Dysport® for the treatment of myofascial back pain: Results from an open-label, Phase II, randomized, multicenter, dose-ranging study
-
Gerhard H.H. Müller-Schwefe
 and
Michael A. Überall
and
Michael A. Überall

Abstract
Background and purpose
Botulinum toxin type A (BoNT-A) has antinociceptive and muscle-relaxant properties. The objectives of this study were to investigate the efficacy and safety of a single BoNT-A (Dysport®) treatment in myofascial back pain.
Methods
In this randomized, open-label, multicenter study, adults with myofascial lower back pain received Dysport® injections at four trigger points (60,80 or 120 units per injection point). Patients were followed for 12 weeks. The a priori primary endpoint was a pooled evaluation, at Week 6, of seven measures of efficacy, including pain intensity (patient diary), modified Pain Disability Index (PDI) score, use of interfering concomitant analgesics, and patient-rated global efficacy. Optional assessments of pressure thresholds and tissue compliance were conducted. Safety was also assessed.
Results
A total of 202 patients were randomized to treatment and 189 patients received a low (n = 57), medium (n = 57), or high (n = 75) total dose of Dysport® at 34 centers in Germany between October 2002 and October 2003. All treated patients were included in the safety population; 8 patients were excluded from the intention-to-treat population. Patients had moderate to severe pain at baseline. At baseline, 120 patients were receiving concomitant analgesic therapy; 6.7%, 74.2% and 19.2% were considered to cause mild, moderate and severe interference with pain measurements, respectively. There was no difference between doses for the a priori combined primary endpoint. Patient-reported pain intensity scores at rest and on movement decreased significantly after treatment for all groups combined (p < 0.0001 at all visits). At Week 6, reductions in pain intensity at rest were 29%, 19% and 26% for the low-, medium- and high-dose groups, respectively; reductions in pain intensity on movement were 27%, 18% and 26%, respectively. Overall, patients who reported pain intensity reductions at Week 6 were evident within 3 weeks of treatment and were maintained for the 12 weeks of the study. In the total population, significant decreases in mean PDI sum scores from baseline were observed from Week 3 and were maintained through to the end of treatment (Week 12); no differences between the dose groups were observed. Pressure thresholds and tissue compliance also increased during the study. Adverse events were generally as expected for BoNT-A; the majority were mild or moderate in severity.
Conclusions
Dysport® treatment was associated with reductions in myofascial back pain and was well tolerated. Nodose-response relationship was observed; treatment with Dysport® using a four-trigger-point injection protocol at 60 units per trigger point was associated with a clinically relevant and statistically significant improvement in pain and pain-related disability; there was no additional benefit from the higher doses.
Implications
Our findings are limited by the lack of a control group and further research is warranted to confirm the value of Dysport® for the treatment of myofascial back pain and confirm the optimum dosing in this indication.
1 Introduction
Myofascial pain syndrome is a chronic musculoskeletal disorder that is characterized by muscles in the shortened or contracted state with increased tone and stiffness [1,2,3]. These muscles contain tender, firm nodules called trigger points, which, on stimulation, transfer pain to surrounding areas [1,2,3]. The pathogenesis of myofascial pain syndrome is unproven but several mechanisms have been proposed, such as muscle spindle hyperactivity or sustained depolarization of post-junctional muscle cells due to excessive acetylcholine release [3,4,5]. Various pharmacological and physical therapies are available for the treatment of myofascial pain syndrome, but the effects of these agents may only be short term (e.g. with therapeutic injections), their efficacies can be unreliable (e.g. non-steroidal anti-inflammatory drugs [NSAIDs], therapeutic injections) and many are limited by toxicities (e.g. NSAIDs, tricyclic antidepressants) [3,6,7,8].
Botulinum toxin type A (BoNT-A) is a neurotoxin complex that is currently used to treat various disorders involving muscle hyperactivity, including focal spasticity, blepharospasm, spasmodic torticollis, and hemifacial spasm. Importantly, BoNT-A also has antinociceptive and muscle-relaxant properties and has been used successfully to treat chronic pain [9,10,11]. Furthermore, evidence suggests that BoNT-A may modulate the activity of muscle spindles [12,13], which are thought to play a role in the pathogenesis of myofascial pain syndrome [3].
Several studies have investigated the use of BoNT-A injections as a potential new treatment option for myofascial pain syndrome and most have shown a positive effect on treatment of pain [14,15,16,17,18,19,20,21,22,23,24,25]. However, the majority of these studies were case studies or small controlled clinical trials [15,18,20,22,24,25] and the two larger controlled clinical trials produced conflicting findings [16,17]. Göbel et al. conducted a large, double-blind, placebo-controlled study to evaluate the efficacy and tolerability of Dysport® in 145 patients with moderate-to-severe myofascial pain in cervical and/or shoulder muscles [17]. In this study, a significant improvement in pain levels was reported 4-6 weeks after injections of 400 units of Dysport® into the 10 most tender individual trigger points (40 units per trigger point). In contrast to the findings of Göbel et al., Ferrante et al. [16] conducted a large, doubleblind, placebo-controlled study in 132 patients with myofascial pain and did not find a significant benefit with BoNT-A (Botox®) treatment. It has, however, been suggested that the low disease severity (patients with more than five active trigger points were excluded) of patients in this study may have influenced the findings [17].
Clostridium botulinum type A toxin-haemagglutinin complex (Dysport®, Ipsen Ltd, Slough, UK) is a highly purified and highly potent form of BoNT-A. Dysport® combines a well-established safety profile with excellent clinical efficacy in a wide range of neuromuscular disorders [26,27,28,29,30,31,32,33]. Although the efficacy of Dysport® for the treatment of myofascial pain has been documented in a prior placebo-controlled trial [17], the effective dose range of Dysport® for myofascial low back pain has not been established. Therefore, the aim of this pilot study was to investigate the efficacy and tolerability of a range of doses of Dysport® in a large number of patients with myofascial low back pain, in order to estimate the lowest effective dose. It should be noted that different BoNT-A products are available and that the units of these different preparations are not equivalent; in our study, dose specification of units refers exclusively to Dysport® and cannot be applied to other BoNT-A treatments.
2 Methods
2.1 Study design
This was an investigator-initiated, prospective, randomized, open-label, multicenter, Phase II study. The aims of the study were to investigate the efficacy and tolerability of a single Dysport® treatment in patients with myofascial back pain in the lower back, and to optimize the therapeutic dose of Dysport®. After a screening visit (Week –2), patients received treatment at Week 0 and were then assessed for 12 weeks, with visits scheduled at Weeks 3, 6 and 12 (±3 days). The study protocol was approved by the local ethics committee or institutional review board, and conducted according to the principles of good clinical practice and the Declaration of Helsinki. All patients gave informed consent prior to the study.
2.2 Patients
Adults aged at least 18 years were enrolled in the study if they had myofascial back pain affecting muscles of the lower back (pain in the region from the thoracic vertebra 7 downwards, including the gluteal muscles). Eligible patients were required to have experienced myofascial back pain for more than 3 weeks; have at least four trigger points (one-sided or two-sided, in at least two different muscles); a neuro-orthopedic basic diagnosis of spine involvement to rule out evidence of fractures, blocking of vertebral bodies, radicular irritation syndrome, or other noticeable problems. Patients were also required to have pain intensity at rest or on movement of at least 2 at baseline, rated using the 5-category Verbal Rating Scale (0 = no pain; 1 = mild pain; 2 = moderate pain; 3 = severe pain; and 4 = very severe pain).
