TGF-β signaling in diabetic nephropathy: An update
-
Li Wang


Abstract
Diabetic nephropathy (DN) is a common complication in patients with diabetes and the leading cause of end-stage renal disease. Accumulating evidence shows that transforming growth factor beta-1 (TGF-β1) is a key mediator in the pathogenesis of DN. TGF-β1 binds to its receptors to activate canonical and noncanonical downstream signaling pathways to exert its biological activities. Among them, canonical Smad signaling is the major pathway responsible for the development of DN. In addition to TGF-β1, many stress molecules, such as advanced glycation end products (AGEs), angiotensin II (Ang II), and C-reactive protein (CRP), can also activate Mothers against decapentaplegic homologs (Smads) via the extracellular signal-regulated kinase (ERK)/p38 mitogen-activated protein kinase (MAPK) cross talk mechanism. Furthermore, TGF-β/Smad signaling can also cross talk with nuclear factor kappa B (NF-κB) signaling to regulate renal inflammation via the induction of IκBα by Smad7. In the context of renal fibrosis, Smad3 is pathogenic, while Smad2 and Smad7 are protective. TGF-β signaling also upregulates the pathogenic microRNAs (miRNAs) (namely, miR-21, miR-192, and miR-377) and long noncoding RNAs (lncRNAs) (namely, Erbb4-IR (intron region, IR), LncRNA9884, and Arid2-IR) but downregulates the protective miRNAs (namely, miR-29a/b and miR-200a) to mediate DN. Thus, targeting TGF-β signaling, either by blocking its ligand, its receptor (i.e., TGF-β receptor-2 [TGFBR2]), Smad3, and downstream miRNAs/lncRNAs or by overexpressing Smad7, has been shown to improve DN. In addition, pharmaceutically targeting TGF-β signaling using chemical inhibitors and traditional Chinese medicine (TCM), including Tangshen formula, Chaihuang-Yishen granule, and herbal extracts (berberine, asiatic acid, and naringenin), also shows renoprotective effect in diabetes. In summary, TGF-β signaling is a critical pathway leading to DN and may be a therapeutic target for combating DN.
1 Introduction
Diabetic nephropathy (DN) is one of the most common complications in both type-1 and type-2 diabetes mellitus. With the prevalence of diabetes, DN has become the primary cause of end-stage renal disease worldwide [1, 2]. Pathologically, DN is characterized by glomerular hypertrophy, mesangial expansion, glomerular basement membrane (GBM) thickening, podocyte injury, glomerulosclerosis, tubulointerstitial fibrosis, and renal inflammation, which is accompanied by proteinuria, increased serum creatinine, and eventually the fall of glomerular filtration rate [2, 3]. Various mediators and signaling mechanisms play pivotal roles in the pathogenesis of DN, which also include the cytokine transforming growth factor beta (TGF-β).
TGF-β1 is a well-known prosclerotic/profibrotic cytokine that functions to induce the expression of extracellular matrix (ECM) components and thus causes tissue fibrosis, including the development of DN [4]. The pathogenic role of TGF-β1 in fibrosis is demonstrated by the findings that mice overexpressing Tgfb1 develop fibrosis in multiple organs, including the kidney, liver, heart, blood vessel, pancreas, and testis [5,6,7,8]. This has also been found in diabetic Akita mice (a rodent model of DN) in which hypermorphic but not hypomorphic expression of Tgfb1 aggravates the typic features of DN, including increased glomerulosclerosis, glomerular mesangial expansion, and proteinuria [9]. Clinically, patients with DN exhibit elevated levels of plasma and urinary TGF-β1, which positively correlates with the severity of disease, including progressive renal fibrosis [10,11,12,13]. It is now well known that after binding to receptors, TGF-β can activate its downstream signaling, including Smad-dependent and -independent pathways, to mediate renal fibrosis [4]. Under diabetic conditions, TGF-β signaling is highly activated in the diabetic kidney in patients and experimental animal models [1, 14,15,16,17]. In this review, we focus on the roles and signaling mechanisms of TGF-β in the pathogenesis of DN. The recent progress in the development of anti-DN therapy by targeting TGF-β signaling is also highlighted.
2 Overview of TGF-β Signaling
TGF-β belongs to the TGF superfamily, which also contains the signaling molecules bone morphogenetic protein (BMP), growth and differentiation factor (GDF), activin, and nodal [18]. TGF-β exerts its functions by binding to the transmembrane type-2 serine/threonine kinase receptor TGFBR2, resulting in the recruitment and activation of the type-1 receptor TGFBR1 by phosphorylation. Then, the activated TGFBR1 can initiate the activation of both Smad-dependent (canonical) and -independent (noncanonical) pathways to exert its biological activities [1, 4]. In the Smad-dependent pathway, the activated TGFBR1 can phosphorylate the cytosolic receptor-regulated Mothers against decapentaplegic homolog (Smad) proteins (R-Smads) Smad2 and Smad3, which then form a complex with Smad4 (common Smad or Co-Smad) and translocate into the nucleus to regulate target gene transcription [1, 4, 18]. Smad7 is an inhibitory Smad protein induced by TGF-β1, but it functions to counterbalance the TGF-β signaling activity by competitively binding to TGFBR1 and inducing its degradation (Figure 1) [19, 20]. In addition to the canonical TGF-β/Smad signaling, TGF-β ligand can also induce Smad-independent signaling, such as MAPK, phosphoinositide 3-kinase (PI3K), and small guanosine triphosphatase (GTPase) pathways [1, 4]. Furthermore, the R-Smads can also be activated through TGF-β ligand-independent pathways by cross talk with MAPK signaling [18]. These diverse regulatory networks add additional complexity and plasticity to TGF-β signaling. The blueprint of TGF-β signaling is illustrated in Figure 1, and the reader can also refer to other review articles for a more comprehensive description [1, 18, 21].
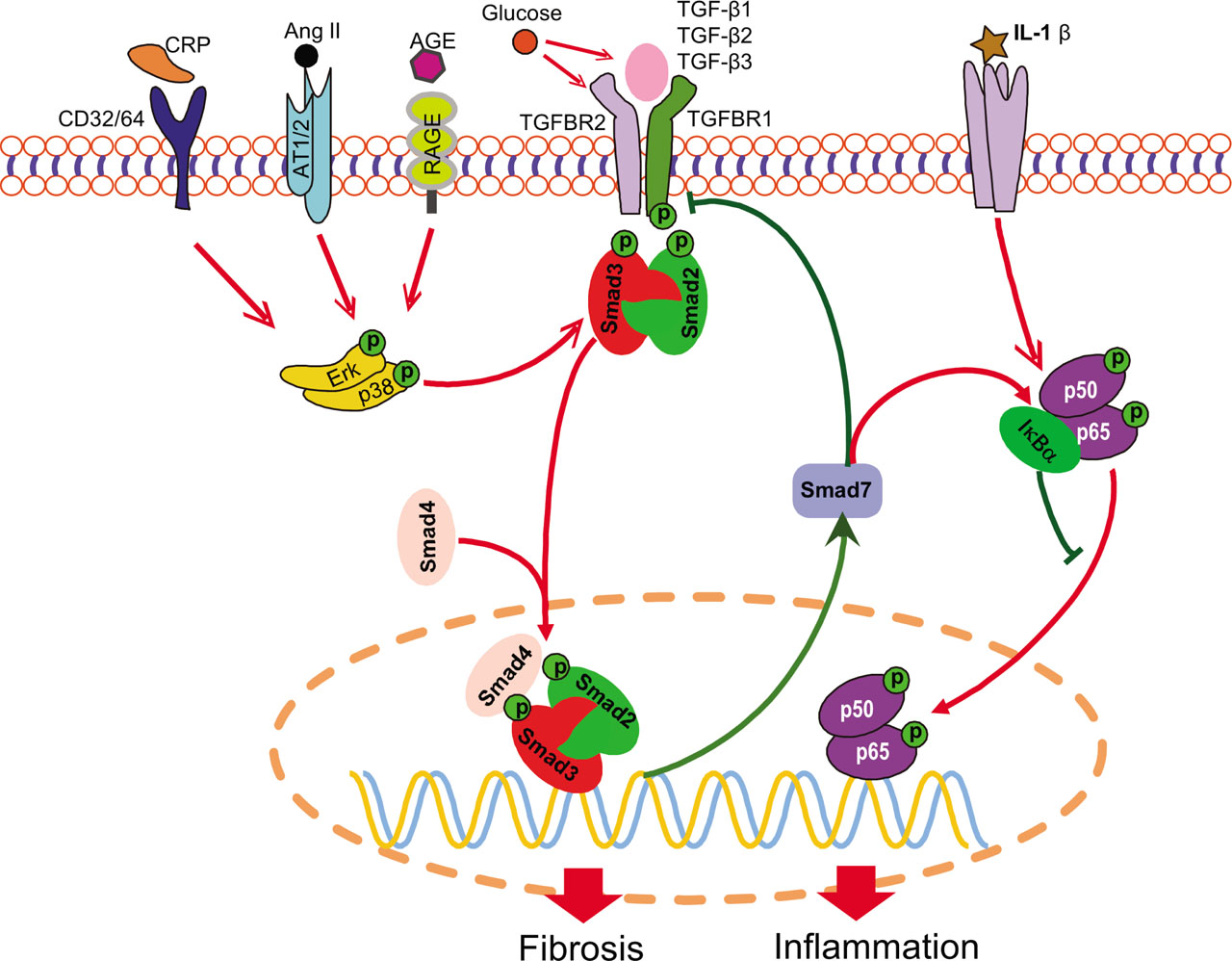
Regulatory pathways of TGF-β signaling in DN. Smad2/3 can be activated by both TGF-β-dependent and -independent mechanisms. Many stress molecules such as Ang II, AGE, and CRP can activate Smad2/3 to exert their biological effects via cross talk with ERK/p38 MAPK pathway. High glucose promotes the activation of latent TGF-β ligand and the transcription of Tgfb1 and Tgfbr2. In addition, Smad7 functions to counteract Smad2/3 signaling via the negative feedback loop and suppresses NF-κB activation by inducing IκBα. The red line indicates pathogenic, whereas the green line implies protective, pathways. The arrow stands for positive regulation, and the blunt line represents negative regulation. AGE, advanced glycation end product; Ang II, angiotensin II; AT1/2, Ang II type 1/2 receptor; CD, cluster of differentiation; CRP, C-reactive protein; DN, diabetic nephropathy; Erk, extracellular signal-regulated kinase; IL, interleukin; MAPK, mitogen-activated protein kinase; NF-κB, nuclear factor kappa B; RAGE, receptor of AGE; TGF-β, transforming growth factor beta; TGFBR, transforming growth factor beta receptor.
