PubMed Central
[188] Specific aspects of cytotoxic and hormonal drug therapiesLeon P. Bignold, in Principles of Tumors (Second Edition), 2020.Search in Google Scholar
[189] Wang F, Liu X, Liu C, Liu Z, Sun L. Effects of antibiotic antitumor drugs on nucleotide levels in cultured tumor cells: an exploratory method to distinguish the mechanisms of antitumor drug action based on targeted metabolomics. Acta Pharm Sin B. 2015;5(3):223–30.10.1016/j.apsb.2015.03.010Search in Google Scholar PubMed PubMed Central
[190] DeLeve LD. Cancer chemotherapy. Drug-induced liver disease. Cambridge, Massachusetts, USA: Academic Press; 2013. p. 541–67.10.1016/B978-0-12-387817-5.00030-3Search in Google Scholar
[191] Truxova I, Hensler M, Skapa P, Halaska MJ, Laco J, Ryska A, et al. Rationale for the combination of dendritic cell-based vaccination approaches with chemotherapy agents. International Review of Cell and Molecular Biology. Vol. 330, Cambridge, Massachusetts, USA: Academic Press; 2017. p. 115–56.10.1016/bs.ircmb.2016.09.003Search in Google Scholar PubMed
[192] Sankaranarayanan K, Sundararajan R. Electrically-enhanced proliferation control of cancer-stem-cells-like adult human mesenchymal stem cells – A novel modality of treatment. Electroporation Based Ther Cancer. 2014;127–59.10.1533/9781908818294.127Search in Google Scholar
[193] Anest J, Inten Care Med. Bleomycin lung injury: A case report and review of the literature. J Anest & Inten Care Med. 2017;2:555607.Search in Google Scholar
[194] Gerson SL, Caimi PF, William BM, Creger RJ. Pharmacology and molecular mechanisms of antineoplastic agents for hematologic malignancies. Hematology. Philadelphia, Pennsylvania, USA: Elsevier; 2018. p. 849–912.10.1016/B978-0-323-35762-3.00057-3Search in Google Scholar
[195] Respiratory Toxicology, Rowland AM, Yost GS. In Comprehensive toxicology. Amsterdam, Netherlands: Elsevier; 2010. 8.25.2.1.2 Reduced enzymatic detoxification.Search in Google Scholar
[196] Hult EM, Warheit-Niemi H, Moore BB. Animal models of fibrotic interstitial lung disease. Amsterdam, Netherlands: Elsevier; 2019.Search in Google Scholar
[197] Forrest L, Cohen M, Cai S. Intralymphatic chemotherapy drug carriers. U.S. Patent Application No. 13/341, 282; 2012.Search in Google Scholar
[198] Verma SP, Subbiah A, Vishwanath VK, Dutta TK. Bleomycin-induced skin toxicity: is it always flagellate erythema? Case Reports. 2016;2016:bcr2014204575.10.1136/bcr-2014-204575Search in Google Scholar PubMed PubMed Central
[199] Gad SE. Mitomycin C. Cambridge, Massachusetts, USA: Academic Press; 2014. p. 354–6.10.1016/B978-0-12-386454-3.00883-6Search in Google Scholar
[200] Kidney GO, Rankin MA. Valentovic, in encyclopedia of toxicology. 3rd edn. Antibiotics. Cambridge, Massachusetts, USA: Academic Press; 2014.10.1016/B978-0-12-386454-3.00326-2Search in Google Scholar
[201] González-Ventosa A, Ariz-Juan J, Sabater-Cruz N. Measures to prevent the risks associated with exposure to cytostatic drugs in glaucoma filtering surgery. Archivos de la Soc Española de Oftalmología (Engl Ed). 2020;95(7):334–44.10.1016/j.oftale.2020.04.004Search in Google Scholar
[202] Kumar S, Bhandari C, Sharma P, Agnihotri N. Role of piperine in chemoresistance. Role of nutraceuticals in cancer chemosensitization. Cambridge, Massachusetts, USA: Academic Press; 2018. p. 259–86.10.1016/B978-0-12-812373-7.00013-9Search in Google Scholar
[203] Chang A. A case of mitoxantrone extravasation. J Oncol Pharm Pract. 2020;26(5):1270–3.10.1177/1078155219893736Search in Google Scholar PubMed
