An improved and scalable synthesis of zolpidem via a CuI/BINOL-mediated tandem reaction of imine and alkyne
-
Bingbing Zhang


Abstract
An improved and scalable method for the synthesis of zolpidem (1), a hypnotic drug, was developed. A two-step sequence involving imine formation and subsequent tandem reaction between an imine and propiolamide in the presence of CuI/BINOL, an efficient promoter for the tandem reaction, is described. Zolpidem was efficiently prepared in a 54% isolated yield and the hemitartrate salt of zolpidem was produced in 37% yield by simple crystallization, without tedious column chromatography. The procedure can be scaled up to >10 g. The yield of 1 increased to 83% following isolation of the intermediate imine 5.
Introduction
Zolpidem (1 in Scheme 1) is a short-acting hypnotic drug. It potentiates the inhibitory neurotransmitter, γ-aminobutyric acid (GABA), by binding to benzodiazepine receptors located on the γ-aminobutyric acid receptors. Compound 1 is clinically used for the treatment of insomnia [1]. In addition, it can also be used for the treatment of anxiety and other brain disorders because of its anxiolytic and anticonvulsant properties [2]. Despite its similar hypnotic effects to those of benzodiazepines, 1 is structurally different and is classified as a non-benzodiazepine hypnotic drug. Drug 1 is usually marketed as a tartrate salt.
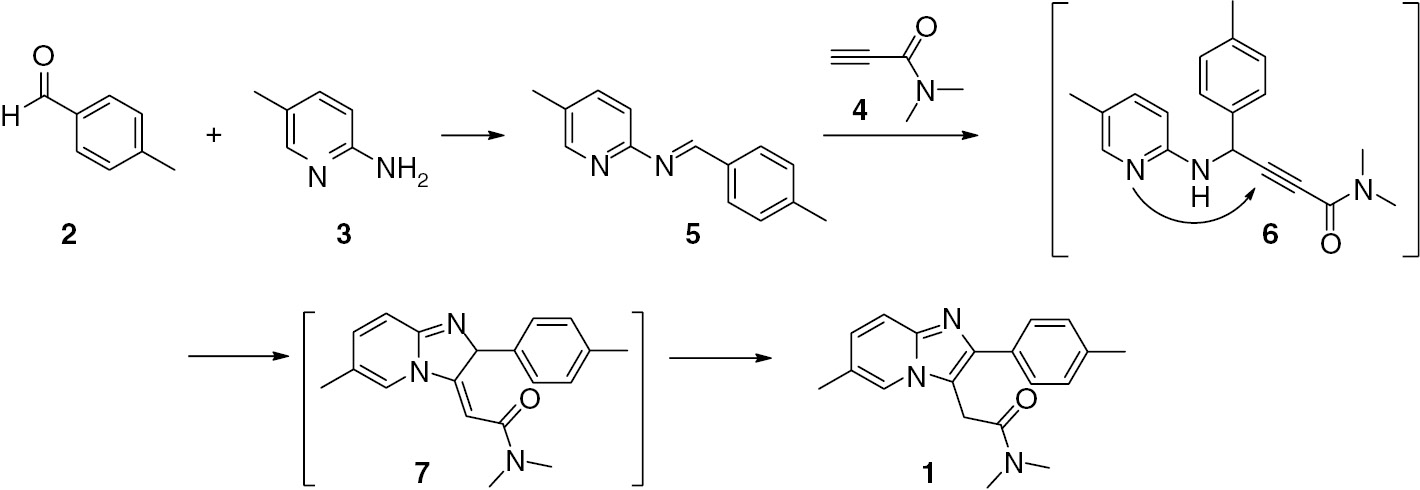
Synthesis of zolpidem.
