Sugar glues for broken neurons
-
Vimal P. Swarup
Vimal P. Swarup is a graduate student in the Department of Bioengineering at the University of Utah. Vimal received his BE from Birla Institute of Technology, India, and his MS from Illinois Institute of Technology, Chicago. Vimal’s current research interest lies in studying the role of glycosaminoglycans in spinal cord injuries and engineering approaches to modify their sulfation pattern at the injury site.
Caitlin P. Mencio is a graduate student in the Neuroscience Program at the University of Utah. Caitlin received her BS in Neuroscience from Texas Christian University in Fort Worth, TX. Caitlin is currently studying the role proteoglycans play in neural development, synapse formation, and plasticity.
Vladimir Hlady is a Professor of Bioengineering at University of Utah. He received BS, MS, and DSc degrees in Chemistry from the University of Zagreb, Croatia. His current research focus is on the role of glycocalyx in axonal pathfinding and platelet activation. He has two patents and published more than 120 scientific papers, reviews, and book chapters.
Kuberan (Kuby) Balagurunathan, PhD, is an Associate Professor of Medicinal Chemistry at the University of Utah. Kuby received his BSc from St. Joseph’s College, Trichy, India, and his MSc from the Indian Institute of Technology, Madras, India, under the guidance of Prof. Duraikkannu Loganathan, and his PhD from the University of Iowa under the guidance of Prof. Robert Linhardt. He did his postdoctoral training at MIT in Prof. Robert Rosenberg’s laboratory where he developed various chemoenzymatic approaches for creating defined structures of heparin and heparan sulfate. Kuby’s laboratory works on exploring the chemical biology of carbohydrates such as heparan, chondroitin, dermatan, and keratan sulfates and to determine their pathological and physiological roles at the molecular level.


Abstract
Proteoglycans (PGs) regulate diverse functions in the central nervous system (CNS) by interacting with a number of growth factors, matrix proteins, and cell surface molecules. Heparan sulfate (HS) and chondroitin sulfate (CS) are two major glycosaminoglycans present in the PGs of the CNS. The functionality of these PGs is to a large extent dictated by the fine sulfation patterns present on their glycosaminoglycan (GAG) chains. In the past 15 years, there has been a significant expansion in our knowledge on the role of HS and CS chains in various neurological processes, such as neuronal growth, regeneration, plasticity, and pathfinding. However, defining the relation between distinct sulfation patterns of the GAGs and their functionality has thus far been difficult. With the emergence of novel tools for the synthesis of defined GAG structures, and techniques for their characterization, we are now in a better position to explore the structure-function relation of GAGs in the context of their sulfation patterns. In this review, we discuss the importance of GAGs on CNS development, injury, and disorders with an emphasis on their sulfation patterns. Finally, we outline several GAG-based therapeutic strategies to exploit GAG chains for ameliorating various CNS disorders.
List of abbreviations
3OST, 3-O-sulfotransferase; 6OST, 6-O-sulfotransferase; 6OST1, 6-O-sulfotransferase 1; AB, amyloid-β; AD, Alzheimer’s disease; APP, amyloid precursor protein; BACE1, β-secretase-1; BDNF, brain-derived neurotrophic factor; ChABC, chondroitinase ABC; ChAC, chondroitinase AC; ChB, chondroitinase B; CNS, central nervous system; CS, chondroitin sulfate; CSPG, chondroitin sulfate proteoglycan; DRG, dorsal root ganglion; DS, dermatan sulfate; ECM, extracellular matrix; FGF, fibroblast growth factor; FGFR, fibroblast growth factor receptor; GAG, glycosaminoglycan; Gal, galactose; GalNAc, N-acetylgalactosamine; GDNF, glial cell-derived neurotrophic factor; GlcA, glucuronic acid; GlcNAc, N-acetylglucosamine; GPI, glycosyl phosphatidyl inositol; HA, hyaluronan; HB-EGF, heparin-binding epidermal growth factor; HBGAM, heparin-binding growth-associated molecule; Hep I, II, III, heparitinase I, II, and III; HS, heparan sulfate; HSPG, heparan sulfate proteoglycan; IdoA, iduronic acid; KS, keratan sulfate; MAPK, mitogen-activated protein kinase; NCAM, neural cell adhesion molecule; NDST, N-deacetylase/N-sulfotransferase; NGF, nerve growth factor; NgR, Nogo receptor; NPCs, neuronal progenitor cells; NT, neutrophin; NFT, neurofibrillary tangles; OST, O-sulfotransferase; PD, Parkinson’s disease; PG, proteoglycan; PKC, protein kinase C; PNN, perineuronal net; PTN, pleiotrophin; RPTPβ, receptor type protein-tyrosine phosphatase beta; RPTPσ, receptor type protein tyrosine phosphatase sigma; SCI, spinal cord injury; SE, status epilepticus; TGF-β, transforming growth factor beta; Trk, tyrosine receptor kinase.
Introduction
Glycosaminoglycans (GAGs) are a family of linear, sulfated polysaccharides that are associated with central nervous system (CNS) development, maintenance, and disorders. For example, the heparan sulfate (HS) chain, a member of the GAG family, regulates receptor-ligand interactions that control neurite outgrowth and pathfinding (1–3 and references therein). Similarly, the chondroitin sulfate (CS) chain, another common GAG, is overexpressed at the scar site in spinal cord injuries (SCIs), and is a major roadblock to regeneration (4, 5). All GAGs but hyaluronan are found associated with a core protein that belongs to a special class of biomolecules called proteoglycans (PGs). HS and CS are the two most common and prominent GAG types found in the CNS. Although these chains have been reported to affect various functions in the CNS, a comprehensive understanding of their structure-function relation is still lacking. Sulfate groups present in these molecules impart negative charge at discrete positions defining not only the fine structure but also the interactions with various signaling factors. Sulfation of GAG chains is a non-template process that occurs in the Golgi apparatus and relatively little is understood about specific contributions of various ‘sulfation patterns’ on GAG functionality (6, 7, and references therein). However, with recent advancements in GAG synthesis and characterization, many critical functions of sugar moieties of HSPGs/CSPGs are being revealed. In this review, we focus on the role of HS and CS chains, in particular their sulfation patterns, in various CNS pathophysiological processes. Finally, we propose various GAG-based therapeutic approaches to combat different CNS disorders for which no cure exists at present.
Molecular perspective of PGs
GAGs can be distinguished into four groups depending on their sugar building blocks and the nature of their glycosidic linkages: HS/heparin, CS/dermatan sulfate (DS/CS-B), keratan sulfate (KS), and hyaluronan (HA). HS is composed of alternating glucosamine (GlcN) and glucuronic acid (GlcA) or iduronic acid (IdoA); CS/DS is composed of alternating N-acetylgalactosamine (GalNAc) and GlcA or IdoA; KS is composed of alternating galactose (Gal) and N-acetylglucosamine (GlcNAc); HA is a non-sulfated GAG, composed of alternating GlcNAc and GlcA residues (Figure 1). Furthermore, unlike other GAG chains, HA is not covalently bound to any protein and exists exclusively in the extracellular matrix (ECM) (8).
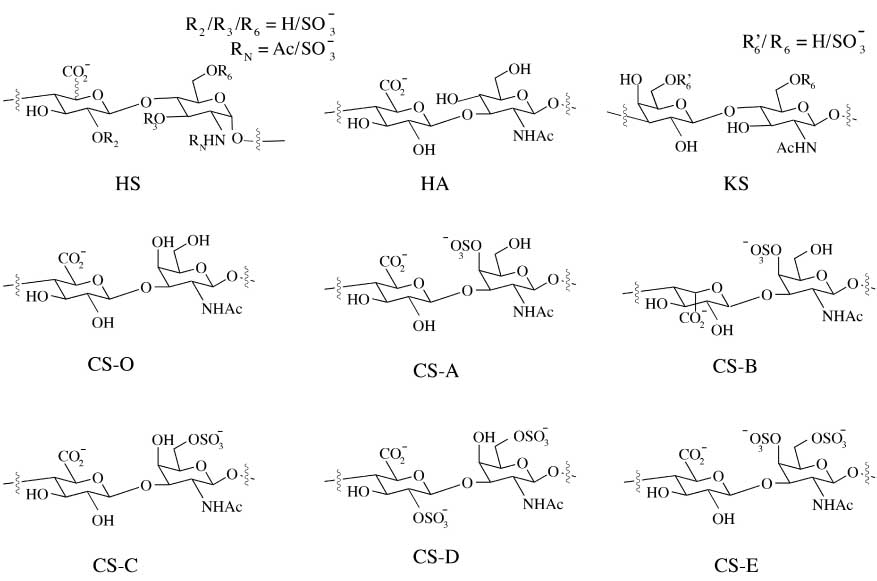
Schematic representation of disaccharide units found in HS, HA, KS, and CS.
The synthesis of HS and CS polysaccharide chains involves two main steps: (i) attachment of linkage tetrasaccharide (GlcA-Gal-Gal-Xyl) to the core proteins through the serine residue, and (ii) subsequent elongation and modification of the GAG chains. GAG chains undergo diverse modifications by the action of various enzymes in a tissue-specific manner. These modifications include epimerization, N-deacetylation/sulfation, and O-sulfation by various O-sulfotransferases (OSTs), which act by sulfating residues at specific positions (Table 1). The review by Sugahara and Kitagawa (6 and references therein) provides a detailed description of HS and CS biosynthesis.
Enzymes associated with HS and CS modifications and their implications in the CNS.
| Enzymes | Role in GAG modification | Known implications | References |
|---|---|---|---|
| HS C-5 epimerase | Epimerization of GlcA to IdoA of HS | Caenorhabditis elegans lacking the enzyme showed axonal and cellular guidance defects | (2, 198) |
| HS NDST (1–4) | Converts GalNAc to GalNS | Initiated sulfation of HS; required for FGF-4 signaling; disruption can impair wingless, FGF, and hedgehog signaling in mice | (199) |
| HS 2-OST | 2-O-sulfation of GlcA/IdoA | (198) | |
| HS endosulfatase | Removes 6S, preference for NS2S6S | Expression regulated Wnt signaling positively and FGF signaling negatively | (200–202) |
| HS 3-OST | 3-O-sulfation | Dramatic changes in its expression were seen in developing zebrafish; expression observed in various locations during mouse development | (139–141) |
| HS 6-OST | 6-O-sulfation | Modified HS chains on syndecan-1; enhanced Slit signaling; dramatic changes in its expression were seen in developing zebrafish; expression reported during mouse development | (139, 142, 198, 203, 204) |
| CS/DS 2-OST | 2-O-sulfation | Knockdown led to failure of neuronal polarization; upregulated in differentiated neuronal cells | (92, 205, 206) |
| CS 6-OST | 6-O-sulfation | Ratio of 4S/6S varied during development and affected neuronal plasticity; upregulated after CNS injury; upregulated in neuronal stem cells | (81, 160, 205, 207) |
| CS 4-OST | 4-O-sulfation | Upregulated in neuronal stem | (205) |
| N-acetylgalactosamine 4-sulfate-6-OST | 6-O-sulfation | Knockdown led to failure of neuronal polarization in mice hippocampal neurons; downregulation reduced CSPG-mediated inhibition in E18 rat cortical neurons | (92, 161) |
| CS/DS C5-epimerase | Epimerization of GlcA to IdoA | Upregulated in astrocytes and neurons as compared with neuronal stem cells | (205, 208) |
| DS 4-OST1 | Mutation associated with adducted thumb-clubfoot syndrome; deficiency resulted in impaired differentiation and proliferation of neural stem cells | (209, 210) |
Two of the most abundant HSPGs in the CNS are syndecans and glypicans. Syndecans belong to transmembrane core protein family composed of four distinct members and carry both HS and CS chains (9, 10). Unlike syndecans, glypicans are attached to the cell membrane through a glycosyl phosphatidyl inositol (GPI) linkage, are composed of six distinct members, and carry the HS chain exclusively (11 and references therein). CS chains are primarily carried by large PGs such as hyalectans or lecticans, the most abundant PGs found in the ECM of the CNS (11). The lectican family consists of four members that have been cloned thus far: versican (also called PG-M) aggrecan, neurocan, and brevican (12–15). It has been suggested that the number of GAG chains per unit length in versican, neurocan, and brevican is fairly constant. However, the number, size, and composition of the GAG chains are not only influenced by the core protein but also by the tissue from which the lectican originates. For example, the number of KS-binding domains of aggrecan varies among different species and tissues. Aggrecan DNA sequence shows the number of domains to be 13 in human, 4 in rat and mouse, and none in chicken (16). PGs other than syndecans, glypicans, and lecticans exist in the CNS; however, many of these are structurally unique. One example of such a PG is the receptor-type protein tyrosine phosphatase RPTPβ. Two of the three identified isoforms of RPTPβ have been found to carry CS chains (17). This article mainly focuses on GAGs, and detailed description of PGs can be found elsewhere (11, 18).
Influence of HS and CS in the CNS
(i) Neural development
HSPGs are expressed in actively mitotic areas of the brain. Glypican-1 transcripts have been reported in the ventricular zone, the area of neurogenesis, during CNS development (19). Most neuronal precursor cells do not express glypican-2, -4, and -5, excluding glypican-4 (or K-glypican) expression in the ventricular zone of the cerebral cell wall (20) and glypican-5 expression in the ganglionic eminence (21). In Drosophila, mutations in the gene associated with glypican (Dally locus) results in alteration of the cell division pattern at the larval stage (22). Loss of Dally protein delays lamina precursor cells from entering into the final round of cell division. Furthermore, in humans, mutations in glypican-3 result in human X-linked Simpson-Golabi-Behmel syndrome (SGBS) (23). SGBS results in prenatal and postnatal overgrowth and has been associated with the high incidence of neuroblastoma. Syndecan-3 is the most prominent syndecan family member expressed in the adult mammalian CNS; its maximal expression in rats was found on postnatal day 7 and corresponded to glial cell differentiation, myelination, and formation of neuronal connections. The expression declined in adult neurons, where it was mainly found in axons (24).
CSPGs have also been found to influence CNS development by regulating cell division, neuronal stem cell proliferation, secondary neurosphere formation, and neurogenesis (25–27). For example, phosphacan, an RPTPβ variant, is upregulated in areas of active cell proliferation during embryonic development of rats (11, 28, 29). Neurosphere-forming cells in rat fetal telencephalons were found to express neurocan, phosphacan, and neuroglycan C. In addition, CS chains have been associated with neural stem cell proliferation through FGF-2 signaling (30).
(ii) Neuronal migration
In addition to affecting various developmental processes, PGs (mainly CS) also affect the signaling properties of the ECM that control neuronal migration. In the CNS, neuronal migration is guided by a radial glial fiber system that acts as a scaffold for migrating neurons. In early cortical neurons, CSPGs such as protein tyrosine phosphatase RPTPβ/phosphacan are localized along radial glial fibers and on migrating neurons (31). RPTPβ/phosphacan binds to several adhesion molecules, including f3/contactin, N-CAM, L1, TAG1, and tenascin (32). The most important factor that interacts with RPTPβ is pleiotrophin (PTN). PTN is a member of the HS-binding proteins that stimulates neurite outgrowth in vitro (33). The CS side chain and protein of RPTPβ form the binding site for pleiotrophin, and several GAGs have been reported to inhibit this binding (34). In the developing cortex, PTN is synthesized by radial glial cells and is deposited along their fibers (35). Antibodies against RPTPβ and treatment with exogenous GAGs were reported to disturb PTN-induced migration of neurons (36). This evidence suggests that a PG-dependent ligand receptor mechanism must play a role in neuronal migration.
Studies also indicate that changes in CSPG expression are inversely correlated with migration pathways of neural crest (NC) cells. Immunohistochemical studies of chick embryos showed the restricted appearance of the CSPG versican into the CNS barrier tissues (37). In addition, surface-immobilized versican does not support the attachment of NC cells in vitro (38). Thus, the avoidance of versican-expressing regions by NC cells should have functional importance in their migration within the CNS. However, not all CSPGs are inhibitory. For example, highly sulfated CS motifs have been shown to enhance neuronal migration. Ishii and Maeda (39) used shRNA constructs to downregulate the production of highly sulfated CS chains, CS-D and CS-E types. The treatment of neuronal progenitor cells with such shRNA constructs in mouse embryos resulted in the accumulation of non-migrated neurons in the subventricular and intermediate zones of the cortex. This evidence further strengthens the argument that GAG type as well as their sulfation influences crucial processes in neuronal development.
(iii) Neurite outgrowth
One of the most widely studied roles of GAGs is their effect on neurite outgrowth and axonal pathfinding. Both HS and CS chains are known to be involved in neurite outgrowth. The expression pattern of HSPGs, such as glypicans and syndecans, is tightly regulated in the developing nervous system and is closely associated with neurite outgrowth (40–42). The study by Wang and Denburg (43) showed that exogenous GAGs can alter axon growth in situ. The involvement of HS was confirmed by using HS-degrading enzymes that led to perturbation of axonal pathfinding. Another study on the developing Xenopus optic pathway showed that HS binding of exogenous FGF-2, but not FGF-1, disrupts target recognition (44).
In vitro studies have indicated that HSPGs support neurite outgrowth by sequestering growth-enhancing molecules such as laminin, NCAM, heparin-binding EGF (HB-EGF), and several other midkine (MK) family members (45, 46). In addition to their growth-promoting effects, glypican-1 has been reported to serve as a receptor for Slit proteins (47). Slit proteins function as chemorepellants and inhibit axonal growth upon binding to their roundabout (Robo) receptors (48). Syndecan-3 has been identified as a possible receptor for HB-GAM (49). Inhibitors of Src family kinases affect HB-GAM-dependent neurite outgrowth of syndecan-3-transfected cells. Therefore, Kinnunen et al. (50) suggested that syndecan-3-mediated neurite growth is associated with the cortactin-Src kinase pathway. The role of HSPGs is dependent on the developmental stage of the CNS (18). Various HS-modifying enzymes (Table 1) regulate the spatiotemporal variations in the sulfations of HS chains. The affinity of HS-signaling complexes is highly dependent on the subtle structural changes of their sulfated motifs. Hence, the diversity in HS-ligand interactions forms the basis of their observed variations in their influence on neurons. An overview of the prominent HSPG-mediated ligand-receptor interactions of the CNS is shown in Figure 2. More information in this regard can be found in the review by Lee and Chien (3) and the references cited therein.
