Synthesis of hydroxylated tryptanthrins as possible metabolites and characterization
-
Dong Hyeon Kim


Abstract
A series of dihydroxytryptanthrins was prepared by the reaction of a suitably substituted isatoic anhydride and isatin as possible metabolites of tryptanthrin. A high-performance liquid chromatography analysis of the synthetic compounds confirmed that 8,9-dihydroxytryptanthrin is the metabolite isolated from rat liver cytosolic metabolites. Although tryptanthrin shows strong cytotoxicity against human cancer cell lines, none of the hydroxylated tryptanthrins exhibit any significant cytotoxicity against selected human cancer cell lines up to 50 μm.
Introduction
Tryptanthrin (1 in Scheme 1, 6,12-dihydro-6,12-dioxoindolo[2,1-b]quinazoline) is an indoloquinazolinone alkaloid first obtained by the sublimation of powdered natural indigo under reduced pressure [1]. Friedländer and Roschdestwensky [2] prepared tryptanthrin chemically from isatin chloride and anthranilic acid and proposed its structure. Tryptanthrin was also isolated from the culture of yeast Candida lipolytica [3] and later from higher plant sources, Couroupita guianensis [4, 5], as well as from fungus, Schizophyllum commune [6].
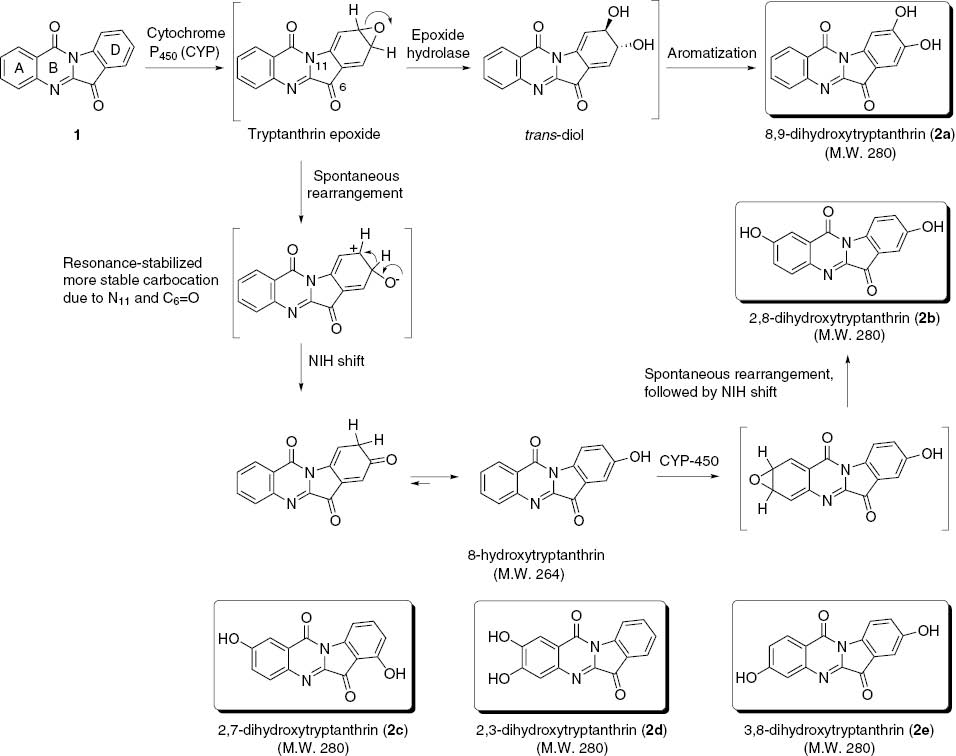
Proposed mechanisms for metabolites of tryptanthrin.
Tryptanthrin shows a variety of biological properties including antimicrobial [3, 7–10] and antifungal activity [7]. The inhibitory activities of tryptanthrin against COX-2 [11], 5-LOX [12], and prostaglandin E(2) expressions at the cellular level [13] led a new vista for the development of new anti-inflammatory agents. Inhibitory activities on hepatocyte growth factor in human fibroblasts [14] and the multidrug resistance gene MDR1 in breast cancer cells [15] and cytotoxicity against selected human cancer cell lines (IC50=10 μm for HT-1376) [6, 16] and antitumor activity [17] of tryptanthrin have also been reported. Such intriguing properties have led to continuous efforts not only to search for similar compounds in other plant sources but also to trigger the development of new methods for total synthesis of tryptanthrin, as recently reviewed [18].
