Preparation of optically active 4-substituted γ-lactones by lipase-catalyzed optical resolution
-
Yasutaka Shimotori
 ,
Masayuki Hoshi
,
Masayuki Hoshi

Abstract
Optically active 4-substituted γ-lactones (3 and 4) were synthesized effectively using lipase-catalyzed optical resolution. N-methyl-4-hydroxyalkanamides (rac-1a–i) as substrates were prepared from N-methylsuccinimide. The alkylation of N-methylsuccinimide using Grignard reagents generated from various alkyl halides followed by reduction resulted in N-methyl-4-hydroxyalkanamides. The optical resolution of rac-1a–g was performed using Novozym 435-catalyzed stereoselective acetylation. The stereoselective preparation of 4-substituted γ-lactones (3 and 4) possessing various side chains such as isopentyl, phenyl, and phenethyl groups was achieved with more than 90% enantiopurity.
Introduction
γ-Lactones are well-known natural flavor and fragrance compounds [1–3], pheromone components [4–7], and useful building blocks [8–11] for pharmaceutical synthesis. These lactones are present in a wide variety of natural products, such as mango [12, 13], peach [14, 15], strawberry [16, 17], Gouda cheese [18, 19], and other dairy products [20, 21]. The preparation of optically active compounds has become very important for the development of new biologically active substances containing one or several chiral centers, considering that many chiral drugs [22, 23] and agrochemicals [24, 25] display quite different activity and toxicity profiles with respect to their absolute configuration. Chiral lactones are important components in the synthesis of natural products and biologically active compounds, such as antitumor, antidepressant, and antiviral agents. Ghosh et al. [26] synthesized (+)-cryptophycin 52, a potent antimitotic antitumor agent, by using chiral 4-phenyl-γ-butyrolactone as a building block. This γ-lactone was also used for the preparation of other biologically active compounds [27, 28]. In addition, Kotkar et al. [29] reported the synthesis of (+)-harzialactone starting with chiral 5-phenyl-γ-pentalactone. This lactone exhibits strong antitumor and cytotoxic activities against cultured P388 cells. Here we report the preparation of various chiral γ-lactones by optical resolution using lipase-catalyzed enantioselective acetylation.
Results and discussion
Preparation of N-methyl-4- hydroxyalkanamides
We have previously reported the preparation of various N-methyl-4-hydroxyalkanamides from N-methylsuccinimide by the Grignard reaction and subsequent reductive reaction [30]. In this paper, the introduction of various functional groups was investigated (Scheme 1, Table 1). Alkylations with primary alkyl halides yielded the corresponding N-methyl-4-hydroxyalkanamides (rac-1a,e,f) in the yields ranging from 61% to 79% (entries 1, 5, and 6). With aryl bromides, N-methyl-4-hydroxyalkanamides (rac-1c,d,g) were synthesized with yields in the range of 87–94% (entries 3, 4, and 7). By contrast, the purification of N-methyl-4-hydroxyalkanamides (rac-1b,h,i) obtained from secondary and tertiary alkyl bromides was not satisfactory, and the crude yields were very low (entries 2, 8, and 9). These decreases in yield result from the side reactions such as the Wurtz reaction and olefin formation, and the low reactivity is caused by steric hindrance [31, 32]. Accordingly, the reactions with organolithium reagents were attempted (Scheme 2). The reactivity of organolithium reagents is higher than that of Grignard reagents and organozinc reagents [33]. Organolithium reagents can generally be used as nucleophiles. Many reports have been published about effective alkylation using organolithium reagents [34–39]. N-methyl-4-hydroxy-5,5-dimethylhexanamide (rac-1b) substituted with a tert-butyl group was synthesized with 44% yield using tert-butyllithium.

Synthesis of N-methyl-4-hydroxyalkanamides with Grignard reagent.
| Entry | R-X | Product | Yield (%) | Entry | R-X | Product | Yield (%) |
|---|---|---|---|---|---|---|---|
| 1 | 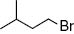 | rac-1a | 68 | 6 |  | rac-1f | 61 |
| 2 |  | rac-1b | 10a | 7 |  | rac-1g | 87 |
| 3 |  | rac-1c | 94 | 8 |  | rac-1h | 6a |
| 4 |  | rac-1d | 94 | 9 |  | rac-1i | 10a |
| 5 |  | rac-1e | 79 |
aCrude product.

Synthesis of N-methyl-4-hydroxy-5,5-dimethylhexanamide rac-1b.
Lipase-catalyzed enantioselective acetylation of N-methyl-4-hydroxyalkanamides
We have previously reported detailed studies of lipase-catalyzed enantioselective acetylation [30, 40]. The lipase-catalyzed acetylation was performed in diethyl ether using N-alkyl-4-hydroxyalkanamides as substrates, vinyl acetate as acyl donor, and Novozym 435 as lipase. These conditions result in high enantioselectivity, and both enantiomers of various γ-lactones have successfully been obtained with more than 99% enantiopurity. In that previous work, lipase screening has been examined using Novozym 435 (immobilized from Candida antarctica), porcine pancreas lipase (PPL) (from porcine pancreas), lipase from pseudomonas fluorescens, immobilized (LPI) (immobilized from Pseudomonas fluorescens), and Lipozym IM (immobilized from Mucor miehei) in the acetylation of racemic N-methyl-4-hydroxyundecanamide that is similar in structure to rac-1. With a notable exception of Novozym 435, these lipases exhibit no reactivity toward racemic N-methyl-4-hydroxyundecanamide.
To determine the optimal amount of Novozym 435, the reaction was conducted in diethyl ether at room temperature with different ratios of Novozym 435–1.0 mmol of the substrate. It was concluded that 0.4 g is the optimal amount of Novozym 435–1.0 mmol of the substrate. The acetylation with Novozym 435 proceeds well for all substrates except rac-1b, although the time required to reach approximately 50% conversion varies for different starting materials (Scheme 3, Table 2). We have previously studied Novozym 435-catalyzed acetylation of racemic N-methyl-4-hydroxynonanamide [40]. The 50% conversion was reached in 2 h, and (R)- and (S)-γ-nonalactone were obtained with 98% and more than 99% enantiopurities, respectively. In this work, Novozym 435-catalyzed acetylation of rac-1a required 4 h despite the structural small difference between n-pentyl and isopentyl groups. In addition, (R)- and (S)-7-methyl-γ-octalactones (3a and 4a) derived from optically active 1 and 2 were obtained with 96% and more than 99% enantiomeric excesses. The enantiopurity of (R)-7-methyl-γ-octalactone was slightly low compared with that of (R)-γ-nonalactone. These results show that Novozym 435 exhibits not only low substrate affinity but also low substrate selectivity toward rac-1a, which has a sterically bulky R group. Therefore, a long reaction time can be predicted because the substrate possesses a sterically bulky side chain. An attempted acetylation failed for rac-1b substituted with a bulky tert-butyl group (entry 2). It appears that the substrate affinity of Novozym 435 is low for a substrate with a large steric hindrance around the asymmetric carbon atom. The reaction times for N-methyl-4-hydroxy-4-phenylbutanamide (rac-1c, entry 3) and N-methyl-4-hydroxy-4-p-tolylbutanamide (rac-1d, entry 4) are longer than those for rac-1a (entry 1) and N-methyl-4-hydroxy-6-phenylhexanamide (rac-1f, entry 6). By contrast, rac-1c with a phenyl group and rac-1d with a tolyl group are acetylated by Novozym 435. Phenyl and tolyl groups are more bulky than the isopentyl group; thus, a long reaction time is required to reach 50% conversion. In the case of rac-1c and rac-1d, both enantiomers of 4-phenyl-γ-butyrolactone (3c and 4c) and 4-(p-tolyl)-γ-butyrolactone (3d and 4d) were obtained with more than 90% enantiopurities. These optical purities are approximately 5–10% lower than that of rac-1a. These results suggest that the substrate selectivity and the affinity of Novozym 435 toward the high steric hindrance substrate around an asymmetric carbon are low. Naoshima et al. [41] explained the enantioselectivity of lipase by using computer modeling. They measured the C-O distance between the carbonyl carbon atom of the acetyl group in the substrate and the oxygen atom at the active center of lipase. The large difference of the C-O distance among each enantiomer correlated with high enantioselectivity. It appears that for the large steric hindrance around asymmetric carbon atom, the hydroxy group in the substrate and the active center in Novozym 435 are difficult to be approached. It can be suggested that the enantioselectivity of Novozym 435 toward the substrate with large steric hindrance decreases. Although approximately 50% conversion was reached at 11 h for N-methyl-4-hydroxy-4-(p-anisyl)butanamide (rac-1g) with a p-anisyl group, both 4-(p-anisyl)-γ-butyrolactones (3g and 4g) exhibit racemic similarities (entry 7). Phenyl, tolyl, and p-anisyl groups are structurally similar. However, only the reaction of rac-1g gave racemic γ-lactones (3g and 4g), and this result can be attributed to the presence of the methoxy group. The reaction times required for rac-1c and rac-1d to reach 50% conversion by Novozym 435-catalyzed acetylation were 12 and 20 h, respectively, and that for rac-1g took 11 h. These substrates require comparably long reaction time to reach 50% conversion compared with rac-1a and rac-1f. As mentioned earlier, it was assumed that Novozym 435 shows higher substrate affinity toward the small R group in substrates such as rac-1a and 1f. The reaction time of rac-1g was similar to rac-1c. These results also show that Novozym 435 exhibits low affinity toward the bulky substrates around the asymmetric carbon atom. By contrast, it can be suggested that the high substrate affinity of Novozym 435 is due to hydrogen bonding between the oxygen at the methoxy group and the amino acid residues that constitute the lipase. Both enantiomers of rac-1g can be incorporated into the active site by this hydrogen bonding, which causes the observed lack of enantioselectivity. Novozym 435-catalyzed acetylation of all rac-1 except rac-1g progressed with more than 90% enantioselectivity. The stereoselectivity of Novozym 435 varies with structural differences of the R group. The absolute configurations of γ-lactones prepared from hydroxyamide 1 are (R)-form for rac-1a with an isopentyl group and rac-1f with a phenethyl group. By contrast, the corresponding γ-lactones derived from 1 have (S)-configuration in the case of rac-1c with a phenyl group, rac-1d with a tolyl group, and rac-1e with a benzyl group. As shown in Scheme 3, the hydroxyl group is acetylated by Novozym 435 in all substrates. Previously, we have reported a synthetic methodology of the chiral γ-lactones, which combines Novozym 435-catalyzed reaction and Mitsunobu reaction [42, 43]. In this work, Novozym 435-catalyzed stereoselective hydrolysis of N-benzyl-4-acetoxyalkanamides was conducted. The hydrolysis was conducted at 60°C in diisopropyl ether; the reaction progressed with more than 90% enantioselectivity. However, the 50% conversion was reached after a relatively long period of 24–30 h. In summary, Novozym 435 shows high enantioselectivity for both acetylation and hydrolysis.