![Figure 2 Receptors of HSPGs.Prominent interactions in the CNS that are mediated by HSPGs are shown. Robo receptors bind to Slit ligands, UNC-5 and DCC (deleted in colorectal cancer) receptors bind to Netrin, FGF binds to FGFR, and ephrin receptors (Eph) bind to ephrin ligands. Box contains the definition for conserved protein domains. Ig, immunoglobulin; EGF, epidermal growth factor; FN3, fibronectin type III domain; TK, tyrosine kinase domain. [The figure has been modified from the review by Lee et al. (3).]](/document/doi/10.1515/bmc-2012-0042/asset/graphic/bmc-2012-0042_fig2.jpg)
Receptors of HSPGs.
Prominent interactions in the CNS that are mediated by HSPGs are shown. Robo receptors bind to Slit ligands, UNC-5 and DCC (deleted in colorectal cancer) receptors bind to Netrin, FGF binds to FGFR, and ephrin receptors (Eph) bind to ephrin ligands. Box contains the definition for conserved protein domains. Ig, immunoglobulin; EGF, epidermal growth factor; FN3, fibronectin type III domain; TK, tyrosine kinase domain. [The figure has been modified from the review by Lee et al. (3).]
Similar to the influence of HSPGs, the roles of CSPGs on axonal growth are multifaceted and have been studied more extensively. CSPGs are well known to inhibit axonal growth in several regions or developmental stages of the CNS (4, 51, 52, and references therein), and in vivo evidences have shown the inability of axons to penetrate a lectican-containing glial scar (5, 53, 54). However, tissues that express higher levels of CSPGs are not always inhibitory to neural proliferation. In fact, in several studies, CS has been shown to coincide with developing axonal pathways (55–57). Neurocan and phosphacan serve as good examples in highlighting the contrasting roles of CSPGs. While phosphacan was reported to promote neurite outgrowth in rat cortical neurons, neurocan inhibited the growth in embryonic chick neurons (34, 58). Evidence suggests that the stimulatory/inhibitory functions of CSPGs depend on their spatiotemporal expression and interactions that are defined by their sulfation patterns.
The interactions of CS chains with various signaling molecules lead to the promotion of neurite growth. RPTPβ is expressed on migrating neurons and binds to growth factors through its CS side chains (34, 59). Several studies also claim that RPTPβ is associated with CSPG-mediated inhibition in the CNS. For example, Shen et al. (60) have shown that after dorsal column injury, sensory axons grow deeper in RPTPβ mutant mice than they do in wild-type mice.
Some mechanistic pathways are emerging from our present knowledge on the roles of CS in controlling neuronal outgrowth. For example, blood-brain barrier leakage of blood protein fibrinogen containing transforming growth factor-β (TGF-β) is thought to induce CS production in reactive astrocytes by activating the TGF-β/Smad signaling pathway (61–64). Fibrinogen, carrying the latent TGF-β complex, is then responsible for phosphorylation of Smad2 in astrocytes, leading to inhibitory scar formation and limiting neurite outgrowth (61). CS has been reported to inhibit axonal growth by interacting with leukocyte common antigen-related phosphatase, Nogo receptors (NgR1 and NgR3), and the EGF receptors (65–67). The converging downstream effector of most CSPG-related inhibitory pathways is the activated Rho, a Ras homologue, which further activates the Rho-associated protein kinase (RhoA/ROCK) pathway. Once activated, ROCK leads to actin depolymerization through Lim kinase and cofflin stimulation. Actin filament degradation leads to immobility or collapse of growth cones present on the axons, leading to termination of axonal outgrowth (Figure 3) (68–70). Sivasankaran et al. (71) have shown that by blocking the RhoA pathway, myelin and CSPG lost their ability to activate Rho and inhibit neurite outgrowth in dorsal column neurons. However, similar growth enhancement was not observed for corticospinal tract neurons in the same animals, suggesting that the effect of CSPG signaling is specific to the neuronal cell type and region of the CNS.
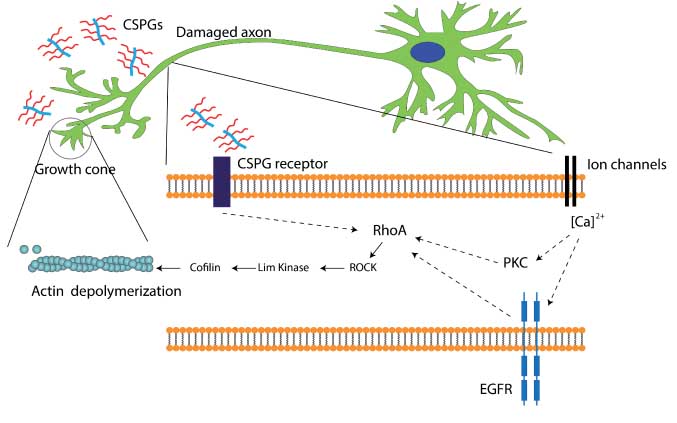
Intracellular signaling mechanism triggered by CSPG present in the glial scar.
CSPGs are thought to be present in axons, although their molecular identity is not well established. Recent studies have shown that CSPG can interact with leukocyte common antigen-related phosphatase, Nogo, or EGF receptors and lead to growth inhibition (65–67). RhoA activation eventually leads to actin depolymerization and the growth cone retraction. RhoA activation has also been shown to be associated with PKC pathway and epidermal growth factor receptor (EGFR) phosphorylation in a calcium-dependent manner (67, 71). However, the process of calcium influx leading to PKC activation or EGFR activation is not well defined. Dashed arrows suggest that mediators of the represented process are yet to be identified.
(iv) CNS plasticity
HS can alter CNS plasticity by interacting with CNS ligands such as NCAM and EGF. Removal of HSPGs can lead to a loss in synaptogenic activity of postsynaptically expressed PSA-NCAM (72). Conversely, loss of the HB-EGF can affect fear and spatial learning as well as alter long-term potentiation (LTP) in the hippocampus (73). Although these studies have documented the importance of GAG chains in plasticity, it remains unclear which sulfation patterns are required for influencing the plasticity. However, some evidence alludes to their importance. For example, mice that are lacking the endosulfatases Sulf-1 and Sulf-2 show shortcomings in spatial learning. Reduction of Sulf-1 alone leads to deficits in synaptic plasticity in the hippocampus (74). As evidences accumulate for the role of HS-ligand interactions in CNS plasticity, it becomes important to delineate how specific sulfation patterns can regulate CNS plasticity.
At the end of the critical period of development, loss of neuroplasticity is seen in the CNS. The critical period refers to the stage until which neuronal properties and connections can be highly modulated by experience. It has been shown that perineuronal net (PNN) formation coincides with the end of the critical period, and other evidences suggest that CSPGs have a critical role in regulating neuronal plasticity (75–78 and references therein). Aggrecan, brevican, and neurocan are major CSPGs that interact with hyaluronan and tenacins to form the PNN, which are areas of condensed ECM that surround neuronal cell bodies and dendrites (79, 80). (The role of PNN in neuroprotection has been widely studied and is discussed under the neurological disorders section.)
A widely studied model for CNS plasticity is the ocular dominance shift in the visual system, whereby the non-deprived eye becomes more represented in the visual cortex as a result of deprivation of the other eye. It has been reported that increase in the 4S/6S sulfation ratio of the CSPGs present in brain ECM leads to the termination of the critical period of ocular dominance in mouse visual cortex (81). Normally, the capability of plastic shift exists only until the termination of the critical period. However, an ocular dominance shift toward the non-deprived eye can also be seen in the mature CNS following treatment with chondroitinase ABC (ChABC), suggesting that CS-mediated loss of plasticity may be reversible (75). CSPGs are furthermore implicated in regulating memory formation in the CNS. The cellular correlates of memory formation are ‘upward’ or ‘downward’ shifts in the synaptic strengths of neurons in response to altered inputs, termed LTP and long-term depression, respectively (82). It was observed that ChABC treatment in the CA1 region of the mice hippocampus led to a reduction in LTP as well as LDP processes at the pyramidal cell synapse (83). Altogether, these findings suggest that, in addition to controlling their growth/migration, GAGs influence synaptic network stabilization and formation of novel synapses, thereby controlling CNS plasticity and memory formation.
GAGs in CNS injury
Even though the contribution of HS and CSPGs in the CNS is evident, in the absence of highly sensitive molecular and analytical techniques, it has been difficult to establish their structure-function relation. HSPGs interact with a diverse array of growth factors and often serve as necessary co-factors stabilizing their signaling complex (3). However, little is known about the role of HS in CNS injury. As noted earlier, as HS chains have various functional implications on CNS development, HS-mediated signaling can possibly be associated with axonal regeneration or CNS healing linked to other neurological disorders. However, the majority of research on the role of GAGs in CNS injury has mostly been focused on CSPGs.
After CNS injury such as SCI, axonal regeneration at the affected site is inhibited owing to the formation of glial scars by the reactive astrocytes (52). Glial scars in the CNS restrict the regenerative capability of injured neurons. CSPGs, which are upregulated at the injury site, are among the major components of the inhibitory scar tissues, functioning as a barrier to neuronal pathfinding (4, 84–86, and references therein). Neurocan and phosphacan are the two major CSPGs present in the scar, whereas BEHAB (brain enriched hyaluronan binding)/brevican expression has also been found to be upregulated by reactive astrocytes (87, 88). Enzymatic removal of CSPGs after injury using ChABC has resulted in partial recovery in several in vivo studies (89–91). Yick et al. (91) found that enzymatic digestion of CS at the lesion site led to the regeneration of Clarke’s nucleus neurons into peripheral nerve grafts implanted at the site. These studies suggest that the upregulation of CS in CNS injuries is the major cause for limited axonal regeneration and therefore is the most thoroughly studied manifestation of CS in the CNS.
In the context of SCI, CS is mostly reported to have an inhibitory effect on neuronal regeneration. However, as described in the sulfation patterns section below, some CS variants were found to promote neurite outgrowth and control polarization (55–57, 92). Therefore, as further discussed in the section on the role of GAGs in sulfation patterns, one cannot draw a simplistic conclusion about the molecular role of CSPGs during CNS injury without a closer examination of their discrete sulfation patterns.
GAGs in neurological disorders
One of the most well-studied neurological disorders associated with GAGs is Alzheimer’s disease (AD), in which patients show signs of cognitive deficits such as memory loss, inability to learn, and confusion (93). PGs appear to play both pathological and neuroprotective roles in AD. The disease is caused by the accumulation of neuritic plaques and neurofibrillary tangles (NFTs) in the cortex. One of the major components of the neuritic plaques is aggregated amyloid-β (AB) peptides in the brain. Highly sulfated HS has been found to be co-localized with these deposits. Less sulfated HS mediates uptake and degradation of AB in non-disease states; however, the highly sulfated HS inhibits this cellular uptake of AB (93–95 and references therein). These deposits of highly sulfated HS can be substantially degraded by Sulf-1 and Sulf-2, suggesting the presence of 6-O sulfates in significant amounts (96). It has also been shown that the source of this highly sulfated HS is the nearby neuronal population and that HS is important for the regulation of Alzheimer’s β-secretase (BACE1) (97). Studies have reported that HS and heparin, which is similar to HS except that it is more heavily charged, bind to BACE-1 and inhibit cleavage of amyloid precursor protein (APP) (97). Other GAG chains, including CS and DS, are also found within these plaques and may contribute to plaque formation and the overall progression of the disease. Previous research also showed that HS, CS, and KS can all interact with AB peptides and enhance the shift of AB42 peptides to the β-sheet confirmation (95). GAGs have been shown to be involved in aggregation and precipitation of amyloid fibrils, leading to increase in neurotoxicity (98). Low molecular weight heparin can compete with endogenous HS for binding to AB and inhibit the formation of fibrils (95). The sulfation patterns present on the HS thus seem to be important for the formation and progression of these neuritic plaques. Non-sulfated GAGs such as hyaluronic acid have no effect on fibril formation and aggregation (95).
Sulfated GAGs have also been implicated in Alzheimer’s-like changes seen in the τ protein. Incubation of this protein with heparin results in the formation of Alzheimer’s-like filaments and promotes the phosphorylation of τ. This phosphorylation prevents the binding of the τ protein to stabilized microtubules and results in the rapid disassembly of microtubules assembled from τ and tubulin. The effects seen from a variety of GAGs is proportional to the extent of sulfation present on the GAG chain (99).
PGs also play a significant role in gliomas. The neural ECM is naturally resistant to cell and neurite motility. As discussed in earlier sections, a major component of these inhibitory regions of the ECM is CS. Upregulation of BEHAB/brevican has been reported in gliomas (100, 101, and references therein). The tumor-specific BEHAB/brevican is differentially glycosylated and is cleaved by metalloproteases promoting cell motility and invasion in glioma (102, 103). Neurocan, versican, and tenascin-c are also implicated in the invasiveness of gliomas (104, 105). HSPGs are also upregulated in some gliomas and interact with factors that promote glioma growth and invasion. Specifically, several HSPGs have been found to promote the signaling and mitogenic properties of FGF-2 (106).
Sulfation of GAGs can alter the behavior of glioma cells. Studies have examined how sulfation density affects glioma proliferation and invasion; for example, reduction of sulfation using sodium chlorate treatment on glioma cells resulted in lower levels of cell proliferation (107). Intracerebral inoculation of glioma cells pretreated with sodium chlorate also lead to a decrease in size and instances of glioma, resulting in longer survival of the treated animals (108). Changes were also seen in specific sulfations on HS. Enhanced FGF-2 activity was correlated with higher sulfation levels in the HS present in gliomas with a higher amount of 2-O and 6-O sulfates (106). Sulf-2 expression was reported to regulate receptor tyrosine kinase signaling pathway through PDGFRα activation in glioblastoma cells as well as primary tumors. Knockdown of Sulf-2 in human glioblastoma cells or generation of gliomas from Sulf2-/- resulted in decreased growth invivo in mice (109). These results indicate that endosulfatases expressing gliomas could respond to various external growth factors, and such factors can serve as potential therapeutic targets in such disorders.
Alterations in GAGs are seen in a variety of neurological disorders, including autism, epilepsy, Parkinson’s disease, and schizophrenia (Table 2). A mouse model for autistic behavior, the BTBR T+tf/J mice, was recently reported to show alterations in HS associated with fractal structures of the subventricular zone (SVZ), specifically the third and lateral ventricles (110–112). This fractone-associated N-sulfated HS is thought to be involved in growth factor sequestering and in modulating glutamatergic synapses (110–112). Furthermore, eliminating HS in postnatal neurons in mice resulted in autistic symptoms. After conditionally inactivating Ext 1, an enzyme involved in HS biosynthesis in the brain after birth, there were no morphological changes; however, these mice showed stereotyped and repetitive behavior and had impaired social interactions (113). In a recent study, Pearson et al. (114) report reduction in N-sulfated HS in the ECM from the SVZ of brain lateral ventricles in postmortem tissue of autistic individuals. These studies implicate that decrease in N-sulfated HS in specific regions of the brain could be contributing to the etiology of autism and may serve as a biomarker for the disorder.
Association of various PGs and GAGs with neurological disorders.
| PG (GAG) | Disorder | Observed effects | References |
|---|---|---|---|
| Neurocan (CS) | Bipolar disorder and schizophrenia | SNP overlap was found in neurocan gene (NCAN) | (211) |
| Brevican (CS) | Episodic falling syndrome, related to human paroxysmal exercise-induced dyskinesia episodic ataxias | Deletion reported in brevican gene (BCAN) | (212) |
| Nauroparin (HS) | AD | Protected neurons against cholinergic lesions; increased arborization and reduced septal caspase 3 and τ immunoreactivity | (181, 182, 213, 214) |
| Perlecan (HS) | AD | Disordered processing was associated with amyloidosis | (215) |
| (HS, DS, CS) | AD | Identified in AD lesions, amyloid deposits, and neurofibrillary tangles | (216–218) |
| Apican (CS) | AD | APP acted as its core protein, was found in human and rat brain | (219) |
| (HS) | AD | O-GlcNAc glycosylation was upregulated | (220) |
| (HS/CS) | AD | Promoted formation of paired helical filaments | (221) |
| CSPG | HIV-1 infection | Facilitated infective entry of virus; PNN damage was observed in AIDS victims | (222, 223) |
| Neurocan, phosphacan, brevican (CS) | Epilepsy | Full-length neurocan was deposited after seizures, associated with axonal sprouting; phosphacan-positive PNN decreased; cleaved brevican increased in the temporal lobe and hippocampal regions of the rat brain | (224, 225) |
| Aggrecan, versican, phosphacan, brevican (CS) | Stroke | Plasticity increased in peri-infarct and remote regions with reduction in aggrecan, versican, and phosphacan, and accumulation of neurocan | (226, 227) |
| HSPGs | Gerstmann-Straussler syndrome, Creutzfeldt-Jakob disease, and scrapie | Sulfated GAGs were found to be present in amyloid plaques | (228) |
| HSPGs/CSPGs | Parkinson’s disease | Lewy bodies were found to contain PGs | (218, 229) |
| Brevican, versican (CS) | Glioma | Overexpressed and promoted tumor growth, vascularization, and invasiveness | (100, 101, 230) |
| CSPGs | Monocular deprivation and amblyopia | ChABC led to complete recovery in rats and moderate recovery in cats | (231, 232) |
| HSPGs | Pick’s disease | Involved in extinction of Pick bodies | (233) |
| HSPGs | Autism | Reduction in N-sulfated HS in SVZ | (114) |
CS chains play an etiological role in epilepsy. This is likely due to the ability of CS to stabilize synapses, reduce plasticity, and interact with GABAergic interneurons. A variety of CSPGs are thought to be involved in the onset and progression of epilepsy. Neurocan, which is primarily expressed during development, shows a renewed expression following status epilepticus (SE) in the adult hippocampus (115). Aggrecan has also been examined in relation to PNN remodeling after SE. After SE, the hippocampus showed a decrease in aggrecan expression 1 week post-SE, specifically in the dentate gyrus, followed by a decrease in aggrecan-labeled PNNs. Additionally, the remaining PNNs show a loss of structural integrity (116).