Our early studies on the two identifiable metabolites from the culture of cytosolic cytochrome P450 (CYP)-mediated metabolism of tryptanthrin have revealed their masses of 264 and 280. The MS2 spectra of protonated tryptanthrin and metabolite (M1) have led us to deduce the position of monohydroxylation as being on the aromatic ring of the indole moiety, and the structure was identified as 8-hydroxytryptanthrin by comparison with a chemically synthesized authentic compound [19]. However, the metabolite (M2) with a mass of 280 has not been characterized as yet. In this study, we synthesized a series of dihydroxytryptanthrins to identify cytosolic metabolite(s) with a mass of 280 and evaluated their biological properties, especially the cytotoxicity against selected human cancer cell lines.
Results and discussion
On the basis of the first metabolite, 8-hydroxytryptanthrin (m/z 264), the isolated but as yet unidentified metabolite with a mass of 280 could be a dihydroxytryptanthrin. Investigation of the first and second metabolites of the related quinazolin-4(3H)-one alkaloid rutaecarpine [20–22] and electronic aspects of indole system versus quinazolin-4(3H)-one moiety indicates that the benzene ring of the indole moiety would be more susceptible to the cytochrome P450 (CYP)-mediated oxidative metabolic process. Thus, the first metabolites of tryptanthrin would be 8-hydroxytryptanthrin and 8,9-dihydroxytryptanthrin (2a in Scheme 1). The subsequent CYP-mediated oxidative metabolism of the first metabolite, 8-hydroxytryptanthrin, would lead to the formation of 2,8-dihydroxytryptanthrin (2b) as the second metabolite (M2).
The isolation of 7-hydroxytryptanthrin (known as phaitanthrin C) [16] from natural sources would point out to 2,7-dihydroxytrptanthrin (2c) as an additionally possible candidate for M2. However, the same metabolic process on the less reactive quinazolin-4(3H)-one ring (ring A) could not be excluded to lead to 2,3-dihydroxytryptanthrin (2d) and less favored 3,8-dihydroxytryptanthrin (2e) as possible metabolites. We thus reasoned that the dihydroxytryptanthrins, 2a–2e, could be possible candidates for the as yet unidentified metabolite with a mass of 280 isolated from rat liver microsomes incubated in the presence of an NADPH-generating system.
Syntheses of hydroxylated tryptanthrins are straightforward by using methods previously described in the literature [23], as shown in Scheme 2. Either reactions of suitable methoxyisatins and isatoic anhydride or reactions of isatin and suitable methoxyisatoic anhydrides afford tryptanthrins with hydroxyl group(s) on only one of the two benzene rings, whereas the use of methoxyisatins and methoxyisatoic anhydrides provide hydroxytryptanthrins with hydroxyl groups on the two benzene rings.
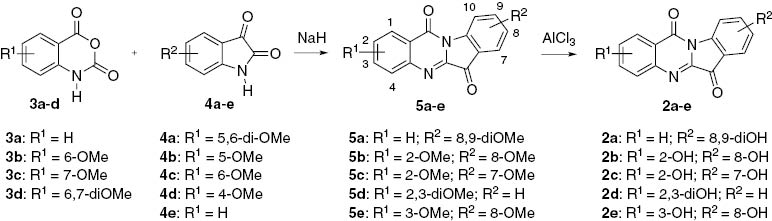
Synthesis of hydroxylated tryptanthrins.
Reactions of isatoic anhydride (3a) with 5,6-dimethoxyisatin (4a) afforded 8,9-dimethoxytryptanthrin (5a) in 35% yield, whereas reactions of 6-methoxyisatoic anhydride (3b) with 5-methoxyisatin (4b) and 4-methoxyisatin (4d) afforded 2,8-dimethoxytryptanthrin (5b) and 2,7-dimethoxytryptanthrin (5c) in 16% and 51% yields, respectively. Similarly, the reactions of 7-methoxyisatoic anhydride (3c) with 4b and 6,7-dimethoxyisatoic anhydride (3d) with isatin (4e) afforded 3,8-dimethoxytryptanthrin (5e) and 2,3-dimethoxytryptanthrin (5d) in 52% and 56% yields, respectively. The AlCl3-mediated O-demethylation of 5 afforded the corresponding hydroxytryptanthrins (2a–e) in 80%–86% yield. The structures of these products were confirmed by spectroscopic methods. Although most of the hydroxytryptanthrins obtained show poor solubility in common organic solvents, the analytically pure samples were easily obtained by using preparative TLC eluting with methanol.