Patients were excluded from the study if they had specific back pain (e.g. tumours, radicular syndromes, spondylolistheses, nerve root irritations due to a discus prolapse, or due to inflammatory processes [hip arthrosis, spondyloptosis, osteomalacia, acute joint inflammations]); back pain in need of another causal therapy; or evidence of specific diseases of the musculoskeletal system (other than myofascial back pain) or diseases of neuromuscular transmission. Other major exclusion criteria included a history of surgery on the spine, fibromyalgia, pain as a primary expression of depression, or chronic respiratory ailments; prior treatment with BoNT-A; a known allergy or antibodies to BoNT-A; bleeding tendency at the time of injection (due to congenital hemorrhagic diathesis or due to drugs); pregnancy, lactation or the lack of a reliable contraceptive method in women of childbearing potential; any severe concomitant disease; alcohol, medication, or other drug abuse; and an inability to work for longer than 6 months.
2.3 Interventions
Patients were randomized to receive a total dose of 240, 320 or 480 units of Dysport® (500 units in 2.5 ml of 0.9% NaCl; Ipsen Pharma GmbH, Ettlingen, Germany). Randomization was carried out according to the random permuted block design, using a block size of 3 (RANCODE Professional Version, IDV, Gauting, Munich, Germany), independent of study center. Treatment was administered by injections at four trigger points (60, 80 or 120 units per injection point, respectively) using a 1-ml tuberculin syringe with a 27-gauge needle. Injections were administered with a safety margin of at least 3 cm lateral to the median line of the spine. The four most troublesome trigger points, in at least two different muscles on one or both sides of the body, were treated. The injection points were documented in a trigger-point scheme (sequential numbering from cranial to caudal in the case of several trigger points in one muscle).
2.4 Concomitant therapy
Concomitant physiotherapy during the study was permissible. However, procedures that could influence the trigger points were not permitted between Weeks –2 and 6. Physiotherapy and manual therapy for the treatment of vertebral blockage were allowed during the study.
Concomitant therapy with aminoglycosides, muscle relaxants, benzodiazepines, or injections of local anaesthetics or corticoids in segments treated with Dysport® was not permitted during the study. Concomitant therapy with anticoagulants or acetylsalicylic acid (>100 mg/day) was not permitted between Weeks –2 and 1, and treatment with opioids or the muscle relaxant tolperisone was not permitted between Weeks –2 and 6. Stable, ongoing therapy with antidepressants was permitted during the study.
Permitted concomitant rescue analgesics included cyclooxygenase (COX) 2 inhibitors, NSAIDs with gastroprotection, and flupirtine (a centrally acting nonopioid, nonsteroidal analgesic with muscle-relaxant properties) if it had already been taken before the study. Opioid analgesics, flupirtine and tolperisone were also permitted from Week 6 onwards.
2.5 Efficacy assessments
Efficacy was assessed using a patient-completed pain diary, the patient-completed modified Pain Disability Index (PDI) [34,35,36], as well as patient and investigator global assessments of efficacy.
Patients used their pain diaries (provided at screening) to assess their average pain at rest and on motion, and their maximum pain at rest and on motion, using the 5-category Verbal Rating Scale, on a daily basis. Patients also used their diaries to record their duration of pain (h).
At each visit, patients also rated their PDI according to the effect (disturbance) of their pain on the seven domains of daily life (family/home responsibilities, recreation, social activity, occupation, sexual behavior, self-supply, and life-support activity) using an 11-point scale (0 = no disturbance; 10 = maximum disturbance). The PDI was calculated as the mean sum of these scores (ranging from 0 to 70). The patient’s pain diary was checked by the investigator at each visit and the patient’s modified PDI score recorded [34,35,36].
Patients and investigators also provided a global assessment of efficacy at Weeks 3, 6 and 12 using a 5-point scale (1 = very good; 2 = good; 3 = moderate; 4 = unsatisfactory; and 5 = poor). At each visit, the physician assessed the ability of the patient to work.
Additional efficacy evaluations included assessments at each visit of the minimum pressure threshold for the sensation of pain and the maximum pressure threshold for just-tolerable pain (assessed using a Pressure Threshold Meter with two repeated measurements made at each of the four trigger points) [37,38]. Tissue compliance (two repeated measurements at the four trigger points made using a Tissue Compliance Meter) was also included as a facultative assessment [39]. Measurement of tissue compliance allows quantitative and objective recording of soft-tissue consistency and can be used to document the effects of treatment of trigger points.
2.6 Safety assessments
A physical examination was conducted and vital signs recorded at each visit. Adverse events (AEs; coded using the World Health Organization-Adverse Reaction Terminology dictionary) were documented at each visit, noting AE type, duration, frequency, intensity, seriousness, relationship to study medication, measures taken, and outcome. Global tolerability was assessed at Weeks 3, 6 and 12 by the physician and the patient using a 5-point scale (1 = very good, 2 = good, 3 = moderate, 4 = unsatisfactory, and 5 = poor).
2.7 Statistical methods
A total sample size of 180 patients was required to detect a significant difference (p = 0.05, two-sided) with 90% power. This calculation was based on using at least five efficacy criteria and assuming an effect size of 0.64 with respect to the reference treatment (320 units of Dysport®). The effect size of 0.64 was derived from the relevant benchmarks for the Mann-Whitney estimator as follows: 0.5 equality, 0.44/0.56 small, 0.36/0.64 medium-sized, 0.29/0.71 large group differences [40]. With a single criterion for the two group comparisons, a medium-sized (relevant) superiority (Mann-Whitney = 0.64) with respect to the reference treatment (320 U Dysport) was assumed.
Data clearing and finalization of the statistical analysis plan were carried out under blinded conditions. This allowed for a fair review of the data and an updated and more detailed explanation of how the statistical analyses of the primary and secondary variables should be executed. In addition, the blind review enabled a quality check on all entries in the patients’ diaries, checking for plausibility and consistency. In doing so, a large day-to-day variability in pain levels was detected.
In order to achieve stable results in the primary efficacy outcomes, the statistical analysis plan was modified, a priori, so that patients’ pain scores were analyzed as weekly means of the daily entries.
The intent-to-treat (ITT) population was the population of primary interest and included all patients who received at least one dose of study medication, attended at least one follow-up visit, and had at least one follow-up week in the patient diary. Missing data in the ITT analysis were input using a last observation carried forward (LOCF) approach; when values for a particular pain dimension during a particular week were missing, the mean score of the available entries for that pain dimension at that week was used to replace the missing values(s). Patients who discontinued at baseline were excluded from all analyses. The per protocol (PP) population included patients in the ITT population who completed the study in accordance with the study protocol, but did not exclude patients with protocol deviations related to lack of efficacy (e.g. discontinuation, rescue medication) or lack of tolerability.
Summary statistics are presented for demographics and efficacy data. The Mann-Whitney statistic was utilized to test the degree of homogeneity (as represented by 90% confidence intervals). In the case of heterogeneities in the ITT population, stratified analyses were conducted using the Wilcoxon test with Cochran-Mantel-Haenszel pooling. Statistical non-parametric within-group comparisons (Wilcoxon-Pratt test, one-sided) were performed for the daily average pain at rest and the daily average pain on movement up to Week 6 and through study endpoint (Week 12). The time taken to a reduction of pain intensity score was analyzed using a Kaplan–Meier plot with the treatment of censored values (patients who did not reduce their score by the specified amount within 12 weeks were entered manually as 13 weeks). For the patient global assessment of efficacy after 6 weeks, the worst rank score technique was used to replace missing values (no global assessment available) for patients with premature discontinuation due to lack of efficacy.