The current methods for the synthesis of 1 require multi-step preparations with low yields and sometimes use toxic reagents or special technological devices [3], [4], [5], [6], [7]. The typical step for the preparation of the imidazo[1,2-a]pyridine scaffold of 1 is condensation of an α-bromoketone or its equivalent with 2-amino-5-methylpyridine. Recently, Gevorgyan has explored an elegant strategy for 1, in which the key step is the CuCl/Cu(OTf)2–catalyzed tandem reaction of terminal alkyne and imine, in situ prepared from a 2-aminopyridine and aldehyde, affording the scaffold of 1 with a good yield on a milligram scale in a microreactor [8].
More recently, many catalysts have been developed for the tandem reaction, namely, nanocopper oxide/sodium ascorbate [9], Cu-Mn spinel oxide [10], CuSO4-glucose [11], CuI/NaHSO4/SiO2 [12], Cu(BDC) [13], InBr3 [14], CuSO4/TsOH·H2O [15], CuI/ZnCl2 [16] and Cu(OAc)2·H2O/air [17]. Unfortunately, these catalysts are ineffective in the synthesis of 1 on a large scale and in one routine flask. In this report we describe a new catalytic system, CuI/BINOL, for the scalable synthesis of 1 using the tandem reaction of the corresponding imine and alkyne.
Results and discussion
According to Gevorgyan’s strategy, compound 1 can be prepared from 4-methylbenzaldehyde (2), 2-amino-5-methylpyridine (3), and the propiolamide 4. The reaction sequence is depicted in Scheme 1. Based on our previous experience [15], [16], during the formation of 5 from 2 and 3, the presence of propiolamide 4, as a good Michael acceptor, would not be tolerated in this step. Thus, we performed the synthesis of 1 in a stepwise manner (Scheme 1). Substrates 4 [18] and 5 [19] were independently prepared and subsequently used in tandem reaction for 1.
The addition reaction of 4 with 5 requires generation of an alkynyl anion of 4. Although many bases can convert 4 into its alkynyl anion [20], [21], [22], the resultant hard anion is too unstable to trap the non-activated imine 5 and undergoes rapid decomposition. Therefore, a softer alkynyl anion is required to complete the alkynylation of the imine. Based on our success with copper salts [9], [10], [11], [12], [13], [14], [15], [16], [17], a variety of copper catalysts were screened. In a typical experiment, a mixture of 5 (0.5 mmol), 4 (1.0 mmol), catalyst (Cu2+ or Cu+ salt, 0.05 mmol) and an additive (0–0.05 mmol) in toluene (10 mL) was heated under reflux in a routine flask instead of a microreactor under argon for 16–36 h (Scheme 2). The progress of the reaction was monitored by thin lipid chromatography (TLC).
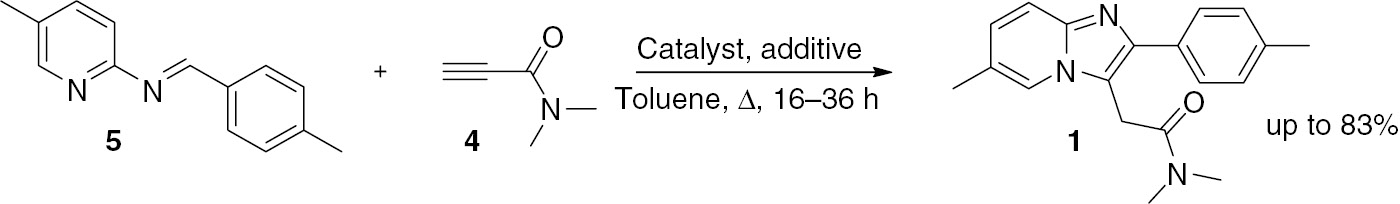
Tandem reaction for zolpidem.
In the presence of CuCl/Cu(OTf)2 compound 1 was furnished in 32% yield but an increased amount of the catalyst (0.5 equiv.) and prolonged time (36 h) were required [8]. The use of CuSO4/TsOH offered a 13% yield [15], and only traces of 1 were produced in the presence of InBr3 [14]. On the other hand, the use of CuI/DIPEA [23] provided a promising yield of 46%. Subsequently, it was found that the yield increased to 70% in the presence of 0.5 equiv. of CuI. The replacement of CuI with CuBr, CuCl or other CuI salts did not improve the yield of 1. It was also found that DIPEA was not necessary and either a Brønsted or Lewis acid as an additive [15] was unable to ameliorate the reaction. However, a large amount of propiolamide was required in these experiments, possibly due to the competition from the Glaser reaction [24].
