Impact of the new European medical device regulation: a two-year comparison
-
Ann-Kathrin Carl

 and
David Hochmann
and
David Hochmann

Abstract
Objectives
In recent years, the European Union has revised its regulatory framework for medical devices, primarily to improve patient safety and public health. The Medical Device Regulation (MDR) is fully applicable since May 2021, strengthening the requirements for all stakeholders. As a result, many companies are facing enormous challenges. The aim of this study was to assess the impact of the MDR on the orthopaedic aids industry.
Methods
Two surveys were conducted: one shortly before the MDR became applicable (146 respondents) and a second survey almost two years later (233 respondents).
Results
Both surveys revealed that all businesses in the orthopaedic aids sector, regardless of size, have difficulty implementing the MDR. Key challenges include additional workload for technical documentation, increased resource expenditure and cost, and lack of clarity regarding the new requirements. Many companies are downsizing their product portfolio, resulting in potential supply shortages and a loss of competitive advantage and innovation for the medical device industry in Europe.
Conclusions
The full extent of the MDR’s impact on clinical practice is still unclear. However, many companies lack the necessary resources. The MDR can potentially be a bottleneck in the availability of medical devices.
Introduction
The European Union has revised its legislation on medical devices, introducing a new regulatory framework in April 2017: the Medical Device Regulation (EU) 2017/745 (MDR). It can be seen as a comprehensive change to the regulation of medical devices in Europe. The MDR replaces the Directive 93/42/EEC (Medical Device Directive, MDD) and the Directive 90/385/EEC (Active Implantable Medical Device Directive, AIMDD) and has been in full force and effect since May 26, 2021. Advances in medical device technology and science have led to the need for a revision of previous regulatory requirements [1]. Scandals involving unsafe medical devices, such as problems with breast implants manufactured by the French company Poly Implant Prothèse (PIP) [2], faulty metal-on-metal total hip prosthesis [3] or defective implantable transvaginal mesh devices [4], highlighted the shortcomings of the previous EU legislation and increased the need for change.
The MDR seeks to harmonize the regulatory framework and the approval process for medical devices across all EU Member States [1]. Unlike directives, regulations are binding legal acts. They are directly applicable and have immediate effect in all EU Member states without having to be transposed into national law [5]. The MDR is therefore intended to improve equality for patients in the EU and to level the playing field for suppliers [1]. Improving patient safety, efficiency and public health through appropriate regulatory oversight is the ultimate goal of the MDR [6, 7].
In general, the MDR retains the core elements and requirements of the previous directives. However, there have been a number of changes and additions to the requirements. With a much broader scope, the MDR addresses the entire lifecycle of a medical device. For all stakeholders in the medical device industry, this inevitably means change. This is due to, among other reasons,
the extended scope of the MDR,
increased requirements for clinical evaluation and clinical investigations,
mandatory implementation of a system for identification and traceability of medical devices: the Unique Device Identification (UDI) system,
entering information into the EUDAMED database, and
strengthened requirements for post market surveillance and post market clinical follow-up.
As a result, many companies are faced with enormous challenges [8]. Small and medium-sized enterprises (SMEs) in particular are struggling to implement the MDR. Yet, the medical device industry in Germany is dominated by SMEs: 93 percent of companies have fewer than 250 employees [9]. In most cases, these companies have limited resources. Implementing the additional effort and cost, including refinancing on the market, is therefore much more difficult.
In the present study, German distributors and manufacturers of orthopaedic aids, primarily exo-prosthetics and orthotics, as well as employees of health care supply stores were surveyed twice on numerous aspects of the implementation of the new regulatory framework. The first survey was conducted shortly before the MDR came into force, and the repeat survey almost two years later. It was designed to assess the current status of the MDR in the orthopaedic aids industry before and after the MDR became effective, including the expected consequences. Simultaneously, the two surveys were compared in order to estimate the changes and the impact of the MDR since the date of its application. The main purpose of the questionnaires was to provide answers to the following questions:
How far along has the orthopaedic aids industry come in implementing the MDR?
How has the transition from MDD to MDR gone so far?
What difficulties have been encountered since the date of application?
Is assistance needed in implementing the MDR, and if so, where?
Does the MDR stifle innovation in the industry?
Materials and methods
General procedure
This non-experimental, observational study was designed as a web-based, cross-sectional, repeated survey to compare experiences and opinions regarding the acceptance and feasibility of the MDR among workers in the orthopaedic aids industry in Germany. Data collection took place in two phases: before the MDR became applicable in 2021 (initial survey, InS) and almost two years after in 2023 (repeat survey, ReS).
The questionnaire was evaluated by two independent external reviewers, based on which the wording of some of the questions was changed and the structure adjusted. Additional minor edits were made after the initial survey was pretested with three orthopaedic technicians.
Data collection
Surveys were completed by workers in the orthopaedic aids sector to investigate the current status of the application of the MDR (mandatory as of May 2021) and the expected consequences. Data were self-reported and collected automatically and anonymously by SoSci Survey. The surveys were completed online. The initial survey was open for approximately six weeks in January and February 2021. The follow up survey was conducted two years later. It was also accessible for about six weeks in February and March 2023. The self-administered, web-based survey was distributed to the approximately 3,000 health care supply stores in Germany.