To identify the metabolite, the natural metabolites and the synthetic compounds 2 were analyzed using high-performance liquid chromatography (HPLC). The retention time of metabolite M2 perfectly matches that of 8,9-dihydroxytryptanthrin (2a) at 6.82 min, whereas the retention times of 2,7-, 2,8-, and 3,8-dihydroxytryptanthrin are quite different at 7.38, 7.38, and 7.63 min, respectively. In addition, the mass spectral fragmentation pattern of the metabolite is identical to that of the synthetic compound: m/z 281.1 (M+H), 253.1 [(M+H)-CO], 178.2, and 162.1.
In vitro cytotoxicities of tryptanthrin (7) against selected human cancer lines, including ductal breast epithelial tumor cell line (T47D), colon rectal adrenocarcinoma tumor (HCT15), prostate tumor (DU145), and embryonic kidney 293 cells (HEK293), have been reported [24], and the activities are comparable with those of camptothecin. In this work, cytotoxicities of the hydroxytryptanthrins against the previously selected cancer cell lines were evaluated. However, none of the compounds showed any significant cytotoxic activity up to 50 μm.
Conclusions
A series of dihydroxytryptanthrins was prepared by the reaction of suitably substituted isatoic anhydride and isatin as possible second metabolites of tryptanthrin. HPLC analyses of the synthetic compounds confirm 8,9-dihydroxytryptanthrin as the second metabolite isolated from rat liver cytosolic metabolites. The hydroxylated tryptanthrins did not show any significant cytotoxicity against selected human cancer cell lines up to 50 μm.
Experimental
Melting points were determined using a Fischer-Jones melting points apparatus and are not corrected. NMR spectra were measured using a Bruker-250 spectrometer operating at 250 MHz for 1H NMR and 62.5 MHz for 13C NMR. Chemicals and solvents were commercial reagent grade and used without further purification. Electrospray ionization (ESI) mass spectrometry (MS) experiments were performed on an LCQ advantage-trap mass spectrometer (Thermo Finnigan, San Jose, CA, USA). The prerequisite isatoic anhydrides (3) were prepared as previously reported in the literature by either oxidation of isatin with CrO3 [25] or reaction of suitably substituted anthranilic acid with triphosgene [26]. 4-Methoxyisatin (4b) and 6-methoxyisatin (4d) were prepared by Friedel-Crafts acylation of 2-(hydroxyimino)-N-(3-methoxyphenyl)acetamide [27]. In the HPLC separations, an Intertsil® ODS-3 column (5 μm, 4.6×150 mm, GL Sciences Inc., Tokyo, Japan) with a Phenomenex® SecurityGuard™ cartridge C18 (3.0×4.0 mm, Torrance, CA, USA) was used, eluting with a pH 4.0 buffer (90% acetonitrile with 10% 20 mm ammonium formate; the flow rate was 1.0 mL/min.
8,9-Dimethoxytryptanthrin (5a)
NaH (170 mg, 7.08 mmol) was added to a mixture of isatoic anhydride (3a, 1.16 g, 7.11 mmol) and 5,6-dimethoxyisatin (4a, 1.45 g, 7.00 mmol) in DMF (20 mL). The mixture was heated at 65°C for 2 h then poured into ice water and extracted with CH2Cl2 (3×30 mL). The solvent was evaporated, and the resulting solid was washed with CH3OH to afford the desired 2,3-dimethoxytryptanthrin (5a, 750 mg, 35%) as a brown solid; mp 283–285°C; 1H NMR (CDCl3): δ 8.37 (1H, dd, J = 7.8, 1.4 Hz, H1), 8.18 (1H, s, H10), 8.00 (1H, dd, J = 8.0, 0.8 Hz, H4), 7.82 (1H, 2t, J = 7.8, 1.5 Hz, H2), 7.64 (1H, 2t, J = 7.8, 1.2 Hz, H3), 7.31 (1H, s, H7), 4.09 (3H, s), 3.93 (3H, s); 13C NMR (CDCl3): δ 180.8, 158.1, 157.8, 148.6, 146.9, 143.8, 135.3, 132.0, 130.8, 130.3, 127.5, 123.8, 114.6, 106.3, 101.3, 57.3, 56.6; MS (ESI): m/z 309 [M + H]+. Anal. Calcd for C17H12N2O4: C, 66.23; H, 3.92; N, 9.09. Found: C, 66.15; H, 3.91; N, 9.11.