[204] Maghzi AH, Borazanci A, McGee J, Alexander JS, Gonzalez-Toledo E, Minagar A. Multiple sclerosis: pathophysiology, clinical features, diagnosis, and management. Neuroinflammation. Philadelphia, Pennsylvania, USA: Elsevier; 2011. p. 1–23.10.1016/B978-0-12-384913-7.00001-0Search in Google Scholar
[205] Edan G. Currently approved injectable disease-modifying drugs: cytotoxic immunosuppressive drugs. Translational neuroimmunology in multiple sclerosis. Cambridge, Massachusetts, USA: Academic Press; 2016. p. 245–57.10.1016/B978-0-12-801914-6.00020-9Search in Google Scholar
[206] O’Connor PW, Oh J. Disease-modifying agents in multiple sclerosis. Handbook of clinical neurology. Vol. 122, Philadelphia, Pennsylvania, USA: Elsevier; 2014. p. 465–501.10.1016/B978-0-444-52001-2.00021-2Search in Google Scholar PubMed
[207] Chang HM, Moudgil R, Scarabelli T, Okwuosa TM, Yeh ET. Cardiovascular complications of cancer therapy: best practices in diagnosis, prevention, and management: part 1. J Am Coll Cardiol. 2017;70(20):2536–51.10.1016/j.jacc.2017.09.1096Search in Google Scholar PubMed PubMed Central
[208] Tsuji W, Plock JA. Breast cancer metastasis. Introduction to cancer metastasis. Cambridge, Massachusetts, USA: Academic Press; 2017. p. 13–31.10.1016/B978-0-12-804003-4.00002-5Search in Google Scholar
[209] Saleem T, Kasi A. Daunorubicin. StatPearls [Internet]. Treasure Island, Florida, USA: StatPearls Publishing LLC; 2020.Search in Google Scholar
[210] Morabito F, Voso MT, Hohaus S, Gentile M, Vigna E, Recchia AG, et al. Panobinostat for the treatment of acute myelogenous leukemia. Expert Opin Invest Drugs. 2016;25(9):1117–31.10.1080/13543784.2016.1216971Search in Google Scholar PubMed
[211] Iwamoto T. Clinical application of drug delivery systems in cancer chemotherapy: review of the efficacy and side effects of approved drugs. Biol Pharm Bull. 2013;36(5):715–8.10.1248/bpb.b12-01102Search in Google Scholar PubMed
[212] Adeniyi AA. Computational studies, synthesis and characterization of ruthenium (II) anticancer complexes. Doctoral dissertation. University of Fort Hare; 2014.Search in Google Scholar
[213] Montgomery B, Lin DW. Toxicities of chemotherapy for genitourinary malignancies. Complications of urologic surgery. Philadelphia, Pennsylvania, USA: WB Saunders; 2010. p. 117–23.10.1016/B978-1-4160-4572-4.00010-8Search in Google Scholar
[214] Smuder AJ. Exercise stimulates beneficial adaptations to diminish doxorubicin-induced cellular toxicity. Am J Physiol-Regulatory, Integr Comp Physiology. 2019;317(5):R662–72.10.1152/ajpregu.00161.2019Search in Google Scholar PubMed PubMed Central
[215] Wierstra I. FOXM1 (Forkhead box M1) in tumorigenesis: overexpression in human cancer, implication in tumorigenesis, oncogenic functions, tumor-suppressive properties, and target of anticancer therapy. Advances in cancer research. Vol. 119, Cambridge, Massachusetts, USA: Academic Press; 2013. p. 191–419.10.1016/B978-0-12-407190-2.00016-2Search in Google Scholar PubMed
[216] Wu J, Xue X, Zhang B, Cao H, Kong F, Jiang W, et al. Enhanced antitumor activity and attenuated cardiotoxicity of Epirubicin combined with Paeonol against breast cancer. Tumor Biol. 2016;37(9):12301–13.10.1007/s13277-016-5088-9Search in Google Scholar PubMed
[217] Ajani JA, D’Amico TA, Bentrem DJ, Chao J, Corvera C, Das P, et al. Esophageal and esophagogastric junction cancers, version 2 2019, NCCN clinical practice guidelines in oncology. J Natl Compr Cancer Netw. 2019;17(7):855–83.10.6004/jnccn.2019.0033Search in Google Scholar PubMed