Novozym 435-catalyzed stereoselective acetylation of rac-1a–g.
Lipase-catalyzed acetylation of rac-1a and lactonization.
| Entry | Substrate | Time (h) | Yield (%) | Yield (%)/enantiomeric excess (% e.e.)b/absolute configuration | ||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | |||
| 1 | rac-1a | 4 | 47 | 47 | >99/96/R | >99/>99/S |
| 2c | rac-1b | 24 | No reaction | – | – | |
| 3 | rac-1c | 12 | 44 | 51 | >99/91/S | >99/94/R |
| 4 | rac-1d | 20 | 38 | 43 | >99/95/S | >99/93/R |
| 5 | rac-1e | 14 | 48 | 36 | >99/>99/S | >99/95/R |
| 6 | rac-1f | 7 | 34 | 46 | >99/>99/R | >99/99/S |
| 7 | rac-1g | 11 | 47 | 50 | >99/racemic/– | >99/racemic/– |
aConditions: rac-1, 1.0 mmol; vinyl acetate, 2.0 mmol; Novozym 435, 0.4 g; Et2O, 20 mL; room temperature.
bDetermined by GC using a Chirasil-Dex CB column.
cAcetylation conducted at 40°C.
Lipase-catalyzed enantioselective hydrolysis of N-methyl-4-acetoxyalkanamides
In the acetylation using Novozym 435, rac-1b was inert (Table 2, entry 2). Enantioselectivity was not observed for rac-1g, although approximately 50% conversion was reached in 11 h (Table 2, entry 7). Novozym 435-catalyzed hydrolysis of rac-2 was investigated to prepare optically active lactones 3b, g and 4b, g (Scheme 4, Table 3). In all cases, the time required for the hydrolysis to reach approximately 50% conversion is much longer than that for acetylation. Novozym 435 is inert toward the hydrolysis of rac-2b as well as the acetylation of rac-1b (entry 2). It appears that the bulky tert-butyl group is not compatible with the substrate specificity of Novozym 435. Although rac-2g is hydrolyzed, the obtained lactone 3g/4g is racemic (entry 7). Because rac-2c and rac-2d are hydrolyzed enantioselectively, it can be suggested that the anisyl group of rac-2g is a factor that does not promote enantioselectivity. The reaction mechanism of lipase was reported [44, 45].

Novozym 435-catalyzed stereoselective hydrolysis of rac-2a–g.
Lipase-catalyzed hydrolysis of rac-2a and lactonization.
| Entry | Substrate | Time (h) | Yield (%) | Yield (%)/enantiomeric excess (% e.e.)b/absolute configuration | ||
|---|---|---|---|---|---|---|
| 1 | 2 | 4 | 3 | |||
| 1 | rac-2a | 33 | 41 | 44 | >99/98/S | >99/79/R |
| 2 | rac-2b | 48 | No reaction | – | – | |
| 3 | rac-2c | 48 | 37 | 56 | >99/91/R | >99/72/S |
| 4 | rac-2d | 48 | 43 | 50 | >99/92/R | >99/75/S |
| 5 | rac-2e | 48 | 42 | 53 | >99/95/R | >99/70/S |
| 6 | rac-2f | 48 | 44 | 46 | >99/91/S | >99/96/R |
| 7 | rac-2g | 48 | 49 | 43 | >99/racemic/– | >99/racemic/– |
aConditions: rac-2, 1.0 mmol; MeOH, 3.0 mmol; Novozym 435, 0.4 g; Et2O, 20 mL; 40°C.
bDetermined by GC using a CycloSil B column.
Conclusions
N-methyl-4-hydroxyalkanamides with various side chains were prepared in a high yield by the addition reaction of organometallic reagents with N-methylsuccinimide. Novozym 435-catalyzed acetylation of all rac-1 except rac-1b and rac-1g the respective products 2 with more than 90% enantioselectivity. Both enantiomers of γ-lactones with various side chains were successfully synthesized with enantiopurities more than 90%.
Experimental
Column chromatography was conducted with silica gel FL60D (Fuji Silysia Chemical Ltd., Aichi, Japan). Thin-layer chromatography was performed with silica gel F-254 on aluminum plates (Merck Ltd., Darmstadt, Germany). 1H NMR (500 MHz) and 13C NMR (126 MHz) spectra were recorded in CDCl3 on a JNM-ECA-500 spectrometer (JEOL, Tokyo, Japan), using an internal standard of tetramethylsilane and the central peak of CDCl3 (77 ppm). Near-infrared spectra were recorded in KBr pellets on an FT-IR 460plus spectrophotometer (JASCO Corp., Tokyo, Japan). Enantiomeric excesses of γ-lactones were determined using a Perkin Elmer Auto System XL gas chromatograph equipped with a chiral capillary column CycloSil B (30 m×0.25 mm i.d.×0.25 μm film thickness; Agilent Technologies, Santa Clara, CA, USA). The carrier gas was helium. Optical rotations were measured on a Jasco P-1010 spectropolarimeter (JASCO Corp.), and the reported data refer to the Na-line value using a 25 mL cuvette. High-resolution mass spectral (HRMS) analyses were performed on an AccuTOF GCv 4G instrument (JEOL, Tokyo, Japan). Novozym 435 immobilized lipase from C. antarctica was obtained as a gift from Novozymes A/S (Paraná, Brazil).
Preparation of racemic N-methyl-4-hydroxyalkanamides rac-1a,c–g
Organomagnesium reagents were freshly prepared by the slow addition of the corresponding bromides or chlorides (16.5 mmol) in Tetrahydrofuran (THF) (50 mL) onto magnesium turnings (0.37 g, 15.0 mmol) previously activated with a crystal of iodine. Under vigorous stirring and cooling with an ice bath, N-methylsuccinimide (1.13 g, 10.0 mmol) dissolved in THF (30 mL) was then added to the suspension, and the mixture was stirred for 8 h at room temperature. The residual Grignard reagent was hydrolyzed by gradual addition of ice (10 g) and saturated aqueous NH4Cl (100 mL). The organic phase was separated, and the aqueous phase was extracted with CHCl3 (4×50 mL). The combined extracts were washed with water and dried with MgSO4. The solvent was evaporated under reduced pressure, and the residue was not purified. NaBH4 (0.76 g, 20 mmol) in methanol (20 mL) was added to the crude product with stirring, and the mixture was stirred for 1 h at room temperature. The solvent was evaporated under reduced pressure, and water (50 mL) was added. The aqueous phase was extracted with CHCl3 (4×50 mL). The combined extracts were washed with water and dried with MgSO4. The solvent was evaporated, and the residue was purified by flash chromatography on silica eluting with EtOAc to give the corresponding N-methyl-4-hydroxyalkanamide 1.
