Stereoselective Michael addition of O-nucleophiles to carbohydrate-based nitro-olefin
-
Ilgvalds Ivanovs
 ,
Sergey Belyakov
,
Sergey Belyakov


Abstract
Michael addition reactions of O-nucleophiles to C(3) exocyclic nitromethylene derivative of diacetone glucose are reported. The reactions with primary alcohols proceed at ambient temperature in the presence of different bases with good yields and give products with excellent diastereoselectivity. The addition of the nucleophile occurs from the β-face of the carbohydrate as shown by single crystal X-ray analysis. The reactions with secondary alcohols give low yields of products while phenolic compounds do not react. Under certain conditions, isomerization of starting material is observed.
Introduction
Conjugated nitro-alkene moieties proved their utility as Michael acceptors [1]. The products, nitro-alkanes, are widely used for the syntheses of hetero- and carbocycles [2, 3]. In the carbohydrate chemistry, nitro-olefins are used for the syntheses of various derivatives. Thus, endocyclic nitro glycal 1 when subjected to Michael addition gives various glycosides, thioglycosides, and N-, P-, and C-glycosyl compounds (Scheme 1) [4–12]. Depending on the stereochemistry and the substituents of the substrate as well as on the reaction conditions both α- and β-anomers can be obtained [9, 11].

2-Nitroglycal 1 as substrate for glycoside syntheses.
On the other hand, there are only few examples in carbohydrate chemistry where exocyclic nitro-olefins are used. Nitro-olefin 3 was originally synthesized by Albrecht and Moffatt from nitro-alcohol 2 (Scheme 2) [13–17]. Recently, we have shown that under modified work-up conditions alkene 3 undergoes a reaction with in situ generated S-nucleophile to form sulfide 4 (Scheme 2) [18]. The adduct 4 and other products of hetero-Michael addition to nitro olefin 3 are used for the syntheses of azaheterocycles, amines, amino acids, and complex glycohybrids [17, 19–23].

Synthesis of nitro-alkene 3.
In order to broaden the scope of the Michael addition reaction and to pave the way for further syntheses of heterocyclic compounds based on this scaffold, we have explored a reactivity of alcohols and phenols as O-nucleophiles towards the addition to the nitro-olefin 3.
Results and discussion
It was observed that relatively small nucleophiles, such as ethoxide anion, undergo a reaction with nitro-olefin 3 in DMF solution (Scheme 3). The adduct 5a was obtained in a crystalline form and its structure was unambiguously established by single crystal X-ray diffraction analysis (Figure 1).
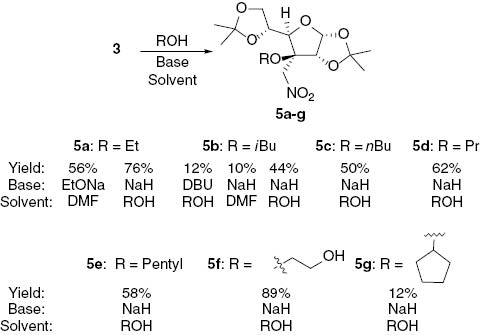
Synthesis of Michael adducts 5a–g.

Crystal structure of compound 5a.
Better yields were achieved with primary alcohols when the reaction was carried out in neat alcohol. To find the optimal reaction conditions, various bases were screened. Stoichiometric amounts of tertiary amines (triethylamine (Et3N), diisopropylethyl amine (DIPEA), 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU)), methyl magnesium chloride, and sodium hydride were tried. In the presence of DBU the adduct 5b was isolated in 12% yield. In the presence of DIPEA the degradation of the substrate with some traces of the needed product were observed. On the other hand, when Et3N in isobutanol was used, full conversion of the alkene under the same conditions was observed in 2 h. However, the analysis of the crude NMR spectrum revealed that the major product was a non-conjugated nitro-alkene 6 (Scheme 4). To establish a synthetically useful procedure for the process 3 → 6 the reaction conditions were varied. Thus, nitro-alkene 2 in the isopropanol solution completely isomerizes to nitro-alkene 6 (68% isolated yield) in the presence of 1 equivalent of triethylamine after 2 h at 50°C.

Conditions for the synthesis of nitro-alkene 6.
