Tertiary formylated amines by microwave irradiation of N,N-dimethyl-N′-(2-pyridyl)formamidines with methyl vinyl ketone
-
Omar Gómez-García
 und
Elena Campos-Aldrete
und
Elena Campos-Aldrete

Abstract
Treatment of N,N-dimethyl-N′-(2-pyridyl)formamidine with methyl vinyl ketone under microwave irradiation yields N-formyl-N-(3-oxobutyl)-2-pyridylamine without decomposition or polymerization of methyl vinyl ketone. The result is an alternative non-oxidative approach to the preparation of N-formyl-substituted aminoketones.
Introduction
The amidine functional group contains an amino nitrogen atom with a free electron pair conjugated with the π-electrons of the C=N double bond. This functional group has been often utilized in the synthesis of heterocycles. For example, N-monosubstituted formamidines readily react with ylidenemalononitriles to give useful pyrimidine intermediates [1]. Amidines have been used as a three- or two-unit synthon in the synthesis of 3- to 6-membered heterocycles [2]. Amidines have also been reacted with methyl-2-dimethoxymethyl-3-methoxypropionate to give dihydropyridopyrimidines [3]. They are also intermediates in the synthesis of [1,2,4]triazolo[1,5-a]pyridines [4]. Evaluation of biological activity of some amidine derivatives has also been reported [5]. Recently, we have utilized the core N,N-dimethyl-N′-heteroarylformamidine in the construction of heteroaryl-N-imidazoles [6].
Herein, we describe the results obtained from the reaction of N,N-dimethyl-N′-(2-pyridyl)formamidine with methyl vinyl ketone (MVK) under microwave irradiation.
Results and discussion
Pyridylformamidines 1 were readily prepared by condensation of a 2-aminopyridine with an excess of N,N-dimethylformamide dimethyl acetal [7]. Initially, we were interested in the possibility that pyridylformamidines 1 would undergo a hetero Diels-Alder type reaction with MVK 2 (Scheme 1), to give pyridopyrimidines 3 by analogy with the reaction of N1,N1-dimethyl-N2-[(1-phenyl-3-methyl)pyrazol-5-yl]formamidine, which undergoes a hetero Diels-Alder reaction with N-phenylmaleimide or N-4′-nitrophenyl maleimide under microwave irradiation to give a tricyclic cycloadduct [8]. First, the reaction was carried out on conventional heating using an excess of MVK in different solvents (DMF, MeCN) or without solvent. In most experiments, mixtures of substrate 1 and polymerized MVK were obtained. Experiments under microwave (mw) irradiation followed, until best conditions (160 W, 140°C) were found that did not cause MVK polymerization. However, the isolated product did not correspond to the expected cycloadduct 3; instead, the tertiary amine N-formyl-N-(3-oxobutyl)-2-pyridylamine (4) was isolated in good yield.
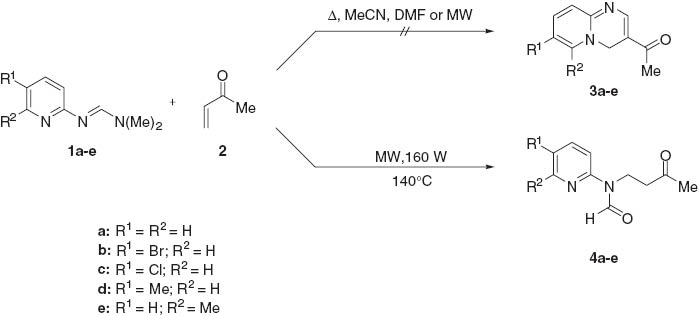
This result, together with the finding that formamidines 1 can be transformed into N-arylimidazoles [6] in a related reaction, support the suggestion that the most nucleophilic nitrogen is the nitrogen of the moiety C=N2. By contrast, in the reaction of these formamidines with phenacyl bromides, it is the endo pyridine nitrogen which reacts first [9]. The most probable mechanism of the synthesis of 4 involves the initial attack of the imine nitrogen C=N2 (assisted by the functionality Me2N1) to the Michael acceptor MVK. Hydrolysis of the dimethyliminium intermediate leads to the N-formyl derivative 4.