2,8-Dimethoxytryptanthrin (5b)
CrO3 (2 g) was added to a mixture of 5-methoxyisatin (4c, 690 mg, 3.90 mmol) in glacial HOAc (10 mL) and acetic anhydride (10 mL) in portions at 80°C. The mixture was heated for10 min then cooled to room temperature, and the resultant solid was filtered. The 1H NMR spectrum of the solid showed a mixture of starting 5-methoxyisatin (4c) and desired 6-methoxyisatoic anhydride (3b) in a 1:1.5 ratio. This mixture was subjected to the reaction described previously for 5a to afford the desired 2,8-dimethoxytryptanthrin (5c, 58 mg, 16%) as a brown solid; mp 230°C (sublimed); 1H NMR (CDCl3): δ 8.48 (1H, d, J = 9.0 Hz, H10), 7.90 (1H, d, J = 9.0 Hz, H4), 7.78 (1H, d, J = 2.8 Hz, H1), 7.36 (1H, dd, J = 8.8, 2.5 Hz, H3), 7.34 (1H, d, J = 2.5 Hz, H7), 7.30 (1H, dd, J = 8.8, 2.5 Hz, H9), 3.96 (3H, s), 3.87 (3H, s); 13C NMR (CDCl3): δ 185.1, 163.8, 161.2, 160.0, 145.5, 143.3, 142.7, 134.9, 127.8, 127.3, 126.7, 125.9, 121.7, 110.8, 110.6, 58.6, 58.5; MS (ESI): m/z 309 [M + H]+. Anal. Calcd for C17H12N2O4·H2O: C, 62.57; H, 4.32; N, 8.59. Found: C, 62.63; H, 4.33; N, 8.61.
2,7-Dimethoxytryptanthrin (5c)
The procedure described previously for 4a was applied to 6-methoxyisatoic anhydride (3b, 750 mg, 3.89 mmol) and 4-methoxyisatin (4b, 680 mg, 3.84 mmol) to give the desired 2,7-dimethoxytryptanthrin (5b, 603 mg, 51%) as a brown solid; mp 270°C (dec); 1H NMR (CDCl3): δ 8.24 (1H, d, J = 8.5 Hz, H10), 7.67 (1H, td, J = 8.5, 1.2 Hz, H9), 7.49 (1H, d, J = 1.5 Hz, H1), 7.33 (1H, dd, J = 8.8, 1.3 Hz, H4), 7.18 (1H, dd, J = 8.5, 1.2 Hz, H3), 7.06 (1H, d, J = 8.5 Hz, H8), 3.93 (3H, s), 3.78 (3H, s); MS (ESI): m/z 309 [M + H]+. Anal. Calcd for C17H12N2O4: C, 66.23; H, 3.92; N, 9.09. Found: C, 66.13; H, 3.93; N, 9.10.
2,3-Dimethoxytryptanthrin (5d)
The procedure described previously for 5a was applied to 6,7-dimethoxyisatoic anhydride (3d, 450 mg, 2.08 mmol) and isatin (4e, 290 mg, 1.97 mmol) to give of the desired 2,3-dimethoxytryptanthrin (5d, 316 mg, 52%) as a brown solid; mp >300°C. Spectral data are virtually identical to those reported in the literature [28].