[218] Petit K, Suwalsky M, Colina JR, Contreras D, Aguilar LF, Jemiola-Rzeminska M, et al. Toxic effects of the anticancer drug epirubicin in vitro assayed in human erythrocytes. Toxicol Vitro. 2020;68:104964.10.1016/j.tiv.2020.104964Search in Google Scholar PubMed
[219] Hohloch K, Zwick C, Ziepert M, Hasenclever D, Kaiser U, Engert A, et al. Significant dose Escalation of Idarubicin in the treatment of aggressive Non-Hodgkin Lymphoma leads to increased hematotoxicity without improvement in efficacy in comparison to standard CHOEP-14: 9-year follow up results of the CIVEP trial of the DSHNHL. SpringerPlus. 2014;3(1):5.10.1186/2193-1801-3-5Search in Google Scholar PubMed PubMed Central
[220] Liang B, Li N, Zhang S, Qi A, Feng J, Jing W, et al. Idarubicin-loaded methoxy poly (ethylene glycol)-b-poly (L-lactide-co-glycolide) nanoparticles for enhancing cellular uptake and promoting antileukemia activity. Int J Nanomed. 2019;14:543.10.2147/IJN.S190027Search in Google Scholar PubMed PubMed Central
[221] Lacetera A, Galante S, Jiménez-Barbero J, Martín-Santamaría S. Glycans in medicinal chemistry. Amsterdam, Netherlands: Elsevier; 2016.10.1016/B978-0-12-409547-2.11712-3Search in Google Scholar
[222] Matsuo T, Konya Y, Hirayama E, Sadzuka Y. 2‑Deoxy‑D‑glucose enhances the anti‑cancer effects of idarubicin on idarubicin‑resistant P388 leukemia cells. Oncol Lett. 2020;20(1):962–6.10.3892/ol.2020.11616Search in Google Scholar PubMed PubMed Central
[223] Turek A, Stoklosa K, Borecka A, Paul-Samojedny M, Kaczmarczyk B, Marcinkowski A, et al. Designing biodegradable wafers based on poly(L-lactide-co-glycolide) and poly(glycolide-co-ε-caprolactone) for the prolonged and local release of idarubicin for the therapy of glioblastoma multiforme. Pharm Res. 2020;37:1–13, Nauta A, et al. Chapter 6-scarless wound healing. Princ Regener Med . 2011:103–27.10.1007/s11095-020-02810-2Search in Google Scholar PubMed PubMed Central
[224] Patel BA, Liu SV. Lung adenocarcinoma. Pulmonary adenocarcinoma: Approaches to treatment. Amsterdam, Netherlands: Elsevier; 2019. p. 103–14. 10.1016/B978-0-323-55433-6.00006-7.Search in Google Scholar
[225] Li M, Liu Y. Topoisomerase I in human disease pathogenesis and treatments. Genomics Proteom Bioinforma. 2016;14(3):166–71.10.1016/j.gpb.2016.02.004Search in Google Scholar PubMed PubMed Central
[226] Shen CH. Diagnostic molecular biology. Cambridge, Massachusetts, USA: Academic Press; 2019.Search in Google Scholar
[227] Bharti AC, Rajan P, Jadli M, Pande D, Singh T, Bhat A. Berberine as an adjuvant and sensitizer to current chemotherapy. Role of nutraceuticals in cancer chemosensitization. Cambridge, Massachusetts, USA: Academic Press; 2018. p. 221–40.10.1016/B978-0-12-812373-7.00011-5Search in Google Scholar
[228] Li F, Jiang T, Li Q, Ling X. Camptothecin (CPT) and its derivatives are known to target topoisomerase I (Top1) as their mechanism of action: did we miss something in CPT analogue molecular targets for treating human disease such as cancer? Am J Cancer Res. 2017;7(12):2350.Search in Google Scholar
[229] Lin J, Yang L, Liao X, Gao C, Yang B. Host–guest systems based on pH-sensitive acyclic cucurbit [n] urils for controlled release of camptothecin. J Incl Phenom Macrocycl Chem. 2019;95(1–2):159–68.10.1007/s10847-019-00935-5Search in Google Scholar