For the transformation 3 → 5 other bases were tried. Sodium hydride was found to be the most suitable base for deprotonation of the neat alcohols, and the highest yields were achieved using this approach (Scheme 3). For example, in the reaction with ethylene glycol the mono adduct 5f was formed exclusively in 89% yield. Other primary alcohols gave the corresponding products in 44–76% yields. The addition of neat cyclopentanol in the presence of NaH gave product 5g with 12% yield. Similarly, in the case of cyclohexanol the product was observed in the crude mixture in a low yield (<10%), but could not be isolated. With MeMgBr used as a base the decomposition of the starting material was observed and no product could be detected by analysis of NMR spectra of crude reaction mixtures. To our surprise, decomposition of the substrate was also observed if phenols were used as the nucleophiles regardless of the applied base.
Conclusions
It was shown that C(3)-quaternary 3-O-alkyl-3-nitromethylglucose derivatives can be synthesized by Michael addition of primary alcohols to glucose-derived exocyclic nitro-olefin in neat alcohols in the presence of sodium hydride. On the other hand, the use of secondary alcohols and phenols, other bases and solvents lowers the yields or prevents the addition reaction, and may result in isomerization of the double bond. The conditions for the synthesis of the non-conjugated nitro-alkene were optimized.
Experimental
IR spectra were recorded on a FT-IR Perkin Elmer Spectrum BX spectrometer in KBr pellets. 1H NMR (300 MHz) and 13C NMR (75 MHz) spectra were recorded in CDCl3 on a Bruker 300 spectrometer. Electrospray ionization high-resolution mass spectra (HRMS) were recorded on a Q-TOF Micromass spectrometer. Melting points were determined on a Fisher Digital Melting Point Analyzer Model 355 and are uncorrected. X-ray crystal structure analysis was done with a Nonius KappaCCD diffractometer (MoKα-radiation, graphite monochromator) at 173 K. Silica gel (60 Å, 40–63 μm, ROCC) was used for flash chromatography. Alcohols, sodium ethoxide, potassium t-butoxide, triethylamine, DIPEA, DBU, and sodium hydride were commercial products.
Isomerization of the nitro-olefin 3 to 3,4-dideoxy-1,2: 5,6-di-O-isopropylidene-3-C-nitromethyl-α-d-erythro-hex-3-enofuranose (6)
A solution of nitro-alkene 3 (110 mg, 0.37 mmol, 1.0 eq.) and triethylamine (50 μL, 0.37 mmol, 1.0 eq.) in isopropanol (2 mL) was stirred for 2 h at 50°C, then treated with hexanes/EtOAc (10 mL, 1:1 v/v). The mixture was washed with 1 m H2SO4 aqueous solution (3×5 mL) and brine (3×5 mL), dried over sodium sulfate, filtered and concentrated. The residue was purified by column chromatography on silica eluting with hexanes/EtOAc. Product 6 was obtained as a colorless oil (74 mg, 68%). The NMR spectra are consistent with those reported earlier [16].
General procedure for synthesis of 5a–g
A suspension of NaH in mineral oil (60%, 16 mg, 0.4 mmol, 1.1 eq.) was added to the selected alcohol (1 mL) at ambient temperature, the mixture was stirred for 10 min and then treated with nitro-olefin 3 (110 mg, 0.37 mmol, 1.0 eq.) in the same alcohol (0.5 mL). The mixture was stirred at ambient temperature for an additional 30 min. After consumption of the starting material 3 (TLC control) the mixture was neutralized with Dowex 50WX8 ion exchange resin (H+ form), filtered and concentrated under reduced pressure. A solution of the oily residue in EtOAc (20 mL) was washed with brine (3×5 mL), dried over sodium sulfate, filtered and concentrated. The product was purified by column chromatography on silica eluting with hexanes/EtOAc.