3,8-Dimethoxytryptanthrin (5e)
The procedure described previously for 5a was applied to 7-methoxyisatoic anhydride (3c, 80 mg, 0.42 mmol) and 5-methoxyisatin (4b, 70 mg, 0.40 mmol) to afford the desired 3,8-dimethoxytryptanthrin (5e, 69 mg, 56%) as a brown solid; mp 250°C (dec); 1H NMR (CDCl3): δ 8.48 (1H, d, J = 9.0 Hz), 8.29 (1H, d, J = 9.0 Hz), 7.40 (1H, d, J = 1.2 Hz), 7.34 (1H, d, J = 1.2 Hz), 7.28 (1H, dd, J = 8.8, 1.2 Hz), 7.18 (1H, dd, J = 8.8, 1.2 Hz), 4.08 (3H, s), 3.89 (3H, s); MS (ESI): m/z 309 [M + H]+. Anal. Calcd for C17H12N2O4: C, 66.23; H, 3.92; N, 9.09. Found: C, 66.16; H, 3.91; N, 9.12.
8,9-Dihydroxytryptanthrin (2a)
A mixture of 8,9-dimethoxytryptanthrin (0.6 g, 1.96 mmol) in freshly distilled CH2Cl2 (100 mL) was treated slowly with AlCl3 (1.50 g, 22.43 mmol). This mixture was heated under reflux for 18 h then cooled to room temperature and carefully treated with water (approximately 200 mL). The resultant precipitate (450 mg) was washed with CH2Cl2 (50 mL, three times) to yield 8,9-dihydroxytryptanthrin (2a, 450 mg, 82%) as a brown solid; mp >300°C; 1H NMR (CDCl3): δ 8.21 (1H, dd, J = 7.5, 0.8 Hz, H1), 7.82 (2H, m, H2 & H3), 7.64 (1H, dd, J = 8.0, 0.8 Hz, H4), 7.46 (1H, s, H7/H10), 7.28 (1H, s, H10/H7), 6.56 (1H, phenolic OH, D2O exchangeable), 6.34 (1H, phenolic OH, D2O exchangeable); MS (ESI): m/z 281 [M + H]+. Anal. Calcd for C15H8N2O4·H2O: C, 60.41; H, 3.38; N, 9.39. Found: C, 60.57; H, 3.37; N, 9.38.
2,8-Dihydroxytryptanthrin (2b)
The procedure described previously for 2a was used for 5b (9 mg, 0.03 mmol) to afford 2b as a brown solid (6.5 mg, 80%); mp >275°C (sublimed); 1H NMR (DMSO-d6): δ 10.69 (s, 1H, OH), 10.18 (1H, s, OH), 8.28 (1H, d, J = 8.8 Hz, H10), 7.78 (1H, d, J = 8.5 Hz, H4), 7.57 (1H, d, J = 1.5 Hz, H1), 7.31 (1H, dd, J = 8.5, 1.5 Hz, H3), 7.20 (1H, dd, J = 8.8, 1.5 Hz, H9), 7.11 (d, 1H, J = 1.5 Hz, H7); MS (ESI): m/z 281 [M + H]+. Anal. Calcd for C15H8N2O4: C, 64.29; H, 2.88; N, 10.00. Found: C, 64.06; H, 2.87; N, 10.03.
2,7-Dihydroxytryptanthrin (2c)
The procedure described previously for 2a was used for 5c (31 mg, 0.10 mmol) to afford 2,7-dihydroxytryptanthrin (2c, 24 mg, 85%) as a brown solid; mp >275°C; 1H NMR (DMSO-d6): δ 8.48 (1H, d, J = 9.0 Hz), 8.29 (1H, d, J = 9.0 Hz), 7.40 (1H, d, J = 1.2 Hz), 7.34 (1H, d, J = 1.2 Hz), 7.28 (1H, dd, J = 8.8, 1.2 Hz), 7.18 (1H, dd, J = 8.8, 1.2 Hz); MS (ESI): m/z 281 [M + H]+. Anal. Calcd for C15H8N2O4: C, 64.29; H, 2.88; N, 10.00. Found: C, 64.36, H, 2.87; N, 10.01.
2,3-Dihydroxytryptanthrin (2d)
The procedure described previously for 2a was used for 5d (25 mg, 0.08 mmol) to afford 2d as a brown solid (20 mg, 86%); mp >275°C; MS (ESI): m/z 281 [M + H]+ [29].