3 Role and Mechanisms of TGF-β Signaling in DN
Patients with DN usually experience hyperglycemia, hyper-lipidemia, and hypertension [22]. It is now clear that many hazardous factors, such as high glucose, advanced glycation end products (AGEs), angiotensin II (Ang II), and C-reactive protein (CRP), can activate Smad signaling via both TGF-β ligand-dependent and -independent mechanisms (Figure 1). Under diabetic conditions, high glucose can activate TGF-β signaling via several mechanisms, including the induction of thrombospondin 1 (TSP1), to convert inactive latent TGF-β1 into the active form and thus leading to upregulation of TGF-β1 and TGFBR2 [16, 17, 23,24,25,26,27]. AGEs are glycation products of proteins or lipids in the condition of hyperglycemia and widely deposited in local tissues, including kidney, in patients with diabetes [28]. AGE can induce the rapid (within 30 min) phosphorylation of Smad2 and Smad3 via the receptor of AGE (RAGE)-mediated extracellular signal-regulated kinase (ERK)/p38 mitogen-activated protein kinase (MAPK) signaling cross talk in tubular epithelial cells (TECs) and mesangial cells (MCs) through a TGF-β-independent pathway (Figure 1) [14, 29]. AGE also induces TGF-β production, leading to the stimulation of classic TGF-β signaling to maintain subsequent long-term activation of downstream Smad signaling in a TGF-β-dependent manner [14].
Ang II is a key mediator of hypertension and can also rapidly phosphorylate Smad2/3 through ERK/p38 MAPK signaling cross talk in vascular smooth muscle cells (VSMCs) and TECs via a TGF-β-independent manner (Figure 1) [30,31,32]. Similar to AGEs, Ang II also induces the long-term activation of Smad2/3 in a TGF-β-dependent way [31]. In addition to MAPK, Ang II also induces de novo TGF-β1 expression via the protein kinase C (PKC)-dependent pathway [33, 34].
CRP is an acute-phase protein that is a biomarker of systemic inflammation. It is reported that elevated blood CRP level is associated with the occurrence of diabetes and DN [35, 36]. Transgenic overexpression of human CRP exaggerates the severity of renal injury in both type-1 and type-2 diabetic mouse models [37, 38]. After binding to its receptor cluster of differentiation-32 (CD32), CRP rapidly activates Smad3 via ERK/p38 MAPK signal cross talk in a TGF-β-independent manner. Similar to AGE and Ang II, CRP also triggers long-term Smad3 activation by inducing TGF-β1 expression (Figure 1) [37].
Free fatty acids can also activate TGF-β signaling as the addition of palmitate (a common saturated free fatty acid) can induce TGF-β1 expression and activate downstream Smad2/3 signaling through the CD36-mediated transient receptor potential cation channel subfamily C member 6/nuclear factor of activated T-cells 2 (TRPC6/NFAT2) pathway in human MCs [39].
As described earlier, TGF-β1 is highly upregulated in patients with DN and is induced by various DN-associated risk factors [10,11,12, 23, 24, 40]. The pathogenic role of TGF-β1 in DN can be perceived from the fact that transgenic Tgfb1 overexpression directly induces renal injury with features of DN, including glomerulosclerosis and protein-uria [5, 8, 41, 42]. Further, in a DN model of Akita mice with a finely modified expression of endogenous Tgfb1, hypermorphic Tgfb1 expression aggravates, while hypomorphic Tgfb1 expression attenuates or even prevents, DN phenotypes [9]. On the other hand, blocking TGF-β by in vivo delivery of a neutralizing antibody, antisense oligodeoxynucleotide, and soluble TGFBR2 ameliorates renal injury in experimental diabetic animals [43,44,45,46,47]. In vitro, high glucose-triggered ECM production can also be reversed by a neutralizing antibody against TGF-βs [48]. These findings confirm the role of TGF-β1 as a critical pathogenic mediator in DN.
TGFBR also plays a vital role in DN. Although the TGFBR2-null mutation is embryonically lethal, heterozygous deletion of TGFBR2 (Tgfbr2+/−) inhibits Smad2/3 signaling and improves renal function by significantly attenuating urinary protein secretion, which is accompanied by substantially alleviated glomerular hypertrophy, mesangial expansion, and glomerulosclerosis in streptozocin (STZ)-induced DN [49].
Although both Smad2 and Smad3 are R-Smads and share high similarity in terms of amino acid sequence, structure, and DNA-binding motif, they play distinct roles in renal fibrosis and inflammation during the development of DN as deletion of Smad3 suppresses, but deletion of Smad2 promotes, renal fibrosis in various mouse models of kidney diseases, including DN [50,51,52,53]. In vitro evidence also confirms this functional diversity, as Smad3—but not Smad2—mediates the fibrosis response induced by TGF-β1, high glucose, AGE, and Ang II [29, 31, 32, 50, 54, 55]. One recent study further demonstrated that deletion of Smad3 from db/db mice is capable of protecting db/db mice from the development of DN [56]. Compared with Smad3 wild-type db/db mice with severe diabetes and DN, age-matched Smad3-null db/db mice show normal levels of urinary albumin and serum creatinine without notable renal inflammation and fibrosis [56].
In contrast to the profibrotic role of Smad3, Smad2 plays an antifibrotic role as specific deletion of Smad2 in tubular cells using KSP-Cre promotes renal fibrosis in a mouse model of unilateral ureteral obstruction (UUO) [50]. However, Loeffler et al [57]. reported an antifibrosis phenotype in STZ-induced DN [56] by specific deletion of Smad2 in fibroblasts driven by fibroblast-specific protein 1 (FSP1)-Cre. This discrepancy may be attributed to the difference in the genetic mouse models used, implying the distinct roles of Smad2 in different cell types during fibrosis.
Smad7 is an inhibitory Smad that functions to counterbalance the activity of TGF-β signaling by suppressing the phosphorylation of R-Smads and inducing ubiquitin-mediated degradation of TGFBR1 [19, 20]. Deletion of Smad7 aggravates, but overexpression of Smad7 inhibits, renal fibrosis and inflammation in various animal models of kidney disease, including UUO, 5/6 nephrectomy, crescentic glomerulonephritis, aristolochic acid nephropathy, hypertensive nephropathy, and DN [32, 58,59,60,61,62,63,64,65,66,67,68,69]. In vitro overexpression of Smad7 in renal tubular cells and VSMCs suppresses fibrosis induced by TGF-β, high glucose, AGE, and Ang II [14, 32, 70, 71]. Mechanistically, the renal protective role of Smad7 is associated with inactivation of both TGF-β and NF-κB signaling [68, 69]. Indeed, as illustrated in Figure 1, overexpression of Smad7 not only inhibits TGF-β/Smad3-mediated renal fibrosis but also inactivates NF-κB signaling by inducing IκBα, thereby inhibiting renal inflammation [62, 72]. In addition, Smad7 is also responsible for the anti-inflammatory effect of TGF-β1, as TGF-β1 can induce the expression of Smad7 [62, 73]. Therefore, Smad7 has both antifibrotic and anti-inflammatory properties and may be a therapeutic agent for various kidney diseases, including DN [74].