3-O-Ethyl-3-C-nitromethyl-1,2;5,6-di-O-isopropylidene-α-d-glucofuranose (5a)
Yield 103 mg (76%); mp 65–66°C; Rf 0.5 (hexanes/EtOAc, 3:1); IR: 2990, 2865, 1560, 1380, 1070, 840 cm-1; 1H NMR: δ 5.86 (d, 1H, J = 3.6, H-C(1)), 5.05 (d, 1H, J = 14.3 Hz, Ha-C(3′)), 5.00 (d, 1H, J = 3.6 Hz, H-C(2)), 4.72 (d, 1H, J = 14.3 Hz, Hb-C(3′)), 4.22 (ddd, 1H, J = 6.1, 5.1, 4.6, Hz, H-C(5)), 4.12 (dd, 1H, J = 8.7, 6.1 Hz, Ha-C(6)), 3.96 (dd, 1H, J = 8.7, 5.1 Hz, Hb-C(6)), 3.79–3.64 (m, 3H, H-C(4), H-C(a)), 1.52, 1.42, 1.35 (3s, 12H, (H3C)2-C-), 1.18 (t, 3H, J = 7.2 Hz, H-C(b)); 13C NMR: δ 112.5, 109.9, 104.8, 84.6, 83.0, 82.6, 74.5, 72.3, 67.9, 61.4, 27.1, 27.0, 26.4, 25.3, 15.9. HRMS. Calcd for [C15H25NO8 + H]+: m/z 348.1653. Found: m/z 348.1655.
The data of the single crystal X-ray diffraction analysis were deposited at the Cambridge Crystallographic Data Centre as supplementary publication with the following deposition number: CCDC 1443541. Copies of the data can be obtained, free of charge, on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK.
3-O-Isobutyl-3-C-nitromethyl-1,2;5,6-di-O-isopropylidene- α-d-glucofuranose (5b)
Yield 63 mg (44%); a colorless oil; Rf 0.65 (hexanes/EtOAc, 3:1); IR: 2985, 2875, 1560, 1420, 1375, 1335, 1165, 1075, 1000 cm-1; 1H NMR: δ 5.86 (d, 1H, J = 3.6, H-C(1)), 5.03 (d, 1H, J = 14.3 Hz, Ha-C(3′)), 5.02 (d, 1H, J = 3.6 Hz, H-C(2)), 4.73 (d, 1H, J = 14.3 Hz, Hb-C(3′)), 4.25 (ddd, 1H, J = 8.7, 6.3, 5.5, Hz, H-C(5)), 4.12 (dd, 1H, J = 8.7, 6.3 Hz, Ha-C(6)), 3.94 (dd, 1H, J = 8.7, 5.1 Hz, Hb-C(6)), 3.75 (d, 1H, J = 8.7 Hz, H-C(4), H-C(a)), 3.39 (d, 2H, J = 6.4 Hz, H2C(a)), 1.81 (sept., 1H, J = 6.4 Hz, HC(b)), 1.53, 1.43, 1.37, 1.35 (4s, 12H, (H3C)2-C-), 0.89 (d, 6H, J = 6.8 Hz, H3C(c)); 13C NMR: δ 112.6, 109.9, 104.9, 84.5, 82.8, 82.6, 74.9, 72.1, 72.0, 68.0, 29.3, 27.1, 27.0, 26.5, 25.3, 19.3 (2C). HRMS. Calcd for [C17H29NO8 + Na]+: m/z 398.1792. Found: m/z 398.1791.
3-O-Butyl-3-C-nitromethyl-1,2;5,6-di-O-isopropylidene-α- d-glucofuranose (5c)
Yield 68 mg (50%); a colorless oil; Rf 0.4 (hexanes/EtOAc, 3:1); IR: 2985, 2875, 1560, 1420, 1375, 1335, 1165, 1075, 1000 cm-1; 1H NMR: δ 5.86 (d, 1H, J = 3.6, H-C(1)), 5.03 (d, 1H, J = 14.3 Hz, Ha-C(3′)), 5.00 (d, 1H, J = 3.6 Hz, H-C(2)), 4.71 (d, 1H, J = 14.3 Hz, Hb-C(3′)), 4.11 (dd, 1H, J = 8.7, 6.2 Hz, Ha-C(6)), 3.91–3.83 (dd, 1H, J = 8.7, 5.3 Hz, Hb-C(6)), 3.73 (d, 1H, J = 8.7 Hz, H-C(4)), 3.65 (dd, 2H, J = 6.3, 2.0 Hz, H2C(a)), 1.54, 1.42, 1.36, 1.35 (4s, 12H, (H3C)2-C-), 1.54–1.34 (m, 4H, H2C(b), H2C(c)) 0.89 (t, 3H, J = 6.6 Hz, H3C(d)); 13C NMR: δ 112.5, 109.9, 105.9, 84.4, 82.8, 82.7, 74.7, 72.2, 68.0, 65.5, 27.1, 27.0, 26.5, 25.5, 19.3, 14.0. HRMS. Calcd for [C17H29NO8 + Na]+: m/z 398.1791. Found: m/z 398.1805.