3,8-Dihydroxytryptanthrin (2e)
The procedure described previously for 2a was used for 5e (16 mg, 0.05 mmol) to afford 2e as a brown solid (11 mg, 81%); mp >275°C (sublimed); 1H NMR (DMSO-d6): δ 10.85 (s, 1H, OH), 10.16 (1H, s, OH), 8.24 (1H, dd, J = 8.0, 1.3 Hz, H10), 7.78 (1H, d, J = 8.0 Hz, H1), 7.20-7.11 (4H, m, H2, H4, H7, H9);1H NMR (CD3OD): δ 8.29 (1H, dd, J = 8.3, 1.3 Hz, H10), 8.14 (1H, d, J = 8.3 Hz, H1), 7.14 (2H, overlapped d, J = 2.5 Hz, H4 & H7), 7.13 (1H, dd, J = 8.3, 1.2 Hz, H2), 7.07 (1H, dd, J = 8.3, 2.5 Hz, H9); MS (ESI): m/z 281 [M + H]+. Anal. Calcd for C15H8N2O4: C, 64.29; H, 2.88; N, 10.00. Found: C, 64.36; H, 2.89; N, 9.96.
Biotransformation of tryptanthrin
Metabolism of tryptanthrin (100 μm, final concentration) was determined with 1 mg/mL of microsomal protein, obtained from rat live using previously reported method [19], in 0.1 m K2PO4 buffer, pH 7.4, at 37°C for 2 h, in a final incubation volume of 500 μL. The reactions were initiated by the addition of an NADPH-generating system containing 0.8 mm NADPH, 10 mm glucose 6-phosphate, and 1 U glucose 6-phosphate dehydrogenase to the reaction mixture. The reaction was stopped by the addition of EtOAc (1 mL). After mixing and centrifugation, the organic layer (850 μL) was separated and concentrated under a stream of nitrogen gas to give a solid material, which was analyzed using liquid chromatography–mass spectrometry.
Acknowledgments
Financial support from Yeungnam University (212A380221) is gratefully appreciated.
References
[1] von Sommaruga, E. Über die Moleculargrösse des Indigos. Ann. 1879, 195, 302–313.Suche in Google Scholar
[2] Friedländer, P.; Roschdestwensky, N. Über ein Oxydationspdukt des Indigblaus. Ber. Chem.1915, 48, 1841–1847.Suche in Google Scholar
[3] Schindler, W.; Zähner, H. Stoffwechselprodukte von Mikroorganismen-91. Mittelung-Tryptanthrin, ein von Tryptophan abzuleittendes Antibioticum aus Candida lipolitica. Arch. Mikrobiol. 1971, 79, 187–203.Suche in Google Scholar
[4] Sen, A. K.; Mahato, S. B.; Dutta, N. L. Couroupitine A, A new alkaloid from Courouita guianensis. Tetrahedron Lett.1974, 609–610.10.1016/S0040-4039(01)82284-XSuche in Google Scholar
[5] Bergman, J.; Egestad, B.; Lindström, J. O. The structure of some indolic constituents in Couroupita guaianesis Abul. Tetrahedron Lett.1977, 2625–2626.10.1016/S0040-4039(01)83838-7Suche in Google Scholar
[6] Hosoe, T.; Nozawa, K.; Kawahara, N.; Fukushima, K.; Nishimura, K.; Miyaji, M.; Kawai, K. Isolation of a new potent cytotoxic pigment along with indigotin from the pathogenic Basicdiomycetous fungus Schzophyllum commune. Mycopathologia1999, 146, 9–12.Suche in Google Scholar
[7] Honda, G.; Tabata, M. Isolation of antifungal principle tryptanthrin, from Strobilanthes cusia O. Kuntze. Planta Med.1979, 36, 85–86.Suche in Google Scholar
[8] Bandekar, P. P.; Roopnarine, K. A.; Parekh, V. J.; Mitchell, T. R.; Novak, M. J.; Sinden, R. R. Antimicrobial activity of tryptanthrins in Escherichia coli. J. Med. Chem. 2010, 53, 3558–3565.Suche in Google Scholar
[9] Mitscher, L. A.; Baker, W. R. Tuberculosis: a search for novel therapy starting with natural products. Med. Res. Rev. 1998, 18, 363–374.Suche in Google Scholar
[10] Hashimoto, T.; Aga, H.; Chaen, H.; Fukuda, S.; Kurimoto, M. Isolation and identification of anti-Helicobacter pylori compounds from Polygonum tinctorium Lour. Nat. Med. (Tokyo), 1999, 53, 27–31.Suche in Google Scholar