4 Role of Smad3-Dependent Noncoding RNAs in DN
In the genome, >98% of genes are noncoding RNAs, with about 1.2% of genes producing proteins [75]. Noncoding RNAs can be classified into transfer RNAs (tRNAs), heterogeneous nuclear RNAs (hnRNAs), small nucleolar RNAs (snoRNAs), ribosomal RNAs (rRNAs), microRNAs (miRNAs), long non-coding RNAs (lncRNAs), and so on [75]. Among them, miRNAs and lncRNAs play pivotal regulatory roles in diverse biological and disease processes. Accumulating evidence indicates that TGF-β/Smad3 signaling regulates a number of miRNAs and lncRNAs to mediate DN (Figure 2).
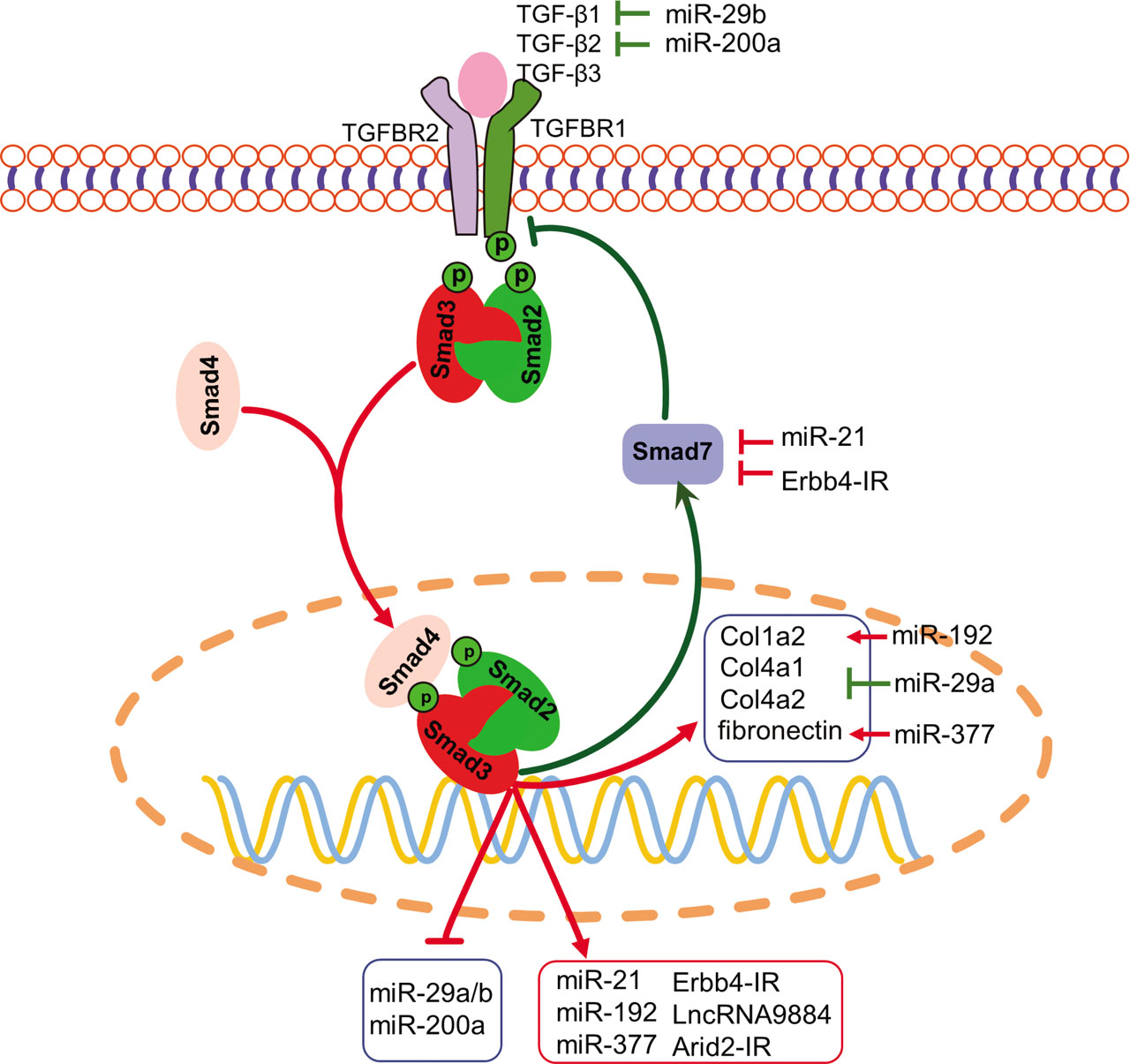
TGF-β/Smad3-dependent miRNAs and lncRNAs in DN. Smad3 can transcriptionally induce miR-21, miR-192, miR-377, Erbb4-IR, LncRNA9884, and Arid2-IR but suppress the miR-29a/b and miR-200a families, contributing to the modulation of renal fibrosis and inflammation in DN. The red line indicates pathogenic, whereas green line implies protective, pathways. The arrow stands for positive regulation, and blunt line represents negative regulation. DN, diabetic nephropathy; lncRNA, long noncoding RNA; miRNA (miR), microRNA; TGF-β, transforming growth factor beta; TGFBR, transforming growth factor beta receptor.
4.1 Role of Smad3-dependent miRNAs in DN
miRNAs are short noncoding RNAs that posttranscriptionally repress gene expression by binding to the 3′-untranslated region (UTR) of their target mRNAs to induce their degradation or translational suppression [76, 77]. Microarray-based screening and subsequent functional studies identified multiple miRNAs that are regulated by TGF-β signaling and that modulate the pathogenesis of kidney disease [76,77,78]. Among them, it is now clear that miR-21, miR-192, and miR-377 are pathogenic, whereas miR-29a/b and miR-200a are protective in DN.
miR-21 is a Smad3-dependent miRNA and plays a pathogenic role in the renal fibrosis of UUO mice [78]. miR-21 is also found to be upregulated in DN of db/db mice, while knockdown of miR-21 ameliorates DN by inhibiting renal fibrosis and inflammation. Mechanistically, miR-21 promotes renal fibrosis and inflammation in db/db mice by suppressing Smad7 by binding to its 3′-UTR [79]. miR-192 is another Smad3-dependent miRNA that shows increased expression in the glomeruli of db/db and STZ-induced diabetic mice [80, 81]. In vitro, silencing miR-192 blocks but overexpression of miR-192 promotes TGF-β1-induced production of ECM [81]. Further analysis revealed that miR-192 targets Smad-interacting protein 1 (SIP1), which is an E-box repressor, to suppress Col1a2 transcription [80]. Therefore, miR-192 mediates TGF-β/Smad3 signaling-driven fibrosis by promoting the collagen gene Col1a2 by decreasing SIP1. miR-377 is increased in STZ-induced DN and in TGF-β- or high glucose-stimulated MCs. miR-377 contributes to fibrosis by promoting fibronectin expression, although the detailed mechanism remains to be elucidated [82].
miR-29b is an antifibrotic miRNA which is expressed in fibrotic diseases of multiple organs, including the heart [83], liver [84], lung, and kidney [85, 86]. In DN, the expression of miR-29b is downregulated by AGE in a Smad3-dependent manner. Overexpression of miR-29b improves DN by attenuating fibrosis and inflammation [87]. Mechanistically, miR-29b suppresses TGF-β signaling by targeting the third exon of Tgfb1 mRNA in heart-derived fibroblasts [83]. In addition to miR-29b, another member of the miR-29 cluster, namely, miR-29a, is also downregulated by TGF-β1 and high glucose in vitro. miR-29a directly targets the 3′-UTR of the collagen genes Col4a1 and Col4a2 to suppress ECM production [88]. Similar to miR-29b, overexpression of miR-29a also shows a renoprotective effect on DN [89]. miR-200a is another miRNA that is downregulated in DN in response to TGF-β1 and TGF-β2 treatment, whereas overexpression of miR-200a attenuates TGF-β/Smad3 signaling and renal fibrosis. Further analysis indicates that miR-200a negatively regulates TGF-β signaling by binding to the 3′-UTR of Tgfb2 [90]. Ahn et al [91] identified a Smad3-binding site in the promoter of miR-200a/b. However, they found that Smad3 activates but does not suppress transcription of miR200a/b in gastric cancer cells. Therefore, Smad3 may regulate miR-200a expression in a cell type-dependent manner.