Both HS and CS appear to play a role in Parkinson’s disease (PD). Agrin, an ECM and transmembrane HSPG, has been implicated in fibril formation in AD and PD (117). NG2, a CSPG, has also been implicated in PD. In a study examining two rat models of PD, NG2-positive cells expressed calcium-binding adaptor molecule 1 and GDNF (118). It has also been seen that subcutaneous injection of a cytokine mixture containing granulocyte macrophage colony-stimulating factor and interleukin-3 ameliorated the loss of dopaminergic neurons in one of these rat PD models. One possible mechanism for this reduction of degradation is thought to be the increase in NG2-positive glia found in treated animals (118, 119). Hence, NG2-positive glia seems important for the survival of dopaminergic neurons and presents NG2 as a possible therapeutic target for PD.
PGs have also been implicated in schizophrenia. Upon examining postmortem brains, researchers found an increase in CSPG-positive glial cells in specific brain nuclei of the schizophrenic patients. The increase was primarily seen in the deep amygdala and entorhinal cortex. A decrease in PNNs was observed in different areas of the amygdala and entorhinal cortex as well (120). However, there was no change in the presence of parvalbumin-positive GABA neurons that are most commonly associated with PNNs in the entorhinal cortex or the amygdala (121). Changes have been seen in fear learning and extinction in schizophrenia patients. This may be directly influenced by alterations in the PNN present in the amygdala, as previous studies have shown that enzymatic degradation of this structure disrupted fear learning (122, 123). This presents evidence that CS could play a key role in development of schizophrenia and its overproduction in certain areas while disruption in others may lead to improper neural functionality.
GAGs are altered in many neurological disease states; however, it remains unclear what is the exact mechanism of their involvement. Although GAGs have been implicated in several CNS pathological processes, there are ample evidences to suggest their neuroprotective role as well. Brain samples from patients with sporadic Creutzfeldt-Jakob disease revealed that PNN surrounding parvalbumin-IR neurons of the cerebral cortex disappear before these cells die. Following their death, this zone becomes occupied with protease-resistant prion protein (PrP), which then spreads into the space and interacts with other GAGs to form protease-resistant aggregates (124, 125). Likewise, cortical neurons associated with PNN were found to be resistant to neurofibrillary changes and τ pathology in AD (126, 127). In vitro experiments suggest that CSPGs protect PNN-associated neurons against glutamate toxicity, which also plays a role in AD (128). CS chains were also found to elicit neuroprotective effects in an in vitro model of calcium-dependent excitotoxicity in a sulfation-dependent manner. Treatment of rat cortical neurons with CS-E reduced cell death by N-methyl-d-aspartate (NMDA), (S)-a-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid, or kainate, whereas other sulfation variants of CS or HS had no such protective effect (129). Dopaminergic neurons are associated with the pathophysiology of PD and undergo significant reorganization in their aggrecan-based ECM, leading to degeneration (130). It has been suggested that polyanionic GAGs associated with PNN contribute to the reduction in local oxidative stress by scavenging redox-active ions (131). Taken together, these studies suggest a paradoxical role of PGs in neurodegenerative diseases. For example, while several evidences suggest strong interaction between GAGs and amyloid deposits and NFTs (132, 133), CSPGs containing PNNs are known to protect cells from degeneration. Therefore, it is evident that GAG structures must play different roles in various states of damaged CNS. Our understanding about the specific roles of PGs in CNS disorders is still growing, and we have summarized observations on their associations with various CNS pathologies in Table 2 and Figure 4.
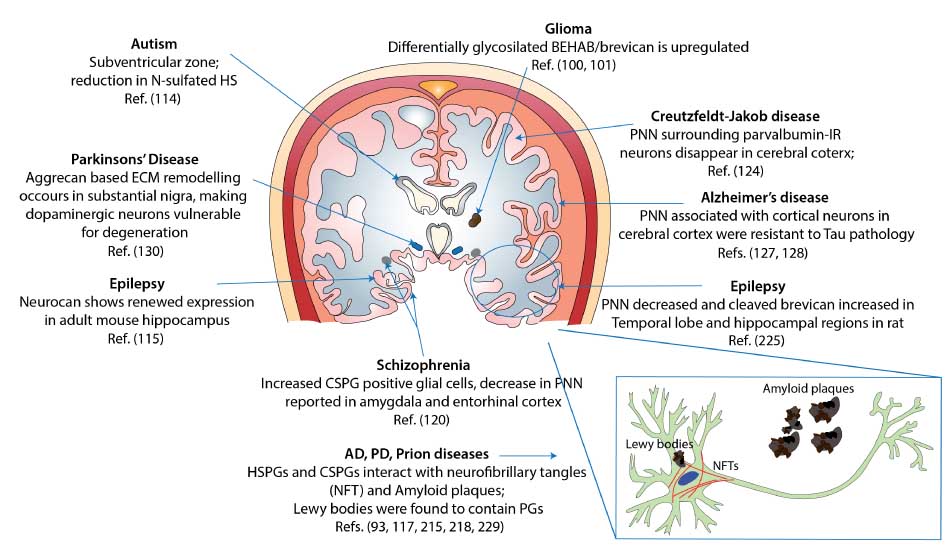
Observed and potential associations of PGs and GAGs with neurological diseases in various regions of the human brain.
This diagram represents a coronal section of the human brain and highlights evidence reported about the involvement of GAGs in various CNS disorders. Observations made in non-human brain samples may suggest that such effects persist in the human brain as well.
The role of GAG sulfation patterns in the CNS
The elaborate molecular interactions of the HSPGs/CSPGs are tightly controlled by their sulfation patterns, which provide their ligands unique selectivity for binding (Table 3). Diverse sulfations are found in both HS and CS (134). Wang et al. (43) reported that highly sulfated HS-like polymers, in comparison with unsulfated HS, increased error in median fiber tract axonal pathfinding of pioneer axons in cockroach embryo. Furthermore, genetic studies done by deleting various sulfotransferase enzymes have shown the importance of sulfation. For example, mutation in the N-deacetylase-N-sulfotransferase gene in Drosophila inhibited activation of the MAPK pathway and led to defects in cell migration during embryogenesis (135). Different sulfotransferase isoforms of CS have been associated with specific regions of the brain (136). With the alternating sulfotransferase activity, the sulfation patterns of HS and CS keep changing during embryonic brain development (137, 138). Therefore, it is understood that sulfated domains of HS and CS are highly regulated by the spatiotemporal expression of various sulfotransferase and endosulfatase enzymes.
Different GAG sulfation patterns and their effects related to the CNS.
| GAG | Sulfation | Observed effects | References |
|---|---|---|---|
| HS | 2S | Binding of FGF1 and FGF8 to several FGFRs. These interactions were associated with neural plate patterning, neurogenesis, gliogenesis, cell migration, axonal pathfinding, and neuronal regeneration | (145, 234, 235) |
| HS | 6S | Binding of FGF1 and FGF8 to several FGFRs, associated with effects stated in the row above | (145, 235) |
| HS | 2S/3S/6S | Associated with Slit protein binding | (236) |
| HS | IdoA(2S)-Glc(NS) | Associated with bFGF binding | (237) |
| HS | NS2S6S | Netrin 1 and semaphorin 5B binding; NS6S was preferred by Slit 2; NS2S was preferred by ephrin A1 and ephrin A2 | (238) |
| HS | Totally desulfated/NS | Failed to bind basic FGF | (239) |
| CS-A | GlcA-GalNAc(4S) | Provided negative guidance cues to cerebellar granule neurons | (163) |
| CS-C | GlcA-GalNAc(6S) | Expressed in tissues acting as barrier to axonal advancement in chick embryo; upregulated after CNS injury; upregulation led to axonal regeneration; associated with Schwann cell motility | (159, 160, 167, 240) |
| CS-E | GlcA-GalNAc(4S,6S) | Stimulated outgrowth in dopaminergic, hippocampal, and DRG neurons; associated with FGF-2, FGF-10, FGF-16, FGF-18, MK, PTN, TNF-α, BDNF, HB-EGF binding; inhibited rat cortical cell binding; inhibited DRG neurite outgrowth through PTPσ pathway; assembled neurotrophin-Trk complex | (148, 150, 151, 154, 156, 162, 166, 241) |
| CS-D/CS-E | GlcA(2S)-GalNAc(4S)/GlcA-GalNAc(4S,6S) | Promoted growth in rat hippocampal neurons | (242, 243) |
| CS-B/CS-E | IdoA-GalNAc(4S)/GlcA-GalNAc(4S,6S) | Interacted with PTN; promoted neurite outgrowth in hippocampal neurons by interacting with PTN | (1, 244) |
| CS-A/CS-D/CS-E | Stimulated neurosphere formation through an EGF-dependent pathway | (29) | |
| CS-B/CS-D/CS-E | Promoted FGF-2-mediated proliferation of rat embryonic neuronal stem cells | (30) |
HS sulfotransferase enzymes are expressed differentially in the cerebrum during neural development. During mouse embryonic development, significant 3OST and NDST expression was observed in the ventricular zone and the cortical plate (139). NDST was also expressed in other brain areas such as the marginal zone and subplate (139). In juveniles, 3OST and 6OST expression was found to be more widespread, being found in the ventricular zone, layers V/VI, intermediate zone, and cortical plate. 3OST expression in adults was found in layers II/III and V of the cortex, and 3OST-2 and 3OST-4 were found abundantly in trigeminal ganglion neurons. 6OST showed expression in layers I and II/III. NDST expression in juveniles was seen in layers II/III and V, whereas expression was seen in a manner similar to that of 3OST in adults (139, 140). In adults, 3OST expression can also be induced by environmental cues. Dramatic changes in the expression of various 3OST and 6OST isoforms were observed in a spatiotemporal manner in developing zebrafish (141, 142). The HS fine structure in the pineal gland showed differences when assessed in light versus dark conditions (143). The differential expression in development as well as changes in sulfotransferase expression due to environmental cues such as light implies that control of specific HS sulfation is important to normal development and neural function.
GAG functions are controlled by their sulfation pattern as well as density. The specificity of HS sulfation in determining their binding affinity for different isoforms of FGF has been investigated extensively. IdoA2S-GlcNS was found to increase the binding affinity of HS with basic FGF, and the affinity increased with increasing sulfation density (144). Sulfates seem to impart a defined structure to the HS microdomains, thus controlling their selectivity for binding to various ligands. Many such ligand receptor interactions, such as FGF-FGFR, play crucial roles in various CNS processes, such as neural plate patterning, neurogenesis, gliogenesis, axonal pathfinding, and regeneration during injury. Further details on the role of FGFs in CNS can be found in the review by Guillemot and Zimmer (145 and references cited therein).
Although several distinct roles have been proposed for HS- and CS-mediated signaling, the existence of overlap in their CNS interactions cannot be excluded completely. In fact, crystallographic analysis of RPTPσ revealed a shared binding site for HS and CS (146). In fact, Coles et al. (146) proposed that both HS and CS can bind to RPTPσ and still mediate their differential response on neurons. The computational model suggested that due to the difference in their sulfation densities, HS and CS are able to differently affect RPTPσ signaling. The discretely sulfated domains of HS may promote RPTPσ clustering, resulting in compromised phosphatase activity and hence enhanced life of neurite-promoting phosphorylated proteins. In contrast, the uniformly sulfated domain of CS results in higher phosphorylase activity mediated by RPTPσ, leading to neuronal inhibition (146). Others have also observed the contrasting roles of HS and CS mediated by RPTPσ. The role of RPTPσ as a CSPG receptor is well known (60). RPTPσ have also been reported to promote growth in chick retinal ganglion cell axons in response to basal lamina, which contains HSPG ligands (147). Although the presence of IdoA imparts very distinct structural features to HS, the presence of IdoA in certain other CS variants (CS-B and CS-D) can likely enhance their binding with HS binding proteins. This suggests that the HS and CS structure may regulate their CNS interactions in two ways. Owing to the gross structural similarity between HS and CS, they may interact with common receptors. However, owing to the distinct, fine differences in their sulfation, the outcomes of such interactions can vary widely.
Distinct sulfated motifs of CS have been shown to control neuronal outgrowth differently. Tully et al. (148) have shown that surface-immobilized CS-E tetrasaccharide can promote outgrowth of hippocampal neurons. Highly sulfated CS structures such as CS-D and CS-E are known to bind many proteins, including FGFs, MK, PTN, EGF, brain-derived neurotrophic factor (BDNF), and chemokines (see Table 3) (149–151). The CS-E motif is enriched in a developing rat brain (136); interacts with several PGs; and acts in association with appican, syndecan-1, syndecan-4, neuroglycan C, and phosphacan (136, 152, 153). In the study by Gama et al. (151), it was shown that BDNF selectively binds to the CS-E motif with a 20-fold preference over those of CS-A and CS-C at physiologically relevant concentrations. Moreover, surface plasmon resonance experiments show that CS-E bound to MK as strongly as heparin, which was followed by other GAGs (154). Similarly, exogenously added CS-E and CS-D were seen to block pleiotrophin-RPTPβ interaction, resulting in abnormal morphogenesis of Purkinje cell dendrites (155). The observations made on binding of highly sulfated CS with HS-binding growth factors such as MK further strengthens the hypothesis that a combination of gross and fine structural features mediates the interactions of HS and CS with common receptors. Furthermore, the high sulfation density of CS-D and CS-E might structurally be more similar to HS in terms of charge distribution and the presence of 2S-containing IdoA (in CS-D). Hence, these subtle features may enhance the binding of CS motifs to HS-binding growth factors, leading to similarity in their growth-promoting outcomes.
In spite of evidences demonstrating the influence of distinct CS motifs on neurons, the underlying mechanistic pathways are not yet fully understood. Rogers et al. (156) demonstrated another aspect of CS-E signaling by modulating the neutrophin (NT)-tyrosine receptor kinase (Trk) interactions. CS-E, but neither CS-A nor CS-C, was shown to assemble the NT-Trk complex, which led to the formation of CS-E-nerve growth factor (NGF)-TrkA and CS-E-BDNF-TrkB complexes (156). NT-Trk binding mediates neurite outgrowth, differentiation, and survival through the extracellular signal-regulated kinase, phosphatidylinositol 3-kinase, and phospholipase Cγ pathways (157, 158). Multiple signaling pathways may regulate CS-mediated neuronal growth or inhibition; thus, CS-E-dependent NT-Trk binding presents another way in which CS-E plays a role in neuronal growth and proliferation. Although NT signaling is mediated by Trk receptor dimerization, the possibility of CS-E in dimerizing Trk itself cannot be excluded completely.
In contrast to highly sulfated units, the monosulfated CS-A and CS-C were shown to have inhibitory influences on neuronal growth. The composition of three barrier tissues between the spinal cord and hind limb of the chick embryo illustrates the expression of peanut agglutinin and CS-C at the time when they are avoided by growing axons (159). In a more recent study, it was shown that CS-C synthesis is upregulated in CNS after injury (160). The study also analyzed mRNA levels of the enzyme chondroitin 6-O-sulfotransferase 1 (CS-6OST1) and showed its upregulation in most glial cells around cortical injuries. Although these results indicate that CS-C acts as an inhibitor for axonal regeneration, others have suggested CS-E or CS-A to be the inhibitory component of the glial scar (161–163).
The role of GAGs in enhancing amyloid aggregation revealed the following order: heparin>HS>CS=DS, suggesting that highly sulfated GAGs such as heparin were most effective in forming plaques. Furthermore, in the case of heparin, the order was found to be heparin>N-desulfated N-acetylated heparin>completely desulfated N-sulfated heparin>completely desulfated N-acetylated heparin. These results clearly show that the sulfate moiety of GAGs plays a critical role in the amyloid-β fibril formation (164, 165). Table 3 summarizes various evidences on the influence of CS and HS sulfation patterns in relation to the CNS.
Although there are a growing number of evidences that suggest distinct roles of different sulfation patterns of CS on neurons, a consensus is still lacking. For example, there is overwhelming evidence on the neurite-promoting activity of CS-E; however, Brown et al. (166) have recently shown that CS-E inhibits neurite outgrowth of dorsal root ganglion (DRG) neurons. Likewise, Karumbaiah et al. (161) reported that CS-E is upregulated in rats after SCI and is inhibitory for the growth of cortical neurons. Similarly, some studies point out the growth-promoting effects of 6-O-sulfation. Lin et al. (167) reported that highly 6-O-sulfated CS (CS-C motifs) in glial scar formed after CNS injury promoted axonal regeneration in nigrostriatal axons. Overall, contradictory evidence on the role of distinct CS motifs have been shown in the recent literature, and it seems plausible that the influence of such motifs depends on the neuronal cell type, site, and developmental stage of the CNS. Therefore, a systematic characterization of a library of CS variants is required to examine the relative influence of various sulfation patterns on neuronal growth and plasticity.
Owing to the variations in the chain length and the sulfation pattern of the CS chains used in different studies that also involved different neuronal models/sources, an understanding on the role of specific sulfation patterns is currently lacking. Although many studies utilize CS structural variants isolated from natural sources, these chains do not contain uniform sulfation patterns. Most of the commercially available CS chains are classified on the basis of the dominant sulfation motif present on them. We therefore characterized the sulfation profiles of five major CS variants found in the mammalian brain: CS-A, dermatan sulfate (CS-B), CS-C, CS-D, and CS-E, and assessed their influence on embryonic day 18 rat hippocampal neurons. Structural analysis of CS chains revealed a vast diversity in their sulfation content. Surface-immobilized CSs were used in the neurite growth assay by conjugating CS chains to the poly-l-lysine-coated surfaces. Single CS plain-field patterns revealed differential inhibitory potential of the CS variants. Interestingly, over the three time points of 24, 48, and 72 h, we observed that the neurite length and the number of neurites per cell were maximum in neurons growing over CS-D surfaces, whereas CS-C surfaces were the most inhibitory (unpublished data). These findings support previous reports on the inhibitory role of CS-C toward neuronal growth (160, 168). As already mentioned, disulfated CS chains have been reported to promote neuronal adhesion, migration, and neurogenesis. Likewise, we observed relatively better neuronal growth for both CS-D and CS-E among the CS chains tested. Interestingly, the neurite length and the number on CS-D-coated surfaces were significantly higher than in the other CS variants used (unpublished data). Most importantly, the study demonstrated that sulfation pattern variation could lead to diverse cell response to the GAGs, which might be the basis of the opposing roles of CSPGs in the CNS.