[11] Danz, H.; Stoyanova, S.; Wippich, P.; Brattstrom, A.; Hamburger, M. Identification and isolation of the cyclooxygenase-2 inhibitory principles in Isatis tinctoria. Planta Med.2001, 67, 411–416.Suche in Google Scholar
[12] Oberthür, C.; Jaeggi, R.; Hamburger, M. HPLC based activity profiling for 5-lipoxygenase inhibitory activity in Isatis tinctoria leaf extracts. Fitoterapia2005, 76, 324–332.Suche in Google Scholar
[13] Ishihara, T.; Kohno, K.; Ushio, S.; Iwaki, K.; Ikeda, M.; Kurimoto, M. Tryptanthrin inhibits nitric oxide and prostaglandin E2 synthesized by murine macrophases. Eur. J. Pharmacol. 2000, 407, 197–204.Suche in Google Scholar
[14] Motoki, T.; Takami, Y.; Yagi, Y.; Tai, A.; Yamamoto, I.; Gohda, E. Inhibition of hepatocyte growth factor induction in human dermal fibroblasts by tryptanthrin. Biol. Pharm. Bull. 2005, 28, 260–266.Suche in Google Scholar
[15] Yu, S. T.; Chen, T. M.; Tseng, S. Y.; Chen, Y. H. Tryptanthrin inhibits MDR1 and reverse doxorubicin resistance in breast cancer cells. Biochem. Biophys. Res. Commun. 2007, 358, 79–84.Suche in Google Scholar
[16] Jao, C.-W.; Lin, W.-C.; Wu, Y.-T.; Wu. P.-L. Isolation, structure elucidation, and synthesis of cytotoxic tryptanthrin analogues from Phaius mishmensis. J. Nat. Prod. 2008, 71, 1275–1279.Suche in Google Scholar
[17] Koya-Miyata, S.; Kimoto, T.; Micallef, M. J.; Hino, K.; Taniguchi, M.; Ushio, S.; Iwaki, K.; Ikeda, M.; Kurimoto, M. Prevention of azoxymethane-induced intestinal tumors by a cute ethyl acetate-extract and tryptanthrin extracted from Polygonum tinctorium lour. Anticancer Res. 2001, 21, 3295–3300.Suche in Google Scholar
[18] Jahng, Y. Progress in the studies on tryptanthrin, an alkaloid of history. Arch. Pharm. Res. 2013, 36, 517–535.Suche in Google Scholar
[19] Lee, S. K.; Kim, G. H.; Kim, D. H.; Kim, D. H.; Jahng, Y.; Jeong, T. C. Identification of a tryptanthrin metabolite in rat liver microsomes by liquid chromatography/electrospray ionization-tandem mass spectrometry. Biol. Pharm. Bull. 2007, 30, 1991–1995.Suche in Google Scholar
[20] Lee, S. K.; Kim, N. H.; Lee, J.; Kim, D. H.; Lee, E. S.; Choi, H. G.; Chang, H. W.; Jahng, Y.; Jeong, T. C. Induction of cytochrome P450s by rutaecarpine and metabolism of rutaecarpine by cytochrome P450s. Planta Med.2004, 70, 753–757.Suche in Google Scholar
[21] Lee, S. K.; Lee, J.; Lee, E. S.; Jahng, Y.; Kim, D. H.; Jeong, T. C. Characterization of in vitro Metabolites of rutaecarpine in rat liver microsomes using liquid chromatography/tandem mass spectrometry. Rapid Commun. Mass Spectrom.2004, 18, 1073–1080.Suche in Google Scholar
[22] Ueng, Y. F.; Yu, H. J.; Lee, C. H.; Peng, C.; Jan, W. C.; Ho, L. K.; Chen, C. F.; Don, M. J. Identification of the microsomal oxidation metabolites of rutaecarpine: a main active alkaloid of the medicinal herb. J. Chromatogr. A2005, 1076, 103–109.Suche in Google Scholar
[23] Mitscher, L. A.; Wong, W.-C.; DeMeulenaere, T.; Sulko, J.; Crake, S. Antimicrobial agents from plants. New synthesis and bioactivity of tryptanthrin (indolo[2,1-b]quinazoline-6,12-dione) and its derivatives. Heterocycles1981, 15, 1017–1021.Suche in Google Scholar
[24] Liang, J. L.; Eom, J.-E.; Kwon, Y.; Jahng, Y. Synthesis of benzo-annulated tryptanthrins and their biological properties. Bioorg. Med. Chem. 2012, 20, 4962–4967.Suche in Google Scholar