4.2 Role of Smad3-dependent lncRNAs in DN
Furthermore, lncRNAs are another group of noncoding RNAs of >200 nucleotides in length. Using RNA sequencing (RNA-Seq), thousands of lncRNAs have been discovered, many of which have been proven to play important roles in various biological processes with distinct mechanisms [92]. In tissue fibrosis, many lncRNAs are dysregulated by TGF-β signaling [93]. With respect to kidney disease, Zhou et al. [94]. found 21 lncRNAs that are tightly regulated by TGF-β/Smad3 signaling in both UUO and anti-GBM glomerulonephritis mouse models. Further studies revealed that some of these lncRNAs execute critical roles in different kidney diseases, including DN. Among them, lncRNA Erbb4-IR (np_5318), which is localized in the intron of Erbb4 gene, is highly expressed in the UUO kidney and in DN in db/db mice [88, 89]. Silencing Erbb4-IR attenuates renal fibrosis in both mouse models of UUO and diabetic kidney diseases, as well as in TGF-β1- and AGE-treated tubular cells [95]. It is now clear that Erbb4-IR can be induced in a Smad3-dependent manner, which promotes renal fibrosis by binding to the genomic locus of miR-29b and the 5′-UTR of Smad7 to suppress their expression [95, 96]. Another lncRNA, LncRNA9884 (na_9884), is intensively upregulated in the nucleus of renal tubular cells and MCs of db/db mice and can also be stimulated in vitro by AGE and high glucose in a Smad3-dependent manner. Silencing LncRNA9884 protects against renal injury in db/db mice by alleviating mesangial expansion, glomerulosclerosis, albuminuria, and serum creatinine levels. Mechanistic studies have revealed that lncRNA9884 promotes inflammation by transcriptionally inducing monocyte chemoattractant protein (MCP)-1 by physically binding to its promoter [97]. In addition, in the UUO kidney, another Smad3-dependent lncRNA, Arid2-IR (np_28496), has been found to promote inflammation via the NF-κB-dependent mechanism [98]. However, the role of Arid2-IR in DN remains unclear.
5 Treatment of DN by Targeting TGF-β Signaling
Considering the pathogenic role of TGF-β signaling in DN, inhibition of this signaling pathway may represent a novel therapy for DN. This is supported by the findings that targeting the TGF-β/Smad signaling by antibody-mediated blockade of TGF-β ligand [44,45,46], genetic deletion of Tgfbr2 or downstream mediator Smad3 [49, 56, 67, 99], and overexpression of Smad7 [68, 69] are capable of alleviating renal injury in both type-1 and type-2 diabetic animal models. However, as TGF-β is also an anti-inflammatory cytokine and because Smad3-null mice develop impaired mucosal immunity, long-termed blockade of TGF-β/Smad signaling may cause unwanted side effects [62, 100]. Thus, treatment by targeting TGF-β/Smad signaling should be focused on rebalancing the downstream Smads by inactivating Smad3 while restoring Smad7 as Smad7 can inhibit both Smad3-driven fibrosis and NF-κB-signaling-driven inflammation in a number of disease models, including DN [74]. As illustrated in Figure 2, targeting the downstream Smad3-dependent miRNAs and lncRNAs may be another promising treatment strategy for DN. This can be achieved by silencing the pathogenic miR-21, miR-192, miR-377, Erbb4-IR, and LncRNA9884 or by overexpressing the beneficial miR-29a/b and miR-200a, as reported in a number of studies [79, 81, 82, 87, 88, 90, 96, 97].
Pharmaceutical inhibition of TGF-β signaling may be another promising therapeutic approach for DN. As shown in Table 1, oral delivery of GW788388, a phosphorylation inhibitor targeting both TGFBR1 and TGFBR2, decreases renal fibrosis and albuminuria in db/db mice [101]. In addition, a novel chemical, halofuginone, which shows notable inhibitory activity on TGF-β signaling by decreasing Smad2 phosphorylation and TGFBR2 expression, has an antifibrotic effect on DN in db/db mice [102]. Furthermore, directly targeting Smad3 with the Smad3 inhibitor SIS3 also inhibits renal injury in STZ-induced DN [103].
List of TGF-β signaling-targeting chemical agents with validated efficacy in the treatment of DN.
| Chemical agent name | Test model | Function and underlying mechanism | Ref. |
|---|---|---|---|
| GW788388 | db/db mice | Decreases renal fibrosis and albuminuria by targeting phosphorylation of TGFBR1 and TGFBR2 | [101] |
| Halofuginone | db/db mice | Attenuates glomerular mesangial expansion and interstitial fibrosis by inhibiting phosphorylation of Smad2 and transcription of Tgfbr2 | [102] |
| SIS3 | STZ-induced DN in mice | Reduces renal fibrosis by suppressing endothelial–mesenchymal transition by targeting Smad3 phosphorylation | [103] |
| Naringenin | db/db mice | Reduces renal fibrosis by functioning as a Smad3 inhibitor | [104] and unpublished data |
| Asiatic acid | db/db mice | Reduces renal fibrosis by functioning as an agonist of Smad7 | [104] and unpublished data |
DN, diabetic nephropathy; STZ, streptozocin; TGF-b, transforming growth factor beta; TGFBR, transforming growth factor beta receptor.
Increasing evidence shows that the use of traditional Chinese medicine (TCM) or herbal medicine could be an alternative preventive or therapeutic treatment for DN. TCM is considered to contain multiple active ingredients/compounds that target multiple mediators or pathways. TCM prescriptions have been widely used in clinical applications in many Asian countries with controllable side effects [105]. Large cohort-based retrospective analysis has proven that TCM improves the long-term survival of patients with chronic kidney disease [106]. TCM prescriptions Tangshen formula and Chaihuang-Yishen granule have been demonstrated to have plausible clinical efficacy in patients with DN [107,108,109]. Animal studies indicate that these two TCM prescriptions substantially inhibit TGF-β signaling and renal fibrosis, with improvement of renal function, in DN [107,108,109]. In addition, a number of compounds extracted from plants, such as berberine (an isoquinoline alkaloid from Cortex Phellodendri Chinensis), asiatic acid (a triterpenoid from Centella asiatica), and naringenin (a flavonoid found abundantly in citrus) also show ability to lower blood glucose, improve metabolic abnormalities–especially insulin insensitivity and glucose tolerance, and decrease renal fibrosis and inflammation in db/db mice [104, 110]. Mechanistically, naringenin has been found to act as a Smad3 inhibitor, while asiatic acid functions as a Smad7 agonist. The combination of naringenin and asiatic acid can effectively rebalance Smad3/Smad7 signaling and therefore synergistically attenuate progressive renal fibrosis and inflammation [104, 111]. Clinical trials to validate the therapeutic efficacy of these herbal extracts on chronic kidney diseases, including DN, are warranted.
6 Conclusion
TGF-β signaling is a major pathway leading to the development of DN. TGF-β signaling can be activated and dysregulated by many diabetic mediators, including high glucose, AGE, Ang II, free fatty acid, and CRP, through TGF-β-dependent and -independent mechanisms. Generally, Smad3 is pathogenic and highly activated, while Smad7 is protective but downregulated, during the pathogenesis of DN. Thus, treatment by rebalancing Smad3/Smad7 signaling could be a better preventive and therapeutic strategy for combatting DN. In addition, Smad3 may mediate DN by transcriptionally upregulating the pathogenic, while inhibiting the renoprotective, miRNAs/lncRNAs, which may also be specific targets for the treatment of DN. Development of pharmaceutical inhibitors or TCM compounds that directly target or balance Smad3/Smad7 signaling could be a better approach for the prevention and treatment of DN.
Source of Funding
This work was supported by a Scientific Research Platform grant from the Research Grants Council of Hong Kong (grants GRF 14163317, 14117418, 14104019, and R4012-18), the Health and Medical Research Fund of Hong Kong (HMRF 05161326, 06173986, 14152321, and 07180516), the Lui Che Woo Institute of Innovative Medicine (CARE program) of the Chinese University of Hong Kong, the Luzhou–Southwest Medical University Science and Technology Strategic Cooperation Project (2021LZXNYD-P04), and the Luzhou Municipal–Southwest Medical University Joint Special Grant for High-Level Talents (HYL Team).
Conflict of Interest
Hui-yao Lan is an Editorial Board Member of the journal. The article was subject to the journal's standard procedures, with peer review handled independently of this editor and his research groups.
Author Contribution
Wang L and Wang HL wrote the manuscript, which was further revised by Lan HY.
Statement
The authors state that the manuscript has been read and approved by all members of the authorship, all the listed authors meet the criteria of authorship and the contents included in the manuscript represent honest work of the authorship.