3-C-Nitromethyl-1,2;5,6-di-O-isopropylidene-3-O-propyl-α- d-glucofuranose (5d)
Yield 60 mg (62%); a colorless oil; Rf 0.65 (hexanes/EtOAc, 3:1); IR: 2985, 1555, 1455, 1420, 1375, 1250, 1165, 1075, 1000 cm-1; 1H NMR: 5.86 (d, 1H, J = 3.6, H-C(1)), 5.03 (d, 1H, J = 14.3 Hz, Ha-C(3′)), 5.01 (d, 1H, J = 3.6 Hz, H-C(2)), 4.73 (d, 1H, J = 14.3 Hz, Hb-C(3′)), 4.24 (ddd, 1H, J = 8.7, 6.3, 5.5, Hz, H-C(5)), 4.12 (dd, 1H, J = 8.7, 6.3 Hz, Ha-C(6)), 3.94 (dd, 1H, J = 8.7, 5.1 Hz, Hb-C(6)), 3.73 (d, 1H, J = 8.7 Hz, H-C(4), H-C(a)), 3.58 (t, 2H, J = 7.3 Hz, H2C(a)), 1.65–1.53 (m, 2H, H2C(b)), 1.52, 1.41, 1.35, 1.34 (4s, 12H, (H3C)2-C-), 0.89 (t, 6H, J = 7.3 Hz, H3C(c)). 13C NMR: δ 112.5, 109.9, 104.8, 84.4, 82.7, 74.7, 72.2, 67.9, 67.3, 62.1, 27.1, 27.0, 26.4, 25.3, 23.6, 10.6. HRMS. Calcd for [C16H27NO8 + Na]+: m/z 384.1634. Found: m/z 384.1632.
3-C-Nitromethyl-1,2;5,6-di-O-isopropylidene-3-O-pentyl-α- d-glucofuranose (5e)
Yield 83 mg (58%); a colorless oil; Rf 0.65 (hexanes/EtOAc, 3:1); IR: 2985, 2875, 1560, 1455, 1420, 1375, 1335, 1250, 1215, 1165, 1075, 1000. 1H NMR: δ 5.85 (d, 1H, J = 3.6, H-C(1)), 5.03 (d, 1H, J = 14.3 Hz, Ha-C(3′)), 5.00 (d, 1H, J = 3.6 Hz, H-C(2)), 4.71 (d, 1H, J = 14.3 Hz, Hb-C(3′)), 4.22 (ddd, 1H, J = 8.7, 6.3, 5.5, Hz, H-C(5)), 4.11 (dd, 1H, J = 8.7, 6.3 Hz, Ha-C(6)), 3.94 (dd, 1H, J = 8.7, 5.1 Hz, Hb-C(6)), 3.73 (d, 1H, J = 8.7 Hz, H-C(4), H-C(a)), 3.62 (dt, 2H, J = 6.4, 1.8 Hz, H2C(a)), 1.62–1.52 (m, 2H, J = 6.4 Hz, HC(b)), 1.51, 1.41, 1.35, 1.34 (4s, 12H, (H3C)2-C), 1.32–1.26 (m, 4H, H2C(c), H2C(d)) 0.88 (t, 3H, J = 6.6 Hz, H3C(e)); 13C NMR: δ 112.6,109.9, 104.8, 84.4, 82.8, 82.7, 74.7, 72.2, 68.0, 65.7, 30.0, 28.2, 27.1, 26.9, 26.5, 25.3. HRMS. Calcd for [C18H31NO8 + Na]+: m/z 412.1947. Found: m/z 412.1951.