Subsequently, CuI complexes with ligands containing oxygen, nitrogen, sulfur or phosphorus atom were screened. The ligands were Me2NCH2CH2NMe2, 2,2′-bipyridine, 1,10-phenanthroline, BINAP, and BINOL. The best result of 83% was obtained with 0.5 equivalents of CuI and BINOL. The yield decreased from 83% to 62% when the amounts of the two additives were reduced from 0.5 to 0.1 equivalents. To simplify the operation for one-pot procedure, the imine 5 formed from 2 and 3 was not isolated but directly allowed to react with 4 in the presence of CuI/BINOL. This procedure provided product 1 in a yield of 54%.
Next, the tartrate salt of 1, the commonly marketed form of the drug, was scaled up to 10 g. After optimization, zolpidem tartrate was obtained in 37% yield directly by crystallization from 2-propanol without using column chromatography (Scheme 3 – equation 1).
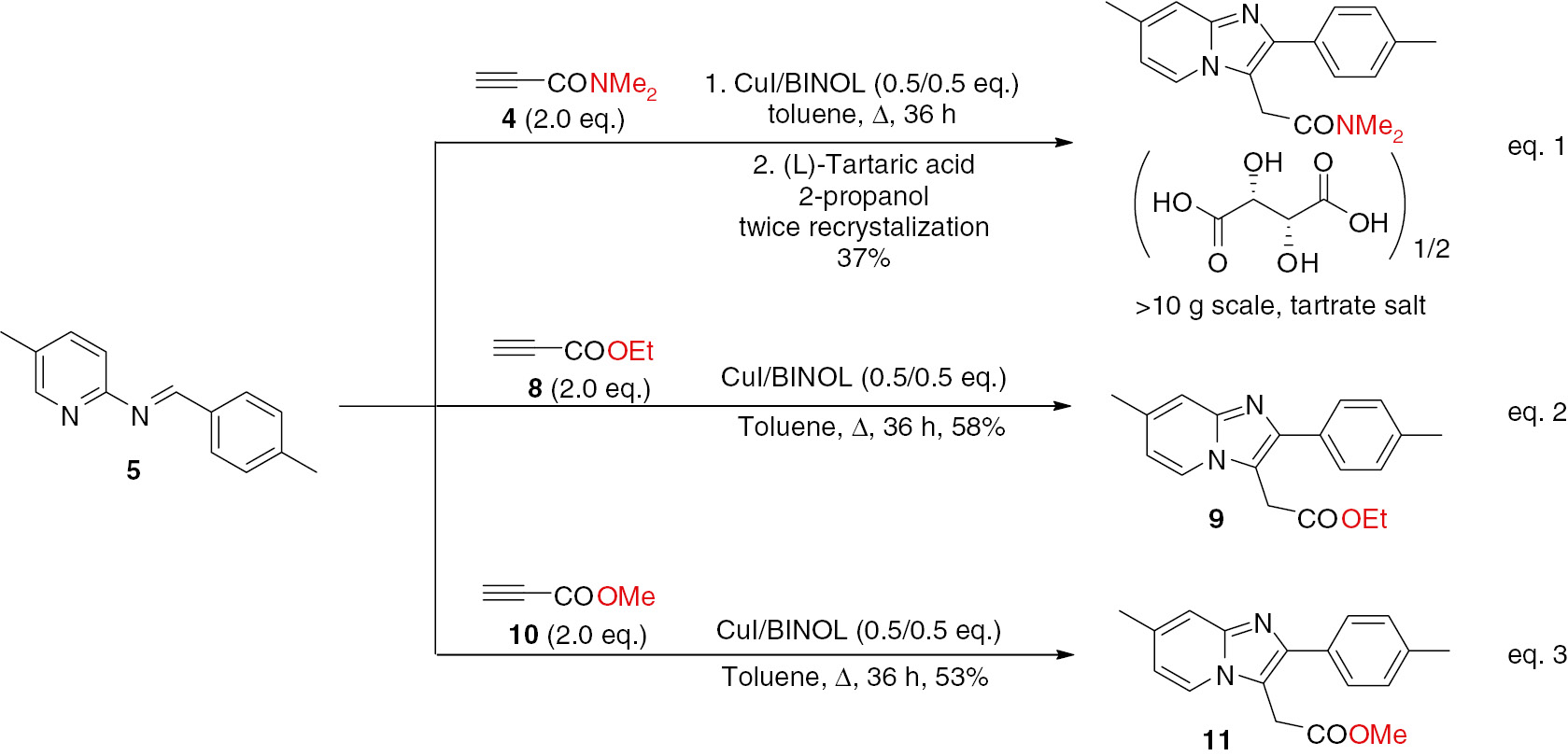
Synthesis of zolpidem tartrate and analogues 9, 11.