Participants
Participants were distributors and manufacturers of orthopaedic aids, primarily exo-prosthetics and orthotics, as well as employees of health care supply stores in Germany. The call for participation was distributed via various media channels, such as the 360° specialist portal of the Bundesinnungsverband für Orthopädie-Technik, the MTD-Instant newsletter and others. In addition, an email containing a hyperlink to the survey and a cover letter explaining the purpose of the study, summarizing the content of the questionnaire, and providing contact information was sent to health care supply stores in Germany. Two reminders with the same structure as the initial email were sent electronically approximately three and five weeks after the first invite. Subjects were informed that participation was voluntary, and that no financial compensation was provided for completing of the survey.
Instrumentation
A web-based questionnaire was developed using the SoSci Survey tool, taking into account previous surveys [10, 11]. Each questionnaire took approximately 5–10 min to complete. In preparation for the repeat survey, the first questionnaire was adapted to the experiences of the initial survey. Some of the questions were omitted or modified as they related to the period before the MDR became applicable. Due to the adjustments made, the repeat survey contained 15 closed-ended questions, five fewer than the initial survey. The questions, which were the same in both surveys, were divided into four main areas, see Table 1.
Survey structure, content of the questions and question types.
| No. | Section | Number of questions | Content of the questions | Survey question types |
|---|---|---|---|---|
| 1 | Socio-demographics | 4 questions | Risk class of the medical device, end-users, job role, and company size | Closed-ended questions (single and multiple choice), some of which had “Others” as an open-ended response option |
| 2 | Implementation of the MDR | 6 questions | Knowledge of changes by the MDR, benefits, risks, perceived difficulties in implementing changed requirements, resulting challenges, and existential threat | Closed-ended questions (single and multiple choice), some of which had “Others” as an open-ended response option & sets of questions using a 5 point Likert scale (from non-existent to excellent & from not challenging to extremely challenging) |
| 3 | Quality management system | 2 questions | Status of compliance with MDR and certification of the quality management system | Closed-ended questions (single choice) |
| 4 | Custom-made devices | 3 questions | Manufacturing custom-made devices, changed requirements due to the MDR and feasibility of implementing the increased requirements | Closed-ended questions (single choice) & set of questions using a 5 point Likert scale (from strongly disagree to strongly agree) |
Data analysis and statistics
Upon completion of the survey, the data were exported from SoSci Survey and then analyzed using IBM® SPSS® Statistics (Version 27) for Windows. Data, which were categorical only, were described using frequencies and percentages to define the proportions of the sample. The median was reported for all Likert scale questions. Open-ended response options were not formally analyzed. Rather, selected quotes are included in this text to support the findings. Responses from the repeat survey (N=179) were then compared to responses from the initial survey (N=105) using Mann–Whitney U tests to assess differences in views of the orthopaedic aids industry since the MDR’s date of application. In order to analyze the effect of company size on different response distributions, Chi-square tests were used to identify possible correlations. When expected cell frequencies were below 5, an exact Fisher test was used instead of a Chi-square test. A probability of p<0.05 was set as being statistically significant.
Results
A total of 146 respondents participated in the initial survey and 233 in the repeat survey. Ninety-five respondents were not included in the study because they did not complete the questionnaire (N=86), were only interested in the results (N=1), or took less than half the average response time[1] to fully complete the survey (N=8).
The majority of respondents in both surveys were manufacturers of class I medical devices (InS: 72 %; ReS: 59 %). The participants’ roles within the company were diverse, ranging from CEOs (InS: 54 %; ReS: 66 %), orthopedic technicians and orthopedic shoe technicians (InS: 17 %; ReS: 47 %) to clinical affairs, regulatory affairs, or quality management staff (InS: 16 %; ReS: 51 %). Most respondents indicated that patients are the end-users of their products (InS: 94 %; ReS: 93 %). Companies of all sizes were represented in the sample. Respondent characteristics are summarized in Table 2.
Overview of the respondents’ characteristics: risk classification, end-user, role, and company size.
| Prior to the date of application; InS (N=105) | After the date of application; ReS (N=179) | |||
|---|---|---|---|---|
| N (%) | N (%) | |||
| Risk class | ||||
|
|
||||
| Class I | 76 | (72) | 105 | (59) |
| Class I* (Is, Im, Ir) | 7 | (7) | 13 | (7) |
| Class IIa | 9 | (9) | 22 | (12) |
| Class IIb | 5 | (5) | 14 | (8) |
| Class III | 7 | (7) | 7 | (4) |
| Not a medical device manufacturer | 20 | (19) | 34 | (19) |
|
|
||||
| End-user | ||||
|
|
||||
| Physicians | 7 | (7) | 12 | (7) |
| Healthcare professionals and caregivers | 21 | (20) | 38 | (21) |
| Patients | 99 | (94) | 167 | (93) |
| Others | 5 | (5) | 6 | (3) |
|
|
||||
| Role | ||||
|
|
||||
| CEO/management level | 55 | (54) | 118 | (66) |
| Product management | 1 | (1) | 18 | (10) |
| Orthopaedic (shoe) technology | 17 | (17) | 84 | (47) |
| Clinical affairs/regulatory affairs/quality management | 16 | (16) | 91 | (51) |
| Sales/marketing | 3 | (3) | 26 | (15) |
| Project management | 2 | (2) | 16 | (9) |
| Others | 7 | (7) | 11 | (6) |
|
|
||||
| Company size | ||||
|
|
||||
| <10 employees | 36 | (35) | 52 | (30) |
| 10–49 employees | 34 | (33) | 83 | (47) |
| 50–249 employees | 21 | (21) | 30 | (17) |
| ≥250 employees | 11 | (11) | 10 | (6) |
State of knowledge
Respondents’ knowledge of the regulatory changes brought about by the MDR has not improved since the date of application, U=10,490.00, Z=−0.420, p=0.675. Both the companies surveyed before the date of application (Mdn=3, N=103) and those in the repeat survey (Mdn=3, N=103) reported average knowledge of the new medical device regulation. Nevertheless, all companies have gained some level of knowledge about the amendments to the regulation since the date of application. In the initial survey, almost one in 10 companies (9 %) reported to have no knowledge, whereas the repeat survey showed that all companies have some level of knowledge, albeit poor.