N-methyl-4-hydroxy-7-methyloctanamide (1a)
Yield 68%; colorless solid; mp 62–63°C (dec); Rf=0.33 (eluent: CHCl3-MeOH, 10:1, v/v); IR: 3293 (N-H, O-H), 2954 (-CH3), 2934 (-CH2-), 2871 (-CH3), 2846 (-CH2-), 1645 cm-1 (-NHC=O); 1H NMR: δ 0.88 (d, 6H, J = 6.9 Hz, -CH(CH3)2), 1.19 (m, 1H, -CH2CH(CH3)2), 1.32 (m, 1H, -CH2CH(CH3)2), 1.46 (m, 2H, -CH2CH2CH(CH3)2), 1.53 (quint, 1H, J = 6.8, 6.4 Hz, -CH2CH(CH3)2), 1.66 (m, 1H, -NHC(=O)CH2CH2-), 1.87 (m, 1H, -NHC(=O)CH2CH2-), 2.36 (m, 2H, -NHC(=O)CH2-), 2.81 (d, 3H, J = 4.6 Hz, -NHCH3), 2.99 (br, 1H, -OH), 3.60 (m, 1H, -CH(OH)-), 5.79 (br, 1H, -NH-); 13C NMR: δ 22.6, 22.6 (-CH(CH3)2), 26.4 (-NHCH3), 28.1 (-CH(CH3)2), 32.5 (-CH2-), 33.2 (-C(=O)CH2-), 34.8 (-CH2CH-), 35.6 (-CH2-), 71.8 (-CHOH), 174.4 (-NHC(=O)-). HRMS (FI). Calcd for C10H22NO2 (M+H)+: m/z 188.1651. Found: m/z 188.1645.
N-methyl-4-hydroxy-4-phenylbutanamide (1c)
Yield 94%; colorless oil; Rf=0.35 (eluent: CHCl3-MeOH, 10:1, v/v); IR: 3328 (O-H, N-H), 3109 (Ar, C-H), 2937 (CH3), 2877 (CH2), 1649 (-NC=O), 1495, 1450 (Ar, C=C), 760, 702 cm-1 (Ar, C-H); 1H NMR: δ 2.02 (m, 2H, -NHC(=O)CH2CH2-), 2.30 (t, 2H, J = 6.9 Hz, -NHC(=O)CH2-), 2.75 (d, 3H, J = 4.6 Hz, -NHCH3), 4.28 (br, 1H, -OH), 4.73 (m, 1H, -CH(OH)-), 6.02 (br, 1H, -NH-), 7.22–7.36 (m, 5H, -Ph); 13C NMR: δ 26.4 (-NHCH3), 32.7 (-C(=O)CH2-), 34.3 (-CH2-), 73.5 (-CHOH), 125.7, 127.3, 128.3, 144.4 (-Ph), 174.3 (-NHC(=O)-). HRMS (FD). Calcd for C11H16NO2 (M)+: m/z 193.1103. Found: m/z 193.1096.
N-methyl-4-hydroxy-4-(p-tolyl)butanamide (1d)
Yield 94%; colorless oil; Rf=0.24 (eluent: CHCl3-MeOH, 10:1, v/v); IR: 3321 (O-H, N-H), 3039 (Ar, C-H), 2937 (CH3), 2905 (CH2), 1645 (-NC=O), 1568, 1515 (Ar, C=C), 816 cm-1 (Ar, C-H); 1H NMR: δ 1.96 (m, 2H, -NHC(=O)CH2CH2-), 2.27 (t, 2H, J = 6.9 Hz, -NHC(=O)CH2-), 2.31 (s, 3H, -PhCH3), 2.70 (d, 3H, J = 4.6 Hz, -NHCH3), 4.55 (br, 1H, -OH), 4.64 (q, 1H, J = 3.7, 4.1 Hz, -CH(OH)-), 6.43 (br, 1H, -NH-), 7.10 (d, 2H, J = 7.8 Hz, -Ph-), 7.19 (d, 2H, J = 8.2 Hz, -Ph-); 13C NMR: δ 21.0 (-PhCH3), 26.2 (-NHCH3), 32.7 (-CH2-), 34.5 (-CH2-), 73.2 (-CHOH), 125.6, 128.9, 136.7, 141.4 (-Ph-), 174.4 (-NHC(=O)-). HRMS (FD). Calcd for C12H18NO2 (M)+: m/z 207.1259. Found: m/z 207.1257.
N-methyl-4-hydroxy-5-phenylpentanamide (1e)
Yield 79%; colorless solid; mp 100–101°C (dec); Rf=0.30 (eluent: CHCl3-MeOH, 10:1, v/v); IR: 3412, 3370 (N-H), 3285 (O-H), 3094, 3058, 3033 (Ar, C-H), 2944 (-CH3), 2926 (-CH2-), 1651 (-NHC=O), 1495, 1454 cm-1 (Ar, C=C); 1H NMR: δ 1.69 (m, 1H, -NHC(=O)CH2CH2-), 1.89 (m, 1H, -NHC(=O)CH2CH2-), 2.34 (t, 2H, J = 6.9 Hz, -NHC(=O)CH2-), 2.73–2.85 (m, 5H, -CH2Ph, -NHCH3), 3.21 (br, 1H, -OH), 3.84 (m, 1H, -CH(OH)-), 5.92 (br, 1H, -NH-), 7.17–7.35 (m, 5H, -Ph); 13C NMR: δ 26.3 (-NHCH3), 31.8 (-CH2-), 33.1 (-C(=O)CH2-), 44.2 (-CH2Ph), 72.3 (-CHOH), 126.4, 128.5, 129.4, 138.4 (-Ph), 174.2 (-NHC(=O)-). HRMS (FD). Calcd for C12H18NO2 (M+H)+: m/z 208.1338. Found: m/z 208.1305.
N-methyl-4-hydroxy-6-phenylhexanamide (1f)
Yield 61%; colorless solid; mp 40–41°C (dec); Rf=0.29 (eluent: CHCl3-MeOH, 10:1, v/v); IR: 3315 (N-H, O-H), 3085, 3064, 3029 (Ar, C-H), 2950 (-CH3), 2929 (-CH2-), 2878 (-CH3), 2861 (-CH2-), 1645 (-NHC=O), 1497, 1456 cm-1 (Ar, C=C); 1H NMR: δ 1.74 (m, 2H, -CH2CH2Ph), 1.86 (m, 2H, -NHC(=O)CH2CH2-), 2.36 (m, 2H, -NHC(=O)CH2-), 2.68–2.88 (m, 2H, -CH2Ph), 2.80 (d, 3H, J = 4.6 Hz, -NHCH3), 3.23 (br, 1H, -OH), 3.66 (m, 1H, -CH(OH)-), 5.70 (br, 1H, -NH-), 7.13–7.36 (m, 5H, -Ph); 13C NMR: δ 26.4 (-NHCH3), 32.1 (-CH2Ph), 32.5 (-CH2-), 33.1 (-C(=O)CH2-), 39.4 (-CH2-), 70.8 (-CHOH), 125.8, 128.3, 128.4, 128.6, 129.0, 142.1 (-Ph), 174.3 (-NHC(=O)-). HRMS (FD). Calcd for C13H20NO2 (M)+: m/z 221.1416. Found: m/z 221.1405.
N-methyl-4-hydroxy-4-(p-anisyl)butanamide (1g)
Yield 87%; colorless oil; Rf=0.24 (eluent: CHCl3-MeOH, 10:1, v/v); IR: 3313 (O-H, N-H), 3106 (Ar, C-H), 2938 (CH3), 2836 (CH2), 1645 (-NC=O), 1563, 1513 (Ar, C=C), 834 cm-1 (Ar, C-H); 1H NMR: δ 2.05 (m, 2H, -NHC(=O)CH2CH2-), 2.32 (t, 2H, J = 6.9 Hz, -NHC(=O)CH2-), 2.81 (d, 3H, J = 4.6 Hz, -NHCH3), 3.61 (br, 1H, -OH), 3.80 (s, 3H, -PhOCH3), 4.72 (t, 1H, J = 6.0 Hz, -CH(OH)-), 5.67 (br, 1H, -NH-), 6.87 (d, 2H, J = 8.2 Hz, -Ph-), 7.28 (d, 2H, J = 9.6 Hz, -Ph-); 13C NMR: δ 26.4 (-NHCH3), 32.9 (-C(=O)CH2-), 34.3 (-CH2-), 55.3 (-PhOCH3), 73.2 (-CHOH), 113.7, 126.9, 136.6, 158.9 (-Ph), 174.1 (-NHC(=O)-). HRMS (FD). Calcd for C12H18NO3 (M)+: m/z 223.1208. Found: m/z 223.1207.