REFERENCES
[1] Lan HY, Chung ACK. Transforming growth factor-beta and Smads. Contrib Nephrol 2011; 170: 75–82.10.1159/000324949Search in Google Scholar PubMed
[2] Lan HY. Transforming growth factor-beta/Smad signalling in diabetic nephropathy. Clin Exp Pharmacol Physiol 2012; 39: 731–8.10.1111/j.1440-1681.2011.05663.xSearch in Google Scholar PubMed
[3] Tervaert TW, Mooyaart AL, Amann K, Cohen AH, Cook HT, Drachenberg CB, et al. Pathologic classification of diabetic nephropathy. J Am Soc Nephrol 2010; 21: 556–63.10.1681/ASN.2010010010Search in Google Scholar PubMed
[4] Meng XM, Tang PM, Li J, Lan HY. TGF-beta/Smad signaling in renal fibrosis. Front Physiol 2015; 6: 82.10.3389/fphys.2015.00082Search in Google Scholar PubMed PubMed Central
[5] Sanderson N, Factor V, Nagy P, Kopp J, Kondaiah P, Wakefield L et al. Hepatic expression of mature transforming growth factor beta 1 in transgenic mice results in multiple tissue lesions. Proc Natl Acad Sci U S A 1995; 92: 2572–6.10.1073/pnas.92.7.2572Search in Google Scholar PubMed PubMed Central
[6] Kanzler S, Lohse AW, Keil A, Henninger J, Dienes HP, Schirmacher P et al. TGF-beta1 in liver fibrosis: an inducible transgenic mouse model to study liver fibrogenesis. Am J Physiol 1999; 276: G1059–68.10.1152/ajpgi.1999.276.4.G1059Search in Google Scholar PubMed
[7] Lee MS, Gu D, Feng L, Curriden S, Arnush M, Krahl T et al. Accumulation of extracellular matrix and developmental dysregulation in the pancreas by transgenic production of transforming growth factor-beta 1. Am J Pathol 1995; 147: 42–52.Search in Google Scholar
[8] Kopp JB, Factor VM, Mozes M, Nagy P, Sanderson N, Bottinger EP et al. Transgenic mice with increased plasma levels of TGF-beta 1 develop progressive renal disease. Lab Invest 1996; 74: 991–1003.Search in Google Scholar
[9] Hathaway CK, Gasim AM, Grant R, Chang AS, Kim HS, Madden VJ et al. Low TGFbeta1 expression prevents and high expression exacerbates diabetic nephropathy in mice. Proc Natl Acad Sci U S A 2015; 112: 5815–20.10.1073/pnas.1504777112Search in Google Scholar PubMed PubMed Central
[10] Ibrahim S, Rashed L. Estimation of transforming growth factor-beta 1 as a marker of renal injury in type II diabetes mellitus. Saudi Med J 2007; 28: 519–23.Search in Google Scholar
[11] Shaker YM, Soliman HA, Ezzat E, Hussein NS, Ashour E, Donia A, et al. Serum and urinary transforming growth factor beta 1 as biochemical markers in diabetic nephropathy patients. Beni-Suef Univ J Basic Appl Sci 2014; 3: 16–23.10.1016/j.bjbas.2014.02.002Search in Google Scholar
[12] Jakus V, Sapak M, Kostolanska J. Circulating TGF-beta1, glycation, and oxidation in children with diabetes mellitus type 1. Exp Diabetes Res 2012; 2012: 510902.10.1155/2012/510902Search in Google Scholar PubMed PubMed Central
[13] Murakami K, Takemura T, Hino S, Yoshioka K. Urinary transforming growth factor-beta in patients with glomerular diseases. Pediatr Nephrol 1997; 11: 334–6.10.1007/s004670050289Search in Google Scholar PubMed
[14] Li JH, Huang XR, Zhu HJ, Oldfield M, Cooper M, Truong LD, et al. Advanced glycation end products activate Smad signaling via TGF-beta-dependent and independent mechanisms: implications for diabetic renal and vascular disease. FASEB J 2004; 18: 176–8.10.1096/fj.02-1117fjeSearch in Google Scholar
[15] Hong SW, Isono M, Chen S, Iglesias-De La Cruz MC, Han DC, Ziyadeh FN. Increased glomerular and tubular expression of transforming growth factor-beta1, its type II receptor, and activation of the Smad signaling pathway in the db/db mouse. Am J Pathol 2001; 158: 1653–63.10.1016/S0002-9440(10)64121-1Search in Google Scholar
[16] Isono M, Chen S, Hong SW, Iglesias-de la Cruz MC, Ziyadeh FN. Smad pathway is activated in the diabetic mouse kidney and Smad3 mediates TGF-beta-induced fibronectin in mesangial cells. Biochem Biophys Res Commun 2002; 296: 1356–65.10.1016/S0006-291X(02)02084-3Search in Google Scholar
[17] Isono M, Mogyorosi A, Han DC, Hoffman BB, Ziyadeh FN. Stimulation of TGF-beta type II receptor by high glucose in mouse mesangial cells and in diabetic kidney. Am J Physiol Renal Physiol 2000; 278: F830–8.10.1152/ajprenal.2000.278.5.F830Search in Google Scholar
[18] Weiss A, Attisano L. The TGFbeta superfamily signaling pathway. Wiley Interdiscip Rev Dev Biol 2013; 2: 47–63.10.1002/wdev.86Search in Google Scholar
[19] Hayashi H, Abdollah S, Qiu Y, Cai J, Xu YY, Grinnell BW, et al. The MAD-related protein Smad7 associates with the TGFbeta receptor and functions as an antagonist of TGFbeta signaling. Cell 1997; 89: 1165–73.10.1016/S0092-8674(00)80303-7Search in Google Scholar
[20] Kavsak P, Rasmussen RK, Causing CG, Bonni S, Zhu H, Thomsen GH, et al. Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF beta receptor for degradation. Mol Cell 2000; 6: 1365–75.10.1016/S1097-2765(00)00134-9Search in Google Scholar
[21] Itoh S, Itoh F. Inhibitory machinery for the TGF-beta family signaling pathway. Growth Factors 2011; 29: 163–73.10.3109/08977194.2011.614236Search in Google Scholar PubMed
[22] Yadav H, Quijano C, Kamaraju AK, Gavrilova O, Malek R, Chen W, et al. Protection from obesity and diabetes by blockade of TGF-beta/Smad3 signaling. Cell Metab 2011; 14: 67–79.10.1016/j.cmet.2011.04.013Search in Google Scholar PubMed PubMed Central
[23] Rocco MV, Chen Y, Goldfarb S, Ziyadeh FN. Elevated glucose stimulates TGF-beta gene expression and bioactivity in proximal tubule. Kidney Int 1992; 41: 107–14.10.1038/ki.1992.14Search in Google Scholar PubMed
[24] Wolf G, Sharma K, Chen Y, Ericksen M, Ziyadeh FN. High glucose-induced proliferation in mesangial cells is reversed by autocrine TGF-beta. Kidney Int 1992; 42: 647–56.10.1038/ki.1992.330Search in Google Scholar PubMed
[25] Han DC, Isono M, Hoffman BB, Ziyadeh FN. High glucose stimulates proliferation and collagen type I synthesis in renal cortical fibroblasts: mediation by autocrine activation of TGF-beta. J Am Soc Nephrol 1999; 10: 1891–9.10.1681/ASN.V1091891Search in Google Scholar PubMed
[26] Wang S, Skorczewski J, Feng X, Mei L, Murphy-Ullrich JE. Glucose up-regulates thrombospondin 1 gene transcription and transforming growth factor-beta activity through antagonism of cGMP-dependent protein kinase repression via upstream stimulatory factor 2. J Biol Chem 2004; 279: 34311–22.10.1074/jbc.M401629200Search in Google Scholar PubMed
[27] Riser BL, Ladson-Wofford S, Sharba A, Cortes P, Drake K, Guerin CJ, et al. TGF-beta receptor expression and binding in rat mesangial cells: modulation by glucose and cyclic mechanical strain. Kidney Int 1999; 56: 428–39.10.1046/j.1523-1755.1999.00600.xSearch in Google Scholar PubMed
[28] Tan AL, Forbes JM, Cooper ME. AGE, RAGE, and ROS in diabetic nephropathy. Semin Nephrol 2007; 27: 130–43.10.1016/j.semnephrol.2007.01.006Search in Google Scholar PubMed
[29] Chung AC, Zhang H, Kong YZ, Tan JJ, Huang XR, Kopp JB, et al. Advanced glycation end-products induce tubular CTGF via TGF-beta-independent Smad3 signaling. J Am Soc Nephrol 2010; 21: 249–60.10.1681/ASN.2009010018Search in Google Scholar PubMed PubMed Central
[30] Rodriguez-Vita J, Sanchez-Lopez E, Esteban V, Ruperez M, Egido J, Ruiz-Ortega M. Angiotensin II activates the Smad pathway in vascular smooth muscle cells by a transforming growth factor-beta-independent mechanism. Circulation 2005; 111: 2509–17.10.1161/01.CIR.0000165133.84978.E2Search in Google Scholar PubMed