Challenges due to the MDR
The implementation of the MDR is perceived as challenging. This assessment was not influenced by the time of the survey, U=3,206.50, Z=−0.893, p=0.372. Prior to the application of the MDR, more than half of those questioned (55 %) described the implementation of the MDR as very or extremely challenging for their company. Almost two years later, 44 percent of the respondents still described the implementation in the same way. Company size did not affect this assessment, Fisher’s exact test: p=0.617, N=88 (InS); p=0.317, N=78 (ReS).
The MDR poses a number of challenges for companies. This is evidenced by the fact that, when asked to identify the greatest challenges posed by the MDR, respondents named an average of 3.5 challenges. Additional workload for technical documentation (InS: 73 %; ReS: 86 %), increased resource expenditure and cost (InS: 64 %; ReS: 71 %), and lack of clarity regarding the new requirements of the MDR (InS: 59 %; ReS: 54 %) are among the top challenges for companies in the orthopaedic aids industry, as shown in Figure 1. Product-specific regulatory requirements (InS: 47 %; ReS: 35 %) and the implementation of a quality management system (InS: 39 %; ReS: 30 %) also cause problems. Difficulties in implementing quality management systems were reported significantly more often by smaller companies with fewer than 50 employees, χ2(1, N=277)=4.38, p=0.036. In particular, the additional workload associated with the Unique Device Identification (UDI) system was cited as a major challenge following the application date of the MDR (InS: 27 %; ReS: 49 %).

Main challenges faced by companies in the orthopaedic aids industry as a result of the MDR; respondents who answered “Others” are not shown.
Risks
The implementation of the MDR poses different types of risks for businesses in the orthopaedic aids industry. Nearly all companies (InS: 90 %; ReS: 89 %) anticipate an increase in costs or have already experienced higher costs, see Figure 2. A reduction in their product portfolio is also feared by many respondents (InS: 46 %; ReS: 33 %). In addition, about one-third of those surveyed (InS: 28 %; ReS: 30 %) indicated that they are having difficulty bringing innovative products to the market because of the MDR. Some companies (InS: 18 %; ReS: 14 %) said they would have to reduce or have already reduced their research and development (R&D) activities in order to meet the increased requirements of the MDR within their operations, shown in Figure 2.
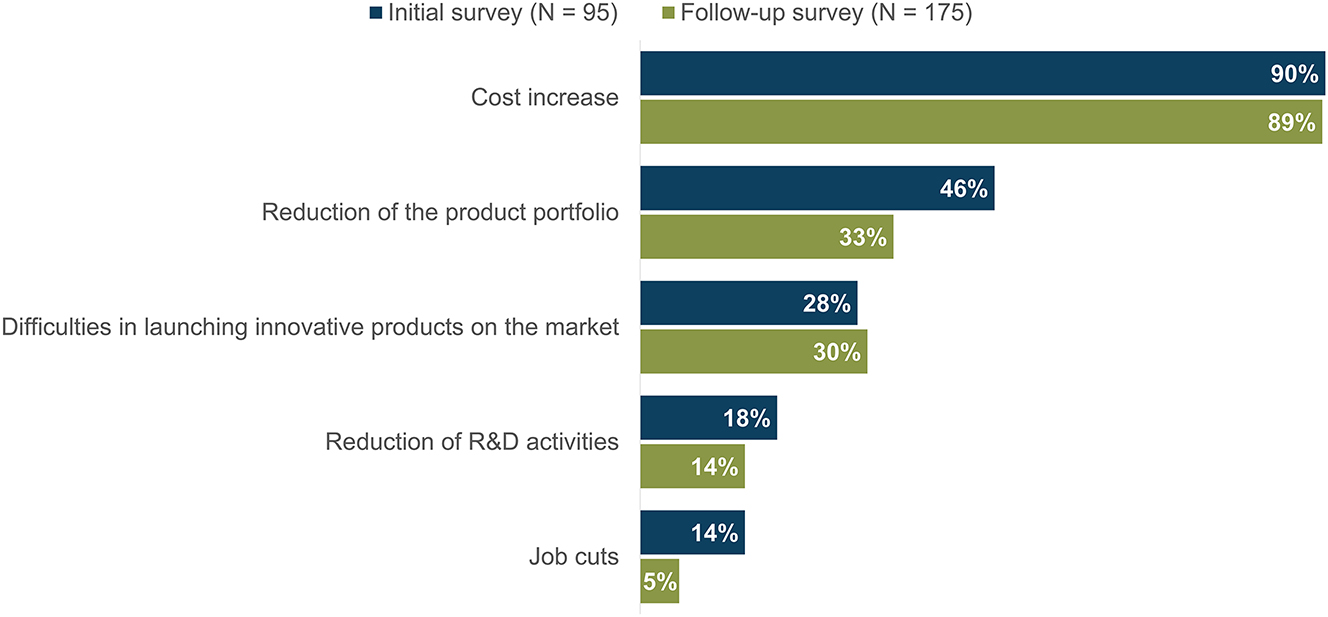
Major risks associated with the MDR according to survey respondents, ranked in descending order; respondents who answered “Others” are not shown.