The seven primary efficacy criteria (as specified in the amended final statistical analysis plan) were: the percentage change from baseline (Week –1 to Week 0) in median pain intensity at rest and on movement at Week 6 (days 35–41); the time to the reduction of the weekly medians of the daily average pain at rest and pain on movement by at least 1 point; percentage change from baseline (Week –2 to Week 0) of the median PDI at Week 6; the change from baseline (Week –1 to Week 0) at Week 6 with respect to the interfering class of concomitant analgesic therapy; and the patient’s global assessment of efficacy after 6 weeks. The a priori primary efficacy endpoint was defined as a pooled evaluation of these seven primary efficacy criteria by means of the summarizing Wei-Lachin directional procedure [41], and analyzed according to pre-post comparisons of efficacy criteria (dosing groups were pooled in the event of small differences between the three doses). If the result of the global test was significant, individual criteria were tested using the modified Bonferroni procedure.
As patients were permitted to receive concomitant analgesic therapy during the study, exploratory sensitivity analyses were conducted to compensate for bias that may have resulted from concomitant medication use. If concomitant analgesic therapy was received within a particular week, then the mean values of the daily pain scores (average pain at rest, average pain on movement, maximum pain at rest, maximum pain on movement) were adjusted for concomitant analgesic therapy based on the strength of the analgesic received as follows: +0.5 points in case of “mild interference”, +1 point in case of “moderate interference” and +1.5 points in case of “severe interference”.
In addition to the main analyses, an exploratory post-hoc analysis was performed, in which changes in mean PDI sum scores from baseline by study week were evaluated by analysis of variance (ANOVA), with a factor for time (baseline vs. study week).
The safety population included all patients who received at least one dose of study medication and attended at least one follow-up visit. Data from this population were analyzed using descriptive statistics.
3 Results
Outpatients were enrolled between 15 October 2002 and 9 October 2003 at 34 participating pain clinics and hospitals in Germany (all centers belong to the Schmerztherapeutisches Kolloquium [STK] pain treatment colloquium). The last patient completed the study in January 2004.
3.1 Patient disposition and baseline characteristics
A total of 202 patients were randomized and 189 patients received a low (n = 57), medium (n = 57) or high (n = 75) total dose of Dysport® (240,320 and 480 units of Dysport®, respectively) (Fig. 1).
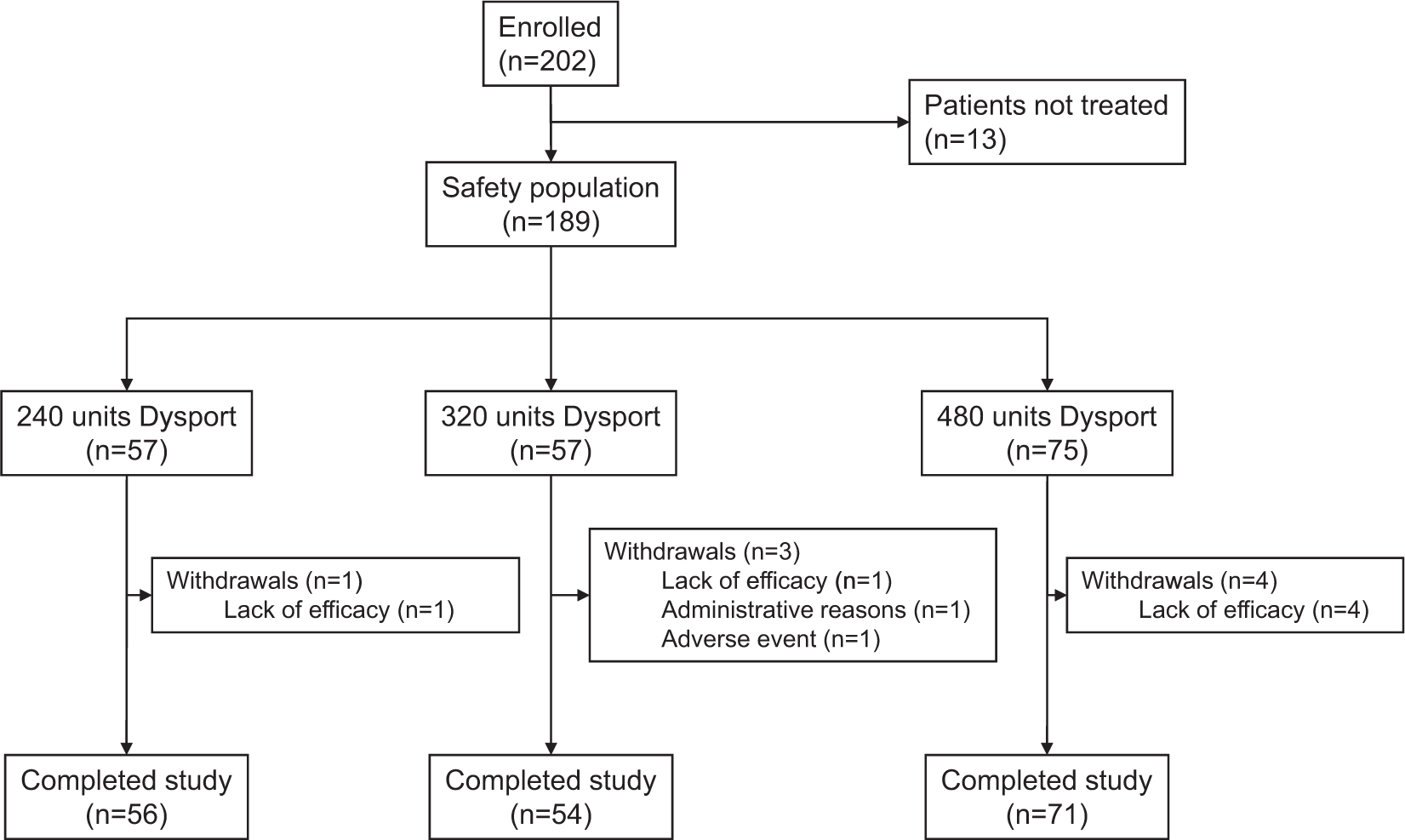
Patient flow through the study.
Overall, 189 and 181 patients were included in the safety and ITT populations, respectively. Eight patients were excluded from the ITT population; seven patients as they had no follow-up entry in their patient diary and one patient as they did not attend any follow-up visits. The PP population comprised 138 patients (76.2% of the ITT population). Of the 43 patients in the ITT population who were excluded from the PP population, two were excluded due to premature discontinuation unrelated to efficacy (one due to an AE and one because of administrative issues), and the remaining 41 patients were excluded because they were found not to meet the inclusion/exclusion criteria during the blind review (low mean pain score of less than 2 at baseline, severe interfering concomitant analgesic at baseline, indication for other disease of the movement system, previous treatment with BoNT-A).
Demographic and clinical characteristics at baseline were generally similar among treatment groups (Table 1). There were differences in body mass index and body weight between the low-and high-dose groups, but the stratified analysis was adjusted for this in efficacy calculations.