Expert opinion
It is clear that GAGs play a major role in CNS development and maintenance. The goal for the future will be to exploit these GAGs as therapeutic agents in CNS disorders. From the reported evidence, we understand that bimolecular interactions mediated by GAGs are influenced by the fine sulfation patterns present on HS and CS chains. Multiple approaches are being explored to utilize these molecules for developing solutions for the neurological injuries and disorders discussed earlier. Presently, the efforts on developing therapies for CNS injury/disorders are headed in three directions: (i) removing inhibitory CSPGs from the injury site (85); (ii) delivering neurotrophic factors for stimulating regeneration (169); and (iii) using stem cells to support neuronal regeneration (170). However, as none of these approaches have shown complete functional restoration independently, there is a need to design combinatorial therapies that modulate the complex neuroinhibitory environment present at the scar/disease site to facilitate neuronal regeneration.
Digestion of CSPGs at the scar site after injury is one of the most widely investigated therapeutic approaches for CNS injuries. Several studies have demonstrated the therapeutic efficacy of combining the digestion of CSPGs with infusions of neurotropic factors (171–174). However, ChABC treatment results in compete degradation of CS chains and may lead to loss of growth-promoting or guiding motifs. Therefore, digestion of specific CS chains using enzymes such as ChAC or ChB should be explored to selectively remove inhibitory motifs and lead to better therapeutic outcomes than ChABC. Such an approach may also be beneficial in AD and schizophrenia, where upregulation of HS or CS leads to disease progression. Partial or complete digestion of such GAG structures with combinations of chondroitinases, heparitinases, or endosulfatases could be explored for functional effective restorations recovery in such disorders.
In spite of promising results of ChABC, delivery of the active enzyme for prolonged periods has practical limitations. ChABC has been reported to be thermally unstable, decreasing its activity significantly at body temperature (175). Therefore, Lee et al. (90) have reported the use of trehalose to thermostabilize ChABC for prolonging its activity. The stabilized enzyme could digest CS chains in vivo up to 2 weeks after injury. Another potential approach to circumvent the limitations of ChABC could be to modulate GAG biosynthesis using xylosides. As xylosides are small molecules, they can be efficiently targeted and delivered at the scar site to alter the production of inhibitory CS chains. Previously, click-xylosides, containing various aglycone residues, were shown to prime a variety of different GAGs in Chinese hamster ovary (CHO) cells (176). Additionally, β-d-xylosides have been used on astrocytes to enhance neuronal growth (177). Moreover, 4-deoxy-4-fluoro xylosides (fluoro-xylosides) could be exploited to inhibit GAG production at the affected region. A number of fluoro-xylosides have been found to inhibit GAG biosynthesis in CHO and endothelial cells (178, 179). Small molecular inhibitors of GAG sulfotransferases are yet another option that can be used to modulate the HS or CS sulfation pattern at the damage site. By selectively targeting one or more sulfotransferase enzymes, highly specific HS or CS motifs can be generated to stimulate neuronal regeneration. As GAGs may enhance formation of protease-resistant plaques, inhibiting this binding may increase the turnover of these pathological aggregates by prolonged exposure to proteases. Polysulfonated GAG mimetics have been used for this purpose and were found to be protective against amyloid fibril-induced effects (180–182). Furthermore, 4-deoxy-N-acetyl glucosamine was shown to attenuate plaque formation and improve effects of AD (183). Therefore, several permutations and combinations of small molecular inhibitors of GAG biosynthesis could be utilized to create clinically applicable solutions for degrading or modifying pathological PGs in the CNS.
GAG-based approaches may pose some limitations as GAGs have multifunctional roles in CNS injury and pathology. First, even though PGs bind to amyloid fibrils through GAGs, an additional role of the protein core of PGs cannot be excluded. Next, although the use of ChABC or fluoro-xylosides may promote short-term neuronal recovery or plasticity by PNN degradation, it is uncertain whether such non-specific degradation or inhibition of CSPGs will be beneficial for the CNS in the long term. It has been shown that scar formation may have a beneficial role of limiting the injury and restricting the infiltration of inflammatory cells (184). Therefore, limited or transient degradation of CSPGs may be required for optimal recovery, thus making the time window of therapeutic intervention an equally important factor.
Various growth-stimulating factors such as neurotrophin-3, cyclic adenosine-mono-phosphate, and sonic hedgehog have been used to stimulate axonal regeneration at scar sites (169, 185). Many such factors target endogenous progenitor cells that can be stimulated to produce neurons and associated cell members at the injury site. In addition, spinal cord-derived neuronal stem/NPC cells can be transplanted to improve the regenerative capacity of the spinal cord, and to facilitate the functional recovery of experimental models (170, 186). To provide a stimulus for these stem cells to differentiate, they are generally seeded in polymeric scaffolds that mimic the architecture of the healthy spinal cord (170). However, instead of using synthetic polymers, GAG-based natural polymers can be developed to enhance the functionality of the scaffolds and to reproduce the molecular signals found in the developing CNS. HA is an excellent candidate for such applications because of its biocompatibility and its ability to maintain tissue organization, facilitate ion transport, and promote cell proliferation and differentiation (187). HS and CS have numerous effects on neurogenesis and neurodifferentiation, which can be exploited in conjunction with growth factor delivery. Heparin-PEG hybrid hydrogel has been used to explore its applicability toward neuronal cell replacement strategies (188). Although the functions of KS are largely unknown, KS chains are shown to be associated with some critical aspects of the CNS (189). KS is downregulated in AD (190), is the predominant GAG in the cornea of eye (191), and is involved in glial scar formation (192). The coming years will reveal more about the biology of KS and it is likely that all GAG types will become indispensible in therapies for many neurological disorders including AD. As discussed earlier, the influence of sulfation patterns of GAG in the CNS is highly dependent on the neuron type and the state of the CNS. Therefore, different sulfation motifs or GAG combinations may lead to enhanced neuronal regeneration or recovery for different sets of disorders and injury conditions.
Besides enhancing growth and regenerating neurons, it is imperative to guide axons across the scar site to reform severed neuronal wiring. One proposed treatment involves creating a neuronal bridging device that can facilitate connections of the spinal cord regions separated by the wound site. For this approach to succeed, a growth-supporting biomaterial must act as a conduit for the directional growth of neurons. To provide guidance cues for neuronal pathfinding, the bridging scaffold can be ‘sugar coated’ with HS and CS chains to create a directional gradient of the growth-promoting ligands. Previously, protein micropatterning techniques have been utilized to create patterns for directing neuronal outgrowth (193, 194). By utilizing an array of differentially sulfated GAG structures, one can create unique signaling combinations that enhance regeneration, functionality, and directed growth of neurons.
It is clear that GAGs can influence neuronal development through their structural heterogeneity; however, obtaining structurally uniform GAG chains has been a challenging task. As we present the concept of exploiting sulfation patterns to modulate the outcome of GAG-neuron interactions, obtaining uniform structures with defined sulfation motifs is a prerequisite. Significant advances have been made recently in this direction with the development of various chemoenzymatic processes to obtain defined oligosaccharide and polysaccharide structures of HS chains (195–197). Tully et al. (148) have utilized chemically synthesized CS structures in various neuronal assays. These processes can further be extended to create several uniform GAG structures of HS, CS, DS, or KS backbones for use in various CNS applications. Figure 5 summarizes various GAG-based therapies that can be utilized to develop clinical solutions for CNS disorders. It is hence clear that the biology of sulfation patterns in HS and CS chains has finally begun to take shape. The structure-function relations of GAG-mediated interactions can now be utilized to understand CNS signaling mechanisms and to design novel therapies for several neurological disorders.
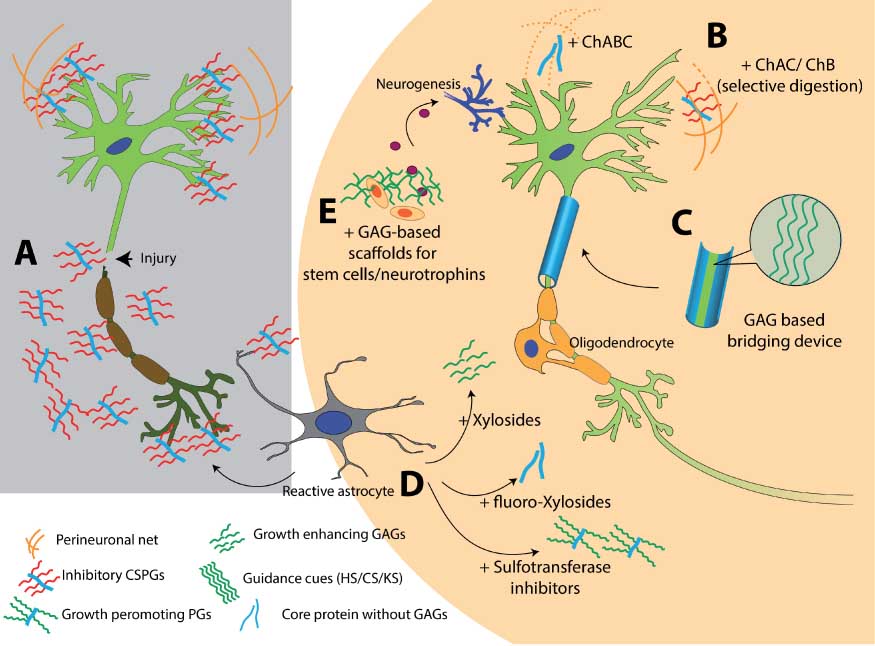
Use of sugar glues for broken neurons and GAG-based therapies for injured CNS.
(A) Neuronal injury results in overexpression of inhibitory CSPGs from reactive astrocytes. (B) Degrading CS chains to stimulate neuronal growth. ChABC is widely studied for this application; however, unspecific digestion of GAGs can damage growth-promoting motifs and other important structures such as PNN. Selective digestion can be done using ChAC and ChB enzymes. (C) Designing GAG-based bridging device to guide regenerating axons. Surface presented HS, CS, DS, KS, or HA domains can be used to direct axonal pathfinding. (D) Using small molecules to modulate GAG production at the scar site. Xylosides would change the composition or sulfation pattern of the GAGs secreted from reactive astrocytes. Addition of fluoro-xylosides would inhibit GAG biosynthesis and result in generation of core protein without inhibitory GAG chains. Sulfotransferase inhibitors can be used to stop production of inhibitory sulfation patterns present in CSPGs released from reactive astrocytes. (E) Bioactive scaffolds can be designed by using GAGs such as HA, HS, CS, DS, or KS. Such scaffolds can be used to deliver stem cells or neurotropic factors at the scar site. A combination of two or more such approaches can be utilized to create effective therapies for neurological disorders.
Highlights
GAGs play pivotal roles in CNS development, injury, and diseases.
Sulfation patterns of GAG chains have a major influence on the regulation of GAG-protein interactions in the CNS.
There are ample evidences that suggest the sulfation pattern of HS and CS chains affect the development and maintenance of CNS.
The presence of remarkable heterogeneity in sulfation patterns of naturally occurring GAG chains is a major impediment in deciphering the role of GAG sulfation patterns in the CNS.
Recent advances in GAG synthesis and analysis have set the stage for defining and exploiting the sulfation pattern of GAG chains to enhance CNS regeneration.
We propose various promising therapeutic approaches to tackle CNS disorders, including the use of GAG-degrading enzymes in combination with molecular scaffolds that modulate GAG biosynthetic pathways to facilitate functional regeneration.
The coming decade is expected to witness novel GAG-based therapeutic approaches for treating several debilitating neurological conditions.
About the authors

Vimal P. Swarup is a graduate student in the Department of Bioengineering at the University of Utah. Vimal received his BE from Birla Institute of Technology, India, and his MS from Illinois Institute of Technology, Chicago. Vimal’s current research interest lies in studying the role of glycosaminoglycans in spinal cord injuries and engineering approaches to modify their sulfation pattern at the injury site.

Caitlin P. Mencio is a graduate student in the Neuroscience Program at the University of Utah. Caitlin received her BS in Neuroscience from Texas Christian University in Fort Worth, TX. Caitlin is currently studying the role proteoglycans play in neural development, synapse formation, and plasticity.

Vladimir Hlady is a Professor of Bioengineering at University of Utah. He received BS, MS, and DSc degrees in Chemistry from the University of Zagreb, Croatia. His current research focus is on the role of glycocalyx in axonal pathfinding and platelet activation. He has two patents and published more than 120 scientific papers, reviews, and book chapters.

Kuberan (Kuby) Balagurunathan, PhD, is an Associate Professor of Medicinal Chemistry at the University of Utah. Kuby received his BSc from St. Joseph’s College, Trichy, India, and his MSc from the Indian Institute of Technology, Madras, India, under the guidance of Prof. Duraikkannu Loganathan, and his PhD from the University of Iowa under the guidance of Prof. Robert Linhardt. He did his postdoctoral training at MIT in Prof. Robert Rosenberg’s laboratory where he developed various chemoenzymatic approaches for creating defined structures of heparin and heparan sulfate. Kuby’s laboratory works on exploring the chemical biology of carbohydrates such as heparan, chondroitin, dermatan, and keratan sulfates and to determine their pathological and physiological roles at the molecular level.
Research in B.K.’s and V.H.’s laboratories are supported by NIH grants (PO1-HL107152, R01-GM075168 and R01-NS057144). V.S. is supported by a Graduate Research Fellowship from the University of Utah, and C.M. is partially supported by a Neuroscience Training Grant.