Fear of having to cut jobs as a result of the MDR has decreased since the date of application. In the initial survey, 14 percent of respondents said they expected job cuts. About two years later, the figure was only 5 percent, see Figure 2. Smaller companies were particularly concerned.
For some companies, the increased requirements resulting from the MDR are so extensive that it is considered to be an existential threat. In the initial survey, one in 10 companies (10 %) stated that their organization’s existence was at risk as a results of the MDR. Two years after the date of application, this figure had risen to a quarter of those surveyed (25 %).
Potential improvement caused by the MDR
The core of the MDR is to better protect public health and patient safety. Prior to the effective start date of the MDR, most respondents (52 %) considered the goal of increased patient safety to be the greatest benefit of the new regulation, see Figure 3. The repeat survey showed that the assessment of the potential for improvement due to the MDR has changed significantly, χ2(5, N=256)=122.94, p<0.001. The effect size Cramér’s V=0.693 is interpreted as a strong association.

Greatest opportunities participants see in the new MDR, ranked in descending order; respondents who answered “Others” are not shown.
Now, almost two years later, the majority of respondents (73 %) said they saw no potential for improvement in the MDR for their company. In fact, respondents spoke of “deterioration”. Participants experience the MDR as “an over-administration in many different areas, which makes smooth working difficult”. In the initial survey, only about one-fifth of the respondents (21 %) were of this opinion. An improvement in patient safety is seen by only 23 percent of the respondents in the repeat survey. Competitive advantages were cited by nearly one in five participants (24 %) in the first survey. Nearly two years later, only 5 percent saw this benefit from the MDR.
Quality management system
For medical device manufacturers, the quality management system is mandatory. With entry into force of the MDR, the importance of quality management has increased. Even manufacturers of class I devices are now obliged to implement a quality management system. According to the MDR, manufacturers are required to “establish, document, implement, maintain, keep up to date and continually improve a quality management system” appropriate to the risk class and type of device (Art. 10, para. 9 MDR).
With the MDR coming into effect, the distribution of MDR-compliant quality management systems has shifted significantly, Fisher’s exact test, p<0.001, N=278, see Figure 4. Before the date of application, most respondents (51 %) had developed a compliance strategy and were in process of making the necessary adjustments. Only a quarter of companies (25 %) had a fully MDR-compliant quality management system at the beginning of 2021. However, two years later, nearly half of the participants (45 %) reported having a fully MDR-compliant quality management system. Overall, more companies in the initial survey reported that they need to develop an appropriate strategy to comply with the quality management system (InS: 8 %; ReS: 2 %), did not plan to change their quality management system (InS: 8 %; ReS: 5 %), or did not have a quality management system at all (InS: 5 %; ReS: 6 %) compared to the repeat survey.
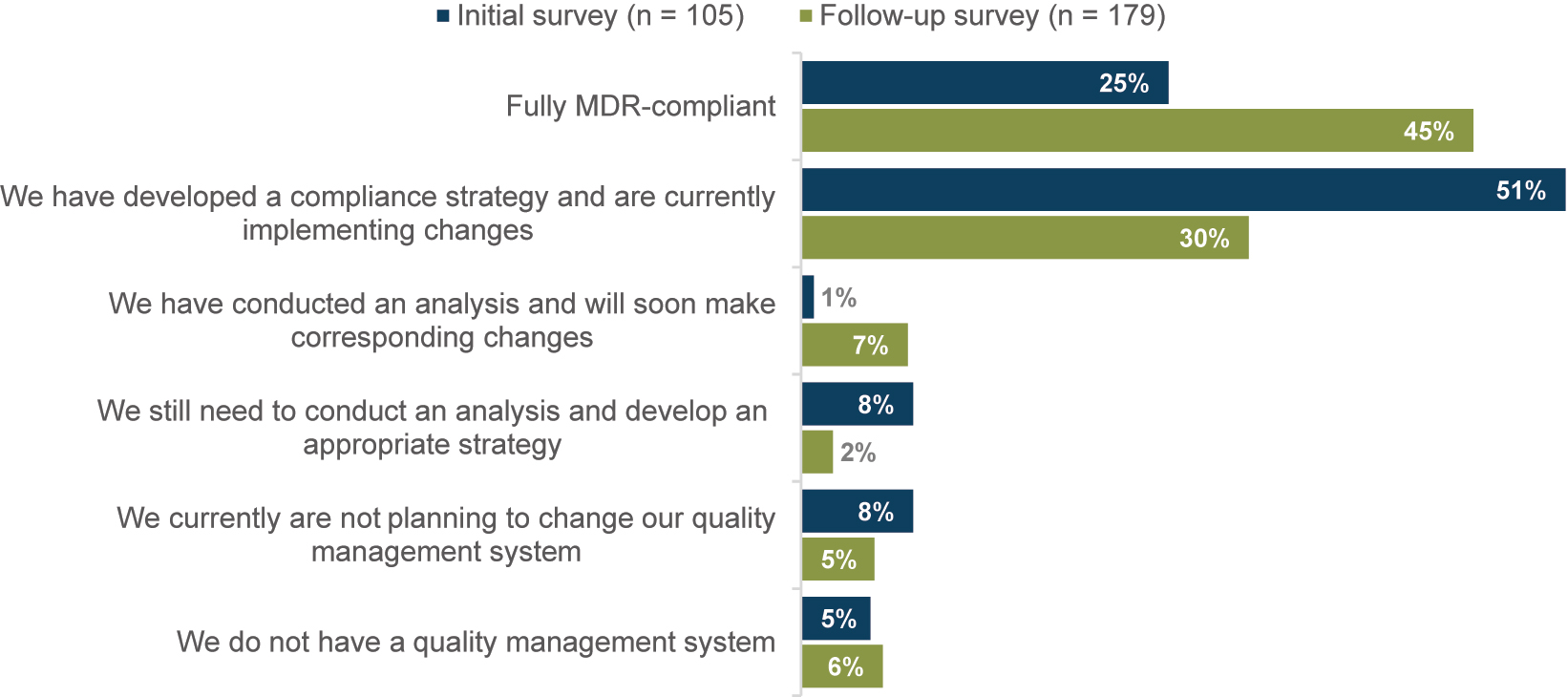
Distribution to the question “To what extent is your quality management system already MDR compliant?”; respondents who answered “Not specified” are not shown.
The introduction of MDR had no effect on whether and how the quality management system was certified, Fisher’s exact test, p=0.667, N=262. The majority of companies with a quality management system had it certified to either DIN EN ISO 13485 (InS: 52 %; ReS: 49 %) or ISO 9001 (InS: 29 %, ReS: 32 %). Approximately one in 10 companies (InS: 10 %; ReS: 11 %) have not yet had their quality management system certified.