Baseline demographic and clinical data for the intent-to-treat population.
| Characteristic | Treatment groups |
||
|---|---|---|---|
| Low-dose (240 units Dysport®) (n = 56) |
Medium-dose (320 units Dysport®) (n = 54) |
High-dose (480 units Dysport®) (n = 71) |
|
| Age (years) | |||
| Mean (SD) | 55.2(11.32) | 55.2(12.76) | 52.7(13.18) |
| Median (range) | 54.0(34,81) | 53.5 (30, 79) | 53.0(26, 80) |
| Women, number (%) | 40(71) | 37 (69) | 46 (65) |
| Body mass index (kg/m2) | |||
| Mean (SD) | 25.6 (3.71) | 25.9 (4.98) | 27.3(4.71) |
| Median (range) | 25.0(19,35) | 24.7(18,41) | 26.5(18,39) |
| Daily pain score at rest | |||
| Mean (SD) | 2.2 (0.77) | 2.3 (0.79) | 2.2 (0.75) |
| Median (range) | 2.0 (1,4) | 2.3 (0, 4) | 2.1 (1,4) |
| Daily pain score on movement | |||
| Mean (SD) | 2.6 (0.78) | 2.5 (0.78) | 2.6 (0.67) |
| Median (range) | 2.7 (1,4) | 2.3 (1,4) | 2.6 (1,4) |
| Modified PDI score | |||
| Mean (SD) | 33.9[a] (12.76) | 33.6[b] (14.10) | 32.9[c] (13.40) |
| Median (range) | 33.0[a] (10,63) | 35.0[b] (4, 70) | 33.0[c] (4, 70) |
-
PDI, Pain Disability Index; SD, standard deviation.
-
Daily pain score at rest or on movement was rated from 0 (no pain) to 4 (very severe pain).
-
The seven items of the modified PDI score were assessed using an 11-point scale from 0 (no disturbance) to 10 (maximum disturbance). The range of the PDI score was therefore 0–70, after the scores were tallied.
3.2 Efficacy
3.2.1 Primary efficacy endpoints
Results for the seven primary efficacy criteria are shown in Table 2 (ITT population). The primary endpoint, pooled analysis of the seven global efficacy criteria, showed no significant difference among the three dose groups (p > 0.1 in the ITT and PP populations). The sensitivity analyses with score correction for interfering concomitant analgesic therapies in the ITT and PP populations were not indicative of greater efficacy with the higher dose than with the low dose.
Overall, the percentage change in weekly median pain intensity scores at rest and on movement decreased after injection in all treatment groups and in all groups combined (Fig. 2). Correcting for interfering concomitant analgesic therapy, the median (range) percentage decrease in weekly median pain intensity score at rest at Week 6 was –17.9% (–100.0% to +425.0%) and on movement –17.6% (–100.0% to +100.0%).
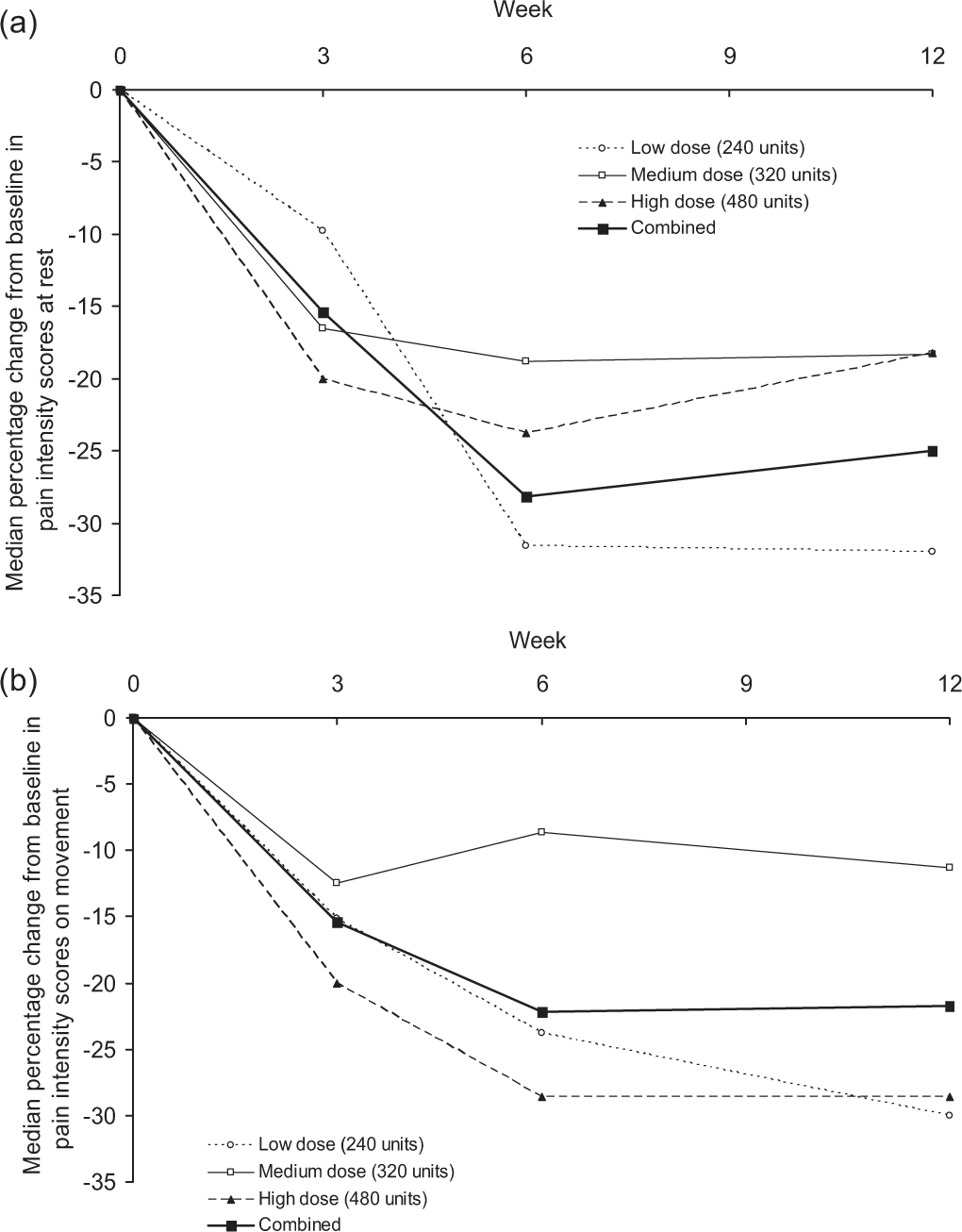
Median percentage change from baseline in pain intensity scores at rest (a) and on movement (b) for all treatment groups combined at Weeks 3, 6, and 12 (intent-to-treat population, n = 181).
For the three groups combined, the median time until reduction of the weekly mean of pain intensity scores by at least one point was similar for pain at rest and pain on movement (Table 2). Correcting for interfering analgesic therapies did not markedly influence the median time to pain reduction by at least one point (increased by 1 week to 8 weeks for pain at rest; decreased by 1 week to 7 weeks for pain on movement).