3-O-(2-Hydroxy)ethyl-3-C-nitromethyl-1,2;5,6-di-O-isopropylidene-α-d-glucofuranose (5f)
Yield 120 mg (89%); a colorless oil; Rf 0.3 (hexanes/EtOAc, 1:1); IR: 3445, 2985, 2940, 2885, 1560, 1455, 1420, 1375, 1335, 1250, 1220, 1165, 1075, 1000 cm-1; 1H NMR: δ 5.89 (d, 1H, J = 3.5, H-C(1)), 5.06 (d, 1H, J = 14.5 Hz, Ha-C(3′)), 5.03 (d, 1H, J = 3.5 Hz, H-C(2)), 4.71 (d, 1H, J = 14.3 Hz, Hb-C(3′)), 4.31 (ddd, 1H, J = 9.0, 6.3, 4.7, Hz, H-C(5)), 4.13 (dd, 1H, J = 9.0, 6.3 Hz, Ha-C(6)), 3.96 (dd, 1H, J = 9.0, 4.7 Hz, Hb-C(6)), 3.84–3.68 (m, 5H, H-C(4), H2C(a), H2C(b)), 2.10 (s, 1H, HO-C(b)), 1.52, 1.42, 1.35 (3s, 12H, (H3C)2-C-); 13C NMR: δ 112.6, 110.0, 104.7, 82.9, 82.4, 81.2, 74.7, 71.9, 68.6, 68.0, 62.0, 30.0, 26.9 (2C), 26.3, 25.2. HRMS. Calcd for [C15H25NO9 + H]+: m/z 364.1653. Found: m/z 364.1655.
3-O-Cyclopentyl-3-C-nitromethyl-1,2;5,6-di-O-isopropylidene- α-d-glucofuranose (5g)
Yield 17 mg (12%); a colorless oil; Rf 0.3 (hexanes/EtOAc, 1:1); IR: 2985, 2940, 2885, 2875, 1555, 1455, 1420, 1375, 1335, 1255, 1215, 1165, 1075, 1005 cm-1; 1H NMR: δ 5.86 (d, 1H, J = 3.5, H-C(1)), 5.04 (d, 1H, J = 3.5, H-C(2)), 4.97 (d, 1H, J = 14.2 Hz, Ha-C(3′)), 4.73 (d, 1H, J = 14.2 Hz, Hb-C(3′)), 4.28 (m, 2H, H-C(5), H-C(a)), 4.10 (dd, 1H, J = 8.7, 6.3 Hz, Ha-C(6)), 3.95 (dd, 1H, J = 8.7, 5.3 Hz, Hb-C(6)), 3.76 (d, 1H, J = 8.3 Hz, H-C(4)), 1.84–1.59 (m, 6H, H2C(b), Ha-C(c)), 1.52 (s, 3H), 1.48 (m, 2H, Hb-C(c)), 1.42 (s, 3H, (H3C)2-C-), 1.37 (s, 3H), 1.35 (s, 3H, (H3C)2-C-); 13C NMR: δ 112.5, 109.7, 104.8, 85.0, 82.8 (2C), 77.6, 75.5, 72.2, 67.8, 35.1, 35.0, 27.1, 26.9, 26.5, 25.3, 23.9 (2C). HRMS. Calcd for [C18H29NO8 + Na]+: m/z 410.1785. Found: m/z 410.1769.