References
1. Bao X, Mikami T, Yamada S, Faissner A, Muramatsu T, Sugahara K. Heparin-binding growth factor, pleiotrophin, mediates neuritogenic activity of embryonic pig brain-derived chondroitin sulfate/dermatan sulfate hybrid chains. J Biol Chem 2005; 280: 9180–91.10.1074/jbc.M413423200Suche in Google Scholar
2. Bülow HE, Hobert O. Differential sulfations and epimerization define heparan sulfate specificity in nervous system development. Neuron 2004; 41: 723–36.10.1016/S0896-6273(04)00084-4Suche in Google Scholar
3. Lee J, Chien C. When sugars guide axons: insights from heparan sulphate proteoglycan mutants. Nat Rev Genet 2004; 5: 923–35.10.1038/nrg1490Suche in Google Scholar
4. Asher RA, Morgenstern DA, Moon LDF, Fawcett JW. Chondroitin sulphate proteoglycans: inhibitory components of the glial scar. Prog Brain Res 2001; 132: 611–9.10.1016/S0079-6123(01)32106-4Suche in Google Scholar
5. Snow DM, Lemmon V, Carrino DA, Caplan AI, Silver J. Sulfated proteoglycans in astroglial barriers inhibit neurite outgrowth in vitro. Exp Neurol 1990; 109: 111–30.10.1016/S0014-4886(05)80013-5Suche in Google Scholar
6. Sugahara K, Kitagawa H. Recent advances in the study of the biosynthesis and functions of sulfated glycosaminoglycans. Curr Opin Struct Biol 2000; 10: 518–27.10.1016/S0959-440X(00)00125-1Suche in Google Scholar
7. Herndon ME, Lander AD. A diverse set of developmentally regulated proteoglycans is expressed in the rat central nervous system. Neuron 1990; 4: 949–61.10.1016/0896-6273(90)90148-9Suche in Google Scholar
8. Weigel PH, Hascall VC, Tammi M. Hyaluronan synthases. J Biol Chem 1997; 272: 13997–4000.10.1074/jbc.272.22.13997Suche in Google Scholar
9. Rapraeger A, Jalkanen M, Endo E, Koda J, Bernfield M. The cell surface proteoglycan from mouse mammary epithelial cells bears chondroitin sulfate and heparan sulfate glycosaminoglycans. J Biol Chem 1985; 260: 11046–52.10.1016/S0021-9258(17)39146-9Suche in Google Scholar
10. David G, Van den Berghe H. Heparan sulfate-chondroitin sulfate hybrid proteoglycan of the cell surface and basement membrane of mouse mammary epithelial cells. J Biol Chem 1985; 260: 11067–74.10.1016/S0021-9258(17)39149-4Suche in Google Scholar
11. Bandtlow C, Zimmermann D. Proteoglycans in the developing brain: new conceptual insights for old proteins. Physiol Rev 2000; 80: 1267–90.10.1152/physrev.2000.80.4.1267Suche in Google Scholar
12. Zimmermann D, Ruoslahti E. Multiple domains of the large fibroblast proteoglycan, versican. EMBO J 1989; 8: 2975–81.10.1002/j.1460-2075.1989.tb08447.xSuche in Google Scholar
13. Doege K, Sasaki M, Kimura T, Yamada Y. Complete coding sequence and deduced primary structure of the human cartilage large aggregating proteoglycan, aggrecan. Human-specific repeats, and additional alternatively spliced forms. J Biol Chem 1991; 266: 894.10.1016/S0021-9258(17)35257-2Suche in Google Scholar
14. Rauch U, Karthikeyan L, Maurel P, Margolis RU, Margolis RK. Cloning and primary structure of neurocan, a developmentally regulated, aggregating chondroitin sulfate proteoglycan of brain. J Biol Chem 1992; 267: 19536–47.10.1016/S0021-9258(18)41808-XSuche in Google Scholar
15. Yamada H, Watanabe K, Shimonaka M, Yamaguchi Y. Molecular cloning of brevican, a novel brain proteoglycan of the aggrecan/versican family. J Biol Chem 1994; 269: 10119–26.10.1016/S0021-9258(17)36998-3Suche in Google Scholar
16. Barry FP, Neame PJ, Sasse J, Pearson D. Length variation in the keratan sulfate domain of mammalian aggrecan. Matrix Biol 1994; 14: 323–8.10.1016/0945-053X(94)90198-8Suche in Google Scholar
17. Barnea G, Grumet M, Sap J, Margolis R, Schlessinger J. Close similarity between receptor-linked tyrosine phosphatase and rat brain proteoglycan. Cell 1994; 76: 205.10.1016/0092-8674(94)90328-XSuche in Google Scholar
18. Bernfield M, Götte M, Park P, Reizes O, Fitzgerald M, Lincecum J, Zako M. Functions of cell surface heparan sulfate proteoglycans. Annu Rev Biochem 1999; 68: 729–77.10.1146/annurev.biochem.68.1.729Suche in Google Scholar
19. Litwack E, Ivins J, Kumbasar A, Paine-Saunders S, Stipp C, Lander A. Expression of the heparan sulfate proteoglycan glypican-1 in the developing rodent. Dev Dyn 1998; 211: 72–87.10.1002/(SICI)1097-0177(199801)211:1<72::AID-AJA7>3.0.CO;2-4Suche in Google Scholar
20. Watanabe K, Yamada H, Yamaguchi Y. K-glypican: a novel GPI-anchored heparan sulfate proteoglycan that is highly expressed in developing brain and kidney. J Cell Biol 1995; 130: 1207–18.10.1083/jcb.130.5.1207Suche in Google Scholar
21. Saunders S, Paine-Saunders S, Lander A. Expression of the cell surface proteoglycan glypican-5 is developmentally regulated in kidney, limb, and brain. Dev Biol 1997; 190: 78–93.10.1006/dbio.1997.8690Suche in Google Scholar PubMed
22. Nakato H, Futch T, Selleck S. The division abnormally delayed (dally) gene: a putative integral membrane proteoglycan required for cell division patterning during postembryonic development of the nervous system in Drosophila. Development 1995; 121: 3687–702.10.1242/dev.121.11.3687Suche in Google Scholar PubMed
23. Pilia G, Hughes-Benzie RM, MacKenzie A, Baybayan P, Chen EY, Huber R, Neri G, Cao A, Forabosco A, Schlessinger D. Mutations in GPC3, a glypican gene, cause the Simpson-Golabi-Behmel overgrowth syndrome. Nat Genet 1996; 12: 241–7.10.1038/ng0396-241Suche in Google Scholar PubMed
24. Hsueh Y, Sheng M. Regulated expression and subcellular localization of syndecan heparan sulfate proteoglycans and the syndecan-binding protein CASK/LIN-2 during rat brain development. J Neurosci 1999; 19: 7415–25.10.1523/JNEUROSCI.19-17-07415.1999Suche in Google Scholar
25. Mizuguchi S, Uyama T, Kitagawa H, Nomura KH, Dejima K, Gengyo-Ando K, Mitani S, Sugahara K, Nomura K. Chondroitin proteoglycans are involved in cell division of Caenorhabditis elegans. Nature 2003; 423: 443–8.10.1038/nature01635Suche in Google Scholar PubMed
26. Hwang HY, Olson SK, Esko JD, Horvitz HR. Caenorhabditis elegans early embryogenesis and vulval morphogenesis require chondroitin biosynthesis. Nature 2003; 423: 439–43.10.1038/nature01634Suche in Google Scholar PubMed
27. Sirko S, von Holst A, Wizenmann A, Götz M, Faissner A. Chondroitin sulfate glycosaminoglycans control proliferation, radial glia cell differentiation and neurogenesis in neural stem/progenitor cells. Development 2007; 134: 2727–38.10.1242/dev.02871Suche in Google Scholar PubMed
28. von Holst A, Sirko S, Faissner A. The unique 473HD-Chondroitinsulfate epitope is expressed by radial glia and involved in neural precursor cell proliferation. J Neurosci 2006; 26: 4082–94.10.1523/JNEUROSCI.0422-06.2006Suche in Google Scholar PubMed PubMed Central
29. Tham M, Ramasamy S, Gan HT, Ramachandran A, Poonepalli A, Yu YH, Ahmed S. CSPG is a secreted factor that stimulates neural stem cell survival possibly by enhanced EGFR signaling. PLoS One 2010; 5: e15341.10.1371/journal.pone.0015341Suche in Google Scholar PubMed PubMed Central
30. Ida M, Shuo T, Hirano K, Tokita Y, Nakanishi K, Matsui F, Aono S, Fujita H, Fujiwara Y, Kaji T. Identification and functions of chondroitin sulfate in the milieu of neural stem cells. J Biol Chem 2006; 281: 5982–91.10.1074/jbc.M507130200Suche in Google Scholar PubMed
31. Maeda N, Hamanaka H, Oohira A, Noda M. Purification, characterization and developmental expression of a brain-specific chondroitin sulfate proteoglycan, 6B4 proteoglycan/phosphacan. Neuroscience 1995; 67: 23–35.10.1016/0306-4522(94)00069-HSuche in Google Scholar
32. Desai CJ, Sun Q, Zinn K. Tyrosine phosphorylation and axon guidance: of mice and flies. Curr Opin Neurobiol 1997; 7: 70–4.10.1016/S0959-4388(97)80122-5Suche in Google Scholar
33. Kretschmer PJ, Fairhurst JL, Decker MM, Chan CP, Gluzman Y, Bohlen P, Kovesdi I. Cloning, characterization and developmental regulation of two members of a novel human gene family of neurite outgrowth-promoting proteins. Growth Factors 1991; 5: 99–114.10.3109/08977199109000275Suche in Google Scholar
34. Maeda N, Nishiwaki T, Shintani T, Hamanaka H, Noda M. 6B4 proteoglycan/phosphacan, an extracellular variant of receptor-like protein-tyrosine phosphatase ζ/RPTPβ, binds pleiotrophin/heparin-binding growth-associated molecule (HB-GAM). J Biol Chem 1996; 271: 21446–52.10.1074/jbc.271.35.21446Suche in Google Scholar
35. Matsumoto K, Wanaka A, Mori T, Taguchi A, Ishii N, Muramatsu H, Muramatsu T, Tohyama M. Localization of pleiotrophin and midkine in the postnatal developing cerebellum. Neurosci Lett 1994; 178: 216–20.10.1016/0304-3940(94)90762-5Suche in Google Scholar
36. Maeda N, Noda M. Involvement of receptor-like protein tyrosine phosphatase /RPTP and its ligand pleiotrophin/heparin-binding growth-associated molecule (HB-GAM) in neuronal migration. J Cell Biol 1998; 142: 203–16.10.1083/jcb.142.1.203Suche in Google Scholar PubMed PubMed Central
37. Landolt RM, Vaughan L, Winterhalter KH, Zimmermann DR. Versican is selectively expressed in embryonic tissues that act as barriers to neural crest cell migration and axon outgrowth. Development 1995; 121: 2303–12.10.1242/dev.121.8.2303Suche in Google Scholar PubMed
38. Perris R, Perissinotto D, Pettway Z, Bronner-Fraser M, Morgelin M, Kimata K. Inhibitory effects of PG-H/aggrecan and PG-M/versican on avian neural crest cell migration. FASEB J 1996; 10: 293–301.10.1096/fasebj.10.2.8641562Suche in Google Scholar PubMed
39. Ishii M, Maeda N. Oversulfated chondroitin sulfate plays critical roles in the neuronal migration in the cerebral cortex. J Biol Chem 2008; 283: 32610–20.10.1074/jbc.M806331200Suche in Google Scholar PubMed
40. Saunders S, Paine-Saunders S, Lander AD. Expression of the cell surface proteoglycan glypican-5 is developmentally regulated in kidney, limb, and brain. Dev Biol 1997; 190: 78–93.10.1006/dbio.1997.8690Suche in Google Scholar PubMed
41. Hsueh YP, Sheng M. Regulated expression and subcellular localization of syndecan heparan sulfate proteoglycans and the syndecan-binding protein CASK/LIN-2 during rat brain development. J Neurosci 1999; 19: 7415–25.10.1523/JNEUROSCI.19-17-07415.1999Suche in Google Scholar
42. Ivins J, Litwack E, Kumbasar A, Stipp C, Lander A. Cerebroglycan, a developmentally regulated cell-surface heparan sulfate proteoglycan, is expressed on developing axons and growth cones. Dev Biol 1997; 184: 320–32.10.1006/dbio.1997.8532Suche in Google Scholar
43. Wang L, Denburg J. A role for proteoglycans in the guidance of a subset of pioneer axons in cultured embryos of the cockroach. Neuron 1992; 8: 701–14.10.1016/0896-6273(92)90091-QSuche in Google Scholar
44. Walz A, McFarlane S, Brickman Y, Nurcombe V, Bartlett P, Holt C. Essential role of heparan sulfates in axon navigation and targeting in the developing visual system. Development 1997; 124: 2421.10.1242/dev.124.12.2421Suche in Google Scholar
45. Zhou Y, Besner GE. Heparin-binding epidermal growth factor-like growth factor is a potent neurotrophic factor for PC12 cells. NeuroSignals 2010; 18: 141–51.10.1159/000319823Suche in Google Scholar
46. Muramatsu T. Midkine, a heparin-binding cytokine with multiple roles in development, repair and diseases. Proc Jpn Acad Ser B Phys Biol Sci 2010; 86: 410–25.10.2183/pjab.86.410Suche in Google Scholar
47. Liang Y, Annan R, Carr S, Popp S, Mevissen M, Margolis R, Margolis R. Mammalian homologues of the Drosophila slit protein are ligands of the heparan sulfate proteoglycan glypican-1 in brain. J Biol Chem 1999; 274: 17885–92.10.1074/jbc.274.25.17885Suche in Google Scholar
48. Hu H. Cell-surface heparan sulfate is involved in the repulsive guidance activities of Slit2 protein. Nat Neurosci 2001; 4: 695–701.10.1038/89482Suche in Google Scholar
49. Rauvala H, Vanhala A, Nolo R, Raulo E, Merenmies J, Panula P. Expression of HB-GAM (heparin-binding growth-associated molecules) in the pathways of developing axonal processes in vivo and neurite outgrowth in vitro induced by HB-GAM. Dev Brain Res 1994; 79: 157–76.10.1016/0165-3806(94)90121-XSuche in Google Scholar
50. Kinnunen T, Kaksonen M, Saarinen J, Kalkkinen N, Peng H, Rauvala H. Cortactin-Src kinase signaling pathway is involved in N-syndecan-dependent neurite outgrowth. J Biol Chem 1998; 273: 10702–8.10.1074/jbc.273.17.10702Suche in Google Scholar PubMed
51. Thon N, Haas CA, Rauch U, Merten T, Fässler R, Frotscher M, Deller T. The chondroitin sulphate proteoglycan brevican is upregulated by astrocytes after entorhinal cortex lesions in adult rats. Eur J Neurosci 2000; 12: 2547–58.10.1046/j.1460-9568.2000.00109.xSuche in Google Scholar
52. Fawcett J, Asher R. The glial scar and central nervous system repair. Brain Res Bull 1999; 49: 377–91.10.1016/S0361-9230(99)00072-6Suche in Google Scholar
53. Oohira A, Matsui F, Katoh-Semba R. Inhibitory effects of brain chondroitin sulfate proteoglycans on neurite outgrowth from PC12D cells. J Neurosci 1991; 11: 822–7.10.1523/JNEUROSCI.11-03-00822.1991Suche in Google Scholar
54. Mckeon RJ, Höke A, Silver J. Injury-induced proteoglycans inhibit the potential for laminin-mediated axon growth on astrocytic scars. Exp Neurol 1995; 136: 32–43.10.1006/exnr.1995.1081Suche in Google Scholar PubMed
55. Bicknese A, Sheppard A, O’Leary D, Pearlman A. Thalamocortical axons extend along a chondroitin sulfate proteoglycan-enriched pathway coincident with the neocortical subplate and distinct from the efferent path. J Neurosci 1994; 14: 3500–10.10.1523/JNEUROSCI.14-06-03500.1994Suche in Google Scholar
56. McAdams B, McLoon S. Expression of chondroitin sulfate and keratan sulfate proteoglycans in the path of growing retinal axons in the developing chick. J Comp Neurol 1995; 352: 594–606.10.1002/cne.903520408Suche in Google Scholar PubMed
57. Sheppard A, Hamilton S, Pearlman A. Changes in the distribution of extracellular matrix components accompany early morphogenetic events of mammalian cortical development. J Neurosci 1991; 11: 3928–42.10.1523/JNEUROSCI.11-12-03928.1991Suche in Google Scholar
58. Friedlander D, Milev P, Karthikeyan L, Margolis R, Margolis R, Grumet M. The neuronal chondroitin sulfate proteoglycan neurocan binds to the neural cell adhesion molecules Ng-CAM/L1/NILE and N-CAM, and inhibits neuronal adhesion and neurite outgrowth. J Cell Biol 1994; 125: 669–80.10.1083/jcb.125.3.669Suche in Google Scholar PubMed PubMed Central
59. Maeda N, Ichihara-Tanaka K, Kimura T, Kadomatsu K, Muramatsu T, Noda M. Receptor-like protein-tyrosine phosphatase PTPζ/RPTPβ binds a heparin-binding growth factor midkine involvement of arginine 78 of midkine in the high affinity binding to PTPζ. J Biol Chem 1999; 274: 12474–9.10.1074/jbc.274.18.12474Suche in Google Scholar PubMed
60. Shen Y, Tenney AP, Busch SA, Horn KP, Cuascut FX, Liu K, He Z, Silver J, Flanagan JG. PTP {σ} is a receptor for chondroitin sulfate proteoglycan, an inhibitor of neural regeneration. Sci Signal 2009; 326: 592.Suche in Google Scholar
61. Schachtrup C, Ryu JK, Helmrick MJ, Vagena E, Galanakis DK, Degen JL, Margolis RU, Akassoglou K. Fibrinogen triggers astrocyte scar formation by promoting the availability of active TGF-β after vascular damage. J Neurosci 2010; 30: 5843–54.10.1523/JNEUROSCI.0137-10.2010Suche in Google Scholar PubMed PubMed Central
62. Logan A, Berry M, Gonzalez AM, Frautschy SA, Sporn MB, Baird A. Effects of transforming growth factor β1, on scar production in the injured central nervous system of the rat. Eur J Neurosci 2006; 6: 355–63.10.1111/j.1460-9568.1994.tb00278.xSuche in Google Scholar PubMed
63. Rimaniol AC, Lekieffre D, Serrano A, Masson A, Benavides J, Zavala F. Biphasic transforming growth factor-[β] production flanking the pro-inflammatory cytokine response in cerebral trauma. Neuroreport 1995; 7: 133–6.10.1097/00001756-199512290-00032Suche in Google Scholar
64. Lagord C, Berry M, Logan A. Expression of TGFβ2 but not TGFβ1 correlates with the deposition of scar tissue in the lesioned spinal cord. Mol Cell Neurosci 2002; 20: 69–92.10.1006/mcne.2002.1121Suche in Google Scholar PubMed
65. Fisher D, Xing B, Dill J, Li H, Hoang HH, Zhao Z, Yang XL, Bachoo R, Cannon S, Longo FM, Sheng M, Silver J, Li S. Leukocyte common antigen-related phosphatase is a functional receptor for chondroitin sulfate proteoglycan axon growth inhibitors. J Neurosci 2011; 31: 14051–66.10.1523/JNEUROSCI.1737-11.2011Suche in Google Scholar PubMed PubMed Central