Results for the seven primary efficacy criteria (intent-to-treat population).
| Efficacy variable | Treatment groups |
|||
|---|---|---|---|---|
| Low-dose (240 units Dysport®) (n = 56) | Medium-dose (320 units Dysport®) (n = 54) | High-dose (480 units Dysport®) (n = 71) | Combined (n =181) | |
| Change from baseline to Week 6 in pain intensity scores at rest (%) | ||||
| Mean (SD) | –28.5 (46.67) | –19.1 (54.43) | –26.2(40.78) | –24.8 (46.90) |
| Median (range) | –31.6 (–100,180) | –18.8 (–100, 250) | –23.8 (–100, 91) | –28.2 (–100, 250) |
| Change from baseline to Week 6 in pain intensity scores on movement (%) | ||||
| Mean (SD) | –27.4 (35.92) | –17.5 (49.63) | –25.8(37.81) | –23.9 (41.15) |
| Median (range) | –23.8 (–100, 62) | –8.6 (–100, 200) | –28.6 (–100, 91) | –22.2 (–100, 200) |
| Time to reduction in pain intensity score at rest, weeks[f] | ||||
| Mean (SD) | 7.8 (4.86) | 8.0 (5.22) | 8.1 (5.19) | 8.0 (5.08) |
| Median (range) | 6.5 (1, 13) | 10.0(1, 13) | 13.0(1,13) | 7.0(1,13) |
| Time to reduction in pain intensity score on movement, weeks[f] | ||||
| Mean (SD) | 7.8 (4.83) | 8.4 (5.21) | 7.4(5.17) | 7.8 (5.07) |
| Median (range) | 7.0 (1,13) | 13.0(1, 13) | 6.0(1,13) | 8.0(1,13) |
| Change from baseline to Week 6 in PDI score (%) | ||||
| Mean (SD) | –23.4 (34.67)[a] | –16.9 (48.55)[b] | –20.9 (79.80)[c] | –20.4 (59.54)[d] |
| Median (range) | –28.0 (–100, 80)[a] | –11.9 (–98,182)[b] | –25.0 (–98, 540)[c] | –23.4 (–100, 540)[d] |
| Patients with a change6"[g] in class of interfering concomitant analgesic therapy (n (%)) | ||||
| Increase | 2(3.6) | 6(11.1) | 8 (11.3) | 16(8.8) |
| Unchanged | 51 (91.1) | 46(85.2) | 58(81.7) | 155(85.6) |
| Decrease | 3 (5.4) | 2(3.7) | 5(7.0) | 10(5.5) |
| Global assessment of efficacy by patient at Week 6 (n (%)) | ||||
| Very good or good | 32(57.1) | 25 (47.2)[b] | 30(42.3) | 87 (48.3)[e] |
| Moderate | 11(19.6) | 8 (15.1)[b] | 25 (35.2) | 44 (24.4)[e] |
| Unsatisfactory or poor | 13 (23.2) | 20 (37.7)[b] | 16(22.5) | 49 (27.2)[e] |
| Summarizing Wei–Lachin analysis7[h] | ||||
| Low vs. high | 0.483 | |||
| Low vs. medium | 0.458 | |||
| Medium vs. high | 0.525 | |||
-
PDI, Pain Disability Index.
At Week 6, the median percent change in PDI scores from baseline was –23.4% for the combined groups and was similar in all treatment groups (Table 2).
Of 120 patients who documented the use of concomitant analgesic therapies at baseline, 8/120 (6.7%) were receiving analgesics considered to cause mild interference, 89/120 (74.2%) moderate interference, and 23/120 (19.2%) severe interference with pain measurements. For the majority of patients (155/181; 85.6%), the class of interfering concomitant analgesic therapy did not change between baseline and Week 6 (Table 2).
Global efficacy was rated as ‘good’ or ‘very good’ by 57.1% of patients in the low-dose group, 47.2% of patients in the medium-dose group, and 42.3% of patients in the high-dose group at Week 6 (Table 2).
3.2.2 Pain Disability Index
In an exploratory analysis, all three dose groups showed significant decreases in mean PDI sum scores from baseline to Week 6 (data not shown). As for all other parameters under investigation, there was no difference between the three dose groups. In the total population (Fig. 3), significant decreases in mean PDI sum scores from baseline were observed from Week 3 and were maintained through to the end of treatment (Week 12), as shown by the 95% confidence intervals.
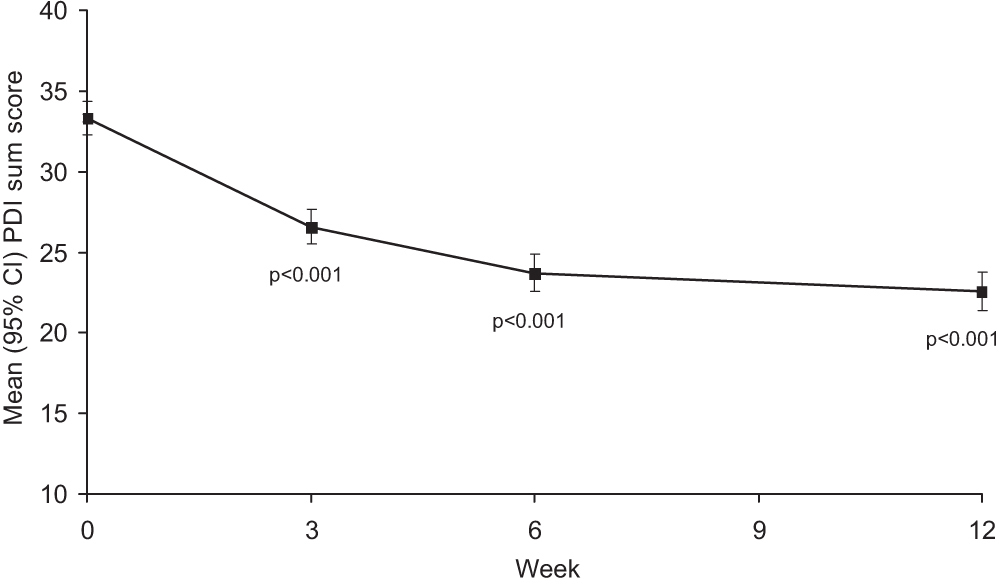
Change in mean (95% CI) PDI sum score from baseline to end of treatment for all treatment groups combined (intent-to-treat population). PDI, Pain Disability Index.
3.2.3 Pain pressure thresholds and tissue compliance
Both the minimum and maximum pressure thresholds increased after treatment, although the improvement was not statistically significant (Fig. 4). For all groups combined, mean (SD) tissue compliance increased from 11.80 (8.345) mm/3 kg at baseline to 12.79 (8.726) mm/3 kg at Week 6 and 13.28 (9.203) mm/3 kg at Week 12, although improvements were small and not statistically significant.
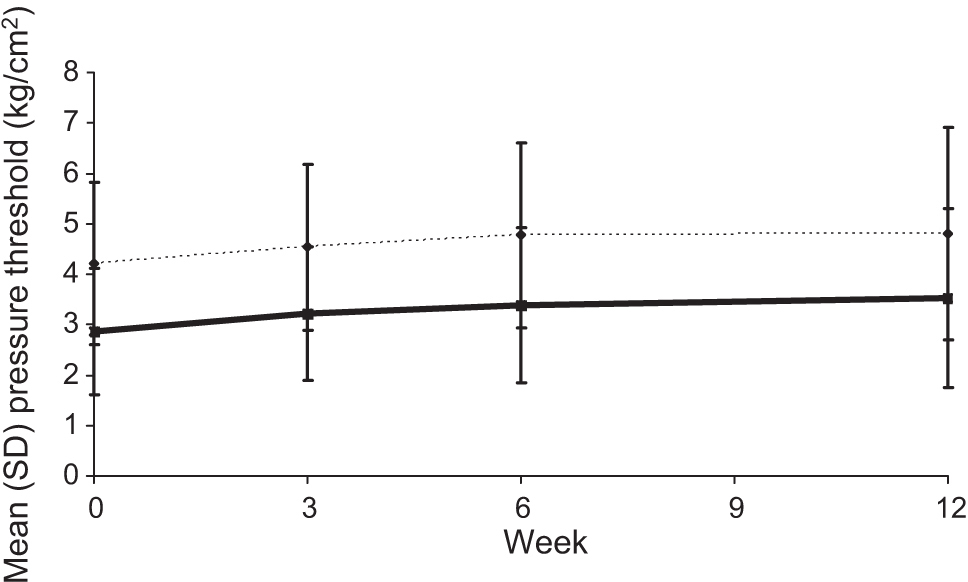
Minimum and maximum pressure thresholds at Weeks 0–12 for all treatment groups combined. n = 144 atWeek0, n = 137 atWeek3, n = 138 atWeek6, and n = 138 at Week 12.