[31] Wang W, Huang XR, Canlas E, Oka K, Truong LD, Deng C, et al. Essential role of Smad3 in angiotensin II-induced vascular fibrosis. Circ Res 2006; 98: 1032–9.10.1161/01.RES.0000218782.52610.dcSearch in Google Scholar PubMed PubMed Central
[32] Yang F, Chung AC, Huang XR, Lan HY. Angiotensin II induces connective tissue growth factor and collagen I expression via transforming growth factor-beta-dependent and -independent Smad pathways: the role of Smad3. Hypertension 2009; 54: 877–84.10.1161/HYPERTENSIONAHA.109.136531Search in Google Scholar PubMed
[33] Gibbons GH, Pratt RE, Dzau VJ. Vascular smooth muscle cell hypertrophy vs. hyperplasia. Autocrine transforming growth factor-beta 1 expression determines growth response to angiotensin II. J Clin Invest 1992; 90: 456–61.10.1172/JCI115881Search in Google Scholar PubMed PubMed Central
[34] Weigert C, Brodbeck K, Klopfer K, Haring HU, Schleicher ED. Angiotensin II induces human TGF-beta 1 promoter activation: similarity to hyperglycaemia. Diabetologia 2002; 45: 890–8.10.1007/s00125-002-0843-4Search in Google Scholar PubMed
[35] Wang C, Yatsuya H, Tamakoshi K, Uemura M, Li Y, Wada K, et al. Positive association between high-sensitivity C-reactive protein and incidence of type 2 diabetes mellitus in Japanese workers: 6-year follow-up. Diabetes Metab Res Rev 2013; 29: 398–405.10.1002/dmrr.2406Search in Google Scholar PubMed
[36] Overgaard AJ, McGuire JN, Hovind P, Parving HH, Rossing P, Pociot F. Serum amyloid A and C-reactive protein levels may predict microalbuminuria and macroalbuminuria in newly diagnosed type 1 diabetic patients. J Diabetes Complications 2013; 27: 59–63.10.1016/j.jdiacomp.2012.06.016Search in Google Scholar PubMed
[37] You YK, Huang XR, Chen HY, Lyu XF, Liu HF, Lan HY. C-reactive protein promotes diabetic kidney disease in db/db mice via the CD32b-Smad3-mTOR signaling pathway. Sci Rep 2016; 6: 26740.10.1038/srep26740Search in Google Scholar PubMed PubMed Central
[38] Liu F, Chen HY, Huang XR, Chung AC, Zhou L, Fu P, et al. C-reactive protein promotes diabetic kidney disease in a mouse model of type 1 diabetes. Diabetologia 2011; 54: 2713–23.10.1007/s00125-011-2237-ySearch in Google Scholar PubMed
[39] Su Y, Chen Q, Ju Y, Li W, Li W. Palmitate induces human glomerular mesangial cells fibrosis through CD36-mediated transient receptor potential canonical channel 6/nuclear factor of activated T cell 2 activation. Biochim Biophys Acta Mol Cell Biol Lipids 2020; 1865: 158793.10.1016/j.bbalip.2020.158793Search in Google Scholar PubMed
[40] Sharma K, Ziyadeh FN. Hyperglycemia and diabetic kidney disease. The case for transforming growth factor-beta as a key mediator. Diabetes 1995; 44: 1139–46.10.2337/diabetes.44.10.1139Search in Google Scholar
[41] Wogensen L, Nielsen CB, Hjorth P, Rasmussen LM, Nielsen AH, Gross K, et al. Under control of the Ren-1c promoter, locally produced transforming growth factor-beta1 induces accumulation of glomerular extracellular matrix in transgenic mice. Diabetes 1999; 48: 182–92.10.2337/diabetes.48.1.182Search in Google Scholar PubMed
[42] Krag S, Osterby R, Chai Q, Nielsen CB, Hermans C, Wogensen L. TGF-beta1-induced glomerular disorder is associated with impaired concentrating ability mimicking primary glomerular disease with renal failure in man. Lab Invest 2000; 80: 1855–68.10.1038/labinvest.3780196Search in Google Scholar PubMed
[43] Han DC, Hoffman BB, Hong SW, Guo J, Ziyadeh FN. Therapy with antisense TGF-beta1 oligodeoxynucleotides reduces kidney weight and matrix mRNAs in diabetic mice. Am J Physiol Renal Physiol 2000; 278: F628–34.10.1152/ajprenal.2000.278.4.F628Search in Google Scholar PubMed
[44] Sharma K, Jin Y, Guo J, Ziyadeh FN. Neutralization of TGF-beta by anti-TGF-beta antibody attenuates kidney hypertrophy and the enhanced extracellular matrix gene expression in STZ-induced diabetic mice. Diabetes 1996; 45: 522–30.10.2337/diabetes.45.4.522Search in Google Scholar
[45] Ziyadeh FN, Hoffman BB, Han DC, Iglesias-De La Cruz MC, Hong SW, Isono M, et al. Long-term prevention of renal insufficiency, excess matrix gene expression, and glomerular mesangial matrix expansion by treatment with monoclonal antitransforming growth factor-beta antibody in db/db diabetic mice. Proc Natl Acad Sci U S A 2000; 97: 8015–20.10.1073/pnas.120055097Search in Google Scholar PubMed PubMed Central
[46] Hill C, Flyvbjerg A, Rasch R, Bak M, Logan A. Transforming growth factor-beta2 antibody attenuates fibrosis in the experimental diabetic rat kidney. J Endocrinol 2001; 170: 647–51.10.1677/joe.0.1700647Search in Google Scholar PubMed
[47] Russo LM, del Re E, Brown D, Lin HY. Evidence for a role of transforming growth factor (TGF)-beta1 in the induction of postglomerular albuminuria in diabetic nephropathy: amelioration by soluble TGF-beta type II receptor. Diabetes 2007; 56: 380–8.10.2337/db06-1018Search in Google Scholar PubMed
[48] Ziyadeh FN, Sharma K, Ericksen M, Wolf G. Stimulation of collagen gene expression and protein synthesis in murine mesangial cells by high glucose is mediated by autocrine activation of transforming growth factor-beta. J Clin Invest 1994; 93: 536–42.10.1172/JCI117004Search in Google Scholar PubMed PubMed Central
[49] Kim HW, Kim BC, Song CY, Kim JH, Hong HK, Lee HS. Heterozygous mice for TGF-betaIIR gene are resistant to the progression of streptozotocin-induced diabetic nephropathy. Kidney Int 2004; 66: 1859–65.10.1111/j.1523-1755.2004.00959.xSearch in Google Scholar PubMed
[50] Meng XM, Huang XR, Chung AC, Qin W, Shao X, Igarashi P, et al. Smad2 protects against TGF-beta/Smad3-mediated renal fibrosis. J Am Soc Nephrol 2010; 21: 1477–87.10.1681/ASN.2009121244Search in Google Scholar PubMed PubMed Central
[51] Sato M, Muragaki Y, Saika S, Roberts AB, Ooshima A. Targeted disruption of TGF-beta1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction. J Clin Invest 2003; 112: 1486–94.10.1172/JCI200319270Search in Google Scholar
[52] Zhou L, Fu P, Huang XR, Liu F, Chung AC, Lai KN, et al. Mechanism of chronic aristolochic acid nephropathy: role of Smad3. Am J Physiol Renal Physiol 2010; 298: F1006–17.10.1152/ajprenal.00675.2009Search in Google Scholar PubMed
[53] Liu Z, Huang XR, Lan HY. Smad3 mediates ANG II-induced hypertensive kidney disease in mice. Am J Physiol Renal Physiol 2012; 302: F986–97.10.1152/ajprenal.00595.2011Search in Google Scholar PubMed
[54] Phanish MK, Wahab NA, Colville-Nash P, Hendry BM, Dockrell ME. The differential role of Smad2 and Smad3 in the regulation of pro-fibrotic TGFbeta1 responses in human proximal-tubule epithelial cells. Biochem J 2006; 393: 601–7.10.1042/BJ20051106Search in Google Scholar
[55] Wang Y, Zhang X, Mao Y, Liang L, Liu L, Peng W, et al. Smad2 and Smad3 play antagonistic roles in high glucose-induced renal tubular fibrosis via the regulation of SnoN. Exp Mol Pathol 2020; 113: 104375.10.1016/j.yexmp.2020.104375Search in Google Scholar
[56] Xu BH, Sheng J, You YK, Huang XR, Ma RCW, Wang Q, et al. Deletion of Smad3 prevents renal fibrosis and inflammation in type 2 diabetic nephropathy. Metabolism 2020; 103: 154013.10.1016/j.metabol.2019.154013Search in Google Scholar
[57] Loeffler I, Liebisch M, Allert S, Kunisch E, Kinne RW, Wolf G. FSP1-specific SMAD2 knockout in renal tubular, endothelial, and interstitial cells reduces fibrosis and epithelial-to-mesenchymal transition in murine STZ-induced diabetic nephropathy. Cell Tissue Res 2018; 372: 115–33.10.1007/s00441-017-2754-1Search in Google Scholar