66. Dickendesher TL, Baldwin KT, Mironova YA, Koriyama Y, Raiker SJ, Askew KL, Wood A, Geoffroy CG, Zheng B, Liepmann CD. NgR1 and NgR3 are receptors for chondroitin sulfate proteoglycans. Nat Neurosci 2012; 15: 703–12.10.1038/nn.3070Suche in Google Scholar PubMed PubMed Central
67. Koprivica V, Cho KS, Park JB, Yiu G, Atwal J, Gore B, Kim JA, Lin E, Tessier-Lavigne M, Chen DF. EGFR activation mediates inhibition of axon regeneration by myelin and chondroitin sulfate proteoglycans. Sci STKE 2005; 310: 106–10.10.1126/science.1115462Suche in Google Scholar PubMed
68. Wong ST, Henley JR, Kanning KC, Huang K, Bothwell M, Poo M. A p75NTR and Nogo receptor complex mediates repulsive signaling by myelin-associated glycoprotein. Nat Neurosci 2002; 5: 1302–8.10.1038/nn975Suche in Google Scholar PubMed
69. Hsieh SHK, Ferraro GB, Fournier AE. Myelin-associated inhibitors regulate cofilin phosphorylation and neuronal inhibition through LIM kinase and Slingshot phosphatase. J Neurosci 2006; 26: 1006–15.10.1523/JNEUROSCI.2806-05.2006Suche in Google Scholar PubMed PubMed Central
70. Lingor P, Koeberle P, Kügler S, Bähr M. Down-regulation of apoptosis mediators by RNAi inhibits axotomy-induced retinal ganglion cell death in vivo. Brain 2005; 128: 550–8.10.1093/brain/awh382Suche in Google Scholar PubMed
71. Sivasankaran R, Pei J, Wang KC, Zhang YP, Shields CB, Xu XM, He Z. PKC mediates inhibitory effects of myelin and chondroitin sulfate proteoglycans on axonal regeneration. Nat Neurosci 2004; 7: 261–8.10.1038/nn1193Suche in Google Scholar PubMed
72. Dityatev A, Dityateva G, Sytnyk V, Delling M, Toni N, Nikonenko I, Muller D, Schachner M. Polysialylated neural cell adhesion molecule promotes remodeling and formation of hippocampal synapses. J Neurosci 2004; 24: 9372–82.10.1523/JNEUROSCI.1702-04.2004Suche in Google Scholar PubMed PubMed Central
73. Oyagi A, Moriguchi S, Nitta A, Murata K, Oida Y, Tsuruma K, Shimazawa M, Fukunaga K, Hara H. Heparin-binding EGF-like growth factor is required for synaptic plasticity and memory formation. Brain Res 2011; 1419: 97–104.10.1016/j.brainres.2011.09.003Suche in Google Scholar PubMed
74. Kalus I, Salmen B, Viebahn C, von Figura K, Schmitz D, D’Hooge R, Dierks T. Differential involvement of the extracellular 6-O-endosulfatases Sulf1 and Sulf2 in brain development and neuronal and behavioural plasticity. J Cell Mol Med 2009; 13: 4505–21.10.1111/j.1582-4934.2008.00558.xSuche in Google Scholar PubMed PubMed Central
75. Pizzorusso T, Medini P, Berardi N, Chierzi S, Fawcett JW, Maffei L. Reactivation of ocular dominance plasticity in the adult visual cortex. Science 2002; 298: 1248–51.10.1126/science.1072699Suche in Google Scholar PubMed
76. Hockfield S, Kalb R, Zaremba S, Fryer H, editors. Expression of neural proteoglycans correlates with the acquisition of mature neuronal properties in the mammalian brain. Cold Spring Harbor Symposia on Quantitative Biology. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press, 1990.10.1101/SQB.1990.055.01.049Suche in Google Scholar
77. Galtrey CM, Fawcett JW. The role of chondroitin sulfate proteoglycans in regeneration and plasticity in the central nervous system. Brain Res Rev 2007; 54: 1–18.10.1016/j.brainresrev.2006.09.006Suche in Google Scholar PubMed
78. Kwok JCF, Dick G, Wang D, Fawcett JW. Extracellular matrix and perineuronal nets in CNS repair. Dev Neurobiol 2011; 71: 1073–89.10.1002/dneu.20974Suche in Google Scholar PubMed
79. Aspberg A, Miura R, Bourdoulous S, Shimonaka M, Heinegård D, Schachner M, Ruoslahti E, Yamaguchi Y. The C-type lectin domains of lecticans, a family of aggregating chondroitin sulfate proteoglycans, bind tenascin-R by protein-protein interactions independent of carbohydrate moiety. Proc Natl Acad Sci USA 1997; 94: 10116–21.10.1073/pnas.94.19.10116Suche in Google Scholar PubMed PubMed Central
80. Matthews RT, Kelly GM, Zerillo CA, Gray G, Tiemeyer M, Hockfield S. Aggrecan glycoforms contribute to the molecular heterogeneity of perineuronal nets. J Neurosci 2002; 22: 7536–47.10.1523/JNEUROSCI.22-17-07536.2002Suche in Google Scholar
81. Miyata S, Komatsu Y, Yoshimura Y, Taya C, Kitagawa H. Persistent cortical plasticity by upregulation of chondroitin 6-sulfation. Nat Neurosci 2012; 15: 414–22.10.1038/nn.3023Suche in Google Scholar PubMed
82. Bliss TVP, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature 1993; 361: 31–9.10.1038/361031a0Suche in Google Scholar
83. Bukalo O, Schachner M, Dityatev A. Modification of extracellular matrix by enzymatic removal of chondroitin sulfate and by lack of tenascin-R differentially affects several forms of synaptic plasticity in the hippocampus. Neuroscience 2001; 104: 359–69.10.1016/S0306-4522(01)00082-3Suche in Google Scholar
84. Moreau-Fauvarque C, Kumanogoh A, Camand E, Jaillard C, Barbin G, Boquet I, Love C, Jones EY, Kikutani H, Lubetzki C. The transmembrane semaphorin Sema4D/CD100, an inhibitor of axonal growth, is expressed on oligodendrocytes and upregulated after CNS lesion. J Neurosci 2003; 23: 9229–39.10.1523/JNEUROSCI.23-27-09229.2003Suche in Google Scholar
85. Bradbury E, Moon L, Popat R, King V, Bennett G, Patel P, Fawcett J, McMahon S. Chondroitinase ABC promotes functional recovery after spinal cord injury. Nature 2002; 416: 636–40.10.1038/416636aSuche in Google Scholar PubMed
86. Silver J, Miller JH. Regeneration beyond the glial scar. Nat Rev Neurosci 2004; 5: 146–56.10.1038/nrn1326Suche in Google Scholar PubMed
87. McKeon RJ, Jurynec MJ, Buck CR. The chondroitin sulfate proteoglycans neurocan and phosphacan are expressed by reactive astrocytes in the chronic CNS glial scar. J Neurosci 1999; 19: 10778–88.10.1523/JNEUROSCI.19-24-10778.1999Suche in Google Scholar
88. Jaworski DM, Kelly GM, Hockfield S. Intracranial injury acutely induces the expression of the secreted isoform of the CNS-specific hyaluronan-binding protein BEHAB/brevican. Exp Neurol 1999; 157: 327–37.10.1006/exnr.1999.7062Suche in Google Scholar PubMed
89. Bradbury EJ, Moon LDF, Popat RJ, King VR, Bennett GS, Patel PN, Fawcett JW, McMahon SB. Chondroitinase ABC promotes functional recovery after spinal cord injury. Nature 2002; 416: 636–40.10.1038/416636aSuche in Google Scholar
90. Lee H, McKeon RJ, Bellamkonda RV. Sustained delivery of thermostabilized chABC enhances axonal sprouting and functional recovery after spinal cord injury. Proc Natl Acad Sci USA 2010; 107: 3340–5.10.1073/pnas.0905437106Suche in Google Scholar PubMed PubMed Central
91. Yick L, Wu W, So K, Yip H, Shum D. Chondroitinase ABC promotes axonal regeneration of Clarke’s neurons after spinal cord injury. Neuroreport 2000; 11: 1063–7.10.1097/00001756-200004070-00032Suche in Google Scholar PubMed
92. Nishimura K, Ishii M, Kuraoka M, Kamimura K, Maeda N. Opposing functions of chondroitin sulfate and heparan sulfate during early neuronal polarization. Neuroscience 2010; 169: 1535–47.10.1016/j.neuroscience.2010.06.027Suche in Google Scholar
93. van Horssen J, Wesseling P, van den Heuvel LPWJ, de Waal RMW, Verbeek MM. Heparan sulphate proteoglycans in Alzheimer’s disease and amyloid-related disorders. Lancet Neurol 2003; 2: 482–92.10.1016/S1474-4422(03)00484-8Suche in Google Scholar
94. Kanekiyo T, Zhang J, Liu Q, Liu C-C, Zhang L, Bu G. Heparan sulphate proteoglycan and the low-density lipoprotein receptor-related protein 1 constitute major pathways for neuronal amyloid-b uptake. J Neurosci 2011; 31: 1644–51.10.1523/JNEUROSCI.5491-10.2011Suche in Google Scholar PubMed PubMed Central
95. Ariga T, Miyatake T, Yu RK. Role of proteoglycans and glycosaminoglycans in the pathogenesis of Alzheimer’s disease and related disorders: amyloidogenesis and therapeutic strategies – a review. J Neurosci Res 2010; 88: 2303–15.10.1002/jnr.22393Suche in Google Scholar PubMed
96. Hosono-Fukao T, Ohtake-Niimi S, Hoshino H, Britschgi M, Akatsu H, Hossain MM, Nishitsuji K, van Kuppevelt TH, Kimata K, Michikawa M. Heparan sulfate subdomains that are degraded by Sulf accumulate in cerebral amyloid β plaques of Alzheimer’s disease: evidence from mouse models and patients. Am J Pathol 2012; 180: 2056–67.10.1016/j.ajpath.2012.01.015Suche in Google Scholar PubMed
97. Scholefield Z, Yates EA, Wayne G, Amour A, McDowell W, Turnbull J. Heparan sulfate regulates precursor protein processing by BACE1 the Alzheimer’s B-secretase. J Cell Biol 2003; 163: 97–107.10.1083/jcb.200303059Suche in Google Scholar PubMed PubMed Central
98. Fraser PE, Nguyen JT, Chin DT, Kirschner DA. Effects of sulfate ions on Alzheimer β/A4 peptide assemblies: implications for amyloid fibril-proteoglycan interactions. J Neurochem 1992; 59: 1531–40.10.1111/j.1471-4159.1992.tb08470.xSuche in Google Scholar PubMed
99. Hasegawa M, Crowther RA, Jakes R, Goedert M. Alzheimer-like changes in microtubule-associated protein τ induced by sulfated glycosaminoglycans. Inhibition of microtubule binding, stimulation of phosphorylation, and filament assembly depend on the degree of sulfation. J Biol Chem 1997; 272: 33118–24.10.1074/jbc.272.52.33118Suche in Google Scholar PubMed
100. Viapiano MS, Matthews RT, Hockfield S. A novel membrane-associated glycovariant of BEHAB/brevican is up-regulated during rat brain development and in a rat model of invasive glioma. J Biol Chem 2003; 278: 33239–47.10.1074/jbc.M303480200Suche in Google Scholar PubMed
101. Nutt CL, Matthews RT, Hockfield S. Glial tumor invasion: a role for the upregulation and cleavage of BEHAB/brevican. Neuroscientist 2001; 7: 113–22.10.1177/107385840100700206Suche in Google Scholar PubMed
102. Viapiano MS, Bi WL, Piepmeier J, Hockfield S, Matthews RT. Novel tumor-specific isoforms of BEHAB/brevican identified in human malignant gliomas. Cancer Res 2005; 65: 6726–33.10.1158/0008-5472.CAN-05-0585Suche in Google Scholar
103. Viapiano MS, Hockfield S, Matthews RT. BEHAB/brevican requires ADAMTS-mediated proteolytic cleavage to promote glioma invasion. J Neurooncol 2008; 88: 261–72.10.1007/s11060-008-9575-8Suche in Google Scholar
104. Varga I, Hutóczki G, Szemcsák CD, Zahuczky G, Tóth J, Adamecz Z, Kenyeres A, Bognár L, Hanzély Z, Klekner A. Brevican, neurocan, tenascin-c and versican are mainly responsible for the invasiveness of low-grade astrocytoma. Pathol Oncol Res 2012; 18: 413–320.10.1007/s12253-011-9461-0Suche in Google Scholar
105. Hu B, Kong LL, Matthews RT, Viapiano MS. The proteoglycan brevican binds to fibronectin after proteolytic cleavage and promotes glioma cell motility. J Biol Chem 2008; 283: 24848–59.10.1074/jbc.M801433200Suche in Google Scholar
106. Su G, Meyer K, Nandini CD, Qiao D, Salamat S, Friedl A. Glypican-1 is frequently overexpressed in human gliomas and enhances FGF-2 signaling in glioma cells. Am J Pathol 2006; 168: 2014–26.10.2353/ajpath.2006.050800Suche in Google Scholar
107. Mendes de Aguiar CBN, Garcez RC, Alvarez-Silva M, Trentin AG. Undersulfation of proteoglycan and proteins alter C6 glioma cells proliferation, adhesion and extracellular matrix organization. Int J Dev Neurosci 2002; 20: 563–71.10.1016/S0736-5748(02)00081-3Suche in Google Scholar
108. Lobao-Soares B, Alvarez-Silva M, Mendes de Aguiar CBN, Nicolau M, Trentin AG. Undersulfation of glycosaminoglycans induced by sodium chlorate treatment affects the progression of C6 rat glioma, in vivo. Brain Res 2007; 1131: 29–36.10.1016/j.brainres.2006.11.014Suche in Google Scholar PubMed
109. Phillips JJ, Huillard E, Robinson AE, Ward A, Lum DH, Polley MY, Rosen SD, Rowitch DH, Werb Z. SULF2 regulates PDGFRα signaling and growth in human and mouse malignant glioma. J Clin Invest 2012; 122: 911–22.10.1172/JCI58215Suche in Google Scholar PubMed PubMed Central
110. Meyza KZ, Blanchard DC, Pearson BL, Pobbe RLH, Blanchard RJ. Fractione-associated N-sulfated heparan sulfate shows reduced quantity in BTBR T+tf/J mice: a strong model of autism. Behav Brain Res 2012; 228: 247–53.10.1016/j.bbr.2011.11.004Suche in Google Scholar PubMed PubMed Central
111. Blanchard DC, Defensor EB, Meyza KZ, Pobbe RL, Pearson BL, Bolivar VJ, Blanchard RJ. BTBR T+tf/J mice: autism-relevant behaviors and reduced fractone-associated heparan sulfate. Neurosci Biobehav Rev 2012; 36: 285–96.10.1016/j.neubiorev.2011.06.008Suche in Google Scholar PubMed PubMed Central
112. Mercier F, Kwon YC, Douet V. Hippocampus/amygdala alterations, loss of heparan sulfates, fractones and ventricle wall reduction in adult BTBR T+tf/J mice, animal model for autism. Neurosci Lett 2012; 506: 208–13.10.1016/j.neulet.2011.11.007Suche in Google Scholar PubMed
113. Irie F, Badie-Mahdavi H, Yamaguchi Y. Autism-like socio-communicative deficits and stereotypies in mice lacking heparan sulfate. Proc Natl Acad Sci USA 2012; 109: 5052–6.10.1073/pnas.1117881109Suche in Google Scholar PubMed PubMed Central
114. Pearson B, Corley M, Vasconcellos A, Blanchard D, Blanchard R. Heparan sulfate deficiency in autistic postmortem brain tissue from the subventricular zone of the lateral ventricles. Behav Brain Res 2013; 243: 138–45.10.1016/j.bbr.2012.12.062Suche in Google Scholar PubMed PubMed Central
115. Heck N, Garwood J, Loeffler JP, Larmet Y, Faissner A. Differential upregulation of extracellular matrix molecules associated with the appearance of granule cell dispersion and mossy fiber sprouting during epileptogenesis in a murine model of temporal lobe epilepsy. Neuroscience 2004; 129: 309–24.10.1016/j.neuroscience.2004.06.078Suche in Google Scholar PubMed
116. McRae PA, Baranov E, Rogers SL, Porter BE. Persistent decrease in multiple components of the perineuronal net following status epilepticus. Eur J Neurosci 2012; 36: 3471–82.10.1111/j.1460-9568.2012.08268.xSuche in Google Scholar PubMed PubMed Central
117. Liu IH, Uversky VN, Munishkina LA, Fink AL, Halfter W, Cole GJ. Agrin binds α-synuclein and modulates α-synuclein fibrillation. Glycobiology 2005; 15: 1320–31.10.1093/glycob/cwj014Suche in Google Scholar PubMed
118. Kitamura Y, Inden M, Minamino H, Abe M, Takata K, Taniguchi T. The 6-hydroxydopamine-induced nigrostriatal neurodegeneration produces microglia-like NG2 glial cells in the rat substantia nigra. Glia 2010; 58: 1686–700.10.1002/glia.21040Suche in Google Scholar PubMed
119. Choudhury ME, Sugimoto K, Kubo M, Nagai M, Nomoto M, Takahashi H, Yano H, Tanaka J. A cytokine mixture of GM-CSF and IL-3 that induces a neuroprotective phenotype of microglia leading to amelioration of (6-OHDA)-induced Parkinsonism of rats. Brain Behav 2011; 1: 26–43.10.1002/brb3.11Suche in Google Scholar PubMed PubMed Central
120. Pantazopoulos H, Woo TU, Lim MP, Lange N, Berretta S. Extracellular matrix-glial abnormalities in the amygdala and entorhinal cortex of subjects diagnosed with schizophrenia. Arch Gen Psychiatry 2010; 67: 155–66.10.1001/archgenpsychiatry.2009.196Suche in Google Scholar PubMed PubMed Central
121. Pantazopoulos H, Lange N, Baldessarini RJ, Berretta S. Parvalbumin neurons in the entorhinal cortex of subjects diagnosed with bipolar disorder or schizophrenia. Biol Psychiatry 2007; 61: 640–52.10.1016/j.biopsych.2006.04.026Suche in Google Scholar PubMed PubMed Central
122. Gogolla N, Caroni P, Lüthi A, Herry C. Perineuronal nets protect fear memories from erasure. Science 2009; 325: 1258–61.10.1126/science.1174146Suche in Google Scholar
123. Holt DJ, Lebron–Milad K, Milad MR, Rauch SL, Pitman RK, Orr SP, Cassidy BS, Walsh JP, Goff DC. Extinction memory is impaired in schizophrenia. Biol Psychiatry 2009; 65: 455–63.10.1016/j.biopsych.2008.09.017Suche in Google Scholar
124. Belichenko PV, Miklossy J, Belser B, Budka H, Celio MR. Early destruction of the extracellular matrix around parvalbumin-immunoreactive interneurons in Creutzfeldt-Jakob disease. Neurobiol Dis 1999; 6: 269–79.10.1006/nbdi.1999.0245Suche in Google Scholar
125. Caughey B, Brown K, Raymond G, Katzenstein G, Thresher W. Binding of the protease-sensitive form of PrP (prion protein) to sulfated glycosaminoglycan and congo red [corrected]. J Virol 1994; 68: 2135–41.10.1128/jvi.68.4.2135-2141.1994Suche in Google Scholar