References
[1] Zard, S. S. Some aspects of the chemistry of nitro compounds. Helv. Chim. Acta2012, 95, 1730–1757.10.1002/hlca.201200324Search in Google Scholar
[2] Halimehjani, A. Z.; Namboothiri, I. N. N.; Hooshmand, S. E. Part I: Nitroalkenes in the synthesis of heterocyclic compounds. RSC Adv. 2014, 4, 48022–48084.10.1039/C4RA08828JSearch in Google Scholar
[3] Halimehjani, A. Z.; Namboothiri, I. N. N.; Hooshmand, S. E. Nitroalkenes in the synthesis of carbocyclic compounds. RSC Adv. 2014, 4, 31261–31299.10.1039/C4RA04069DSearch in Google Scholar
[4] For review on 2-nitro glycals, see: Delaunay, T.; Poisson, T.; Jubault, P.; Pannecoucke, X. 2-Nitroglycals: Versatile Building Blocks for the Synthesis of 2-Aminoglycosides. Eur. J. Org. Chem. 2014, 2014, 7525–7546.10.1002/ejoc.201402805Search in Google Scholar
[5] Takamoto, T.; Sudoh, R.; Nakagawa, T. Studies on nitro sugars: Part II. Synthesis of benzyl 3,5-DI-O-benzyl-2-deoxy-2-Nitro-α-D-xylofuranoside and its transglycosylation with alkali. Carbohydr. Res. 1973, 27, 135–140.10.1016/S0008-6215(00)82432-5Search in Google Scholar
[6] Sakakibara, T.; Tachimori, Y.; Sudoh, R. Stereoselective nucleophilic addition reactions to nitro sugars. Tetrahedron Lett. 1982, 23, 5545–5548.10.1016/S0040-4039(00)85890-6Search in Google Scholar
[7] Holzapfel, C. W.; Marais. C. F.; van Dyk, M. S. 2-Nitroglycals preparation and nucleophilic addition reactions. Synth. Commun. 1988, 18, 97–114.10.1080/00397918808057825Search in Google Scholar
[8] Winterfeld, G. A.; Das, J.; Schmidt, R. R. Convenient synthesis of nucleosides of 2-Deoxy-2-nitro-D-galactose andN-Acetyl-D-galactosamine. Eur. J. Org. Chem. 2000, 2000, 3047–3050.10.1002/1099-0690(200009)2000:17<3047::AID-EJOC3047>3.0.CO;2-6Search in Google Scholar
[9] Schmidt, R. R.; Behrendt, M.; Toepfer, A. Nitriles as solvents in glycosylation reactions: highly selective β-Glycoside synthesis. Synlett1990, 1990, 694–696.10.1055/s-1990-21214Search in Google Scholar
[10] Pachamuthu, K.; Gupta, A.; Das, J.; Schmidt, R. R.; Vankar, Y. D. An easy route to 2-Amino-β-C-Glycosides by conjugate addition to 2-Nitroglycals. Eur. J. Org. Chem. 2002, 2002, 1479–1483.10.1002/1099-0690(200205)2002:9<1479::AID-EJOC1479>3.0.CO;2-PSearch in Google Scholar
[11] Pachamuthu, K.; Figueroa-Perez, I.; Ali, I.; Schmidt, R. R. Synthesis of glycosyl phosphonates by michael-Type addition to 2-Nitroglycals. Eur. J. Org. Chem. 2004, 2004, 3959–3961.10.1002/ejoc.200400266Search in Google Scholar
[12] Delaunay, T.; Poisson, T.; Jubault, P.; Pannecoucke, X. Stereoselective access to β-C-Glycosamines by nitro-michael addition of organolithium reagents. Eur. J. Org. Chem., 2014, 2014, 3341–3345.10.1002/ejoc.201402001Search in Google Scholar
[13] Albrecht, H. P.; Moffatt, J. G. Synthesis of a branched chain aminosugar nucleoside. Tetrahedron Lett. 1970, 11, 1063–1066.10.1016/S0040-4039(01)97908-0Search in Google Scholar PubMed
[14] Hart, D. J.; Patterson, S.; Unch, J. P. Lasonolide A: synthesis of A and B rings via a cycloetherification strategy. Synlett. 2003, 2003, 1334–1338.10.1055/s-2003-40323Search in Google Scholar
[15] Turks, M.; Rodins, V.; Rolava, E.; Ostrovskis, P.; Belyakov, S. A practical access to glucose- and allose-based (5+5) 3-spiropseudonucleosides from a common intermediate. Carbohydr. Res. 2013, 375, 5–15.10.1016/j.carres.2013.04.008Search in Google Scholar PubMed
[16] Turks, M.; Vēze, K.; Kiseļovs, G.; Mackeviča, J.; Lugiņina, J.; Mishnev, A.; Marković, D. Synthesis and X-ray studies of novel 3-C-nitromethyl-hexofuranoses. Carbohydr. Res. 2014, 391, 82–88.10.1016/j.carres.2014.03.003Search in Google Scholar PubMed