Proper assignment of protons and carbons of molecules 4 was achieved with the aid of GHMBC and GHSQC NMR experiments (examples can be found in the supplementary material). The N-formyl proton was readily identified in the 1H NMR spectra as a singlet at δ 8.93–9.07 and the formyl carbon atom in the 13C NMR spectra at δ 161.5–162.2.
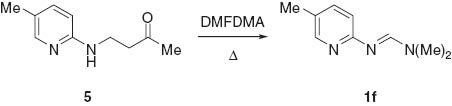
Treatment of the N-formylated aminoketone 4 with acid causes deformylation to give the product of Michael addition of a 2-pyridylamine with MVK, which is exemplified by 5 in Scheme 2. This Michael addition reaction has been reported elsewhere [10]. The treatment of 4 with ammonium acetate causes a retro Michael addition giving the starting MVK and 2-aminopyridine. By contrast, the reaction of sodium borohydride in either neutral or acidic solution gives the deformylated product, such as 5. Interestingly, treatment of 5 with an excess of dimethylformamide dimethyl acetal under mw conditions gave the starting formamidine 1f in 90% yield (Scheme 2).
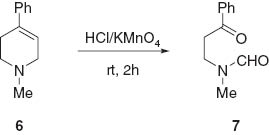
It has been previously demonstrated that treatment of 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine (hydrochloride) 6 with potassium permanganate in hydrochloric acid solution leads to the open structure N-formyl-N-methyl-2-benzoylethylamine 7 (Scheme 3) [11]. Therefore, the reaction described here may be considered as a non- oxidative alternative approach to the preparation of the analogs 4.
Experimental
Melting points were measured on an electrothermal melting point apparatus and are reported without correction. 1H and 13C NMR spectral data were recorded in CDCl3 at 300 and 75 MHz, respectively, using a Varian Mercury 300 MHz NMR spectrometer or at 500 and 125 MHz, respectively, using a Varian NMR System 500 MHz spectrometer. The microwave irradiation was performed in a Discovery microwave apparatus.
2-Aminopyridines, N,N-dimethylformamide dimethyl acetal and MVK were purchased from Aldrich Chemical Co.
General procedure for the synthesis of N-formyl-N-(2-pyridyl)-3-oxobutylamines 4a–e
An N,N-dimethyl-N′-(2-pyridyl)formamidine (1, 0.96 g, 5 mmol) was placed under nitrogen atmosphere in a sealed microwave tube containing a magnetic stirrer. Immediately, MVK (2, 2.8 g, 40 mmol) was added, nitrogen flow was suspended and the tube was sealed. The microwave apparatus was set up at 160 W, 140°C, ramp time 5 min, and hold time 10 min keeping a constant stirring. Progress of the reaction was monitored by TLC indicating the starting material. After completion of the reaction, the mixture was allowed to cool to room temperature and the crude product was extracted with dichloromethane (3×15 mL). The extract was dried (Na2SO4) and concentrated under reduced pressure. The crude product was purified by flash chromatography on silica gel eluting with a mixture of hexanes/ethyl acetate (9:1) to furnish 4 as clear yellow oil or solid.
N-Formyl-N-(3-oxobutyl)-2-pyridylamine (4a)
An oil; yield 89%; 1H NMR (500 MHz): δ 2.14 (3H, s, Me), 2.68 (2H, t, J = 7.5 Hz), 4.18 (2H, t, J = 7.5 Hz), 7.07 (1H, d, J = 8.5 Hz), 7.11 (1H, dq, J = 8.5 Hz, J = 2.5 Hz, J = 1 Hz), 7.7 (1H, dt, J = 8.5 Hz, J = 1 Hz), 8.38 (d, 1H, J = 8.5 Hz), 9.06 (s, 1H); 13C NMR (125 MHz): δ 29.9, 37.6, 41.4, 112.6, 120.5, 138.5, 148.8, 153.3, 162.1, 206.7. Anal. Calcd for C10H12N2O2: C, 62.48; H, 6.29. Found: C, 62.02; H, 6.53.