Custom-made devices
People with disabilities are often treated with individualized devices tailored to their needs – so-called custom-made devices, such as orthoses, prostheses or functional aids. Patient-specific care is therefore essential in the provision of orthopaedic aids. According to Article 2(3) of the MDR, a custom-made device is “any device specifically made in accordance with a written prescription of any person authorised by national law by virtue of that person’s professional qualifications which gives, under that person’s responsibility, specific design characteristics, and is intended for the sole use of a particular patient exclusively to meet their individual conditions and needs” [12]. The importance of individualized care in the orthopaedic aids industry is reflected in the high number of custom-made device manufacturers. 68 percent of those initially questioned and 75 percent of repeat survey respondents were manufacturers of custom-made devices.
With the introduction of the MDR, there are more stringent regulatory requirements for custom-made devices. The Medical Device Coordination Group (MDCG) states in its Q&A-document that “custom-made device manufacturers must meet nearly all of the MDR requirements” [13]. These mainly relate to quality management, risk management, post-market surveillance as well as clinical evaluation and post-market clinical follow-up. Almost all custom-made device manufacturers (InS: 89 %; ReS: 93 %) were aware of these changes.
To assess the feasibility of implementing the increased requirements, respondents were asked to rate several statements. Comparing the results of the two surveys, it is clear that manufacturers of custom-made devices rate the required regulatory changes as significantly more difficult following the application of the MDR, U=3,836.00, Z=−2.653, p=0.008. In 2021, 66 percent of respondents agreed with the statement “We can meet the increased requirements for custom-made devices under the MDR”. Nearly two years later, only 42 percent agreed.
Because healthcare providers who manufacture custom-made devices are the legal manufacturer, they are required to comply with the MDR. Adequate resources are needed. SMEs, which are common in the orthopaedic aids industry, are often ill-equipped to implement the detailed specifications of the MDR. Financial and human resources are lacking. In the initial survey, many custom-made device manufacturers were convinced that their existing resources were sufficient. Therefore, compared to the repeat survey, significantly more respondents in the 2021 survey disagreed with the statement “We lack financial resources to implement the changed requirements” (Mdn=4 equivalent to “disagree”, N=63), U=2,645.50, Z=−3.683, p<0.001. Almost two years after the date of application, most of the participants stated that they lacked financial resources (Mdn=3 equivalent to “undecided”, N=124). The situation is similar for human resources. Again, significantly more respondents in the repeat survey reported a lack of personnel resources (Mdn=2 equivalent to “agree”, N=142), U=3,519.00, Z=−2.656, p=0.008.
The survey results indicated that most manufacturers of custom-made devices do not have the resources for clinical evaluation. This was confirmed by respondents both before (Mdn=5 equivalent to “strongly disagree”, N=59) and after (Mdn=5 equivalent to “strongly disagree”, N=130) the MDR became applicable, see Figure 5. This may be one reason why most participants reported a need for assistance with clinical evaluation and post-market clinical follow-up (InS: 64 %; ReS: 67 %).