[17] Turks, M.; Rolava, E.; Stepanovs, D.; Mishnev, A.; Marković, D. Novel 3-C-aminomethyl-hexofuranose-derived thioureas and their testing in asymmetric catalysis. Tetrahedron: Asymmetry2015, 26, 952–960.10.1016/j.tetasy.2015.07.003Search in Google Scholar
[18] Lugiņina, J.; Rjabovs, V.; Belyakov, S.; Turks, M. On moffatt dehydration of glucose-derived nitro alcohols. Carbohydr. Res. 2012, 350, 86–89.10.1016/j.carres.2011.12.020Search in Google Scholar PubMed
[19] Review on nitro sugars: Estevez, A. M.; Wessel, H. P. Molecular recognition, syntheses and reactions of nitro sugars. Curr. Org. Chem., 2014, 18, 1846–1877.10.2174/1385272819666140527232115Search in Google Scholar
[20] Lugiņina, J.; Rjabovs, V.; Belyakov, S.; Turks, M. A concise synthesis of sugar isoxazole conjugates. Tetrahedron Lett. 2013, 54, 5328–5331.10.1016/j.tetlet.2013.07.103Search in Google Scholar
[21] Rjabovs, V.; Ostrovskis, P.; Posevins, D.; Kiseļovs, G.; Kumpiņš, V.; Mishnev, A.; Turks, M. Synthesis of building blocks for carbopeptoids and their triazole isoster assembly. Eur. J. Org. Chem. 2015, 2015, 5572–5584.10.1002/ejoc.201500695Search in Google Scholar
[22] Uzuleņa, J.; Rjabovs, V.; Moreno Vargas, A. J.; Turks, M. Synthesis of 1,2,3-triazole-linked glycohybrids in the gluco-, gulo-, and allopyranose series. Chem. Heterocycl. Compd. 2015, 51, 664–671.10.1007/s10593-015-1754-xSearch in Google Scholar
[23] Grigorjeva, J.; Uzuleņa, J.; Rjabovs, V.; Turks, M. Synthesis of monomeric methylene-linked 1,2,3-triazole glycoconjugates from allo- and glucofuranoses. Chem. Heterocycl. Compd. 2015, 51, 883–890.10.1007/s10593-015-1791-5Search in Google Scholar
©2016 by De Gruyter
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Articles in the same Issue
- Frontmatter
- Preliminary Communication
- A facile one-pot synthesis of aryl-substituted fused pyrimidinones
- Research Articles
- Three-component synthesis of new o-hydroxyphenyl-substituted pyrazolo[3,4-b]pyridines promoted by FeCl3
- Reactions of ferrocenyl chalcones with hydrazines and active methylene compounds
- Synthesis of 3-alkyl-5-allylamino-2-benzoylimino-1,3,4-thiadiazoles via Dimroth rearrangement
- Chiral oxazoline ligands with two different six-membered azaheteroaromatic rings – synthesis and application in the Cu-catalyzed nitroaldol reaction
- Stereoselective Michael addition of O-nucleophiles to carbohydrate-based nitro-olefin
- The synthesis of hydrophobic 1-alkyl-1H,1′H-2,2′-bibenzo[d]imidazoles
- Novel synthesis and reactions of pyrazolyl-substituted tetrahydrothieno[2,3-c]isoquinoline derivatives
- Acid-base properties and keto-enol equilibrium of a 5-substituted derivative of 1,3-diethyl-2-thiobarbituric acid
Articles in the same Issue
- Frontmatter
- Preliminary Communication
- A facile one-pot synthesis of aryl-substituted fused pyrimidinones
- Research Articles
- Three-component synthesis of new o-hydroxyphenyl-substituted pyrazolo[3,4-b]pyridines promoted by FeCl3
- Reactions of ferrocenyl chalcones with hydrazines and active methylene compounds
- Synthesis of 3-alkyl-5-allylamino-2-benzoylimino-1,3,4-thiadiazoles via Dimroth rearrangement
- Chiral oxazoline ligands with two different six-membered azaheteroaromatic rings – synthesis and application in the Cu-catalyzed nitroaldol reaction
- Stereoselective Michael addition of O-nucleophiles to carbohydrate-based nitro-olefin
- The synthesis of hydrophobic 1-alkyl-1H,1′H-2,2′-bibenzo[d]imidazoles
- Novel synthesis and reactions of pyrazolyl-substituted tetrahydrothieno[2,3-c]isoquinoline derivatives
- Acid-base properties and keto-enol equilibrium of a 5-substituted derivative of 1,3-diethyl-2-thiobarbituric acid