5-Bromo-N-Formyl-N-(3-oxobutyl)-2-pyridylamine (4b)
This compound was obtained after 60 min of mw irradiation as yellow crystals; yield 83%; mp 102–103°C; 1H NMR (300 MHz): δ 2.09 (3H, s), 2.83 (2H, t, J = 7.5 Hz), 4.16 (2H, t, J = 7.5 Hz), 7 (1H, d, J = 8.5 Hz), 7.82 (1H, dd, J = 8.5 Hz, J = 2.5 Hz), 8.43 (1H, d, J = 2.5 Hz), 9,07 (1H, s); 13C NMR (75 MHz): δ 29.8, 37.4, 41.1, 113.4, 115.9, 140.8, 149, 151.7, 161.5, 206.3. Anal. Calcd for C10H11N2BrO2: C, 44.3; H, 4.08. Found: C, 44.19; H, 3.98.
5-Chloro-N-Formyl-N-(3-oxobutyl)-2-pyridylamine (4c)
This compound was obtained after 55 min of mw irradiation as dark yellow crystals; yield 81%; mp 92–93°C; 1H NMR (500 MHz): δ 2.09 (1H, s), 2.77 (2H, t, J = 7.5 Hz), 4.09 (2H, t, J = 7.5 Hz), 7.02 (1H, d, J = 8.5 Hz), 7.63 (1H, dd, J = 8.5 Hz, J = 2.5 Hz), 8.26 (1H, d, J = 2.5 Hz), 8.98 (1H, s); 13C NMR (125 MHz): δ 29.9, 37.5, 41.1, 113.1, 128, 138.1, 147.1, 151.4, 161.7, 206.6. Anal. Calcd for C10H11N2ClO2: C, 52.99; H, 4.89. Found: C, 52.77; H, 5.04.
N-Formyl-5-methyl-N-(3-oxobutyl)-2-pyridylamine (4d)
This compound was obtained after 20 min of mw irradiation as clear yellow oil; yield 88%; 1H NMR (300 MHz): δ 2.13 (3H, s), 2.28 (3 H, s), 2.57 (2H, t, J = 7.5 Hz), 4.15 (3H, t, J = 7.5 Hz), 6.95 (1H, d, J = 8.5 Hz), 7.51 (1H, dt, J = 8.5 Hz, J = 1.0 Hz), 8.20 (1H, dd, J = 2.5 Hz, J = 1.0 Hz), 8.94 (1H, s); 13C NMR (75 MHz): δ 17.7, 29.7, 37.9, 41.6, 112.63, 130.8, 139.1, 148.9, 151.2, 162.1, 206.8. Anal. Calcd for C11H14N2O2: C, 64.06; H, 6.84. Found: C, 63.64; H, 7.29.
N-Formyl-6-methyl-N-(3-oxobutyl)-2-pyridylamine (4e)
This compound was obtained after 22 min of mw irradiation as brown oil; yield 82%; 1H NMR (300 MHz): δ 2.03 (3H, s), 2.36 (3H, s), 2.71 (2H, t, J = 7.5 Hz), 4.04 (2H, t, J = 7.5 Hz), 6.76 (1H, d, J = 8.5 Hz), 6.87 (1H, d, J = 8.5 Hz), 7.49 (1H, t, J = 8.5 Hz), 8.93 (1H, s); 13C NMR (75 MHz): δ 24.2, 30, 37.5, 41.4, 109.4, 119.9, 138.6, 152.4, 158.1, 162.2, 206.9. Anal. Calcd for C11H14N2O2: C, 64.06; H, 6.84. Found: C, 64.50; H, 7.21.