Finally, the use of the catalytic system was extended to the reaction of 5 with ethyl propiolate (8, Scheme 3 – equation 2) and methyl propiolate (10, Scheme 3 – equation 3), because the corresponding products 9 and 11 can also serve as the precursor to 1. Compounds 9 or 11 were obtained in moderate yields of 53–58%. These results, together with the previous research [8], [17], suggest that the discussed tandem reaction is highly dependent on the electronic properties of imine and propiolic acid substrates, and efficient copper catalyst must be fine-tuned on a case-by-case basis.
Conclusion
An efficient catalytic system, CuI/BINOL, for the tandem reaction of N-(4-methylbenzylidene)pyridin-2-amine and N,N-dimethylpropiolamide towards the synthesis of the anxiolytic drug zolpidem was developed. Unlike the previously reported processes, the present protocol avoids the use of a microreactor and uses easily available reagents such as CuI and BINOL to prepare zolpidem in a simple, efficient, and scalable manner.
Experimental
The progress of the reactions was monitored by TLC on silica-coated glass plates and compounds were visualized under ultra violet (UV) light in the presence of iodine or phosphomolybdic acid. Flash column chromatography was performed on silica gel (300–400 mesh). Melting points were measured on a SGWX-4 microscopy melting point apparatus without correction. Unless stated otherwise, nuclear magnetic resonance (NMR) spectra were recorded in CDCl3 at 400 MHz (1H) and 100 MHz (13C). Mass spectra were recorded on a Finnigan-Mat-95 mass spectrometer equipped with an electrospray ionization (ESI) source.
A one-pot two-step synthetic procedure for zolpidem (1)
A round-bottom flask was equipped with a rubber septum, a condenser and a stirring bar. The vessel was charged with 5-methylpyridin-2-amine (0.11 g, 1.0 mmol), 4-methylbenzaldehyde (0.12 mL, 0.12 g, 1.0 mmol), 4Å molecular sieves (0.20 g) and dry toluene (10 mL) under argon, and the mixture was heated to reflux on an oil bath overnight. After cooling, the flask was charged with propiolamide 4 (0.20 g, 2.0 mmol), CuI (0.10 g, 0.5 mol) and BINOL (0.14 g, 0.5 mmol), and the mixture was heated to reflux on an oil bath for 36 h. After cooling, the mixture was quenched with 5% ammonium hydroxide (20 mL), diluted with CH2Cl2 (20 mL) and filtered through a plug of Celite with the aid of CH2Cl2. The filtrate was concentrated under reduced pressure and the residue of 1 was purified by column chromatography on silica gel eluting with EtOAc/petroleum ether/Et3N (1:1:0.1); yield 0.17 g, (54%); a white solid; mp 195–197°C (Lit. [8] mp 194–196°C); 1H NMR: δ 8.00 (s, 1H), 7.54 (m, 3H), 7.26 (d, J=6.2 Hz, 2H), 7.04 (d, J=9.2 Hz, 1H), 4.09 (s, 2H), 2.95 (s, 3H), 2.88 (s, 3H), 2.40 (s, 3H), 2.35 (s, 3H); ESI-MS: m/z 308.2, [M+H]+. Zolpidem (1) was obtained in 83% yield following isolation of the intermediate imine 5.