[25] Castle, R. N.; Adachi, K.; Guither, W. D. Cinnoline chemistry. XII. The synthesis of 6-fluoro-4-methylcinnoline and other cinnolines as potential antitumor agents (1). J. Heterocycl. Chem.1965, 2, 459–462.Suche in Google Scholar
[26] Peet, N. P.; Sunder, S. Phosgenation of methyl anthranilate. J. Org. Chem.1974, 39, 1931–1935.Suche in Google Scholar
[27] Sandmeyer, T. On isonitroacetanilides and their condensation to isatins. Hev. Chim. Acta1919, 2, 234–242.10.1002/hlca.19190020125Suche in Google Scholar
[28] Baker, W. R.; Mitscher, L. A. Preparation of indolo[2,1-b]quinazoline-6,12-dione tuberculostatics. US Patent 5,441,955; 1995.Suche in Google Scholar
[29] Mason, J. J.; Janosik, T.; Bergman, J. A new approach to methoxyisatins leading to the total synthesis of ophiuridine and other hydroxytryptanthrins. Synthesis2009, 21, 3642–3648.Suche in Google Scholar
©2015 by De Gruyter
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Artikel in diesem Heft
- Frontmatter
- Preliminary Communication
- Preparation of cysteine adducts by regioselective ring-opening reactions of phenyloxirane
- Research Articles
- Synthesis of hydroxylated tryptanthrins as possible metabolites and characterization
- Tributylhexadecylphosphonium bromide: an efficient reagent system for the one-pot synthesis of 2,4,5-trisubstituted imidazoles
- Synthesis and properties of multifunctional nitroxyl radical hindered-amine light stabilizers
- Photochemical oxidation of N,N-bis- (tert-butoxycarbonyl)-1,4-dihydropyrazine derivatives
- ZrOCl2·SiO2-catalyzed synthesis of bis(indoles) via conjugate addition of indole with electron-deficient alkenes in water
- Synthesis of polysubstituted pyridines as potential multidrug resistance modulators
- Catalyst-free tandem Knoevenagel-Michael reaction of aldehydes and pyrazolin-5-one: fast and convenient approach to medicinally relevant 4,4′-(arylmethylene)bis(1H-pyrazol-5-ol)s
- Synthesis of mono- and bis-1,2,3-triazole derivatives containing 4H-pyran-4-one moiety by 1,3-dipolar cycloaddition reaction
- Synthesis and biological evaluation of novel 2-(1H-imidazol-2-ylmethylidene)hydrazinyl- 1,3-thiazoles as potential antimicrobial agents
Artikel in diesem Heft
- Frontmatter
- Preliminary Communication
- Preparation of cysteine adducts by regioselective ring-opening reactions of phenyloxirane
- Research Articles
- Synthesis of hydroxylated tryptanthrins as possible metabolites and characterization
- Tributylhexadecylphosphonium bromide: an efficient reagent system for the one-pot synthesis of 2,4,5-trisubstituted imidazoles
- Synthesis and properties of multifunctional nitroxyl radical hindered-amine light stabilizers
- Photochemical oxidation of N,N-bis- (tert-butoxycarbonyl)-1,4-dihydropyrazine derivatives
- ZrOCl2·SiO2-catalyzed synthesis of bis(indoles) via conjugate addition of indole with electron-deficient alkenes in water
- Synthesis of polysubstituted pyridines as potential multidrug resistance modulators
- Catalyst-free tandem Knoevenagel-Michael reaction of aldehydes and pyrazolin-5-one: fast and convenient approach to medicinally relevant 4,4′-(arylmethylene)bis(1H-pyrazol-5-ol)s
- Synthesis of mono- and bis-1,2,3-triazole derivatives containing 4H-pyran-4-one moiety by 1,3-dipolar cycloaddition reaction
- Synthesis and biological evaluation of novel 2-(1H-imidazol-2-ylmethylidene)hydrazinyl- 1,3-thiazoles as potential antimicrobial agents