Preparation of N-methyl-4-hydroxy- 5,5-dimethylhexanamide (1b)
A solution of tert-butyllithium in pentane (6.29 mL, 10.0 mmol) was added dropwise to a solution of N-methylsuccinimide (1.13 g, 10.0 mmol) in THF (20 mL) at -78°C under argon atmosphere, and the mixture was stirred at the same temperature for 2 h. The reaction mixture was poured onto saturated NH4Cl at 0°C and extracted with EtOAc (5×50 mL). The combined organic layers were washed with water, dried with MgSO4, and concentrated in vacuo. The crude product was not purified. NaBH4 (0.76 g, 20.0 mmol) was added to a solution of the crude product in MeOH (20 mL) at 0°C, and the mixture was stirred for 1 h. The solvent was evaporated, and water (50 mL) was added to the residue. The aqueous phase was extracted with EtOAc (5×50 mL), and the combined organic layers were washed with water, dried with MgSO4, and concentrated in vacuo. The crude product was purified by crystallization from n-hexane to give N-methyl-4-hydroxy-5,5-dimethylhexanamide (1b, 0.76 g, 44%) as a colorless solid; mp 117–118°C (dec); Rf=0.30 (eluent: CHCl3-MeOH, 10:1, v/v); IR: 3292 (N-H, O-H), 2954 (-CH3), 2932 (-CH2-), 2868 (-CH3), 2846 (-CH2-), 1645 cm-1 (-NHC=O); 1H NMR: δ 0.91 (s, 9H, -C(CH3)3), 1.60 (m, 1H, -NHC(=O)CH2CH2-), 1.89 (m, 1H, -NHC(=O)CH2CH2-), 2.37 (m, 2H, -NHC(=O)CH2-), 2.63 (br, 1H, -OH), 2.82 (d, 3H, J = 4.6 Hz, -NHCH3), 3.20 (m, 1H, -CH(OH)-), 5.66 (br, 1H, -NH-); 13C NMR: δ 25.6 (-C(CH3)3), 26.4 (-NHCH3), 27.0 (-CH2-), 34.1 (-C(=O)CH2-), 35.0 (-C(CH3)3), 79.7 (-CHOH), 174.6 (-NHC(=O)-). HRMS (ESI). Calcd for C9H20NO2 (M+H)+: m/z 174.1494. Found: m/z 174.1513.
General procedure for Novozym 435-catalyzed acetylation and lactonization
Novozym 435 (0.4 g) was added to a racemic mixture of N-methyl-4-hydroxyalkanamides (1, 1.0 mmol) and vinyl acetate (2.0 mmol) in diethyl ether (20 mL). After stirring at room temperature for a period specified in Table 2, the reaction mixture was filtered. Concentration under a reduced pressure followed by flash chromatography of the residue on silica gel (eluent: EtOAc) afforded the corresponding optically active N-methyl-4-hydroxyalkanamide 1a–g and N-methyl-4-acetoxyalkanamide 2a–g. Then NaOH (2.0 g) was added to a solution of 1a–g or 2a–g in methanol (20 mL), and the mixture was heated under reflux for 3 h. After cooling, methanol was removed under reduced pressure and water (50 mL) was added. The aqueous phase was acidified with 10% HCl to pH 3.0, and the mixture was stirred for 8 h then extracted with EtOAc (4×20 mL). The combined organic layers were washed with water, dried with MgSO4, and concentrated under reduced pressure. The residue was purified by flash chromatography eluting with n-hexane-EtOAc, 4:1, to give the corresponding γ-lactone (3 or 4) as a colorless oil. The enantiomeric excess was measured by using the chiral GC analysis. The absolute configurations of 3 and 4 were determined by optical activity compared with the literature data.
N-methyl-4-acetoxy-7-methyloctanamide (2a)
Colorless oil; Rf=0.33 (eluent: CHCl3-MeOH, 10:1, v/v); IR: 3298 (N-H), 2957 (-CH3), 2933 (-CH2-), 2871 (-CH3), 1738 (-OC=O), 1649 (-NHC=O), 1242 cm-1 (-OC=O);1H NMR: δ 0.87 (d, 6H, J = 6.3 Hz, -CH(CH3)2), 1.18 (m, 2H, -CH2CH2CH(CH3)2), 1.53 (m, 3H, -CH2CH2CH(CH3)2), 1.85 (m, 1H, -NHC(=O)CH2CH2-), 1.94 (m, 1H, -NHC(=O)CH2CH2-), 2.06 (s, 3H, -OC(=O)CH3), 2.18 (m, 2H, -NHC(=O)CH2-), 2.80 (d, 3H, J = 4.6 Hz, -NHCH3), 4.84 (quint, 1H, J = 3.4, 9.2 Hz, -CHOC(=O)CH3), 5.87 (s, 1H, -NH-); 13C NMR: δ 21.2 (-C(=O)CH3), 22.4, 22.5 (-CH(CH3)2), 26.3 (-NHCH3), 27.8 (-CH2CH(CH3)2), 30.2 (-CH2-), 32.1 (-C(=O)CH2-), 32.5 (-CH2-), 34.3 (-CH2CH(CH3)2), 74.1 (-CHOC(=O)CH3), 171.3 (-OC(=O)CH3), 172.9 (-NHC(=O)-). HRMS (FI). Calcd for C12H24NO3 (M)+: m/z 229.1678. Found: m/z 229.1662.
N-methyl-4-acetoxy-4-phenylbutanamide (2c)
Colorless oil; Rf=0.33 (eluent: CHCl3-MeOH, 10:1, v/v); IR: 3302 (N-H), 3035 (Ar, C-H), 2941 (CH3, CH2), 1736 (-OC=O), 1651 (-NHC=O), 1556, 1493 (Ar, C=C), 1240 (C-O-C), 760, 700 cm-1 (Ar, C-H); 1H NMR: δ 2.05 (s, 3H, -OC(=O)CH3), 2.16 (m, 2H, -NHC(=O)CH2CH2-), 2.24 (m, 2H, -NHC(=O)CH2-), 2.76 (d, 3H, J = 4.6 Hz, -NHCH3), 5.75 (m, 1H, -CHOC(=O)CH3), 7.20–7.41 (m, 5H, -Ph); 13C NMR: δ 21.2 (-C(=O)CH3), 26.3 (-NHCH3), 32.0 (-CH2-), 32.3 (-C(=O)CH2-), 75.3 (-CHOC(=O)CH3), 126.3, 128.0, 128.5, 139.8 (-Ph), 170.5 (-OC(=O)CH3), 172.6 (-NHC(=O)-). HRMS (FD). Calcd for C13H18NO3 (M)+: m/z 235.1208. Found: m/z 235.1194.
N-methyl-4-acetoxy-4-(p-tolyl)butanamide (2d)
Colorless oil; Rf=0.33 (eluent: CHCl3-MeOH, 10:1, v/v); IR: 3300 (N-H), 3029 (Ar, C-H) 2943 (CH3, CH2), 1738 (-OC=O), 1651 (-NHC=O), 1556, 1520 (Ar, C=C), 1240 (C-O-C), 818 cm-1 (Ar, C-H); 1H NMR: δ 2.02 (s, 3H, -OC(=O)CH3), 2.13 (m, 2H, -NHC(=O)CH2CH2-), 2.24 (m, 2H, -NHC(=O)CH2-), 2.31 (s, 3H, -PhCH3), 2.72 (d, 3H, J = 4.6 Hz, -NHCH3), 5.69 (m, 1H, -CHOC(=O)CH3), 6.18 (br, 1H, -NH-), 7.12 (d, 2H, J = 7.8 Hz, -Ph-), 7.19 (d, 2H, J = 7.8 Hz, -Ph-); 13C NMR: δ 20.9 (-OC(=O)CH3), 21.0 (-PhCH3), 26.1 (-NHCH3), 31.7 (-C(=O)CH2-), 32.1 (-CH2-), 75.1 (-CHOC(=O)CH3), 126.2, 129.0, 136.7, 137.6 (-Ph-), 170.3 (-OC(=O)CH3), 172.7 (-NHC(=O)-). HRMS (FD). Calcd for C14H20NO3 (M)+: m/z 249.1365. Found: m/z 249.1368.