[230] Mohapatra A, Uthaman S, Park IK. Polyethylene glycol nanoparticles as promising tools for anticancer therapeutics. Polymeric nanoparticles as a promising tool for anti-cancer therapeutics. Cambridge, Massachusetts, USA: Academic Press; 2019. p. 205–31.10.1016/B978-0-12-816963-6.00010-8Search in Google Scholar
[231] Chemotherapy in chronic kidney disease and dialysis SABINE KARAM,. ILYA GLEZERMAN, in Onco-Nephrology, 2020 Irinotecan.Search in Google Scholar
[232] Fujita KI, Kubota Y, Ishida H, Sasaki Y. Irinotecan, a key chemotherapeutic drug for metastatic colorectal cancer. World J Gastroenterol. 2015;21(43):12234.10.3748/wjg.v21.i43.12234Search in Google Scholar PubMed PubMed Central
[233] Kciuk M, Marciniak B, Kontek R. Irinotecan – still an important player in cancer chemotherapy: a comprehensive overview. Int J Mol Sci. 2020;21(14):4919.10.3390/ijms21144919Search in Google Scholar PubMed PubMed Central
[234] Kneuertz PJ, Kooby DA. Systemic chemotherapy for hepatic colorectal cancer: Impact on surgical management. Blumgart’s surgery of the liver, pancreas and biliary tract. Amsterdam, Netherlands: WB Saunders; 2012. p. 1434–43.10.1016/B978-1-4377-1454-8.00087-4Search in Google Scholar
[235] Tanoue LT, Detterbeck FC. Lung cancer: a practical approach to evidence-based clinical evaluation and management. Philadelphia, Pennsylvania, USA: Elsevier Health Sciences; 2018.Search in Google Scholar
[236] Mittal B, Tulsyan S, Kumar S, Mittal RD, Agarwal G. Cytochrome P450 in cancer susceptibility and treatment. Advances in clinical chemistry. Vol. 71, Philadelphia, Pennsylvania, USA: Elsevier; 2015. p. 77–139.10.1016/bs.acc.2015.06.003Search in Google Scholar PubMed
[237] Huang H, Wang X, Zhang X, Zhang G, Jinbo M, Wang H, et al. Ganciclovir reduces irinotecan-induced intestinal toxicity by inhibiting NLRP3 activation. Cancer Chemother Pharmacol. 2020;85(1):195–204.10.1007/s00280-019-03996-ySearch in Google Scholar PubMed
[238] Desai D. Evaluating the dopaminergic neurotoxic effects of chemotherapeutics. Master’s thesis, Auburn University; 2018.Search in Google Scholar
[239] Kuehl PJ, Grimes MJ, Dubose D, Burke M, Revelli DA, Gigliotti AP, et al. Inhalation delivery of topotecan is superior to intravenous exposure for suppressing lung cancer in a preclinical model. Drug Delivery. 2018;25(1):1127–36.10.1080/10717544.2018.1469688Search in Google Scholar PubMed PubMed Central
[240] Jeong SH, Jang JH, Lee YB. Pharmacokinetic comparison of three different administration routes for topotecan hydrochloride in rats. Pharmaceuticals. 2020;13(9):231.10.3390/ph13090231Search in Google Scholar PubMed PubMed Central
[241] Sharma S, Chowdhury G, Chakrabarty A. A concise overview of chemotherapeutic drugs in gastrointestinal cancers: mechanisms of action and resistance. J Cancer Sci Clin Ther. 2021;5(1):11–35.Search in Google Scholar
[242] Uusküla-Reimand L, Hou H, Samavarchi-Tehrani P, Rudan MV, Liang M, Medina-Rivera A, et al. Topoisomerase II beta interacts with cohesin and CTCF at topological domain borders. Genome Biol. 2016;17(1):1–22.10.1186/s13059-016-1043-8Search in Google Scholar PubMed PubMed Central
[243] Montecucco A, Zanetta F, Biamonti G. Molecular mechanisms of etoposide. EXCLI J. 2015;14:95–108.Search in Google Scholar
[244] Reyhanoglu G, Tadi P. Etoposide. StatPearls [Internet]. Treasure Island, Florida, USA: StatPearls Publishing; 2020.Search in Google Scholar
[245] Yoneda KY, Cross CE. The pulmonary Toxicity of anticancer agents. Respiratory toxicology. Philadelphia, Pennsylvania, USA: Elsevier Inc; 2010. p. 477–510.10.1016/B978-0-08-046884-6.00924-6Search in Google Scholar