5-Methyl-N-(3-oxobutyl)-2-pyridylamine (5)
[10] This compound was obtained as reported previously by the reaction of 2-amino-5-methylpyridine (1.08 g, 10 mmol) with MVK (0.77 g, 11 mmol); yield 80% of yellow crystals; mp 124–125.5°C; 1H NMR (300 MHz): δ 2.13 (6H, s), 2.73 (2H, t, J = 6.0 Hz), 3.54 (2H, t, J = 6.0 Hz), 6.29 (1H, d, J = 8.4 Hz), 7.19 (1H, dd, J = 8.4 Hz, J = 2.4 Hz), 7.85 (1H, dd, J = 2.4 Hz, J = 0.9 Hz); 13C NMR (75 MHz): δ 17.3, 30.26, 36.4, 43, 107.4, 121.5, 138.4, 147.2, 156.4, 208.4.
N1,N1-Dimethyl-N2-(5-methyl-2-pyridyl)formamidine (1f)
[6] Dark yellow crystals; yield 90%; mp 68–70°C; 1H NMR (300 MHz): δ 2.14 (3H, s), 2.96 (6H, s), 6.77 (1H, d, J = 8.1 Hz), 7.25 (1H, dq, J = 8.1 Hz, J = 3 Hz, J = 0.9 Hz), 7.96 (1H, dd, J = 3 Hz, J = 0.9 Hz), 8.26 (1H, s); 13C NMR (75 MHz): δ 17.4, 17.5, 34.3, 117, 126.5, 138.3, 148, 155, 159.6.
References
[1] Roveb, S. K. Synthesis of heterocycles based on N-substituted amidines and ylidenemalononitriles. Chem. Heterocycl. Compounds1981, 17, 1153–1158.10.1007/BF00506981Suche in Google Scholar
[2] Aly, A. A.; Nour, A. M. Functionality of amidines and amidrazones. ARKIVOC2008, i, 153–194.10.3998/ark.5550190.0009.106Suche in Google Scholar
[3] Nishino, T.; Miichi, Y.; Tokuyama, K. The reaction of methyl 2-dimethoxymethyl-3-methoxypropionate with amidines. Bull. Chem. Soc. Jpn. 1973, 46, 580–583.Suche in Google Scholar
[4] Huntsman, E.; Balsells, J. New method for the general synthesis of [1,2,4]triazolo[1,5-a]pyridines. Eur. J. Org. Chem. 2005, 3761–3765.10.1002/ejoc.200500247Suche in Google Scholar
[5] Sondhi, S. M.; Dwivedi, A. D.; Singh, J.; Gupta, P. P. Synthesis and anti-inflammatory evaluation of some sulfonamide and amidine derivatives of 4-aryl-3-(2 or 4-picolyl)-2-imino-4-thiazolines. Indian J. Chem. 2010, 49, 1076–1082.Suche in Google Scholar
[6] Gómez-García, O.; Salgado-Zamora, H.; Reyes-Arellano, A.; Campos-Aldrete, M.; Peralta-Cruz, J. Reaction of tosylmethylisocyanide with N-heteroaryl formamidines: an alternative approach to the synthesis of N-heteroaryl tosylimidazoles. Bull. Korean Chem. Soc. 2013, 34, 2807–2810.Suche in Google Scholar
[7] Cunningham, I. D.; Blanden, J. S.; Kior, J.; Muñoz, L.; Sharratt, A. P. Chemistry of amidines. Part 1. Determination of the site of initial protonation in N-pyridylformamidines. J. Chem. Soc. Perkin Trans. 1991, 2, 1747–1750.Suche in Google Scholar
[8] Nascimento-Junior, N. M.; Mendes, T. C. F.; Leal, D. M.; Correa, C. M. N.; Sudo, R.T.; Zapata-Sudo, G.; Barreiro E. J.; Fraga, C. A. M. Microwave assisted synthesis and structure-activity relationships of neuroactive pyrazolo[3,4-b]pyrrolo[3,4-d]pyridine derivatives. Bioorg. Med. Chem. Lett.2010, 20, 74–77.Suche in Google Scholar
[9] Podergajs, S.; Stanovnik, B.; Tisler, M. A new approach for the synthesis of fused imidazoles: the synthesis of 3-acyl substituted imidazo[1,2-x]azines. Synthesis1984, 263.10.1055/s-1984-30802Suche in Google Scholar
[10] Jiang, R.; Li, D.-H.; Jiang, J.; Xu. X.-P.; Chen, T.; Ji, S. Green, efficient and practical Michael addition of arylamines to α,β-unsaturated ketones. Tetrahedron2011, 67, 3631–3637.Suche in Google Scholar