N-methyl-4-acetoxy-5-phenylpentanamide (2e)
Colorless oil; Rf=0.33 (eluent: CHCl3-MeOH, 10:1, v/v). IR: 3308 (N-H), 3086, 3062, 3029 (Ar, C-H), 2938 (-CH3), 1735 (-OC=O), 1646 (-NHC=O), 1496, 1455 (Ar, C=C), 1242 cm-1 (-OC=O); 1H NMR: δ 1.88 (m, 1H, -NHC(=O)CH2CH2-), 2.13 (m, 1H, -NHC(=O)CH2CH2-), 1.98 (s, 3H, -OC(=O)CH3), 2.21 (m, 2H, -NHC(=O)CH2-), 2.77 (d, 3H, J = 4.6 Hz, -NHCH3), 2.87 (m, 2H, -CH2Ph), 5.07 (m, 1H, -CHOC(=O)CH3), 5.79 (s, 1H, -NH-), 7.15–7.39 (m, 5H, -Ph); 13C NMR: δ 21.0 (-OC(=O)CH3), 26.2 (-NHCH3), 29.7 (-CH2-), 32.6 (-C(=O)CH2-), 40.7 (-CH2Ph), 74.4 (-CHOC(=O)CH3), 126.5, 128.3, 129.3, 137.0 (-Ph), 170.9 (-OC(=O)CH3), 172.7 (-NHC(=O)-). HRMS (FD). Calcd for C14H20NO3 (M+H)+: m/z 250.1443. Found: m/z 250.1444.
N-methyl-4-acetoxy-6-phenylhexanamide (2f)
Colorless oil; Rf=0.33 (eluent: CHCl3-MeOH, 10:1, v/v); IR: 3297 (N-H), 3087, 3062, 3026 (Ar, C-H), 2943, 2864 (-CH3), 1736 (-OC=O), 1644 (-NHC=O), 1496, 1454 (Ar, C=C), 1242 cm-1 (-OC=O); 1H NMR: δ 1.79–2.01 (m, 4H, -CH2CH(OC(=O)CH3)CH2-), 2.04 (s, 3H, -OC(=O)CH3), 2.19 (m, 2H, -NHC(=O)CH2-), 2.63 (m, 2H, -CH2Ph), 2.78 (d, 3H, J = 4.6 Hz, -NHCH3), 4.92 (m, 1H, -CHOC(=O)CH3), 5.94 (s, 1H, -NH-), 7.11–7.35 (m, 5H, -Ph); 13C NMR: δ 21.1 (-OC(=O)CH3), 26.3 (-NHCH3), 30.2 (-CH2Ph), 31.6 (-CH2-), 32.4 (-C(=O)CH2-), 35.9 (-CH2CH2Ph), 73.5 (-CHOC(=O)CH3), 125.9, 128.2, 128.4, 141.8 (-Ph), 171.3 (-OC(=O)CH3), 173.0 (-NHC(=O)-). HRMS (FD). Calcd for C15H22NO3 (M)+: m/z 263.1521. Found: m/z 263.1517.
N-methyl-4-acetoxy-4-(p-anisyl)butanamide (2g)
Colorless oil; Rf=0.33 (eluent: CHCl3-MeOH, 10:1, v/v); IR: 3306 (N-H), 3094 (Ar, C-H), 2938 (CH3), 2838 (CH2), 1736 (-OC=O), 1648 (-NHC=O), 1613, 1516 (Ar, C=O), 1241 (C-O-C), 832 cm-1 (Ar, C-H); 1H NMR: δ 2.05 (s, 3H, -OC(=O)CH3), 2.14 (m, 2H, -NHC(=O)CH2CH2-), 2.24 (m, 2H, -NHC(=O)CH2-), 2.79 (d, 3H, J = 4.6 Hz, -NHCH3), 3.80 (s, 3H, -PhOCH3), 5.54 (s, 1H, -NH-), 5.71 (t, 1H, J = 5.0 Hz, -CHOC(=O)CH3), 6.87 (d, 2H, J = 8.7 Hz, -Ph-), 7.26 (d, 2H, J = 6.9 Hz, -Ph-); 13C NMR: δ 21.3 (-OC(=O)CH3), 26.3 (-NHCH3), 31.8 (-C(=O)CH2-), 32.6 (-CH2-), 55.3 (-PhOCH3), 75.1 (-CHOC(=O)CH3), 113.9, 127.9, 131.9, 159.4 (-Ph-), 170.5 (-OC(=O)CH3), 172.4 (-NHC(=O)-). HRMS (FD). Calcd for C14H20NO4 (M)+: m/z 265.1314. Found: m/z 265.1313.
7-Methyl-γ-octalactone (3a, 4a)
Colorless oil; Rf=0.25 (eluent: n-Hexane-EtOAc, 4:1, v/v); IR: 2951 (CH3), 2868 (CH2), 1776 (-OC=O), 1184 cm-1 (C-O-C); 1H NMR: δ 0.90 (d, 6H, J = 6.3 Hz, -CH(CH3)2), 1.24 (m, 1H, -CH2CH(CH3)2), 1.36 (m, 1H, -CH2CH(CH3)2), 1.52–1.66 (m, 2H, -CH2CH2CH(CH3)2), 1.74 (m, 1H, -CH2CH2CH(CH3)2), 1.86 (m, 1H, -OC(=O)CH2CH2-), 2.33 (m, 1H, -OC(=O)CH2CH2-), 2.54 (t, 2H, J = 8.0 Hz, -OC(=O)CH2CH2-), 4.47 (quint, 1H, J = 6.3, 7.4 Hz, -OCH(CH2-)CH2-); 13C NMR: δ 22.4 (-CH(CH3)2), 27.8 (-CH(CH3)2), 28.0 (-C(=O)CH2CH2-), 28.8 (-C(=O)CH2CH2-), 33.4 (-CHCH2CH2-), 34.1 (-CH2CH(CH3)2-), 81.3 (-OCHCH2-), 177.26 (-C(=O)O-). HRMS (FI). Calcd for C9H17O2 (M+H)+: m/z 157.1229. Found: m/z 157.1202.
4-Phenyl-γ-butyrolactone (3c, 4c)
Colorless oil; Rf=0.15 (eluent: n-hexane-EtOAc, 4:1, v/v); IR: 3033 (Ar, C-H), 2950 (CH2), 1776 (-OC=O), 1606, 1496 (Ar, C=C), 1176 (C-O-C), 760, 700 cm-1 (Ar, C-H); 1H NMR: δ 2.10 (m, 1H, -OC(=O)CH2CH2-), 2.63–2.72 (m, 3H, -OC(=O)CH2CH2-), 5.52 (t, 1H, J = 6.9 Hz, -OCH(CH2-)Ph), 7.31–7.46 (m, 5H, -Ph); 13C NMR: δ 28.9 (-C(=O)CH2CH2-), 31.0 (-C(=O)CH2CH2-), 81.2 (-OCHPh), 125.2, 128.4, 128.7, 139.3 (-Ph), 176.9 (-C(=O)O-). HRMS (ESI). Calcd for C10H11O2 (M)+: m/z 162.0681. Found: (M)+, 162.0653.
4-(p-Tolyl)-γ-butyrolactone (3d, 4d)
Colorless oil; Rf=0.18 (eluent: n-hexane-EtOAc, 4:1, v/v); IR: 3025 (Ar, C-H), 2985 (CH3), 2949 (CH2), 1774 (-OC=O), 1616, 1518 (Ar, C=C), 1176 (C-O-C), 806 cm-1 (Ar, C-H); 1H NMR: δ 2.18 (m, 1H, -OC(=O)CH2CH2-, 2.35 (s, 3H, -PhCH3), 2.56–2.69 (m, 3H, -OC(=O)CH2CH2-), 5.47 (t, 1H, J = 7.3 Hz, -OCH(CH2-)Ph-), 7.11–7.25 (m, 4H, -Ph-); 13C NMR: δ 21.1 (-PhCH3), 29.0 (-C(=O)CH2CH2-), 30.9 (-C(=O)CH2CH2-), 81.3 (-OCHPh-), 125.3, 129.3, 132.2, 138.3 (-Ph-), 177.0 (-C(=O)O-). HRMS (FI). Calcd for C11H13O2 (M)+: m/z 176.0837. Found: m/z 176.0834.