3.3 Safety
In the post-treatment period, 89 AEs were reported in 60/189 (31.7%) patients in the safety population. The proportion of patients experiencing at least one AE was higher in the medium-dose group (21/57, 36.8%) than in the low-dose (16/57, 28.1%) and high-dose groups (23/75, 30.7%). The most frequently reported AE was influenza-like symptoms, which occurred in eight patients (Table 3). The majority of AEs were mild or moderate in severity (56/85, 65.9%; data not available for four AEs). A total of 16 AEs (18% of all reported AEs) in 13 patients (7%) were considered possibly or probably related to treatment and included back pain, dizziness, eye irritation, headache, infection, influenza-like symptoms, ischial neuralgia, lumbo-sacral pain (n = 2), nausea, pain, pain legs, photophobia, tiredness, vision blurred and vomiting. The majority of events were considered not or unlikely to be related to treatment (73/89; 82.0%). No clear pattern suggesting a dose effect on AE intensity or relation to treatment was evident; only one of the four cases of influenza-like symptoms in the high-dose group was considered possibly related to study medication.
Treatment–emergent adverse events (reported in ≥2 patients during the study; safety population; listed in order of decreasing frequency for all groups combined).
| Adverse event (WHO preferred term) | Treatment groups |
|||
|---|---|---|---|---|
| Low–dose (240 units Dysport®) (n = 57) | Medium–dose (320 units Dysport®) (n = 57) | High–dose (480 units Dysport®) (n = 75) | All (n = 189) | |
| Total | 12 | 21 | 12 | 45 |
| Influenza–like symptoms | 1 | 3 | 4 | 8 |
| Inflicted injury | 2 | 4 | 6 | |
| Arthrosis | 2 | 3 | 5 | |
| Back pain | 2 | 2 | 4 | |
| Lumbosacral pain | 3 | 3 | ||
| Pain neck/shoulder | 1 | 1 | 1 | 3 |
| Bronchitis | 2 | 2 | ||
| Cystitis | 2 | 2 | ||
| Head pain | 2 | 2 | ||
| Ischial neuralgia | 2 | 2 | ||
| Joint ache | 1 | 1 | 2 | |
| Nausea | 1 | 1 | 2 | |
| Pain | 2 | 2 | ||
| Tonsillitis | 1 | 1 | 2 | |
-
WHO, World Health Organization.
Three patients experienced a serious AE during the study. Two were considered to be unlikely to be related to study treatment (severe inflicted injury [fracture of the leg with ruptured tendon] in the medium-dose group; and severe arthrosis in the high-dose group), but which led to withdrawal from the study. One serious AE (severe lumbosacral pain in the medium-dose group) was considered to be possibly related to study treatment. There were no deaths during the study. No notable changes in vital signs were observed during the study in any of the three dose groups.
Dysport® was well tolerated at all doses. At Week 12, global tolerability was rated as ‘good’ or ‘very good’ by 49/57 (86.0%) of patients in the low-dose group, 37/57 (64.9%) of patients in the medium-dose group, and 60/75 (80.0%) of patients in the high-dose group. Similarly, a high proportion of investigators considered global tolerability to be good or very good in the low-dose (49/57, 86.0%), the medium-dose (38/57, 66.7%) and the high-dose groups (62/75, 82.7%).
4 Discussion
Although the efficacy of Dysport® for the treatment of myofascial back pain has been shown previously [17], the optimal dose of Dysport® in this indication has not been determined. Thus, we conducted the largest study to date to investigate the therapeutic effects of a range of doses of Dysport® in patients with myofascial back pain in order to estimate the lowest effective dose. Our results showed a significant reduction in patient-reported pain intensity after injections at four trigger points, for all three doses investigated (4 × 60, 4 × 80 or 4 × 120 units of Dysport®). Overall, reductions in pain of approximately 20%, at rest and on movement, were reported by patients 6 weeks after treatment assessed using the pain diary. Reductions in pain were evident after the first week of dosing, with the positive effect of treatment maintained until Week 6 and up to study endpoint (Week 12). However, no dose-response relationship was observed and the efficacy results exhibited a similar treatment response between the low and high doses. Thus, although the lowest dose of Dysport® used in this study (60 units at 4 trigger points) appears to be effective for the treatment of myofascial pain, we cannot conclude that it is the optimum dose in this indication as lower doses may also be effective.
It is interesting that reductions in pain were also associated with clinically and statistically significant reductions in pain-related impairment of daily activities, as assessed by the PDI sum scores. This is particularly important as pain interventions should always aim to reduce the impact of pain on activities of daily living and increase an individual’s independence. Treatments that can improve pain-related impairment of daily activities have the potential to break the bio-psycho-social disease cycle in chronic back pain. However, improvements in pressure thresholds and tissue compliance in the 12 weeks after dosing were small, not statistically significant from baseline and unlikely to be of clinical significance.
Although our results lend further support to the beneficial effects of Dysport® treatment in patients with myofascial back pain seen in a previously conducted, placebo-controlled trial, the open- label study design and lack of a placebo control group are significant limitations and, as such, we cannot be sure that the beneficial effects seen were specific to the BoNT-A injection. Thus, this study must be considered to be a pilot study and can guide the design of further, large, placebo-controlled, double-blind studies that are needed before the true value of this treatment can be ascertained. As well as establishing the optimal Dysport® dose needed per trigger point, further studies will also need to establish the number of trigger points that should be treated.
It should also be considered that, although Dysport® has been previously shown to be effective for the treatment of myofascial back pain, several studies with BOTOX have failed to show significant benefit from treatment. However, the lack of observed effect in some studies may be due to insufficient dosing. Although the units of Dysport® and Botox are not equivalent, data suggest that the recommended dose equivalence ratio of 2.5 or fewer units of Dysport® to 1 unit of Botox® [42,43,44,45]. Therefore, dose differences between studies may be important. For example, in two studies that failed to show a significant benefit of Botox treatment, a total dose of up to 50 units of Botox was used [21,23], which is low compared with the doses investigated in this study (total dose of 240–480 units of Dysport®).
Treatment was well tolerated by the patients in our study, with the majority of side-effects being mild or moderate in nature. The most commonly reported AE was influenza-like symptoms. The majority of patients and investigators (89.1% in each case) rated the tolerability of the lowest dose as ‘good’ or ‘very good’.
In conclusion, the results of this dose-finding study showed that treatment with BoNT-A (Dysport®) using a four-trigger-point injection protocol at 60 units per trigger point was associated with a clinically relevant and statistically significant improvement in pain and pain-related disability, and was well tolerated in patients with myofascial low back pain. Higher doses of 80 and 120 units per trigger point did not increase the pain-relieving effect. Our findings are limited by the lack of a control group and further research is warranted.