[246] Finkel KW, Perazella MA, Cohen EP. Onco-Nephrology E-Book. Philadelphia, Pennsylvania, USA: Elsevier Health Sciences; 2019.Search in Google Scholar
[247] Alrooqi M, Khan S, Alhumaydhi FA, Asiri SA, Alshamrani M, Mashraqi MM, et al. A therapeutic journey of pyridine-based heterocyclic compounds as potent anti- cancer agents: A review (From 2017 to 2021). Basel, Switzerland: MDPI; 2022. p. 2775–87.10.2174/1871520622666220324102849Search in Google Scholar PubMed
[248] Izuchukwu D, Conradie J. Anticancer properties of complexes derived from bidentate ligands. J Inorg Biochem. 2023;246:112268.10.1016/j.jinorgbio.2023.112268Search in Google Scholar PubMed
[249] Laamari Y, Bimoussa A, Fawzi M, Oubella A, Rohand T, Van Meervelt L, et al. Synthesis, crystal structure and evaluation of anticancer activities of some novel heterocyclic compounds based on thymol. J Mol Struct. 2023;1278:134906.Search in Google Scholar
[250] Hu S, Li Y, Kan W, Ding T, Gu H, Zhang T, et al. Novel orthodiphenyl fi ve-member N-heteroaromatic compounds as potent anticancer cell agents. Medicinal chemistry research. Vol. 31. Berlin, Germany: Springer Nature; 2022. p. 936–48.10.1007/s00044-022-02894-ySearch in Google Scholar
[251] Dabhi RA, Dhaduk MP, Bhatt VD, Bhatt BS. Materials Today: Proceedings Green approach for the synthesis of novel spiro quinoxaline-pyrimidone based heterocyclic compounds as anticancer agents. Mater Today Proc. 2022;65:367–74.10.1016/j.matpr.2022.06.374Search in Google Scholar
[252] Laamari Y, Bimoussa A, Fawzi M, Oubella A, Rohand T, Van Meervelt L, et al. Synthesis, crystal structure and evaluation of anticancer activities of some novel heterocyclic compounds based on thymol. J Mol Structure. 2023;1278:134906.10.1016/j.molstruc.2023.134906Search in Google Scholar
[253] Xu X, Lan J, Huang H, Dai W, Peng X, Liu S. Bioorganic & Medicinal Chemistry Synthesis, biological activity and mechanism of action of novel allosecurinine derivatives as potential antitumor agents. Bioorg Med Chem. 2023;82:117234.10.1016/j.bmc.2023.117234Search in Google Scholar PubMed
[254] Kuzu B, Ergüç A, Karakuş F, Arzuk E Design, synthesis, and antiproliferative activities of novel thiazolyl-pyrazole hybrid derivatives. Med Chem Res. 2023;32(8):1690–700.10.1007/s00044-023-03090-2Search in Google Scholar
© 2025 the author(s), published by De Gruyter
This work is licensed under the Creative Commons Attribution 4.0 International License.
Articles in the same Issue
- Research Articles
- Synthesis, activity exploration, molecular docking, and affinity assessment of theophylline derivatives as inhibitors targeting IDO1
- Synthesis of novel triazole hybrids with pyrene, fluorene, and biphenyl groups and evaluation of their antimycobacterial activity
- Synthesis and antiproliferative evaluation of novel triazolothiadiazine compounds
- Review Article
- Development of heterocyclic-based anticancer agents: A comprehensive review
Articles in the same Issue
- Research Articles
- Synthesis, activity exploration, molecular docking, and affinity assessment of theophylline derivatives as inhibitors targeting IDO1
- Synthesis of novel triazole hybrids with pyrene, fluorene, and biphenyl groups and evaluation of their antimycobacterial activity
- Synthesis and antiproliferative evaluation of novel triazolothiadiazine compounds
- Review Article
- Development of heterocyclic-based anticancer agents: A comprehensive review