5-Phenyl-γ-pentalactone (3e, 4e)
Colorless oil; Rf=0.15 (eluent: n-hexane-EtOAc, 4:1, v/v). IR: 3030 (Ar, C-H), 2943 (CH2), 1774 (-OC=O), 1603, 1496 (Ar, C=C), 1178 (C-O-C), 750, 702 cm-1 (Ar, C-H); 1H NMR: δ 1.96 (m, 1H, -OC(=O)CH2CH2-), 2.25 (m, 1H, -OC(=O)CH2CH2-), 2.42 (m, 2H, -OC(=O)CH2CH2-), 2.93 (q, 1H, J = 6.0, 6.4 Hz, -CHCH2Ph), 3.08 (q, 1H, J = 6.0, 6.4 Hz, -CHCH2Ph), 4.74 (quint, 1H, J = 6.9, 6.4 Hz, -OCH(CH2-)CH2Ph), 7.18–7.38 (m, 5H, -Ph); 13C NMR: δ 27.1 (-C(=O)CH2CH2-), 28.6 (-C(=O)CH2CH2-), 41.3 (-CHCH2Ph), 80.8 (-OCHCH2Ph), 127.0, 128.6, 129.4, 135.8 (-Ph), 177.0 (-C(=O)O-). HRMS (FI). Calcd for C11H13O2 (M)+: m/z 176.0837. Found: m/z 176.0812.
6-Phenyl-γ-hexalactone (3f, 4f)
Colorless oil; Rf=0.18 (eluent: n-hexane-EtOAc, 4:1, v/v); IR: 3028 (Ar, C-H), 2943 (CH2), 1770 (-OC=O), 1603, 1495 (Ar, C=C), 1180 (C-O-C), 750, 702 cm-1 (Ar, C-H); 1H NMR: δ 1.82–1.96 (m, 2H, -C(=O)CH2CH2CH(O-)CH2-), 2.05 (m, 1H, -CHCH2 CH2Ph), 2.31 (m, 1H, -OC(=O)CH2CH2-), 2.53 (m, 2H, -OC(=O)CH2CH2-), 2.73 (m, 1H, -CH2Ph), 2.83 (m, 1H, -CH2Ph), 4.47 (quint, 1H, J = 6.9, 6.9 Hz, -C(=O)CH2CH2CH(O-)CH2-), 7.16–7.36 (m, 5H, -Ph); 13C NMR: δ 27.9 (-C(=O)CH2CH2-), 28.8 (-C(=O)CH2CH2-), 31.6 (-CHPh), 37.3 (-OCHCH2-), 79.8 (-OCHCH2-), 126.1, 128.4, 128.5, 140.7 (-Ph), 177.1 (-C(=O)O-). HRMS (FI). Calcd for C12H15O2 (M)+: m/z 190.0994. Found: m/z 190.0977.
4-(p-Anisyl)-γ-butyrolactone (3g, 4g)
Colorless oil; Rf=0.10 (eluent: n-hexane-EtOAc, 4:1, v/v); IR: 3037 (Ar, C-H), 2956 (CH3), 2937 (CH2), 1773 (-OC=O), 1613, 1517 (Ar, C=C), 1176 (C-O-C), 837 cm-1 (Ar, C-H); 1H NMR: δ 2.20 (m, 1H, -OC(=O)CH2CH2-), 2.57–2.71 (m, 3H, -OC(=O)CH2CH2-), 3.28 (s, 3H, -PhOCH3), 5.47 (t, 1H, J = 7.4 Hz, -C(=O)CH2CH2CH(O-)Ph-), 6.92 (d, 2H, J = 8.6 Hz, -Ph-), 7.27 (d, 2H, J = 7.4 Hz, -Ph-); 13C NMR: δ 29.2 (-C(=O)CH2CH2-), 30.9 (-C(=O)CH2CH2-), 55.3 (-PhOCH3), 81.3 (-OCHPh-), 114.1, 126.9, 131.3, 165.3 (-Ph-), 177.1 (-C(=O)O-). HRMS (FI). Calcd for C11H13O3 (M)+: m/z 192.0786. Found: m/z 192.0758.
General procedure for Novozym 435-catalyzed hydrolysis
Racemic N-methyl-4-acetoxyalkanamides rac-2a–g were prepared almost quantitatively from N-methyl-4-hydroxyalkanamides rac-1a–g by acetylation using acetic anhydride [30]. Briefly, a mixture of racemic N-methy-5-acetoxyalkanamide (rac-2a–g, 1.0 mmol), methanol (3.0 mmol, 0.10 g), Novozym 435 (0.4 g), and diethyl ether (20 mL) was stirred at 40°C for a period specified in Table 3, then filtered to remove Novozym 435, and concentrated. The purification of the crude product by silica gel column chromatography eluenting with EtOAc gave optically active N-methyl-4-hydroxyalkanamide 1a–g and N-methyl-4-acetoxyalkanamide 2a–g. Lactonization is described earlier.
Determination of enantiomeric excess
Enantiomeric excesses of optically active γ-lactone 3 and 4 were measured by chiral GC. General GC conditions: chiral column, CycloSil B; injector temperature, 250°C; detector temperature, 250°C; He gas, 2.0 mL/min.
7-Methyl-γ-octalactone 3a and 4a: Oven temperature, 140°C (isothermal); retention time, 8.5 min for (R)-3a, 8.8 min for (S)-4a.
4-Phenyl-γ-butyrolactone 3c and 4c: Oven temperature, 150°C (isothermal); retention time, 17.2 min for (S)-3c, 19.0 min for (R)-4c.
4-p-Tolyl-γ-butyrolactone 3d and 4d: Oven temperature, 150°C (isothermal); retention time, 27.0 min for (S)-3d, 30.4 min for (R)-4d.
5-Phenyl-γ-pentalactone 3e and 4e: Oven temperature, 140°C (isothermal); retention time, 27.3 min for (S)-3e, 28.2 min for (R)-4e.
6-Phenyl-γ-hexalactone 3f and 4f: Oven temperature, 140°C (isothermal); retention time, 76.0 min for (R)-3f, 77.8 min for (S)-4f.
4-p-Anisyl-γ-butyrolactone 3g and 4g: Oven temperature, 160°C (isothermal); retention time, 39.0 min for 3f, 42.6 min for 4f.
Specific rotation of optically active amides 1 and 2
The absolute configuration and the enantiomeric excesses were determined by using the values of the corresponding lactones 3 and 4.
(R)-N-methyl-4-hydroxy-7-methyloctanamide [(R)-1a]: [α]25D=-4.8 (c 1.0, MeOH, 96% e.e.).
(S)-N-methyl-4-hydroxy-4-phenylbutanamide [(S)-1c]: [α]25D=-40.7 (c 1.0, CHCl3, 91% e.e.).
(S)-N-methyl-4-hydroxy-4-(p-tolyl)butanamide [(S)-1d]: [α]25D= -48.6 (c 1.0, CHCl3, 95% e.e.).
(S)-N-methyl-4-hydroxy-5-phenylpentanamide [(S)-1e]: [α]25D= +4.7 (c 1.0, CHCl3, >99% e.e.).
(R)-N-methyl-4-hydroxy-6-phenylhexanamide [(R)-1f]: [α]25D=+8.4 (c 1.0, MeOH, >99% e.e.).
(S)-N-methyl-4-acetoxy-7-methyloctanamide [(S)-2a]: [α]25D=+8.0 (c 1.0, MeOH, >99% e.e.).
(R)-N-methyl-4-acetoxy-4-phenylbutanamide [(R)-2c]: [α]25D=+58.8 (c 1.0, CHCl3, 94% e.e.).
(R)-N-methyl-4-acetoxy-4-(p-tolyl)butanamide [(R)-2d]: [α]25D=+65.7 (c 1.0, CHCl3, 93% e.e.).
(R)-N-methyl-4-acetoxy-5-phenylpentanamide [(R)-2e]: [α]25D=+13.1 (c 1.0, CHCl3, 95% e.e.).
(S)-N-methyl-4-acetoxy-6-phenylhexanamide [(S)-2f]: [α]25D=+8.1 (c 1.0, MeOH, 99% e.e.).
Assignment of absolute configuration for lactones 3 and 4
The absolute configuration of γ-lactones 3 and 4 was determined by comparison of the sign of the measured specific rotation with that in the literature.
(S)-7-Methyl-γ-octalactone 4a: [α]25D=-46.7 (c 1.0, MeOH, >99% e.e); Lit. [α]20D=-38.6° (c 0.26, MeOH) [46].
(R)-4-Phenyl-γ-butyrolactone 4c: [α]25D=+32.8 (c 1.0, CHCl3, 94% e.e.); Lit. [α]25D=+20.3° (62% e.e.) [47].
(R)-4-p-Tolyl-γ-butyrolactone 4d: [α]25D=+25.7 (c 1.0, CHCl3, 93% e.e.); Lit. [α]25D=+10.8° (91% e.e.) [48].