126. Brückner G, Hausen D, Härtig W, Drlicek M, Arendt T, Brauer K. Cortical areas abundant in extracellular matrix chondroitin sulphate proteoglycans are less affected by cytoskeletal changes in Alzheimer’s disease. Neuroscience 1999; 92: 791–805.10.1016/S0306-4522(99)00071-8Suche in Google Scholar
127. Morawski M, Brückner G, Jäger C, Seeger G, Arendt T. Neurons associated with aggrecan-based perineuronal nets are protected against τ pathology in subcortical regions in Alzheimer’s disease. Neuroscience 2010; 169: 1347–63.10.1016/j.neuroscience.2010.05.022Suche in Google Scholar
128. Mattson MP. Degenerative and protective signaling mechanisms in the neurofibrillary pathology of AD. Neurobiol Aging 1995; 16: 447–57.10.1016/0197-4580(94)00182-ZSuche in Google Scholar
129. Sato Y, Nakanishi K, Tokita Y, Kakizawa H, Ida M, Maeda H, Matsui F, Aono S, Saito A, Kuroda Y. A highly sulfated chondroitin sulfate preparation, CS-E, prevents excitatory amino acid-induced neuronal cell death. J Neurochem 2008; 104: 1565–76.10.1111/j.1471-4159.2007.05107.xSuche in Google Scholar PubMed
130. Brückner G, Morawski M, Arendt T. Aggrecan-based extracellular matrix is an integral part of the human basal ganglia circuit. Neuroscience 2008; 151: 489–504.10.1016/j.neuroscience.2007.10.033Suche in Google Scholar PubMed
131. Morawski M, Brückner MK, Riederer P, Brückner G, Arendt T. Perineuronal nets potentially protect against oxidative stress. Exp Neurol 2004; 188: 309–15.10.1016/j.expneurol.2004.04.017Suche in Google Scholar PubMed
132. Goedert M, Jakes R, Spillantini M, Hasegawa M, Smith M, Crowther R. Assembly of microtubule-associated protein τ into Alzheimer-like filaments induced by sulphated glycosaminoglycans. Nature 1996; 383: 550–3.10.1038/383550a0Suche in Google Scholar PubMed
133. Pérez M, Valpuesta JM, Medina M, Montejo de Garcini E, Avila J. Polymerization of τ into filaments in the presence of heparin: the minimal sequence required for τ-τ interaction. J Neurochem 1996; 67: 1183–90.10.1046/j.1471-4159.1996.67031183.xSuche in Google Scholar PubMed
134. Gama C, Hsieh-Wilson L. Chemical approaches to deciphering the glycosaminoglycan code. Curr Opin Chem Biol 2005; 9: 609–19.10.1016/j.cbpa.2005.10.003Suche in Google Scholar PubMed
135. Lin X, Buff EM, Perrimon N, Michelson AM. Heparan sulfate proteoglycans are essential for FGF receptor signaling during Drosophila embryonic development. Development 1999; 126: 3715–23.10.1242/dev.126.17.3715Suche in Google Scholar PubMed
136. Shuo T, Aono S, Matsui F, Tokita Y, Maeda H, Shimada K, Oohira A. Developmental changes in the biochemical and immunological characters of the carbohydrate moiety of neuroglycan C, a brain-specific chondroitin sulfate proteoglycan. Glycoconj J 2003; 20: 267–78.10.1023/B:GLYC.0000025821.22618.33Suche in Google Scholar
137. Brickman Y, Ford M, Gallagher J, Nurcombe V, Bartlett P, Turnbull J. Structural modification of fibroblast growth factor-binding heparan sulfate at a determinative stage of neural development. J Biol Chem 1998; 273: 4350–9.10.1074/jbc.273.8.4350Suche in Google Scholar PubMed
138. Kitagawa H, Tsutsumi K, Tone Y, Sugahara K. Developmental regulation of the sulfation profile of chondroitin sulfate chains in the chicken embryo brain. J Biol Chem 1997; 272: 31377–81.10.1074/jbc.272.50.31377Suche in Google Scholar PubMed
139. Yabe T, Hata T, He J, Maeda N. Developmental and regional expression of heparan sulfate sulfotransferase genes in the mouse brain. Glycobiology 2005; 15: 982–93.10.1093/glycob/cwi090Suche in Google Scholar PubMed
140. Lawrence R, Yabe T, HajMohammadi S, Rhodes J, McNeely M, Liu J, Lamperti ED, Toselli PA, Lech M, Spear PG. The principal neuronal gD-type 3-O-sulfotransferases and their products in central and peripheral nervous system tissues. Matrix Biol 2007; 26: 442–55.10.1016/j.matbio.2007.03.002Suche in Google Scholar PubMed PubMed Central
141. Cadwallader AB, Yost HJ. Combinatorial expression patterns of heparan sulfate sulfotransferases in zebrafish: I. The 3-O-sulfotransferase family. Dev Dyn 2006; 235: 3423–31.10.1002/dvdy.20991Suche in Google Scholar PubMed
142. Cadwallader AB, Yost HJ. Combinatorial expression patterns of heparan sulfate sulfotransferases in zebrafish: II. The 6-O-sulfotransferase family. Dev Dyn 2006; 235: 3432–7.10.1002/dvdy.20990Suche in Google Scholar PubMed
143. Kuberan B, Lech M, Borjigin J, Rosenberg RD. Light-induced 3-O-sulfotransferase expression alters pineal heparan sulfate fine structure. J Biol Chem 2004; 279: 5053–4.10.1074/jbc.C300492200Suche in Google Scholar PubMed
144. Habuchi H, Suzuki S, Saito T, Tamura T, Harada T, Yoshida K, Kimata K. Structure of a heparan sulphate oligosaccharide that binds to basic fibroblast growth factor. Biochem J 1992; 285: 805–13.10.1042/bj2850805Suche in Google Scholar PubMed PubMed Central
145. Guillemot F, Zimmer C. From cradle to grave: the multiple roles of fibroblast growth factors in neural development. Neuron 2011; 71: 574–88.10.1016/j.neuron.2011.08.002Suche in Google Scholar PubMed
146. Coles CH, Shen Y, Tenney AP, Siebold C, Sutton GC, Lu W, Gallagher JT, Jones EY, Flanagan JG, Aricescu AR. Proteoglycan-specific molecular switch for RPTP {σ} clustering and neuronal extension. Sci STKE 2011; 332: 484–8.Suche in Google Scholar
147. Aricescu AR, McKinnell IW, Halfter W, Stoker AW. Heparan sulfate proteoglycans are ligands for receptor protein tyrosine phosphatase σ. Mol Cell Biol 2002; 22: 1881–92.10.1128/MCB.22.6.1881-1892.2002Suche in Google Scholar PubMed PubMed Central
148. Tully S, Mabon R, Gama C, Tsai S, Liu X, Hsieh-Wilson L. A chondroitin sulfate small molecule that stimulates neuronal growth. J Am Chem Soc 2004; 126: 7736–7.10.1021/ja0484045Suche in Google Scholar PubMed
149. Kawashima H, Atarashi K, Hirose M, Hirose J, Yamada S, Sugahara K, Miyasaka M. Oversulfated chondroitin/dermatan sulfates containing GlcAβ1/IdoAα1-3GalNAc (4, 6-O-disulfate) interact with L-and P-selectin and chemokines. J Biol Chem 2002; 277: 12921–30.10.1074/jbc.M200396200Suche in Google Scholar PubMed
150. Deepa S, Umehara Y, Higashiyama S, Itoh N, Sugahara K. Specific molecular interactions of oversulfated chondroitin sulfate E with various heparin-binding growth factors. J Biol Chem 2002; 277: 43707–16.10.1074/jbc.M207105200Suche in Google Scholar PubMed
151. Gama CI, Tully SE, Sotogaku N, Clark PM, Rawat M, Vaidehi N, Goddard WA, Nishi A, Hsieh-Wilson LC. Sulfation patterns of glycosaminoglycans encode molecular recognition and activity. Nat Chem Biol 2006; 2: 467–73.10.1038/nchembio810Suche in Google Scholar PubMed
152. Tsuchida K, Shioi J, Yamada S, Boghosian G, Wu A, Cai H, Sugahara K, Robakis NK. Appican, the proteoglycan form of the amyloid precursor protein, contains chondroitin sulfate E in the repeating disaccharide region and 4-O-sulfated galactose in the linkage region. J Biol Chem 2001; 276: 37155–60.10.1074/jbc.M105818200Suche in Google Scholar PubMed
153. Deepa SS, Yamada S, Zako M, Goldberger O, Sugahara K. Chondroitin sulfate chains on syndecan-1 and syndecan-4 from normal murine mammary gland epithelial cells are structurally and functionally distinct and cooperate with heparan sulfate chains to bind growth factors. J Biol Chem 2004; 279: 37368–76.10.1074/jbc.M403031200Suche in Google Scholar
154. Ueoka C, Kaneda N, Okazaki I, Nadanaka S, Muramatsu T, Sugahara K. Neuronal cell adhesion, mediated by the heparin-binding neuroregulatory factor midkine, is specifically inhibited by chondroitin sulfate E. J Biol Chem 2000; 275: 37407–13.10.1074/jbc.M002538200Suche in Google Scholar
155. Tanaka M, Maeda N, Noda M, Marunouchi T. A chondroitin sulfate proteoglycan PTPζ/RPTPβ regulates the morphogenesis of Purkinje cell dendrites in the developing cerebellum. J Neurosci 2003; 23: 2804–14.10.1523/JNEUROSCI.23-07-02804.2003Suche in Google Scholar
156. Rogers CJ, Clark PM, Tully SE, Abrol R, Garcia KC, Goddard WA, Hsieh-Wilson LC. Elucidating glycosaminoglycan-protein-protein interactions using carbohydrate microarray and computational approaches. Proc Natl Acad Sci USA 2011; 108: 9747–52.10.1073/pnas.1102962108Suche in Google Scholar
157. Huang EJ, Reichardt LF. Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci 2001; 24: 677–736.10.1146/annurev.neuro.24.1.677Suche in Google Scholar
158. Kaplan DR, Miller FD. Neurotrophin signal transduction in the nervous system. Curr Opin Neurobiol 2000; 10: 381–91.10.1016/S0959-4388(00)00092-1Suche in Google Scholar
159. Oakley RA, Tosney KW. Peanut agglutinin and chondroitin-6-sulfate are molecular markers for tissues that act as barriers to axon advance in the avian embryo. Dev Biol 1991; 147: 187–206.10.1016/S0012-1606(05)80017-XSuche in Google Scholar
160. Properzi F, Carulli D, Asher RA, Muir E, Camargo LM, Van Kuppevelt TH, Ten Dam GB, Furukawa Y, Mikami T, Sugahara K, Toida T, Geller HM, Fawcett JW. Chondroitin 6-sulphate synthesis is up-regulated in injured CNS, induced by injury-related cytokines and enhanced in axon growth-inhibitory glia. Eur J Neurosci 2005; 21: 378–90.10.1111/j.1460-9568.2005.03876.xSuche in Google Scholar PubMed
161. Karumbaiah L, Anand S, Thazhath R, Zhong Y, Mckeon RJ, Bellamkonda RV. Targeted downregulation of N-acetylgalactosamine 4-sulfate 6-O-sulfotransferase significantly mitigates chondroitin sulfate proteoglycan mediated inhibition. Glia 2011; 59: 981–96.10.1002/glia.21170Suche in Google Scholar PubMed PubMed Central
162. Gilbert RJ, McKeon RJ, Darr A, Calabro A, Hascall VC, Bellamkonda RV. CS-4, 6 is differentially upregulated in glial scar and is a potent inhibitor of neurite extension. Mol Cell Neurosci 2005; 29: 545–58.10.1016/j.mcn.2005.04.006Suche in Google Scholar PubMed
163. Wang H, Katagiri Y, McCann TE, Unsworth E, Goldsmith P, Yu ZX, Tan F, Santiago L, Mills EM, Wang Y. Chondroitin-4-sulfation negatively regulates axonal guidance and growth. J Cell Sci 2008; 121: 3083–91.10.1242/jcs.032649Suche in Google Scholar
164. Castillo GM, Cummings JA, Yang W, Judge ME, Sheardown MJ, Rimvall K, Hansen JB, Snow AD. Sulfate content and specific glycosaminoglycan backbone of perlecan are critical for perlecan’s enhancement of islet amyloid polypeptide (amylin) fibril formation. Diabetes 1998; 47: 612–20.10.2337/diabetes.47.4.612Suche in Google Scholar
165. Castillo GM, Lukito W, Wight TN, Snow AD. The sulfate moieties of glycosaminoglycans are critical for the enhancement of β-amyloid protein fibril formation. J Neurochem 1999; 72: 1681–7.10.1046/j.1471-4159.1999.721681.xSuche in Google Scholar
166. Brown JM, Xia J, Zhuang BQ, Cho KS, Rogers CJ, Gama CI, Rawat M, Tully SE, Uetani N, Mason DE, Tremblay ML, Peters EC, Habuchi O, Chen DF, Hsieh-Wilson LC. A sulfated carbohydrate epitope inhibits axon regeneration after injury. Proc Natl Acad Sci USA 2012; 109: 4768–73.10.1073/pnas.1121318109Suche in Google Scholar
167. Lin R, Rosahl TW, Whiting PJ, Fawcett JW, Kwok JCF. 6-Sulphated chondroitins have a positive influence on axonal regeneration. PLoS One 2011; 6: e21499.10.1371/journal.pone.0021499Suche in Google Scholar
168. Shum DKY, Chau CH. Changes in glycosaminoglycans during regeneration of post crush sciatic nerves of adult guinea pigs. J Neurosci Res 1996; 46: 465–76.10.1002/(SICI)1097-4547(19961115)46:4<465::AID-JNR8>3.0.CO;2-ESuche in Google Scholar
169. Lu P, Yang H, Jones LL, Filbin MT, Tuszynski MH. Combinatorial therapy with neurotrophins and cAMP promotes axonal regeneration beyond sites of spinal cord injury. J Neurosci 2004; 24: 6402–9.10.1523/JNEUROSCI.1492-04.2004Suche in Google Scholar
170. Teng YD, Lavik EB, Qu X, Park KI, Ourednik J, Zurakowski D, Langer R, Snyder EY. Functional recovery following traumatic spinal cord injury mediated by a unique polymer scaffold seeded with neural stem cells. Proc Natl Acad Sci USA 2002; 99: 3024–9.10.1073/pnas.052678899Suche in Google Scholar
171. Massey JM, Amps J, Viapiano MS, Matthews RT, Wagoner MR, Whitaker CM, Alilain W, Yonkof AL, Khalyfa A, Cooper NGF. Increased chondroitin sulfate proteoglycan expression in denervated brainstem targets following spinal cord injury creates a barrier to axonal regeneration overcome by chondroitinase ABC and neurotrophin-3. Exp Neurol 2008; 209: 426–45.10.1016/j.expneurol.2007.03.029Suche in Google Scholar
172. Fouad K, Schnell L, Bunge MB, Schwab ME, Liebscher T, Pearse DD. Combining Schwann cell bridges and olfactory-ensheathing glia grafts with chondroitinase promotes locomotor recovery after complete transection of the spinal cord. J Neurosci 2005; 25: 1169–78.10.1523/JNEUROSCI.3562-04.2005Suche in Google Scholar
173. Houle JD, Tom VJ, Mayes D, Wagoner G, Phillips N, Silver J. Combining an autologous peripheral nervous system “bridge” and matrix modification by chondroitinase allows robust, functional regeneration beyond a hemisection lesion of the adult rat spinal cord. J Neurosci 2006; 26: 7405–15.10.1523/JNEUROSCI.1166-06.2006Suche in Google Scholar
174. Karimi-Abdolrezaee S, Eftekharpour E, Wang J, Schut D, Fehlings MG. Synergistic effects of transplanted adult neural stem/progenitor cells, chondroitinase, and growth factors promote functional repair and plasticity of the chronically injured spinal cord. J Neurosci 2010; 30: 1657–76.10.1523/JNEUROSCI.3111-09.2010Suche in Google Scholar
175. Tester NJ, Plaas AH, Howland DR. Effect of body temperature on chondroitinase ABC’s ability to cleave chondroitin sulfate glycosaminoglycans. J Neurosci Res 2007; 85: 1110–8.10.1002/jnr.21199Suche in Google Scholar
176. Victor XV, Nguyen TKN, Ethirajan M, Tran VM, Nguyen KV, Kuberan B. Investigating the elusive mechanism of glycosaminoglycan biosynthesis. J Biol Chem 2009; 284: 25842–53.10.1074/jbc.M109.043208Suche in Google Scholar
177. Smith-Thomas LC, Stevens J, Fok-Seang J, Faissner A, Rogers JH, Fawcett JW. Increased axon regeneration in astrocytes grown in the presence of proteoglycan synthesis inhibitors. J Cell Sci 1995; 108: 1307–15.10.1242/jcs.108.3.1307Suche in Google Scholar
178. Garud D, Tran V, Victor X, Koketsu M, Kuberan B. Inhibition of heparan sulfate and chondroitin sulfate proteoglycan biosynthesis. J Biol Chem 2008; 283: 28881–7.10.1074/jbc.M805939200Suche in Google Scholar
179. Raman K, Ninomiya M, Nguyen TKN, Tsuzuki Y, Koketsu M, Kuberan B. Novel glycosaminoglycan biosynthetic inhibitors affect tumor-associated angiogenesis. Biochem Biophys Res Commun 2011; 404: 86–9.10.1016/j.bbrc.2010.11.069Suche in Google Scholar
180. Kisilevsky R, Szarek WA. Novel glycosaminoglycan precursors as anti-amyloid agents part II. J Mol Neurosci 2002; 19: 45–50.10.1007/s12031-002-0009-3Suche in Google Scholar
181. Rose M, Dudas B, Cornelli U, Hanin I. Protective effect of the heparin-derived oligosaccharide C3, on AF64A-induced cholinergic lesion in rats. Neurobiol Aging 2003; 24: 481–90.10.1016/S0197-4580(02)00093-3Suche in Google Scholar
182. Rose M, Dudas B, Cornelli U, Hanin I. Glycosaminoglycan C3 protects against AF64A-induced cholinotoxicity in a dose-dependent and time-dependent manner. Brain Res 2004; 1015: 96–102.10.1016/j.brainres.2004.04.048Suche in Google Scholar PubMed
183. Kisilevsky R, Szarek WA, Ancsin JB, Elimova E, Marone S, Bhat S, Berkin A. Inhibition of amyloid A amyloidogenesis in vivo and in tissue culture by 4-deoxy analogues of peracetylated 2-acetamido-2-deoxy-α- and β-d-glucose: implications for the treatment of various amyloidoses. Am J Pathol 2004; 164: 2127–37.10.1016/S0002-9440(10)63771-6Suche in Google Scholar
184. Okada S, Nakamura M, Katoh H, Miyao T, Shimazaki T, Ishii K, Yamane J, Yoshimura A, Iwamoto Y, Toyama Y. Conditional ablation of Stat3 or Socs3 discloses a dual role for reactive astrocytes after spinal cord injury. Nat Med 2006; 12: 829–34.10.1038/nm1425Suche in Google Scholar PubMed
185. Lowry N, Goderie SK, Lederman P, Charniga C, Gooch MR, Gracey KD, Banerjee A, Punyani S, Silver J, Kane RS. The effect of long-term release of Shh from implanted biodegradable microspheres on recovery from spinal cord injury in mice. Biomaterials 2012; 33: 2892–901.10.1016/j.biomaterials.2011.12.048Suche in Google Scholar PubMed
186. Parr A, Kulbatski I, Zahir T, Wang X, Yue C, Keating A, Tator C. Transplanted adult spinal cord-derived neural stem/progenitor cells promote early functional recovery after rat spinal cord injury. Neuroscience 2008; 155: 760–70.10.1016/j.neuroscience.2008.05.042Suche in Google Scholar PubMed