A 10-g scale synthetic procedure for the hemitartrate salt of N,N-dimethyl-2-(2-(p-tolyl)imidazo[1,2-a]pyridin-3-yl)acetamide (zolpidem hemitartrate)
A round-bottom flask was equipped with a rubber septum, a condenser, and a stirring bar. The vessel was charged with the imine 5 (10.5 g, 0.05 mol), propiolamide 4 (9.7 g, 0.10 mol), CuI (4.8 g, 0.025 mol), BINOL (7.2 g, 0.025 mol) and anhydrous toluene (100 mL) under argon and the mixture was heated to reflux on an oil bath for 36 h. The mixture was charged with ammonium hydroxide (5%, 100 mL), water and CH2Cl2 (100 mL) and then filtered through a plug of Celite with the aid of CH2Cl2. Then the aqueous phase was extracted with CH2Cl2 (2×100 mL). The combined organic phases were washed with water (200 mL) and brine (200 mL), dried with MgSO4 and concentrated under reduced pressure. The dark residue was dissolved in 2-propanol (200 mL), tartaric acid (7.5 g, 0.05 mol) was added, and then the mixture was heated under reflux. After cooling, the resultant brown solid was filtered and rinsed with ethyl acetate. The crude product (about 11.7 g) was crystallized from 2-propanol ( about 160 mL) to afford zolpidem hemitartrate (8.5 g, 37%) as a white powder; mp 196–197°C (the authentic sample: mp 196–197°C [8], [17]); 1H NMR (DMSO-d6): δ 8.04 (s, 1H), 7.5–7.4 (m, 3H), 7.26 (d, J=7.2 Hz, 2H), 7.12 (d, J=8.8 Hz, 1H), 4.29 (s, 2H), 4.14 (s, 2H), 3.13 (s, 3H), 2.89 (s, 3H), 2.34 (s, 3H), 2.30 (s, 3H); 13C NMR (DMSO-d6): δ 172.9, 167.9, 142.6, 142.2, 136.4, 131.6, 129.0, 127.4, 127.0, 122.2, 120.6, 115.6, 115.1, 71.9, 36.8, 35.1, 28.7, 20.6, 17.6. ESI-MS: m/z 308.2, [M+H]+.
Ethyl 2-(2-(p-tolyl)imidazo[1,2-a]pyridin-3-yl)acetate (9)
A round-bottom flask was equipped with a rubber septum, a condenser, and a stirring bar. The vessel was charged with imine 5 (0.11 g, 0.5 mmol), ethyl propiolate 8 (102 μL, 1.0 mmol), CuI (0.05 g, 0.25 mmol), BINOL (0.07 g, 0.25 mmol) and anhydrous toluene (10 mL) under argon, and heated to reflux on an oil bath for 36 h. After cooling, the mixture was filtered through a plug of Celite with the aid of CH2Cl2. The filtrate was concentrated under reduced pressure to give crude product, which was purified by column chromatography eluting with petroleum ether/acetone/Et3N 4:1:0.25) to give 9 (0.09 g, 58%) as a white solid; mp 127–128°C (Lit. [8], [17] mp 128°C); 1H NMR: δ 7.88 (s, 1 H), 7.72 (d, J=7.9 Hz, 2 H), 7.56 (d, J=9.0 Hz, 1 H), 7.28 (d, J=7.9 Hz, 2H), 7.07 (d, J=9.0 Hz, 1 H), 4.23 (q, J=7.0 Hz, 2 H), 4.01 (s, 2H), 2.41 (s, 3 H), 2.37 (s, 3 H), 1.29 (t, J=7.0 Hz, 3 H), MS (ESI) m/z 309.2 [M+H]+.