(S)-5-Phenyl-γ-pentalactone 3e: [α]25D=+19.0 (c 1.0, CHCl3, >99% e.e.); Lit. [α]25D=+24.7° (c 1, CHCl3, 97% e.e.) [29].
(R)-6-Phenyl-γ-hexalactone 3f: [α]25D=+65.8 (c 1.0, MeOH, >99% e.e.); Lit. [α]25D=+39.2° (c 0.2–1.0, MeOH, >99% e.e.) [46].
Acknowledgments
We are grateful to Novozymes A/S for the generous gift of Novozym 435.
References
[1] Cooke, R. C.; Leeuwen, K. A. V.; Capone, D. L.; Gawel, R.; Elsey, G. M.; Sefton, M. A. Odor detection thresholds and enantiomeric distributions of several 4-alkyl substituted γ-lactones in Australian red wine. J. Agric. Food Chem. 2009, 57, 2462–2467.10.1021/jf802866fSearch in Google Scholar
[2] Tamogami, S.; Awano, K.; Kitahara, T. Analysis of the enantiomeric ratios of chiral compounds in absolute jasmine. Flavour Fragr. J. 2001, 16, 161–163.10.1002/ffj.969Search in Google Scholar
[3] Sarrazin, E.; Frerot, E.; Bagnoud, A.; Aeberhardt, K.; Rubin, M. Discovery of new lactones in sweet cream butter oil. J. Agric. Food Chem. 2011, 59, 6657–6666.10.1021/jf201380uSearch in Google Scholar
[4] Leal, W. S.; Wojtasek, H.; Kuwahara, J. F. P. S.; Saito, H.; Hasegawa, M. Perireceptor events in pheromone perception in scarab beetles. J. Asia Pac. Entomol. 1998, 1, 1–8.10.1016/S1226-8615(08)60001-1Search in Google Scholar
[5] Sabitha, G.; Yadagiri, K.; Yadav, J. S. Synthesis of the Junus integer pheromone (4R,9Z)-9-octadecen-4-olide. Tetrahedron Lett. 2007, 48, 1651–1652.10.1016/j.tetlet.2006.12.115Search in Google Scholar
[6] Sabitha, G.; Fatima, N.; Reddy, E. V.; Yadav, J. S. Prins and RCM protocols for the synthesis of the pheromones of the giant white butterfly Idea leuconoe. Tetrahedron Lett. 2008, 49, 6087–6089.10.1016/j.tetlet.2008.08.010Search in Google Scholar
[7] Brown, H. C.; Kulkarni, S. V.; Racherla, U. S. Chiral synthesis via organoboranes. 39. A facile synthesis of γ-substituted-γ-butyrolactones in exceptionally high enantiomeric purity. J. Org. Chem. 1994, 59, 365–369.10.1021/jo00081a014Search in Google Scholar
[8] Gogoi, S.; Argade, N. P. An efficient PS-catalyzed chemo-, regio- and enantioselective hydrolysis of (±)-2,3-di-O-acetyl-2-C-methyl-d-erythrono-1,4-lactone: a facile preparation of bioactive natural products (-)-saccharinic acid lactone and potassium (2R,3R)-2,3,4-trihydroxy-2-methylbutanoate. Tetrahedron Asymmetry 2006, 17, 927–932.10.1016/j.tetasy.2006.02.025Search in Google Scholar
[9] Zoute, L.; Lemonnier, G.; Nguyen, T. M.; Quirion, J. C.; Jubault, P. Convenient preparation of α-bromo-α-fluoro-β-hydroxyesters building blocks from aldehydes, ketones and lactones. Tetrahedron Lett. 2011, 52, 2473–2475.10.1016/j.tetlet.2011.03.014Search in Google Scholar
[10] Smith, N. D.; Goodman, M. Enantioselective synthesis of α-methyl-d-cysteine and lanthionine building blocks via α-methyl-d-serine-β-lactone. Org. Lett. 2003, 5, 1035–1037.10.1021/ol034025pSearch in Google Scholar
[11] Stangeland, E. L.; Sammakia, T. Use of thiazoles in the halogen dance reaction: application to the total synthesis of WS75624 B. J. Org. Chem. 2004, 69, 2381–2385.10.1021/jo0351217Search in Google Scholar
[12] Chidley, H. G.; Kulkarni, R. S.; Pujari, K. H.; Giri, A. P.; Gupta, V. S. Spatial and temporal changes in the volatile profile of Alphonso mango upon exogenous ethylene treatment. Food Chem. 2013, 136, 585–594.10.1016/j.foodchem.2012.08.029Search in Google Scholar
[13] Macleod, A. J.; Macleod, G.; Snyder, C. H. Volatile aroma constituents of mango (cv Kensington). Phytochemistry 1988, 27, 2189–2193.10.1016/0031-9422(88)80124-9Search in Google Scholar
[14] Wang, Y.; Yang, C.; Li, S.; Yang, L.; Wang, Y.; Zhao, J.; Jiang, Q. Volatile characteristics of 50 peaches and nectarines evaluated by HP-SPME with GC-MS. Food Chem. 2009, 116, 356–364.10.1016/j.foodchem.2009.02.004Search in Google Scholar
[15] Visai, C.; Vanoli, M. Volatile compound production during growth and ripening of peaches and nectarines. Sci. Hortic. 1997, 70, 15–24.10.1016/S0304-4238(97)00032-0Search in Google Scholar
[16] Boishebert, V.; Giraudel, J. L.; Montury, M. Characterization of strawberry varieties by SPME-GC-MS and Kohonen self-organizing map. Chemom. Intell. Lab. Syst. 2006, 80, 13–23.10.1016/j.chemolab.2005.05.003Search in Google Scholar
[17] Lambert, Y.; Demazeau, G.; Largeteau, A.; Bouvier, J. M. Changes in aromatics volatile composition of strawberry after high pressure treatment. Food Chem. 1999, 67, 7–16.10.1016/S0308-8146(99)00084-9Search in Google Scholar
[18] Alewijn, M.; Smit, B. A.; Sliwinski, E. L.; Wouters, J. T. M. The formation mechanism of lactones in Gouda cheese. Int. Dairy J. 2007, 17, 59–66.10.1016/j.idairyj.2006.01.002Search in Google Scholar
[19] Leuven, I. V.; Caelenberg, T. V.; Dirinck, P. Aroma characterization of Gouda-type cheeses. Int. Dairy J. 2008, 18, 790–800.10.1016/j.idairyj.2008.01.001Search in Google Scholar
[20] Rincon-Delgadillo, M. I.; Lopez-Hernandez, A.; Wijaya, I.; Rankin, S. A. Diacetyl levels and volatile profiles of commercial starter distillates and selected dairy foods. J. Dairy Sci. 2012, 95, 1128–1139.10.3168/jds.2011-4834Search in Google Scholar
[21] Cagno, R. D.; Banks, J.; Sheehan, L.; Fox, P. F.; Brechany, E. Y.; Corsetti, A.; Gobbetti, M. Comparison of the microbiological, compositional, biochemical, volatile profile and sensory characteristics of three Italian PDO ewes’ milk cheeses. Int. Dairy J. 2003, 13, 961–972.10.1016/S0958-6946(03)00145-6Search in Google Scholar
[22] Bredikhin, A. A.; Bredikhina, Z. A.; Zakharychev, D. V.; Pashagin, A. V. Chiral drugs related to guaifenesin: synthesis and phase properties of methocarbamol and mephenoxalone. Tetrahedron Asymmetry 2007, 18, 1239–1244.10.1016/j.tetasy.2007.05.019Search in Google Scholar
[23] Patel, R. N. Microbial/enzymatic synthesis of chiral intermediated for pharmaceuticals. Enzyme Microb. Technol. 2002, 31, 804–826.10.1016/S0141-0229(02)00186-2Search in Google Scholar
[24] Tajik, H.; Dadras, A.; Aghabeygi, S. A facile synthesis of novel optically active R,R-2-{4-[2-(4-(5-chloro-3-halopyridin-2-yloxy)phenoxy)propionyloxy]phenoxy}propionic acid esters using cyanuric chlorides as potential herbicide. Chin. Chem. Lett. 2011, 22, 535–538.10.1016/j.cclet.2010.12.001Search in Google Scholar
[25] Itoh, H.; Furukawa, Y.; Tsuda, M.; Takeshiba, H. Synthesis and fungicidal activity of enantiomerically pure (R)- and (S)-silicon-containing azole fungicides. Bioorg. Med. Chem. 2004, 12, 3561–3567.10.1016/j.bmc.2004.04.027Search in Google Scholar
[26] Ghosh, A. K.; Swanson, L. Enantioselective synthesis of (+)-cryptophycin 52 (LY355703), a potent antimitotic antitumor agent. J. Org. Chem. 2003, 68, 9823–9826.10.1021/jo035077vSearch in Google Scholar
[27] Ghosh, A. K.; Shurrush, K.; Kulkarni, S. Asymmetric synthesis of anti-aldol segments via a nonaldol route: synthetic applications to statines and (-)-tetrahydrolipstatin. J. Org. Chem. 2009, 74, 4508–4518.10.1021/jo900642fSearch in Google Scholar