187. Hou S, Xu Q, Tian W, Cui F, Cai Q, Ma J, Lee IS. The repair of brain lesion by implantation of hyaluronic acid hydrogels modified with laminin. J Neurosci Methods 2005; 148: 60–70.10.1016/j.jneumeth.2005.04.016Suche in Google Scholar PubMed
188. Freudenberg U, Hermann A, Welzel PB, Stirl K, Schwarz SC, Grimmer M, Zieris A, Panyanuwat W, Zschoche S, Meinhold D. A star-PEG-heparin hydrogel platform to aid cell replacement therapies for neurodegenerative diseases. Biomaterials 2009; 30: 5049–60.10.1016/j.biomaterials.2009.06.002Suche in Google Scholar PubMed
189. Funderburgh JL. Mini Review. Keratan sulfate: structure, biosynthesis, and function. Glycobiology 2000; 10: 951–8.10.1093/glycob/10.10.951Suche in Google Scholar PubMed
190. Lindahl B, Eriksson L, Spillmann D, Caterson B, Lindahl U. Selective loss of cerebral keratan sulfate in Alzheimer’s disease. J Biol Chem 1996; 271: 16991–4.10.1074/jbc.271.29.16991Suche in Google Scholar PubMed
191. Quantock AJ, Young RD, Akama TO. Structural and biochemical aspects of keratan sulphate in the cornea. Cell Mol Life Sci 2010; 67: 891–906.10.1007/s00018-009-0228-7Suche in Google Scholar PubMed
192. Zhang H, Uchimura K, Kadomatsu K. Brain keratan sulfate and glial scar formation. Ann NY Acad Sci 2006; 1086: 81–90.10.1196/annals.1377.014Suche in Google Scholar PubMed
193. Hlady V, Hodgkinson G. The effects of proteoglycan surface patterning on neuronal pathfinding. Materwiss Werksttech 2007; 38: 975–82.10.1002/mawe.200700224Suche in Google Scholar PubMed PubMed Central
194. Hodgkinson GN, Tresco PA, Hlady V. The role of well-defined patterned substrata on the regeneration of DRG neuron pathfinding and integrin expression dynamics using chondroitin sulfate proteoglycans. Biomaterials 2012; 33: 4288–97.10.1016/j.biomaterials.2012.02.046Suche in Google Scholar PubMed PubMed Central
195. Lindahl U, Li J, Kusche-Gullberg M, Salmivirta M, Alaranta S, Veromaa T, Emeis J, Roberts I, Taylor C, Oreste P. Generation of “neoheparin” from E. coli K5 capsular polysaccharide. J Med Chem 2005; 48: 349–52.10.1021/jm049812mSuche in Google Scholar PubMed
196. Kuberan B, Beeler DL, Lech M, Wu ZL, Rosenberg RD. Chemoenzymatic synthesis of classical and non-classical anticoagulant heparan sulfate polysaccharides. J Biol Chem 2003; 278: 52613–21.10.1074/jbc.M305029200Suche in Google Scholar PubMed
197. Liu R, Xu Y, Chen M, Weïwer M, Zhou X, Bridges AS, DeAngelis PL, Zhang Q, Linhardt RJ, Liu J. Chemoenzymatic design of heparan sulfate oligosaccharides. J Biol Chem 2010; 285: 34240–9.10.1074/jbc.M110.159152Suche in Google Scholar PubMed PubMed Central
198. Maeda N, Ishii M, Nishimura K, Kamimura K. Functions of chondroitin sulfate and heparan sulfate in the developing brain. Neurochem Res 2011; 36: 1228–40.10.1007/s11064-010-0324-ySuche in Google Scholar PubMed
199. Lanner F, Lee KL, Sohl M, Holmborn K, Yang H, Wilbertz J, Poellinger L, Rossant J, Farnebo F. Heparan sulfation-dependent fibroblast growth factor signaling maintains embryonic stem cells primed for differentiation in a heterogeneous state. Stem Cells 2009; 28: 191–200.10.1002/stem.265Suche in Google Scholar PubMed
200. Morimoto-Tomita M, Uchimura K, Werb Z, Hemmerich S, Rosen S. Cloning and characterization of two extracellular heparin-degrading endosulfatases in mice and humans. J Biol Chem 2002; 277: 49175–85.10.1074/jbc.M205131200Suche in Google Scholar PubMed PubMed Central
201. Dhoot GK, Gustafsson MK, Ai X, Sun W, Standiford DM, Emerson Jr CP. Regulation of Wnt signaling and embryo patterning by an extracellular sulfatase. Sci STKE 2001; 293: 1663–6.10.1126/science.293.5535.1663Suche in Google Scholar PubMed
202. Ai X, Do AT, Lozynska O, Kusche-Gullberg M, Lindahl U, Emerson Jr CP. QSulf1 remodels the 6-O sulfation states of cell surface heparan sulfate proteoglycans to promote Wnt signaling. J Cell Biol 2003; 162: 341–51.10.1083/jcb.200212083Suche in Google Scholar PubMed PubMed Central
203. Rhiner C, Gysi S, Fröhli E, Hengartner MO, Hajnal A. Syndecan regulates cell migration and axon guidance in C. elegans. Development 2005; 132: 4621–33.10.1242/dev.02042Suche in Google Scholar PubMed
204. Kitagawa H, Fujita M, Ito N, Sugahara K. Molecular cloning and expression of a novel chondroitin 6-O-sulfotransferase. J Biol Chem 2000; 275: 21075–80.10.1074/jbc.M002101200Suche in Google Scholar PubMed
205. Yamauchi S, Kurosu A, Hitosugi M, Nagai T, Oohira A, Tokudome S. Differential gene expression of multiple chondroitin sulfate modification enzymes among neural stem cells, neurons and astrocytes. Neurosci Lett 2011; 493: 107–11.10.1016/j.neulet.2011.02.019Suche in Google Scholar PubMed
206. Kobayashi M, Sugumaran G, Liu J, Shworak N, Silbert J, Rosenberg R. Molecular cloning and characterization of a human uronyl 2-sulfotransferase that sulfates iduronyl and glucuronyl residues in dermatan/chondroitin sulfate. J Biol Chem 1999; 274: 10474–80.10.1074/jbc.274.15.10474Suche in Google Scholar PubMed
207. Fukuta M, Uchimura K, Nakashima K, Kato M, Kimata K, Shinomura T, Habuchi O. Molecular cloning and expression of chick chondrocyte chondroitin 6-sulfotransferase. J Biol Chem 1995; 270: 18575.10.1074/jbc.270.31.18575Suche in Google Scholar PubMed
208. Pacheco B, Malmström A, Maccarana M. Two dermatan sulfate epimerases form iduronic acid domains in dermatan sulfate. J Biol Chem 2009; 284: 9788.10.1074/jbc.M809339200Suche in Google Scholar PubMed PubMed Central
209. Dündar M, Müller T, Zhang Q, Pan J, Steinmann B, Vodopiutz J, Gruber R, Sonoda T, Krabichler B, Utermann G. Loss of dermatan-4-sulfotransferase 1 function results in adducted thumb-clubfoot syndrome. Am J Hum Genet 2009; 85: 873–82.10.1016/j.ajhg.2009.11.010Suche in Google Scholar PubMed PubMed Central
210. Bian S, Akyüz N, Bernreuther C, Loers G, Laczynska E, Jakovcevski I, Schachner M. Dermatan sulfotransferase Chst14/D4st1, but not chondroitin sulfotransferase Chst11/C4st1, regulates proliferation and neurogenesis of neural progenitor cells. J Cell Sci 2011; 124: 4051–63.10.1242/jcs.088120Suche in Google Scholar PubMed
211. Mühleisen TW, Mattheisen M, Strohmaier J, Degenhardt F, Priebe L, Schultz CC, Breuer R, Meier S, Hoffmann P, Rivandeneira F. Association between schizophrenia and common variation in neurocan (NCAN), a genetic risk factor for bipolar disorder. Schizophr Res 2012; 138: 69–73.10.1016/j.schres.2012.03.007Suche in Google Scholar PubMed
212. Gill JL, Tsai KL, Krey C, Noorai RE, Vanbellinghen JF, Garosi LS, Shelton GD, Clark LA, Harvey RJ. A canine BCAN microdeletion associated with episodic falling syndrome. Neurobiol Dis 2012; 45: 130–6.10.1016/j.nbd.2011.07.014Suche in Google Scholar PubMed PubMed Central
213. Dudas B, Cornelli U, Lee J, Hejna M, Walzer M, Lorens S, Mervis R, Fareed J, Hanin I. Oral and subcutaneous administration of the glycosaminoglycan C3 attenuates Aβ (25–35)-induced abnormal τ protein immunoreactivity in rat brain. Neurobiol Aging 2002; 23: 97–104.10.1016/S0197-4580(01)00255-XSuche in Google Scholar
214. Dudas B, Lemes A, Cornelli U, Hanin I. Prospective role of glycosaminoglycans in apoptosis associated with neurodegenerative disorders. Adv Alzheimers Parkinsons Dis 2008: 57: 247–54.10.1007/978-0-387-72076-0_24Suche in Google Scholar
215. Snow AD, Sekiguchi R, Nochlin D, Fraser P, Kimata K, Mizutani A, Arai M, Schreier WA, Morgan DG. An important role of heparan sulfate proteoglycan (perlecan) in a model system for the deposition and persistence of fibrillar aβ-amyloid in rat brain. Neuron 1994; 12: 219–34.10.1016/0896-6273(94)90165-1Suche in Google Scholar
216. Snow A, Mar H, Nochlin D, Sekiguchi R, Kimata K, Koike Y, Wight T. Early accumulation of heparan sulfate in neurons and in the β-amyloid protein-containing lesions of Alzheimer’s disease and Down’s syndrome. Am J Pathol 1990; 137: 1253–70.Suche in Google Scholar
217. Snow A, Mar H, Nochlin D, Kresse H, Wight T. Peripheral distribution of dermatan sulfate proteoglycans (decorin) in amyloid-containing plaques and their presence in neurofibrillary tangles of Alzheimer’s disease. J Histochem Cytochem 1992; 40: 105–13.10.1177/40.1.1370306Suche in Google Scholar
218. Dewitt DA, Richey P, Praprotnik D, Silver J, Perry G. Chondroitin sulfate proteoglycans are a common component of neuronal inclusions and astrocytic reaction in neurodegenerative diseases. Brain Res 1994; 656: 205–9.10.1016/0006-8993(94)91386-2Suche in Google Scholar
219. Shioi J, Pangalos MN, Ripellino JA, Vassilacopoulou D, Mytilineou C, Margolis RU, Robakis NK. The Alzheimer amyloid precursor proteoglycan (appican) is present in brain and is produced by astrocytes but not by neurons in primary neural cultures. J Biol Chem 1995; 270: 11839–44.10.1074/jbc.270.20.11839Suche in Google Scholar
220. Griffith LS, Schmitz B. O-linked N-acetylglucosamine is upregulated in Alzheimer brains. Biochem Biophys Res Commun 1995; 213: 424–31.10.1006/bbrc.1995.2149Suche in Google Scholar
221. Arrasate M, Perez M, Valpuesta JM, Avila J. Role of glycosaminoglycans in determining the helicity of paired helical filaments. Am J Pathol 1997; 151: 1115.Suche in Google Scholar
222. Argyris EG, Acheampong E, Nunnari G, Mukhtar M, Williams KJ, Pomerantz RJ. Human immunodeficiency virus type 1 enters primary human brain microvascular endothelial cells by a mechanism involving cell surface proteoglycans independent of lipid rafts. J Virol 2003; 77: 12140–51.10.1128/JVI.77.22.12140-12151.2003Suche in Google Scholar
223. Belichenko PV, Miklossy J, Celio MR. HIV-I induced destruction of neocortical extracellular matrix components in AIDS victims. Neurobiol Dis 1997; 4: 301–10.10.1006/nbdi.1997.0143Suche in Google Scholar
224. Okamoto M, Sakiyama J, Mori S, Kurazono S, Usui S, Hasegawa M, Oohira A. Kainic acid-induced convulsions cause prolonged changes in the chondroitin sulfate proteoglycans neurocan and phosphacan in the limbic structures. Exp Neurol 2003; 184: 179–95.10.1016/S0014-4886(03)00251-6Suche in Google Scholar
225. Yuan W, Matthews R, Sandy J, Gottschall P. Association between protease-specific proteolytic cleavage of brevican and synaptic loss in the dentate gyrus of kainate-treated rats. Neuroscience 2002; 114: 1091–101.10.1016/S0306-4522(02)00347-0Suche in Google Scholar
226. Carmichael ST, Archibeque I, Luke L, Nolan T, Momiy J, Li S. Growth-associated gene expression after stroke: evidence for a growth-promoting region in peri-infarct cortex. Exp Neurol 2005; 193: 291–311.10.1016/j.expneurol.2005.01.004Suche in Google Scholar
227. Deguchi K, Takaishi M, Hayashi T, Oohira A, Nagotani S, Li F, Jin G, Nagano I, Shoji M, Miyazaki M. Expression of neurocan after transient middle cerebral artery occlusion in adult rat brain. Brain Res 2005; 1037: 194–9.10.1016/j.brainres.2004.12.016Suche in Google Scholar
228. Snow A, Wight T, Nochlin D, Koike Y, Kimata K, DeArmond S, Prusiner S. Immunolocalization of heparan sulfate proteoglycans to the prion protein amyloid plaques of Gerstmann-Straussler syndrome, Creutzfeldt-Jakob disease and scrapie. Lab Invest 1990; 63: 601.Suche in Google Scholar
229. Perry G, Richey P, Siedlak SL, Galloway P, Kawai M, Cras P. Basic fibroblast growth factor binds to filamentous inclusions of neurodegenerative diseases. Brain Res 1992; 579: 350.10.1016/0006-8993(92)90074-JSuche in Google Scholar
230. Paulus W, Baur I, Dours-Zimmermann MT, Zimmermann DR. Differential expression of versican isoforms in brain tumors. J Neuropathol Exp Neurol 1996; 55: 528.10.1097/00005072-199605000-00005Suche in Google Scholar
231. Pizzorusso T, Medini P, Landi S, Baldini S, Berardi N, Maffei L. Structural and functional recovery from early monocular deprivation in adult rats. Proc Natl Acad Sci USA 2006; 103: 8517–22.10.1073/pnas.0602657103Suche in Google Scholar
232. Vorobyov V, Kwok JCF, Fawcett JW, Sengpiel F. Effects of digesting chondroitin sulfate proteoglycans on plasticity in cat primary visual cortex. J Neurosci 2013; 33: 234–43.10.1523/JNEUROSCI.2283-12.2013Suche in Google Scholar
233. Odawara T, Iseki E, Li F, Kosaka K, Ikeda K. Heparan sulfate proteoglycans recognize ghost Pick bodies. Neurosci Lett 1998; 242: 120–2.10.1016/S0304-3940(98)00027-5Suche in Google Scholar
234. Merry CLR, Bullock SL, Swan DC, Backen AC, Lyon M, Beddington RSP, Wilson VA, Gallagher JT. The molecular phenotype of heparan sulfate in the Hs2st-/- mutant mouse. J Biol Chem 2001; 276: 35429–34.10.1074/jbc.M100379200Suche in Google Scholar PubMed
235. Allen B, Rapraeger A. Spatial and temporal expression of heparan sulfate in mouse development regulates FGF and FGF receptor assembly. J Cell Biol 2003; 163: 637–48.10.1083/jcb.200307053Suche in Google Scholar PubMed PubMed Central
236. Ronca F, Andersen J, Paech V, Margolis R. Characterization of Slit protein interactions with glypican-1. J Biol Chem 2001; 276: 29141–7.10.1074/jbc.M100240200Suche in Google Scholar
237. Turnbull J, Fernig D, Ke Y, Wilkinson M, Gallagher J. Identification of the basic fibroblast growth factor binding sequence in fibroblast heparan sulfate. J Biol Chem 1992; 267: 10337–41.10.1016/S0021-9258(19)50023-0Suche in Google Scholar
238. Shipp EL, Hsieh-Wilson LC. Profiling the sulfation specificities of glycosaminoglycan interactions with growth factors and chemotactic proteins using microarrays. Chem Biol 2007; 14: 195–208.10.1016/j.chembiol.2006.12.009Suche in Google Scholar PubMed
239. Ishai-Michaeli R, Svahn C, Weber M, Chajek-Shaul T, Korner G, Ekre H, Vlodavsky I. Importance of size and sulfation of heparin in release of basic fibroblast growth factor from the vascular endothelium and extracellular matrix. Biochemistry 1992; 31: 2080–8.10.1021/bi00122a027Suche in Google Scholar PubMed
240. Liu J, Chau CH, Liu H, Jang BR, Li X, Chan YS, Shum DKY. Upregulation of chondroitin 6-sulphotransferase-1 facilitates Schwann cell migration during axonal growth. J Cell Sci 2006; 119: 933–42.10.1242/jcs.02796Suche in Google Scholar PubMed
241. Tully S, Rawat M, Hsieh-Wilson L. Discovery of a TNF- antagonist using chondroitin sulfate microarrays. J Am Chem Soc 2006; 128: 7740–1.10.1021/ja061906tSuche in Google Scholar PubMed
242. Sugahara K, Yamada S. Structure and function of oversulfated chondroitin sulfate variants: unique sulfation patterns and neuroregulatory activities. Trends Glycosci Glycotechnol 2000; 12: 321–50.10.4052/tigg.12.321Suche in Google Scholar
243. Nadanaka S, Clement A, Masayama K, Faissner A, Sugahara K. Characteristic hexasaccharide sequences in octasaccharides derived from shark cartilage chondroitin sulfate D with a neurite outgrowth promoting activity. J Biol Chem 1998; 273: 3296–307.10.1074/jbc.273.6.3296Suche in Google Scholar PubMed
244. Bao X, Muramatsu T, Sugahara K. Demonstration of the pleiotrophin-binding oligosaccharide sequences isolated from chondroitin sulfate/dermatan sulfate hybrid chains of embryonic pig brains. J Biol Chem 2005; 280: 35318–28.10.1074/jbc.M507304200Suche in Google Scholar PubMed
©2013 by Walter de Gruyter Berlin Boston
Artikel in diesem Heft
- Masthead
- Masthead
- Reviews
- Free radical-mediated cytosine C-5 methylation triggers epigenetic changes during carcinogenesis
- Neurokinin receptors in the gastrointestinal muscle wall: cell distribution and possible roles
- Sugar glues for broken neurons
- Programmed cell death with a necrotic-like phenotype
- The nucleolus: a raft adrift in the nuclear sea or the keystone in nuclear structure?
- Papain-like peptidases: structure, function, and evolution
- Short Conceptual Overview
- Alternative genetic code for amino acids and transfer RNA revisited
Artikel in diesem Heft
- Masthead
- Masthead
- Reviews
- Free radical-mediated cytosine C-5 methylation triggers epigenetic changes during carcinogenesis
- Neurokinin receptors in the gastrointestinal muscle wall: cell distribution and possible roles
- Sugar glues for broken neurons
- Programmed cell death with a necrotic-like phenotype
- The nucleolus: a raft adrift in the nuclear sea or the keystone in nuclear structure?
- Papain-like peptidases: structure, function, and evolution
- Short Conceptual Overview
- Alternative genetic code for amino acids and transfer RNA revisited