[11] Soldatenkov, A. T.; Temesgen, A. W.; Bekro, I. A.; Khristoforova, T. P.; Soldatova, S. A.; Anissimov, B. N. New oxidative reactions of 1,2,3,6-tetrahydropyridines: imination, lactamination and decyclisation in the presence of potassium permanganate. Mendeleev Commun. 1997, 7, 243–244.Suche in Google Scholar
©2014 by Walter de Gruyter Berlin Boston
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Artikel in diesem Heft
- Frontmatter
- Preliminary Communication
- One-pot three-component synthesis of substituted 2-(1,2,3-triazol-1-yl)pyrimidines from pyrimidin-2-yl sulfonates, sodium azide and active methylene ketones
- Research Articles
- A convenient synthesis of 5,5′-bi-1,2,4-triazines via direct S-arylation and its application in the synthesis of 2,2′-bipyridines
- An efficient approach to the key intermediate of rosuvastatin
- Synthesis and properties of multifunctional hindered amine light stabilizers
- Tertiary formylated amines by microwave irradiation of N,N-dimethyl-N′-(2-pyridyl)formamidines with methyl vinyl ketone
- Synthesis and antimicrobial activity of some novel 2-thienyl substituted heterocycles
- Synthesis of 2-amino-5-mercapto-1,3,4-thiadiazole derivatives
- An easy and efficient protocol for the condensation reaction of isatin and N-substituted isatins with 1,2-diaminobenzene using low cost reusable clay catalyst
- Synthesis and antimicrobial activities of novel 6-(1,3-thiazol-4-yl)-1,3-benzoxazol-2(3H)-one derivatives
- A concise and efficient synthesis of (+)-preussin
- Synthesis, X-ray structural characterization, NLO, MEP, NBO and HOMO-LUMO analysis using DFT study of Zn(II)bis(3,4 dimethoxybenzoate)bis(nicotinamide) dihydrate
Artikel in diesem Heft
- Frontmatter
- Preliminary Communication
- One-pot three-component synthesis of substituted 2-(1,2,3-triazol-1-yl)pyrimidines from pyrimidin-2-yl sulfonates, sodium azide and active methylene ketones
- Research Articles
- A convenient synthesis of 5,5′-bi-1,2,4-triazines via direct S-arylation and its application in the synthesis of 2,2′-bipyridines
- An efficient approach to the key intermediate of rosuvastatin
- Synthesis and properties of multifunctional hindered amine light stabilizers
- Tertiary formylated amines by microwave irradiation of N,N-dimethyl-N′-(2-pyridyl)formamidines with methyl vinyl ketone
- Synthesis and antimicrobial activity of some novel 2-thienyl substituted heterocycles
- Synthesis of 2-amino-5-mercapto-1,3,4-thiadiazole derivatives
- An easy and efficient protocol for the condensation reaction of isatin and N-substituted isatins with 1,2-diaminobenzene using low cost reusable clay catalyst
- Synthesis and antimicrobial activities of novel 6-(1,3-thiazol-4-yl)-1,3-benzoxazol-2(3H)-one derivatives
- A concise and efficient synthesis of (+)-preussin
- Synthesis, X-ray structural characterization, NLO, MEP, NBO and HOMO-LUMO analysis using DFT study of Zn(II)bis(3,4 dimethoxybenzoate)bis(nicotinamide) dihydrate