[28] Hilborn, J. W.; Lu, Z. H.; Jurgens, A. R.; Fang, Q. K.; Byers, P.; Wald, S. A.; Senanayake, C. H. A practical asymmetric synthesis of (R)-fluoxetine and its major metabolite (R)-norfluoxetine. Tetrahedron Lett. 2001, 42, 8919–8921.10.1016/S0040-4039(01)01877-9Search in Google Scholar
[29] Kotkar, S. P.; Suryavanshi, G. S.; Sudalai, A. A short synthesis of (+)-harzialactone A and (R)-(+)-4-hexanolide via proline-catalyzed sequential α-aminooxylation and Horner-Wadsworth-Emmons olefination of aldehydes. Tetrahedron Asymmetry 2007, 18, 1795–1798.10.1016/j.tetasy.2007.07.031Search in Google Scholar
[30] Shimotori, Y.; Nakahachi, Y.; Inoue, K.; Miyakoshi, T. Synthesis of optically active γ-lactones via lipase-catalyzed resolution. Flavour Fragr. J. 2007, 22, 421–429.10.1002/ffj.1816Search in Google Scholar
[31] Garst, J. F.; Lawrence, K. E.; Batlaw, R.; Boone, J. R.; Ungváry, F. Magnesium bromide in Grignard reagent formation. Inorg. Chim. Acta 1994, 222, 365–375.10.1016/0020-1693(94)03928-3Search in Google Scholar
[32] Tuulmets, A.; Panov, D. Solvation effects in partially solvated Grignard reagents. J. Organometal. Chem. 1999, 575, 182–186.10.1016/S0022-328X(98)00991-7Search in Google Scholar
[33] Chalk, A. J. Ring metalation of toluene by butyllithium in the presence of N,N,N′,N′-tetramethylethylenediamine. J. Organometal. Chem. 1968, 11, 615–618.10.1016/0022-328X(68)80091-9Search in Google Scholar
[34] Lenoir, D.; Dauner, H.; Frank, R. M. Dehydratisierung von aliphatischen Di-tert-butylalkylcarbinolen. Kraftfeld-Rechnungen von sterisch gehinderten Ethylenen. Chem. Ber. 1980, 113, 2636–2647.10.1002/cber.19801130807Search in Google Scholar
[35] Hellmann, S.; Beckhaus, H. D.; Rüchardt, C. Synthesen, Spektren, Struktur und Spannung hochverzweigter Pentane. Chem. Ber. 1983, 116, 2219–2237.10.1002/cber.19831160614Search in Google Scholar
[36] Uemura, M.; Yagi, K.; Iwasaki, M.; Nomura, K.; Yorimitsu, H.; Oshima, K. Pentamethylcyclopentadienide in organic synthesis: nucleophilic addition of lithium pentamethylcyclopentadienide to carbonyl compounds and carbon-carbon bond cleavage of the adducts yielding the parent carbonyl compounds. Tetrahedron 2006, 62, 3523–3535.10.1016/j.tet.2006.01.097Search in Google Scholar
[37] Pace, V.; Castoldi, L.; Hoyos, P.; Sinisterra, J. V.; Pregnolato, M.; MaSánchez-Montero, J. Highly regioselective control of 1,2-addition of organolithiums to α,β-unsaturated compounds promoted by lithium bromide in 2-methyltetrahydrofuran: a facile and eco-friendly access to allylic alcohols and amines. Tetrahedron 2011, 67, 2670–2675.10.1016/j.tet.2011.01.067Search in Google Scholar
[38] Basak, D.; Versek, C.; Toscano, D. T.; Christensen, S.; Tuominen, M. T.; Venkataraman, D. Anhydrous proton conductivities of squaric acid derivatives. Chem. Commun. 2012, 48, 5922–5924.10.1039/c2cc31283bSearch in Google Scholar
[39] Harrowven, D. C.; Mohamed, M.; Gonçalves, T. P.; Whitby, R. J.; Bolien, D.; Sneddon, H. F. An efficient flow-photochemical synthesis of 5H-furanones leads to an understanding of torquoselectivity in cyclobutenone rearrangements. Angew. Chem. Int. Ed. 2012, 51, 4405–4408.10.1002/anie.201200281Search in Google Scholar
[40] Shimotori, Y.; Miyakoshi, T.; Synthesis of optically active γ-lactones with lipase catalyst. J. Oleo Sci. 2006, 55, 629–635.10.5650/jos.55.629Search in Google Scholar
[41] Naoshima, Y.; Mori, Y.; Yagi, Y. Proceeding of the 15th International Conference on Genome Informatics Poster and Software Demonstrations, 2004; P129-1–P129-2.Search in Google Scholar
[42] Shimotori, Y.; Miyakoshi, T. Synthesis of (S)-γ-lactones with a combination of lipase-catalyzed resolution and Mitsunobu reaction. Synth. Commun. 2009, 39, 1570–1582.10.1080/00397910802546177Search in Google Scholar
[43] Shimotori, Y.; Miyakoshi, T. Combination of Novozym 435-catalyzed hydrolysis and Mitsunobu reaction for production of (R)-γ-lactones. Synth. Commun. 2010, 40, 1607–1613.10.1080/00397910903134618Search in Google Scholar
[44] Ema, T.; Kobayashi, J.; Maeno, S.; Sakai, T.; Utaka, M. Origin of the enantioselectivity of lipases explained by a stereo-sensing mechanism operative at the transition state. Bull. Chem. Soc. Jpn. 1998, 71, 443–453.10.1246/bcsj.71.443Search in Google Scholar
[45] Raza, S.; Fransson, L.; Hult, K. Enantioselectivity in Candida antarctica lipase B: a molecular dynamics study. Protein Sci. 2001, 10, 329–338.10.1110/ps.33901Search in Google Scholar
[46] Ishihara, M.; Tsuneya, T.; Shiota, H.; Shiga, M.; Yokoyama, Y. The absolute configurations of marmelo lactones. Agric. Biol. Chem. 1983, 47, 2121–2122.10.1271/bbb1961.47.2121Search in Google Scholar
[47] Patil, S. T.; Karnik, A. V. Aluminum chloride-mediated kinetic resolution of racemic γ-substituted-γ-lactones. Chirality 2004, 16, 336–338.10.1002/chir.20036Search in Google Scholar
[48] Fukuzawa, S.; Seki, K.; Tatsuzawa, M.; Mutoh, K. A facile synthesis of chiral γ-butyrolactones in extremely high enantioselectivity mediated by samarium (II) iodide. J. Am. Chem. Soc. 1997, 119, 1482–1483.10.1021/ja962965hSearch in Google Scholar
©2015 by De Gruyter
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Articles in the same Issue
- Frontmatter
- Reviews
- Synthesis of quinazolines and quinazolinones via palladium-mediated approach
- Media with photoinduced irreversible fluorescence
- Synthesis of quinolines and acridines by the reaction of 2-(perfluoroalkyl)anilines with lithium and Grignard reagents
- Preliminary Communications
- Synthesis of tricyclic indolizidines from ethyl isocyanoacetate
- Ligand- and catalyst-free intramolecular C-S bond formation: direct access to indalothiochromen- 4-ones
- Research Articles
- Preparation of optically active 4-substituted γ-lactones by lipase-catalyzed optical resolution
- One-pot synthesis of 4-alkyl-2-amino-4H-chromene derivatives
- Ring transformation and antimicrobial activity of indolyl-substituted 2(3H)-furanones
Articles in the same Issue
- Frontmatter
- Reviews
- Synthesis of quinazolines and quinazolinones via palladium-mediated approach
- Media with photoinduced irreversible fluorescence
- Synthesis of quinolines and acridines by the reaction of 2-(perfluoroalkyl)anilines with lithium and Grignard reagents
- Preliminary Communications
- Synthesis of tricyclic indolizidines from ethyl isocyanoacetate
- Ligand- and catalyst-free intramolecular C-S bond formation: direct access to indalothiochromen- 4-ones
- Research Articles
- Preparation of optically active 4-substituted γ-lactones by lipase-catalyzed optical resolution
- One-pot synthesis of 4-alkyl-2-amino-4H-chromene derivatives
- Ring transformation and antimicrobial activity of indolyl-substituted 2(3H)-furanones