[58] Hou CC, Wang W, Huang XR, Fu P, Chen TH, Sheikh-Hamad D, et al. Ultrasound-microbubble-mediated gene transfer of inducible Smad7 blocks transforming growth factor-beta signaling and fibrosis in rat remnant kidney. Am J Pathol 2005; 166: 761–71.10.1016/S0002-9440(10)62297-3Search in Google Scholar
[59] Ka SM, Huang XR, Lan HY, Tsai PY, Yang SM, Shui HA, et al. Smad7 gene therapy ameliorates an autoimmune crescentic glomerulonephritis in mice. J Am Soc Nephrol 2007; 18: 1777–88.10.1681/ASN.2006080901Search in Google Scholar PubMed
[60] Lan HY, Mu W, Tomita N, Huang XR, Li JH, Zhu HJ, et al. Inhibition of renal fibrosis by gene transfer of inducible Smad7 using ultrasound-microbubble system in rat UUO model. J Am Soc Nephrol 2003; 14: 1535–48.10.1097/01.ASN.0000067632.04658.B8Search in Google Scholar
[61] Ng YY, Hou CC, Wang W, Huang XR, Lan HY. Blockade of NFkappaB activation and renal inflammation by ultrasound-mediated gene transfer of Smad7 in rat remnant kidney. Kidney Int Suppl 2005; S83–91.10.1111/j.1523-1755.2005.09421.xSearch in Google Scholar PubMed
[62] Wang W, Huang XR, Li AG, Liu F, Li JH, Truong LD, et al. Signaling mechanism of TGF-beta1 in prevention of renal inflammation: role of Smad7. J Am Soc Nephrol 2005; 16: 1371–83.10.1681/ASN.2004121070Search in Google Scholar PubMed
[63] Dai XY, Zhou L, Huang XR, Fu P, Lan HY. Smad7 protects against chronic aristolochic acid nephropathy in mice. Oncotarget 2015; 6: 11930–44.10.18632/oncotarget.3718Search in Google Scholar PubMed PubMed Central
[64] Liu GX, Li YQ, Huang XR, Wei L, Chen HY, Shi YJ, et al. Disruption of Smad7 promotes ANG II-mediated renal inflammation and fibrosis via Sp1-TGF-beta/Smad3-NF.kappaB-dependent mechanisms in mice. PLoS One 2013; 8: e53573.10.1371/journal.pone.0053573Search in Google Scholar PubMed PubMed Central
[65] Liu GX, Li YQ, Huang XR, Wei LH, Zhang Y, Feng M, et al. Smad7 inhibits AngII-mediated hypertensive nephropathy in a mouse model of hypertension. Clin Sci (Lond) 2014; 127: 195–208.10.1042/CS20130706Search in Google Scholar PubMed
[66] Chung AC, Huang XR, Zhou L, Heuchel R, Lai KN, Lan HY. Disruption of the Smad7 gene promotes renal fibrosis and inflammation in unilateral ureteral obstruction (UUO) in mice. Nephrol Dial Transplant 2009; 24: 1443–54.10.1093/ndt/gfn699Search in Google Scholar PubMed
[67] Wang A, Ziyadeh FN, Lee EY, Pyagay PE, Sung SH, Sheardown SA, et al. Interference with TGF-beta signaling by Smad3-knockout in mice limits diabetic glomerulosclerosis without affecting albuminuria. Am J Physiol Renal Physiol 2007; 293: F1657–65.10.1152/ajprenal.00274.2007Search in Google Scholar PubMed
[68] Ka SM, Yeh YC, Huang XR, Chao TK, Hung YJ, Yu CP, et al. Kidney-targeting Smad7 gene transfer inhibits renal TGF-beta/MAD homologue (SMAD) and nuclear factor kappaB (NF-kappaB) signalling pathways, and improves diabetic nephropathy in mice. Diabetologia 2012; 55: 509–19.10.1007/s00125-011-2364-5Search in Google Scholar PubMed
[69] Chen HY, Huang XR, Wang W, Li JH, Heuchel RL, Chung AC, et al. The protective role of Smad7 in diabetic kidney disease: mechanism and therapeutic potential. Diabetes 2011; 60: 590–601.10.2337/db10-0403Search in Google Scholar PubMed PubMed Central
[70] Li JH, Zhu HJ, Huang XR, Lai KN, Johnson RJ, Lan HY. Smad7 inhibits fibrotic effect of TGF-Beta on renal tubular epithelial cells by blocking Smad2 activation. J Am Soc Nephrol 2002; 13: 1464–72.10.1097/01.ASN.0000014252.37680.E4Search in Google Scholar PubMed
[71] Li JH, Huang XR, Zhu HJ, Johnson R, Lan HY. Role of TGF-beta signaling in extracellular matrix production under high glucose conditions. Kidney Int 2003; 63: 2010–9.10.1046/j.1523-1755.2003.00016.xSearch in Google Scholar PubMed
[72] Lin N, Ji Z, Huang C. Smad7 alleviates glomerular mesangial cell proliferation via the ROS-NF-kappaB pathway. Exp Cell Res 2017; 361: 210–6.10.1016/j.yexcr.2017.10.003Search in Google Scholar PubMed
[73] Xiao X, Gaffar I, Guo P, Wiersch J, Fischbach S, Peirish L, et al. M2 macrophages promote beta-cell proliferation by up-regulation of SMAD7. Proc Natl Acad Sci U S A 2014; 111: E1211–20.10.1073/pnas.1321347111Search in Google Scholar PubMed PubMed Central
[74] Lan HY. Smad7 as a therapeutic agent for chronic kidney diseases. Front Biosci 2008; 13: 4984–92.10.2741/3057Search in Google Scholar PubMed
[75] Jarroux J, Morillon A, Pinskaya M. History, discovery, and classification of lncRNAs. Adv Exp Med Biol 2017; 1008: 1–46.10.1007/978-981-10-5203-3_1Search in Google Scholar PubMed
[76] Chung AC, Yu X, Lan HY. MicroRNA and nephropathy: emerging concepts. Int J Nephrol Renovasc Dis 2013; 6: 169–79.10.2147/IJNRD.S37885Search in Google Scholar PubMed PubMed Central
[77] Chung AC, Lan HY. MicroRNAs in renal fibrosis. Front Physiol 2015; 6: 50.10.3389/fphys.2015.00050Search in Google Scholar PubMed PubMed Central
[78] Zhong X, Chung AC, Chen HY, Meng XM, Lan HY. Smad3-mediated upregulation of miR-21 promotes renal fibrosis. J Am Soc Nephrol 2011; 22: 1668–81.10.1681/ASN.2010111168Search in Google Scholar PubMed PubMed Central
[79] Zhong X, Chung AC, Chen HY, Dong Y, Meng XM, Li R, et al. miR-21 is a key therapeutic target for renal injury in a mouse model of type 2 diabetes. Diabetologia 2013; 56: 663–74.10.1007/s00125-012-2804-xSearch in Google Scholar PubMed
[80] Kato M, Zhang J, Wang M, Lanting L, Yuan H, Rossi JJ, et al. MicroRNA-192 in diabetic kidney glomeruli and its function in TGF-beta-induced collagen expression via inhibition of E-box repressors. Proc Natl Acad Sci U S A 2007; 104: 3432–7.10.1073/pnas.0611192104Search in Google Scholar PubMed PubMed Central
[81] Chung AC, Huang XR, Meng X, Lan HY. miR-192 mediates TGF-beta/Smad3-driven renal fibrosis. J Am Soc Nephrol 2010; 21: 1317–25.10.1681/ASN.2010020134Search in Google Scholar PubMed PubMed Central
[82] Wang Q, Wang Y, Minto AW, Wang J, Shi Q, Li X, et al. MicroRNA-377 is up-regulated and can lead to increased fibronectin production in diabetic nephropathy. FASEB J 2008; 22: 4126–35.10.1096/fj.08-112326Search in Google Scholar PubMed PubMed Central
[83] Zhang Y, Huang XR, Wei LH, Chung AC, Yu CM, Lan HY. miR-29b as a therapeutic agent for angiotensin II-induced cardiac fibrosis by targeting TGF-beta/Smad3 signaling. Mol Ther 2014; 22: 974–85.10.1038/mt.2014.25Search in Google Scholar PubMed PubMed Central
[84] Wang J, Chu ES, Chen HY, Man K, Go MY, Huang XR, et al. microRNA-29b prevents liver fibrosis by attenuating hepatic stellate cell activation and inducing apoptosis through targeting PI3K/AKT pathway. Oncotarget 2015; 6: 7325–38.10.18632/oncotarget.2621Search in Google Scholar PubMed PubMed Central
[85] Qin W, Chung AC, Huang XR, Meng XM, Hui DS, Yu CM, et al. TGF-beta/Smad3 signaling promotes renal fibrosis by inhibiting miR-29. J Am Soc Nephrol 2011; 22: 1462–74.10.1681/ASN.2010121308Search in Google Scholar PubMed PubMed Central
[86] Xiao J, Meng XM, Huang XR, Chung AC, Feng YL, Hui DS, et al. miR-29 inhibits bleomycin-induced pulmonary fibrosis in mice. Mol Ther 2012; 20: 1251–60.10.1038/mt.2012.36Search in Google Scholar PubMed PubMed Central
[87] Chen HY, Zhong X, Huang XR, Meng XM, You Y, Chung AC, et al. MicroRNA-29b inhibits diabetic nephropathy in db/db mice. Mol Ther 2014; 22: 842–53.10.1038/mt.2013.235Search in Google Scholar PubMed PubMed Central