DOI of refers to article: 10.1016/j.sjpain.2010.12.002.
-
Conflict of interest
Conflict of interest statement: The authors have no commercial associations (e.g. consultancies, stock ownership, equity interest, and patent-licensing arrangements) that might pose a conflict of interest in connection with this article.
Acknowledgements
Data management and statistical analyses were conducted by Stefan Jauch at IT-Solutions for Clinical Research. The authors take full responsibility for the content of the paper but thank Caudex Medical (supported by Ipsen) for their assistance in preparing the initial draft of the manuscript and collating the comments of authors. Further editorial support was also provided by Ogilvy Healthworld Medical Education (supported by Ipsen). This was an investigator-initiated study, with financial support provided by Ipsen Pharma GmbH, Germany.
References
[1] FischerAA. New developments in diagnosis of myofascial pain and fibromyalgia. Phys Med Rehabil Clin NorthAm 1997;8:1–22.Search in Google Scholar
[2] Gerwin RD. Classification, epidemiology, and natural history of myofascial pain syndrome. Curr Pain Headache Rep 2001;5:412–20.Search in Google Scholar
[3] Wheeler AH. Myofascial pain disorders: theory to therapy. Drugs 2004;64:45–62.Search in Google Scholar
[4] Simons DG. Clinical and etiological update of myofascial pain from trigger points. J Musculoskeletal Pain 1996;4:93–121.Search in Google Scholar
[5] KuanTS. Current studies on myofascial painsyndrome. Curr Pain Headache Rep 2009;13:365–9.Search in Google Scholar
[6] Baldry P. Management of myofascial trigger point pain. Acupunct Med 2002;20:2–10.Search in Google Scholar
[7] Hong CZ. Treatment of myofascial pain syndrome. Curr Pain Headache Rep 2006;10:345–9.Search in Google Scholar
[8] Rudin NJ. Evaluation of treatments for myofascial pain syndrome and fibromyalgia. Curr Pain Headache Rep 2003;7:433–42.Search in Google Scholar
[9] Casale R, Tugnoli V. Botulinum toxin for pain. Drugs R D 2008;9:11–27.Search in Google Scholar
[10] Jeynes LC, Gauci CA. Evidence forthe use ofbotulinum toxin inthe chronic pain setting - a review ofthe literature. Pain Pract 2008;8:269–76.Search in Google Scholar
[11] Qerama E, Fuglsang-Frederiksen A, Jensen TS. The role ofbotulinum toxin in management of pain: an evidence-based review. Curr Opin Anaesthesiol 2010;23:602–10.Search in Google Scholar
[12] Filippi GM, Errico P, Santarelli R, Bagolini B, Manni E. BotulinumAtoxin effects on rat jaw muscle spindles. Acta Otolaryngol 1993;113:400–4.Search in Google Scholar
[13] Rosales RL, Arimura K, Takenaga S, Osame M. Extrafusal and intrafusal muscle effects in experimental botulinum toxin-A injection. Muscle Nerve 1996;19:488–96.Search in Google Scholar
[14] Acquadro MA, Borodic GE. Botulinum toxin efficacy forthe treatment of pain. J Clin Anesth 2005;17:328–30.Search in Google Scholar
[15] Cheshire WP, Abashian SW, Mann JD. Botulinum toxin in the treatment of myofascial pain syndrome. Pain 1994;59:65–9.Search in Google Scholar
[16] Ferrante FM, Bearn L, Rothrock R, King L. Evidence againsttriggerpoint injection technique for the treatment ofcervicothoracic myofascial pain with botulinum toxin type A. Anesthesiology 2005;103:377–83.Search in Google Scholar
[17] Göbel H, Heinze A, Reichel G, Hefter H, Benecke R. Efficacy and safety of a single botulinum type A toxin complex treatment (Dysport) for the relief of upperback myofascial pain syndrome: results from a randomized double-blind placebo-controlled multicentre study. Pain 2006;125:82–8.Search in Google Scholar
[18] Graboski CL, Gray DS, Burnham RS. Botulinum toxin A versus bupivacaine trigger point injections for the treatment of myofascial pain syndrome: a randomised double blind crossover study. Pain 2005;118:170–5.Search in Google Scholar
[19] Ho KY, Tan KH. Botulinum toxin A for myofascial trigger point injection: a qualitative systematic review. Eur J Pain 2007;11:519–27.Search in Google Scholar
[20] Kamanli A, Kaya A, Ardicoglu O, Ozgocmen S, Zengin FO, Bayik Y. Comparison of lidocaine injection, botulinum toxin injection, and dry needling to trigger points in myofascial pain syndrome. Rheumatol Int 2005;25:604–11.Search in Google Scholar
[21] Ojala T, Arokoski JP, Partanen J. The effect of small doses of botulinum toxin A on neck-shoulder myofascial pain syndrome: a double-blind, randomized, and controlled crossover trial. Clin J Pain 2006;22:90–6.Search in Google Scholar
[22] Porta M. A comparative trial of botulinum toxin type A and methylprednisolone for the treatment of myofascial pain syndrome and pain from chronic muscle spasm. Pain 2000;85:101–5.Search in Google Scholar
[23] Qerama E, Fuglsang-Frederiksen A, Kasch H, Bach FW, Jensen TS. A double-blind, controlled study of botulinum toxin A in chronic myofascial pain. Neurology 2006;67:241–5.Search in Google Scholar
[24] Wheeler AH, Goolkasian P, Gretz SS. A randomized, double-blind, prospective pilot study of botulinum toxin injection for refractory, unilateral, cervicothoracic, paraspinal, myofascial pain syndrome. Spine (Phila Pa 1976) 1998;23:1662–6, discussion 7.Search in Google Scholar
[25] Yue SK. Initial experience in the use of botulinum-toxin A for the treatment of myofascial related muscle dysfunctions .J Musculoskelet Pain 1995;3:22.Search in Google Scholar
[26] Bakheit AM, Fedorova NV, Skoromets AA, Timerbaeva SL, Bhakta BB, Coxon L. The beneficial antispasticity effect of botulinum toxin type A is maintained after repeated treatment cycles.J Neurol Neurosurg Psychiatry 2004;75:1558–61.Search in Google Scholar
[27] Hyman N, Barnes M, Bhakta B, Cozens A, Bakheit M, Kreczy-Kleedorfer B, et al. Botulinum toxin (Dysport) treatment of hip adductor spasticity in multiple sclerosis: a prospective, randomised, double blind, placebo controlled, dose ranging study. J Neurol Neurosurg Psychiatry 2000;68:707–12.Search in Google Scholar
[28] Jitpimolmard S, Tiamkao S, Laopaiboon M. Long term results of botulinum toxin type A (Dysport) in the treatment of hemifacial spasm: a report of 175 cases. J Neurol Neurosurg Psychiatry 1998;64:751–7.Search in Google Scholar
[29] Pittock SJ, Moore AP, Hardiman O, Ehler E, Kovac M, Bojakowski J, et al. A double-blind randomised placebo-controlled evaluation of three doses of botulinum toxin type A (Dysport) in the treatment of spastic equinovarus deformityafter stroke. Cerebrovasc Dis 2003;15:289–300.Search in Google Scholar
[30] Truong D, Duane DD, Jankovic J, Singer C, Seeberger LC, Comella CL, et al. Efficacy and safety of botulinum type A toxin (Dysport) in cervical dystonia: results of the first US randomized, double-blind, placebo-controlled study. Mov Disord 2005;20:783–91.Search in Google Scholar