Methyl 2-(2-(p-tolyl)imidazo[1,2-a]pyridin-3-yl)acetate (11)
By using the procedure described above, compound 11 was prepared in 53% yield as a white solid; mp 134–136°C; 1H NMR: δ 7.88 (s, 1 H), 7.70 (d, J=8.0 Hz, 2 H), 7.54 (d, J=9.2 Hz, 1 H), 7.27 (d, J=8.0 Hz, 2H), 7.07 (d, J=9.2 Hz, 1 H), 4.01 (s, 2 H), 3.75 (s, 3H), 2.39 (s, 3 H), 2.35 (s, 3 H); 13C NMR: δ 169.4, 143.8, 143.4, 136.9, 130.7, 128.7, 127.7, 126.8, 121.3, 120.5, 116.2, 111.5, 51.8, 29.9, 20.6, 17.8. ESI-MS: m/z 295.1, [M+H]+.
Acknowledgments
We gratefully acknowledge National Natural Science Foundation of China (Grant 21472024) for the research financial support. We are grateful to Prof. Bing Wang and Dr. Yanlong Kang for their helpful discussion.
References
[1] Lemmer, B. The sleep-wake cycle and sleeping pills. Physiol. Behav.2007, 90, 285–293.10.1016/j.physbeh.2006.09.006Search in Google Scholar PubMed
[2] Holm, K. J.; Goa, K. L. Zolpidem. Drugs2000, 59, 865–889.10.2165/00003495-200059040-00014Search in Google Scholar PubMed
[3] Sumalatha, Y.; Reddy, T. R.; Reddy, P. R.; Satyanarayana, B. A simple and efficient synthesis of hypnotic agent, zolpidem and its related substances. ARKIVOC2009, 2009, 315–320.10.3998/ark.5550190.0010.230Search in Google Scholar
[4] Trapani, G.; Franco, M.; Ricciardi, L.; Latrofa, A.; Genchi, G.; Sanna, E.; Tuveri, F.; Cagetti, E.; Biggio, G.; Liso, G. Synthesis and binding affinity of 2-phenylimidazo[1,2-a]pyridine derivatives for both central and peripheral benzodiazepine receptors: a new series of high-affinity and selective ligands for the peripheral type. J. Med. Chem.1997, 40, 3109–3118.10.1021/jm970112+Search in Google Scholar PubMed
[5] Patil, S. S.; Patil, S. V.; Bobade, V. D. An efficient synthesis of zolpidem. Org. Prep. Proc. Int.2011, 43, 260–264.10.1080/00304948.2011.564558Search in Google Scholar
[6] Guetzoyan, L.; Nikbin, N.; Baxendale, I. R.; Ley, S. V. Flow chemistry synthesis of zolpidem, alpidem and other GABAA agonists and their biological evaluation through the use of in-line frontal affinity chromatography. Chem. Sci.2013, 4, 764–769.10.1039/C2SC21850JSearch in Google Scholar
[7] Nair, D. K.; Mobin, S. M.; Namboothiri, I. N. N. Synthesis of imidazopyridines from the Morita-Baylis-Hillman acetates of nitroalkenes and convenient access to Alpidem and Zolpidem. Org. Lett.2012, 14, 4580–4583.10.1021/ol3020418Search in Google Scholar PubMed
[8] Chernyak, N.; Gevorgyan, V. General and efficient copper-catalyzed three-component coupling reaction towards imidazoheterocycles: one-pot synthesis of alpidem and zolpidem. Angew. Chem. Int. Ed.2010, 49, 2743–2746.10.1002/anie.200907291Search in Google Scholar PubMed PubMed Central
[9] Bagdi, P. R.; Basha, R. S.; Khan, A. T. Synthesis of 2-triazolyl-imidazo[1,2-a]pyridine through a one-pot three-component reaction using a nano copper oxide assisted click-catalyst. RSC Adv.2015, 5, 61337–61344.10.1039/C5RA09671ESearch in Google Scholar