[88] Du B, Ma LM, Huang MB, Zhou H, Huang HL, Shao P, et al. High glucose down-regulates miR-29a to increase collagen IV production in HK-2 cells. FEBS Lett 2010; 584: 811–6.10.1016/j.febslet.2009.12.053Search in Google Scholar PubMed
[89] Tung CW, Ho C, Hsu YC, Huang SC, Shih YH, Lin CL. MicroRNA-29a attenuates diabetic glomerular injury through modulating cannabinoid receptor 1 signaling. Molecules 2019; 24: 264.10.3390/molecules24020264Search in Google Scholar PubMed PubMed Central
[90] Wang B, Koh P, Winbanks C, Coughlan MT, McClelland A, Watson A, et al. miR-200a Prevents renal fibrogenesis through repression of TGF-beta2 expression. Diabetes 2011; 60: 280–7.10.2337/db10-0892Search in Google Scholar PubMed PubMed Central
[91] Ahn SM, Cha JY, Kim J, Kim D, Trang HT, Kim YM, et al. Smad3 regulates E-cadherin via miRNA-200 pathway. Oncogene 2012; 31: 3051–9.10.1038/onc.2011.484Search in Google Scholar PubMed
[92] Guttman M, Rinn JL. Modular regulatory principles of large non-coding RNAs. Nature 2012; 482: 339–46.10.1038/nature10887Search in Google Scholar PubMed PubMed Central
[93] Tang PM, Zhang YY, Lan HY. LncRNAs in TGF-beta-driven tissue fibrosis. Noncoding RNA 2018; 4: 26.10.3390/ncrna4040026Search in Google Scholar
[94] Zhou Q, Chung AC, Huang XR, Dong Y, Yu X, Lan HY. Identification of novel long noncoding RNAs associated with TGF-beta/Smad3-mediated renal inflammation and fibrosis by RNA sequencing. Am J Pathol 2014; 184: 409–17.10.1016/j.ajpath.2013.10.007Search in Google Scholar PubMed
[95] Feng M, Tang PM, Huang XR, Sun SF, You YK, Xiao J, et al. TGF-beta mediates renal fibrosis via the Smad3-Erbb4-IR long non-coding RNA axis. Mol Ther 2018; 26: 148–61.10.1016/j.ymthe.2017.09.024Search in Google Scholar
[96] Sun SF, Tang PMK, Feng M, Xiao J, Huang XR, Li P, et al. Novel lncRNA Erbb4-IR promotes diabetic kidney injury in db/db mice by targeting miR-29b. Diabetes 2018; 67: 731–44.10.2337/db17-0816Search in Google Scholar
[97] Zhang YY, Tang PM, Tang PC, Xiao J, Huang XR, Yu C, et al. LRNA9884, a novel Smad3-dependent long noncoding RNA, promotes diabetic kidney injury in db/db mice via enhancing MCP-1-dependent renal inflammation. Diabetes 2019; 68: 1485–98.10.2337/db18-1075Search in Google Scholar
[98] Zhou Q, Huang XR, Yu J, Yu X, Lan HY. Long noncoding RNA Arid2-IR is a novel therapeutic target for renal inflammation. Mol Ther 2015; 23: 1034–43.10.1038/mt.2015.31Search in Google Scholar
[99] Fujimoto M, Maezawa Y, Yokote K, Joh K, Kobayashi K, Kawamura H, et al. Mice lacking Smad3 are protected against streptozotocin-induced diabetic glomerulopathy. Biochem Biophys Res Commun 2003; 305: 1002–7.10.1016/S0006-291X(03)00885-4Search in Google Scholar
[100] Yang X, Letterio JJ, Lechleider RJ, Chen L, Hayman R, Gu H, et al. Targeted disruption of SMAD3 results in impaired mucosal immunity and diminished T cell responsiveness to TGF-beta. EMBO J 1999; 18: 1280–91.10.1093/emboj/18.5.1280Search in Google Scholar PubMed PubMed Central
[101] Petersen M, Thorikay M, Deckers M, van Dinther M, Grygielko ET, Gellibert F, et al. Oral administration of GW788388, an inhibitor of TGF-beta type I and II receptor kinases, decreases renal fibrosis. Kidney Int 2008; 73: 705–15.10.1038/sj.ki.5002717Search in Google Scholar PubMed
[102] Sato S, Kawamura H, Takemoto M, Maezawa Y, Fujimoto M, Shimoyama T, et al. Halofuginone prevents extracellular matrix deposition in diabetic nephropathy. Biochem Biophys Res Commun 2009; 379: 411–6.10.1016/j.bbrc.2008.12.088Search in Google Scholar PubMed
[103] Li J, Qu X, Yao J, Caruana G, Ricardo SD, Yamamoto Y, et al. Blockade of endothelial-mesenchymal transition by a Smad3 inhibitor delays the early development of streptozotocin-induced diabetic nephropathy. Diabetes 2010; 59: 2612–24.10.2337/db09-1631Search in Google Scholar PubMed PubMed Central
[104] Chung JYF, Tang PMK, Chan MKK, Wang L, Huang XR, To KF, et al. AANG prevents Smad3-dependent diabetic nephropathy by restoring pancreatic β-Cell development in db/db mice. Int J Biol Sci (In press).10.7150/ijbs.72977Search in Google Scholar PubMed PubMed Central
[105] Zhang W, Huai Y, Miao Z, Qian A, Wang Y. Systems pharmacology for investigation of the mechanisms of action of traditional Chinese medicine in drug discovery. Front Pharmacol 2019; 10: 743.10.3389/fphar.2019.00743Search in Google Scholar PubMed PubMed Central
[106] Huang KC, Su YC, Sun MF, Huang ST. Chinese herbal medicine improves the long-term survival rate of patients with chronic kidney disease in Taiwan: a nationwide retrospective population-based cohort study. Front Pharmacol 2018; 9: 1117.10.3389/fphar.2018.01117Search in Google Scholar PubMed PubMed Central
[107] Zhao T, Sun S, Zhang H, Huang X, Yan M, Dong X, et al. Therapeutic effects of tangshen formula on diabetic nephropathy in rats. PLoS One 2016; 11: e0147693.10.1371/journal.pone.0147693Search in Google Scholar PubMed PubMed Central
[108] Li P, Chen Y, Liu J, Hong J, Deng Y, Yang F, et al. Efficacy and safety of tangshen formula on patients with type 2 diabetic kidney disease: a multicenter double-blinded randomized placebo-controlled trial. PLoS One 2015; 10: e0126027.10.1371/journal.pone.0126027Search in Google Scholar PubMed PubMed Central
[109] Zhao TT, Zhang HJ, Lu XG, Huang XR, Zhang WK, Wang H, et al. Chaihuang-Yishen granule inhibits diabetic kidney disease in rats through blocking TGF-beta/Smad3 signaling. PLoS One 2014; 9: e90807.10.1371/journal.pone.0090807Search in Google Scholar PubMed PubMed Central
[110] Sun SF, Zhao TT, Zhang HJ, Huang XR, Zhang WK, Zhang L, et al. Renoprotective effect of berberine on type 2 diabetic nephropathy in rats. Clin Exp Pharmacol Physiol 2015; 42: 662–70.10.1111/1440-1681.12402Search in Google Scholar PubMed
[111] Meng XM, Zhang Y, Huang XR, Ren GL, Li J, Lan HY. Treatment of renal fibrosis by rebalancing TGF-beta/Smad signaling with the combination of asiatic acid and naringenin. Oncotarget 2015; 6: 36984–97.10.18632/oncotarget.6100Search in Google Scholar PubMed PubMed Central
© 2022 Li Wang et al., published by Sciendo
This work is licensed under the Creative Commons Attribution-NonCommercial-NoDerivatives 3.0 License.
Articles in the same Issue
- Perspective
- Podocyte developmental pathways in diabetic nephropathy: A spotlight on Notch signaling
- Review
- TGF-β signaling in diabetic nephropathy: An update
- Original Article
- The role of vitamin D receptor agonist on podocyte injury induced by high glucose
- The application effect of the trans-theoretical model of behavior change in diabetic kidney disease patients treated with maintenance hemodialysis
- Correlation analysis between Tervaert glomerular classification and clinical indicators in patients with type 2 diabetic nephropathy
Articles in the same Issue
- Perspective
- Podocyte developmental pathways in diabetic nephropathy: A spotlight on Notch signaling
- Review
- TGF-β signaling in diabetic nephropathy: An update
- Original Article
- The role of vitamin D receptor agonist on podocyte injury induced by high glucose
- The application effect of the trans-theoretical model of behavior change in diabetic kidney disease patients treated with maintenance hemodialysis
- Correlation analysis between Tervaert glomerular classification and clinical indicators in patients with type 2 diabetic nephropathy