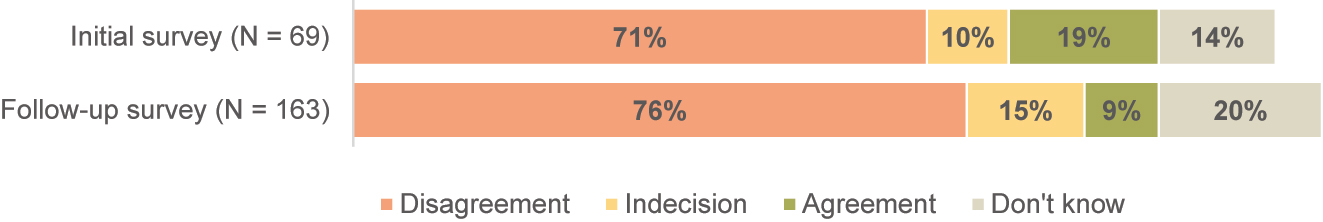
Response distribution to the statement “We have the resources to perform a clinical evaluation”.
The MDR has also increased the documentation requirements of custom-made devices. In order to comply with these additional requirements, manufacturers will have to invest an additional amount of effort. In the initial survey, more than half of the custom-made device manufacturers (51 %) indicated that they could handle the additional workload for documentation (Mdn=2 equivalent to “agree”, N=65). Almost two years later, the number was significantly lower, U=3,360.00, Z=−3.400, p=0.001. In the 2023 survey, half of the custom-made device manufacturers questioned disagreed with the statement regarding the additional workload for documentation (Mdn=3 equivalent to “undecided”, N=145). This suggests that many manufacturers are unable to meet the increased documentation requirements. Automatic generation of some aspects of the documentation could address these practical difficulties. That this would be helpful was confirmed by respondents both before (Mdn=1 equivalent to “strongly agree”, N=64) and after (Mdn=2 equivalent to “agree”, N=128) the MDR became applicable, see Figure 6.

Response distribution to the statement “Automatic generation of some aspects of the documentation is helpful”.
Discussion
Both surveys revealed that the MDR, which has been mandatory since 26 May 2021, is seen as a challenge for all businesses in the orthopaedic aids sector, regardless of size. The introduction of the MDR causes different challenges. Without external support, many companies struggle to overcome these challenges. Especially SMEs are often overwhelmed by the necessary additional expenditure. The results show that frustration with the new regulatory requirements is high in the sector characterized by class I medical devices and custom-made devices. Distributors and manufacturers of orthopaedic aids as well as employees of health care supply stores described many parts of the MDR as not suitable in practice. Many businesses do not see the regulatory change as an improvement. Instead, the MDR was described as a “deterioration” and “over-administration in many different areas, making it difficult to work smoothly”. Respondents indicated that “the MDR is more of an obstacle, that, in the case of class I medical devices, has a negative impact on the quality of life of patients”. However, it is not uncommon for new regulations or regulatory changes to be met with initial scepticism or resistance [14]. The disruption to established practices and the need to adapt existing processes, systems, or practices is often seen as an additional burden. When the MDR was adopted, quality and safety aspects were at the forefront [12]. The European consensus was that stricter regulations were synonymous with increased patient and user safety. The survey results showed that these aspects are often not seen in practice. Prior to the date of application, more than half of respondents agreed that increased patient safety was a benefit of the new regulation. Just two years later, this figure has dropped to 23 percent.
The MDR addresses the entire lifecycle of a medical device to improve patient safety. This includes the establishment of a quality management system. The survey results showed that smaller companies are particularly challenged. Of the approximately 16 percent of companies surveyed that have not yet implemented an MDR-compliant quality management system, over 90 percent of the responses came from businesses with fewer than 50 employees. Nevertheless, the study showed that many companies have a quality management system in place. In the repeat survey nearly half of the participants reported having a fully MDR-compliant quality management system. Prior to the date of application of the MDR, only 25 percent of respondents did. Almost all companies with a quality management system have it certified according to either DIN EN ISO 13485 or ISO 9001. This also applies to almost 90 percent of manufacturers of class I medical devices, although the MDR does not require certification of the quality management system for these devices. However, even two years after the MDR became applicable, one in 20 companies reported that it did not have a quality management system in place. The extent to which this will have an impact on patient care cannot be determined at this time.
The implementation of the MDR is often associated with an increased resource expenditure and therefore increased costs. This is not least due to the additional amount of work required for technical documentation. The MDR prescribes the structure of the technical documentation in more detail and obliges manufacturers to keep documentation continuously up to date. The survey results indicate that many companies did not realize the full extent of the changed requirements until after the MDR went into effect, as the additional workload for technical documentation was more frequently cited as a challenge in the repeat survey. However, longer approval times may also explain the higher costs. In order to assess devices under the new MDR, all notified bodies that have been operating under the device directives will need to be redesignated. As of May 2023, only 38 notified bodies are fully accredited under the MDR, compared to 58 notified bodies under the MDD and AIMDD [15]. This leads to substantial bottlenecks [16], [17], [18]. Many of the companies surveyed fear that they will have to downsize their product portfolio as a result of the MDR, or have already done so. As the participants in both surveys were mainly manufacturers of class I medical devices, these issues were not confirmed by the study conducted. Accordingly, conflicts related to notified bodies were rarely mentioned. Manufacturers of class I medical devices have almost no points of contact with notified bodies, as the conformity assessment of these devices does not require notified body involvement. In addition, as mentioned above, the quality management system for class I devices does not need to be certified by a notified body. Consequently, the survey results are limited in their ability to draw concrete conclusions about difficulties with notified bodies.