[10] Bharate, J. B.; Guru, S. K.; Jain, S. K.; Meena, S.; Singh, P. P.; Bhushan, S.; Singh, B.; Bharate, S. B.; Vishwakarma, R. A. Cu–Mn spinel oxide catalyzed synthesis of imidazo[1,2-a]pyridines through domino three-component coupling and 5-exo-dig cyclization in water. RSC Adv.2013, 3, 20869–20876.10.1039/c3ra42046aSearch in Google Scholar
[11] Guchhait, S. K.; Chandgude, A. L.; Priyadarshani, G. CuSO4-glucose for in situ generation of controlled Cu(I)-Cu(II) bicatalysts: multicomponent reaction of heterocyclic azine and aldehyde with alkyne, and cycloisomerization toward synthesis of N-fused imidazoles. J. Org. Chem.2012, 77, 4438–4444.10.1021/jo3003024Search in Google Scholar PubMed
[12] Mishra, S.; Ghosh, R. Mechanistic studies on a new catalyst system (CuI-NaHSO4×SiO2) leading to the one-pot synthesis of imidazo[1,2-a]pyridines from reactions of 2-aminopyridines, aldehydes, and terminal alkynes. Synthesis2011, 2011, 3463–3470.10.1002/chin.201211135Search in Google Scholar
[13] Luz, I; Xamena, F. X. L. I.; Corma, A. Bridging homogeneous and heterogeneous catalysis with MOFs: Cu-MOFs as solid catalysts for three-component coupling and cyclization reactions for the synthesis of propargylamines, indoles and imidazopyridines. J. Catal.2012, 285, 285–91.10.1016/j.jcat.2011.10.001Search in Google Scholar
[14] Reddy, B. V. S.; Reddy, P. S.; Reddy, Y. J.; Yadav, J. S. InBr3-catalyzed three-component, one-pot synthesis of imidazo[1,2-a]pyridines. Tetrahedron Lett.2011, 52, 5789–5793.10.1016/j.tetlet.2011.08.110Search in Google Scholar
[15] Liu, P.; Fang, L.-S.; Lei, X.; Lin, G.-Q. Synthesis of imidazo[1,2-a]pyridines via three-component reaction of 2-aminopyridines, aldehydes and alkynes. Tetrahedron Lett.2010, 51, 4605–4608.10.1002/chin.201051146Search in Google Scholar
[16] Liu, P.; Deng, C.-L.; Lei, X.; Lin, G.-Q. Tandem amination/cycloisomerization of aryl propargylic alcohols with 2-aminopyridines as an expedient route to imidazo[1,2-a]pyridines. Eur. J. Org. Chem.2011, 2011, 7308–7316.10.1002/ejoc.201101053Search in Google Scholar
[17] Rassokhina, I. V.; Shirinian, V. Z.; Zavarzin, I. V.; Gevorgyan, V.; Volkova, Y. A. Copper(II)-mediated aerobic synthesis of imidazo[1,2-a]pyridines via cascade aminomethylation/cycloisomerization of alkynes. J. Org. Chem.2015, 80, 11212–11218.10.1021/acs.joc.5b02102Search in Google Scholar PubMed
[18] Oakdale, J. S.; Sit, R. K.; Valery, V.; Fokin, V. V. Ruthenium-catalyzed cycloadditions of 1-haloalkynes with nitrile oxides and organic azides: synthesis of 4-haloisoxazoles and 5-halotriazoles. Chem. Eur. J.2014, 20, 11101–11110.10.1002/chem.201402559Search in Google Scholar PubMed PubMed Central
[19] Burkhouse, D. W. W.; Zimmer, H. New synthetic methodology for the preparation of substituted 1-aryl-1-pyridylaminomethanephosphonic acid ester. Synth. Commun.1988, 18, 1437.10.1080/00397918808078815Search in Google Scholar
[20] Zani, L.; Alesi, S.; Cozzi, P. G.; Bolm, C. Dimethylzinc-mediated alkynylation of imines. J. Org. Chem.2006, 71, 1558–1562.10.1021/jo052273oSearch in Google Scholar PubMed