[31] Truong D, Comella C, Fernandez HH, Ondo WG. Dysport Benign Essential Blepharospasm Study Group. Parkinsonism Relat Disord 2008;14:407–14.Search in Google Scholar
[32] Van den Bergh P, Francart J, Mourin S, Kollmann P, Laterre EC. Five-year experience in the treatment of focal movement disorders with low-dose Dysport botulinum toxin. Muscle Nerve 1995;18:720–9.Search in Google Scholar
[33] Wissel J, Kanovsky P, Ruzicka E, Bares M, Hortova H, Streitova H, et al. Efficacy and safety of a standardised 500 unit dose of Dysport (clostridium botulinum toxin type A haemaglutinin complex) in a heterogeneous cervical dystonia population: results of a prospective, multicentre, randomised, double-blind, placebo-controlled, parallel group study. J Neurol 2001;248:1073–8.Search in Google Scholar
[34] Chibnall JT, Tait RC. The Pain Disability Index: factor structure and normative data. Arch Phys Med Rehabil 1994;75:1082–6.Search in Google Scholar
[35] Pollard CA. The relationship of family environment to chronic pain disability. Diss Abstr Int 1981:42 (2077B).Search in Google Scholar
[36] Tait RC, Pollard CA, Margolis RB, Duckro PN, Krause SJ. The Pain Disability Index: psychometric and validity data. Arch Phys Med Rehabil 1987;68: 438–41.Search in Google Scholar
[37] Fischer AA. Pressure threshold meter: its use for quantification of tender spots. Arch Phys Med Rehabil 1986;67:836–8.Search in Google Scholar
[38] Fischer AA. Pressure tolerance over muscles and bones in normal subjects. Arch Phys Med Rehabil 1986;67:406–9.Search in Google Scholar
[39] Fischer AA. Tissue compliance meter for objective, quantitative documentation of soft tissue consistency and pathology. Arch Phys Med Rehabil 1987;68:122–5.Search in Google Scholar
[40] Colditz GA, Miller JN, Mosteller F. Measuring gain in the evaluation of medical technology. The probability of a better outcome. Int J Technol Assess Health Care 1988;4:637–42.Search in Google Scholar
[41] Wei L, Lachin J. Two-sample asymptotically distribution-free tests for incomplete multivariate observations. J Am Stat Assoc 1984;79:653–61.Search in Google Scholar
[42] Grosse J, Kramer G, Stohrer M. Success of repeat detrusor injections of botulinum A toxin in patients with severe neurogenic detrusor overactivity and incontinence. Eur Urol 2005;47:653–9.Search in Google Scholar
[43] Rosales RL, Bigalke H, Dressler D. Pharmacology of botulinum toxin: differences between type A preparations. Eur J Neurol 2006;13(Suppl. 1):2–10.Search in Google Scholar
[44] Straughan D. Progress in applying the three Rs to the potency testing of botulinum toxin type A. Altern Lab Anim 2006;34:305–13.Search in Google Scholar
[45] Van den Bergh PY, Lison DF. Dose standardization of botulinum toxin. Adv Neurol 1998;78:231–5.Search in Google Scholar
Appendix A
Members of the Dysport® in Myofascial Back Pain Study Group are: Dr Djamschid Akbarpour (Köln), Dr Wolfgang Bartel (Halberstadt), Dr Ulrich Bickel (Bocholt), Prof. Dr Klaus Borchert (Greifswald), Dr Fridolin Braig (Ulm), Dr Anette Delbruck-Schneider (Celle), Dr Thomas Eberbach (Osnabrück), Dr Oliver Emrich (Ludwigshafen), Dr Nana Finkelstein-Conea (Dortmund), Dr Thomas Flöter (Frankfurt), Dr Stephan Grunert (Eichstätt), Dr Olaf Günther (Magdeburg), Dr Johannes Horlemann (Kevelaer), Dr Uwe Junker (Remscheid), Dr Edwin Klaus (Würzburg), Dr Knut Kolitsch (Katzhutte), Dr Axel Krau (Bielefeld), Dr Thomas Krohn (Wismar), Dr Thomas Lange (Rudolstadt), Dr Klaus Langes (Würzburg), Dr Uwe Meckbach (Worms), Dr Gerhard Müller-Schwefe (Göppingen), Dr Thomas Nolte (Wiesbaden), Dr Achim Refisch (Krefeld), Dr Johannes Michael Ribbat (Itzehoe), Dr Marie Schlegel (Mühlhausen), Dr André Seeliger (Köln), Dr Ingrid Steigertahl-Liu (Osnabrück), Prof. Dr Hans-Detlev Stober (Berlin), Dr Adrian Stoenescu (Wuppertal), Dr Rudolf Tamm (Hannover), Dr Liliana Tarau (Wiesbaden), Dr Achim Thater (Krefeld), Dr Georgi Tontschev (Bernau), Dr Norbert Vehreschild (Ludwigshafen), Dr Edgar Weller (Dresden).
Abbreviations
- ANOVA
-
analysis of variance
- BoNT-A
-
botulinum toxin type A
- ITT
-
intent-to-treat
- LOCF
-
last observation carried forward
- NSAIDs
-
non-steroidal anti-inflammatory drugs
- PDI
-
Pain Disability Index
- PP
-
per protocol
- SD
-
standard deviation.
© 2010 Scandinavian Association for the Study of Pain
Articles in the same Issue
- Editorial comment and review
- Redheads, pain mechanisms and genetics: Lessons learned from inconclusive studies
- Clinical pain research
- Pain sensitivity and experimentally induced sensitisation in red haired females
- Editorial comment
- Assessment and mechanisms of mechanical allodynia
- Clinical pain research
- The perception threshold counterpart to dynamic and static mechanical allodynia assessed using von Frey filaments in peripheral neuropathic pain patients
- Editorial comment
- Pain during pharmacologically induced termination of pregnancy
- Clinical pain research
- The level of unpleasantness of pain influences the choice of home treatment during medical abortion
- Editorial comment
- Botulinum toxin for the treatment of pain?
- Original experimental
- Dysport® for the treatment of myofascial back pain: Results from an open-label, Phase II, randomized, multicenter, dose-ranging study
- Editorial comment
- Trends in analgesic drug use evaluated by national prescription data bases: Differences between immigrants and native citizens of Norway
- Observational studies
- Dispensing of prescribed analgesics in Norway among young people with foreign-or Norwegian-born parents
Articles in the same Issue
- Editorial comment and review
- Redheads, pain mechanisms and genetics: Lessons learned from inconclusive studies
- Clinical pain research
- Pain sensitivity and experimentally induced sensitisation in red haired females
- Editorial comment
- Assessment and mechanisms of mechanical allodynia
- Clinical pain research
- The perception threshold counterpart to dynamic and static mechanical allodynia assessed using von Frey filaments in peripheral neuropathic pain patients
- Editorial comment
- Pain during pharmacologically induced termination of pregnancy
- Clinical pain research
- The level of unpleasantness of pain influences the choice of home treatment during medical abortion
- Editorial comment
- Botulinum toxin for the treatment of pain?
- Original experimental
- Dysport® for the treatment of myofascial back pain: Results from an open-label, Phase II, randomized, multicenter, dose-ranging study
- Editorial comment
- Trends in analgesic drug use evaluated by national prescription data bases: Differences between immigrants and native citizens of Norway
- Observational studies
- Dispensing of prescribed analgesics in Norway among young people with foreign-or Norwegian-born parents