In the orthopaedic aids sector, the highly individual needs of people with disabilities often imply the provision of custom-made devices. As a result, the majority of medical supply stores in Germany are manufacturers of such devices, as confirmed by the survey findings. The results show that the MDR proved to be more difficult to implement than manufacturers of custom-made devices expected. Before the date of application, the majority of respondents stated to be able to fulfil the changed requirements for custom-made devices, although resources were partly lacking. The repeat survey presents a different image. More companies reported not being able to implement the increased requirements for custom-made devices. There is often a lack of financial and human resources. The biggest problems manufacturers of custom-made devices face are linked to clinical evaluation and post-market surveillance. This was the case before the MDR became applicable and still is almost two years later. Companies lack the necessary resources. An appropriate form of support is required for manufacturers of custom-made devices.
At present, there is a trend towards centralization in the orthopaedic aids industry, with small companies being increasingly abandoned. The stricter requirements of the new regulatory framework are unlikely to improve the situation, although there is still too little information on the exact role of the MDR.
Limitations
While this study provides a comprehensive overview of the impact of the MDR on the orthopaedic aids industry, it also has certain limitations. The use of an unvalidated, non-standardized questionnaire may have introduced bias, even though it was developed based on previous surveys. The recruitment process is also a limiting factor. While online surveys have the potential for high response rates, there are biases regarding the audience reached and therefore the adequacy of the information obtained [19]. For example, participants were limited to those who had access to appropriate technology and the technical skills necessary to submit the information correctly. In addition, respondents in this study were limited to the subset of individuals who actively chose to be part of the surveys and were motivated to provide input on the MDR. Another limitation is that the surveys were conducted only in Germany. The results are therefore relevant to the German medical device industry. In order to confirm (or not) the results for Europe, future research should also survey different European member states. Furthermore, only distributors and manufacturers of orthopaedic aids and employees of health care supply stores were surveyed. The orthopaedic aids industry is characterized by small, owner-operated craft businesses that are primarily manufactures of class I and custom-made medical devices. Future surveys should include other sectors of the medical device industry to identify potential differences between them and to gain an overall view of the industry’s experiences and opinions regarding the acceptance and feasibility of the MDR.
Conclusions
The results of the surveys conducted point to the fact that the MDR can be a bottleneck in the availability of medical devices, as manufacturers face significant financial barriers and additional costs. As a result, respondents indicated a reduction in their product portfolio and withdrawal of medical devices from the EU market. However, a detailed impact assessment of the MDR is beyond the scope of this article, as the results presented here only reflect the experiences of the survey participants. Nevertheless, the authors conclude that the implementation and realization of the increased regulatory requirements resulting from the MDR represent an extremely challenging period for the medical device sector in Europe.
-
Research ethics: Not applicable.
-
Informed consent: Informed consent was obtained from all individuals included in the surveys.
-
Author contributions: The authors have accepted responsibility for the entire content of this manuscript and approved its submission.
-
Competing interests: The authors state no conflict of interest.
-
Research funding: None declared.
-
Data availability: The raw data can be obtained on request from the corresponding author.
References
1. Opinion of the European Economic and Social Committee on the ‘Proposal for a regulation of the European Parliament and of the Council on medical devices and amending Directive 2001/83/EC and Regulations (EC) No 178/2002 and (EC) No 1223/2009’. C 133/52; 2013. https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:52012AE2185 [Accessed 12 May 2023].Search in Google Scholar
2. Greco, C. The poly implant prothèse breast prostheses scandal: embodied risk and social suffering. Soc Sci Med 2015;147:150–7. https://doi.org/10.1016/j.socscimed.2015.10.068.Search in Google Scholar PubMed
3. Cohen, D. Faulty hip implant shows up failings of EU regulation. BMJ 2012;345:e7163. https://doi.org/10.1136/bmj.e7163.Search in Google Scholar PubMed
4. Heneghan, C, Aronson, JK, Goldacre, B, Mahtani, KR, Plüddemann, A, Onakpoya, I. Transvaginal mesh failure: lessons for regulation of implantable devices. BMJ 2017;359:j5515. https://doi.org/10.1136/bmj.j5515.Search in Google Scholar PubMed
5. Types of legislation. https://european-union.europa.eu/institutions-law-budget/law/types-legislation_en [Accessed 12 May 2023].Search in Google Scholar