[21] Zhou, Y.; Lecourt, T.; Micouin, L. Room temperature Lewis base-catalyzed alumination of terminal alkynes. Adv. Syn. Catal.2009, 351, 2595–2598.10.1002/adsc.200900414Search in Google Scholar
[22] Spitz, C.; Lohier, J.-F.; Reboul, V.; Metzner, P. Catalytic generation of cesium acetylide by CsF: synthesis of 1,3-benzothiazines from cyclic sulfenamides. Org. Lett.2009, 11, 2776–2779.10.1021/ol9009333Search in Google Scholar PubMed
[23] Sun, Z.; Yu, S.; Ding, Z.; Ma, D. Enantioselective addition of activated terminal alkynes to 1-acylpyridinium salts catalyzed by Cu−bis(oxazoline) complexes. J. Am. Chem. Soc.2007, 129, 9300–9301.10.1021/ja0734849Search in Google Scholar PubMed
[24] Siemsen, P.; Livingston, R. C.; Diederich, F. Acetylenic coupling: a powerful tool in molecular construction. Angew. Chem. Int. Ed.2000, 39, 2633–2657.10.1002/1521-3773(20000804)39:15<2632::AID-ANIE2632>3.0.CO;2-FSearch in Google Scholar
©2017 Walter de Gruyter GmbH, Berlin/Boston
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Articles in the same Issue
- Frontmatter
- Preliminary Communications
- One-pot synthesis of benzopyrans catalyzed by silica supported dual acidic ionic liquid under solvent-free conditions
- An efficient synthesis of 5-halo-6-trifluoromethylpyridine-3-carbonitriles and carboxylic acids
- Research Articles
- Novel 2H-1,3-benzoxazine ring formation by intramolecular heterocyclization of N-(α-aryloxyalkyl)imidoyl chlorides
- Green synthesis of novel 2-pyrazolyl-1,3-thiazolidine-4-ones using 2-oxoimidazolidine-1,3-disulfonic acid
- A facile synthesis of (E)-2-(aryl/hetaryl)vinyl-4-phenylquinoline-3-carboxylic acids
- Efficient synthesis of new pyrano[3,2-b]pyran derivatives via Fe3O4@SiO2-IL-Fc catalyzed three-component reaction
- An improved and scalable synthesis of zolpidem via a CuI/BINOL-mediated tandem reaction of imine and alkyne
- Scalable synthesis and properties of 7-methyl- 4-azaindole
- Synthesis and insecticidal activities of novel 1H-pyrazole-5-carboxylic acid derivatives
Articles in the same Issue
- Frontmatter
- Preliminary Communications
- One-pot synthesis of benzopyrans catalyzed by silica supported dual acidic ionic liquid under solvent-free conditions
- An efficient synthesis of 5-halo-6-trifluoromethylpyridine-3-carbonitriles and carboxylic acids
- Research Articles
- Novel 2H-1,3-benzoxazine ring formation by intramolecular heterocyclization of N-(α-aryloxyalkyl)imidoyl chlorides
- Green synthesis of novel 2-pyrazolyl-1,3-thiazolidine-4-ones using 2-oxoimidazolidine-1,3-disulfonic acid
- A facile synthesis of (E)-2-(aryl/hetaryl)vinyl-4-phenylquinoline-3-carboxylic acids
- Efficient synthesis of new pyrano[3,2-b]pyran derivatives via Fe3O4@SiO2-IL-Fc catalyzed three-component reaction
- An improved and scalable synthesis of zolpidem via a CuI/BINOL-mediated tandem reaction of imine and alkyne
- Scalable synthesis and properties of 7-methyl- 4-azaindole
- Synthesis and insecticidal activities of novel 1H-pyrazole-5-carboxylic acid derivatives