6. Thienpont, E, Quaglio, G, Karapiperis, T, Kjaersgaard-Andersen, P. Guest editorial: new medical device regulation in Europe: a collaborative effort of stakeholders to improve patient safety. Clin Orthop Relat Res 2020;478:928–30. https://doi.org/10.1097/CORR.0000000000001154.Search in Google Scholar PubMed PubMed Central
7. Klar, E. Medical Device Regulation als aktuelle Herausforderung für die rechtssichere Einführung neuer Technologien. Chirurg 2018;89:755–9. https://doi.org/10.1007/s00104-018-0705-3.Search in Google Scholar PubMed
8. Ergebnisse der BVMed-Herbstumfrage 2019. https://www.bvmed.de/de/branche/standort-deutschland/zusammenfassung-der-ergebnisse-der-bvmed-herbstumfrage-2019 [Accessed 11 March 2021].Search in Google Scholar
9. Branchenbericht Medizintechnologien 2020. https://www.bvmed.de/download/bvmed-branchenbericht-medtech.pdf [Accessed 15 May 2023].Search in Google Scholar
10. Wien, P, Kuhlmann, M. Auswirkungen der neuen EU-Medizinprodukte-Verordnung (MDR) sowie der neuen Verordnung für In-vitro-Diagnostika (IVDR) auf die Hersteller in Deutschland: die Entwicklung innovativer Medizinprodukte wird schwieriger; 2019. https://www.dihk.de/resource/blob/4788/144b1478aebed3e71f6f25a1e1dbab9f/dihk-spectaris-unternehmensumfrage-data.pdf [Accessed 23 Febr 2021].Search in Google Scholar
11. EU-MDR Readiness Check 2020: Stand der MedTech-Branche. https://climedo.de/blog/ergebnisse-jetzt-verfuegbar-eu-mdr-umfrage-zum-stand-der-umsetzung/ [Accessed 11 Mar 2021].Search in Google Scholar
12. Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017 on medical devices amending Directive 2001/83/EC, Regulation (EC) No 178/2002 and Regulation (EC) No 1223/2009 and repealing Council Directives 90/385/EEC and 93/42/EEC: MDR.Search in Google Scholar
13. Medical Device Coordination Group. Questions and answers on custom-made Devices: & considerations on adaptable medical devices and patient-matched medical devices; 2021.Search in Google Scholar
14. Erwin, DG, Garman, AN. Resistance to organizational change: linking research and practice. Leader Organ Dev J 2010;31:39–56. https://doi.org/10.1108/01437731011010371.Search in Google Scholar
15. List of Bodies notified under directive: Regulation (EU) 2017/745 on medical devices. https://webgate.ec.europa.eu/single-market-compliance-space/#/notified-bodies/notified-body-list?filter=legislationId:34,notificationStatusId:1 [Accessed 22 May 2023].Search in Google Scholar
16. Medical Device Survey 2022: data from all 33 members (end 2022). https://www.team-nb.org/wp-content/uploads/members/M2023/Survey-2022-20230411.pdf [Accessed 6 Jul 2023].Search in Google Scholar
17. Melvin, T, Kenny, D, Gewillig, M, Fraser, AG. Orphan medical devices and pediatric cardiology – what interventionists in Europe need to know, and what needs to be done. Pediatr Cardiol 2023;44:271–9. https://doi.org/10.1007/s00246-022-03029-1.Search in Google Scholar PubMed PubMed Central
18. European Academy of Paediatrics (EAP). Open letter: urgent action needed to secure continued access to essential medical devices for children and for patients with orphan diseases; 2023. https://www.eapaediatrics.eu/wp-content/uploads/2023/06/Letter-Kyriakides_Med-Devices-signed-270627.pdf [Accessed 7 Jul 2023].Search in Google Scholar
19. Scott, DM, Berg, MJ, Tolhurst, BA, Chauvenet, ALM, Smith, GC, Neaves, K, et al.. Changes in the distribution of red foxes (Vulpes vulpes) in urban areas in Great Britain: findings and limitations of a media-driven nationwide survey. PLoS One 2014;9:e99059. https://doi.org/10.1371/journal.pone.0099059.Search in Google Scholar PubMed PubMed Central
Supplementary Material
This article contains supplementary material (https://doi.org/10.1515/bmt-2023-0325).
© 2023 the author(s), published by De Gruyter, Berlin/Boston
This work is licensed under the Creative Commons Attribution 4.0 International License.
Articles in the same Issue
- Frontmatter
- Review
- Computational models of bone fracture healing and applications: a review
- Research Articles
- Assessing standing balance with MOTI: a validation study
- Effect of non-thermal plasma treatment and resin cements on the bond strength of zirconia ceramics with different yttria concentrations
- A new approach towards extracorporeal gas exchange and first in vitro results
- Estimation of heart rate and respiratory rate by monitoring cardiopulmonary signals with flexible sensor
- Hand gesture recognition with deep residual network using Semg signal
- Crowdsourcing image segmentation for deep learning: integrated platform for citizen science, paid microtask, and gamification
- Image segmentation of mouse eye in vivo with optical coherence tomography based on Bayesian classification
- Impact of the new European medical device regulation: a two-year comparison
Articles in the same Issue
- Frontmatter
- Review
- Computational models of bone fracture healing and applications: a review
- Research Articles
- Assessing standing balance with MOTI: a validation study
- Effect of non-thermal plasma treatment and resin cements on the bond strength of zirconia ceramics with different yttria concentrations
- A new approach towards extracorporeal gas exchange and first in vitro results
- Estimation of heart rate and respiratory rate by monitoring cardiopulmonary signals with flexible sensor
- Hand gesture recognition with deep residual network using Semg signal
- Crowdsourcing image segmentation for deep learning: integrated platform for citizen science, paid microtask, and gamification
- Image segmentation of mouse eye in vivo with optical coherence tomography based on Bayesian classification
- Impact of the new European medical device regulation: a two-year comparison