Hormones and endometrial carcinogenesis
-
Areege Kamal


Abstract
Endometrial cancer (EC) is the commonest gynaecological cancer in the Western World with an alarmingly increasing incidence related to longevity and obesity. Ovarian hormones regulate normal human endometrial cell proliferation, regeneration and function therefore are implicated in endometrial carcinogenesis directly or via influencing other hormones and metabolic pathways. Although the role of unopposed oestrogen in the pathogenesis of EC has received considerable attention, the emerging role of other hormones in this process, such as androgens and gonadotropin-releasing hormones (GnRH) is less well recognised. This review aims to consolidate the current knowledge of the involvement of the three main endogenous ovarian hormones (oestrogens, progesterone and androgens) as well as the other hormones in endometrial carcinogenesis, to identify important avenues for future research.
Introduction
Endometrial cancer (EC) is the commonest gynaecological cancer in Europe and has an alarmingly increasing incidence related to longevity and obesity [1, 2]. The European estimates suggest, by 2025, an incidence increase of EC in the range of 50%–100% will occur, relative to the observed incidence in 2005 [3]. Many women with EC are postmenopausal (PM) and not suitable for standard surgical treatment because of other commonly coexisting medical problems, such as obesity, hypertension and diabetes. Latest national figures in the UK show that one in four women undergoing major gynaecological surgery for cancer experience serious surgical complications [4]. The 5-year survival rates for advanced EC are 23%, worse than for other common gynaecological cancers, and similar to that of ovarian cancer [5]. Current therapeutics fail to treat late stage disease, and preventing advanced disease and distant metastasis needs early diagnosis of ECs. Identification of those ECs likely to metastasise and treating them with adjuvant therapies is imperative to tackle the impending economic and social burden caused by the increasing incidence of EC predicted over the next decade [3].
Ovarian hormones regulate normal human endometrial cell proliferation, regeneration and function, therefore are implicated in endometrial carcinogenesis. Furthermore, recent evidence suggests that all types of ECs may share common etiological factors, including their response to/stimulation by oestrogen and other ovarian steroid hormones [6]. Although many reviews previously describe the role of oestrogen and progesterone in endometrial carcinogenesis, they have largely disregarded the involvement of androgens and other hormones. Recent reports however, describe a role for androgens and its receptor [7, 8], gonadotropin-releasing hormone (GnRH) and luteinizing hormones (LH) in EC [9, 10]. This review aims to explore the current knowledge on how endogenous and exogenous hormones, influence endometrial carcinogenesis.
Method
We carried out PubMed (Medline) and Ovid searches systematically for publications from November 2000 until November 2015. Keywords used included: EC with hormones, risk factors for EC, obesity, adipose, polycystic ovarian syndrome (PCOS), lynch syndrome (LS), diabetes, Parkinson’s disease (PD), pregnancy, menarche, menopause, combined oral contraceptive pills (COCPs), oestrogens, progestogens, selective oestrogen receptor modulators (SERMs), tamoxifen, selective progesterone receptor modulators (SPRMs), aromatase inhibitors, fulvestrant, hormone replacement therapy (HRT), GnRH analogues/antagonists, LH, danazol and androgens. All studies investigating hormonal influences in EC in women, animals and endometrial cell lines, either primary cells or tissue explants in culture were considered. Further manuscripts published before November 2000 were also reviewed for specific topic areas and included as appropriate.
Human endometrium as a hormone receptive organ
Anatomy/histology
Embryonic Mullerian ducts fuse in the midline to form the human uterus and the inner layer of its mesenchyme forms the endometrium. Initially, the superficial endometrial glands originate from the fetal undifferentiated, single columnar epithelial cells in utero. The histological architecture of the endometrium at birth is analogous to the PM endometrium yet the glands remain to be shallow and superficial [11]. Humans (and other upper order primates) menstruate, and have two distinct layers of the endometrium, making them unique, amongst other mammals. The transient superficial stratum functionalis exists only during the reproductive life of the woman whilst the structurally permanent deeper stratum basalis adjacent to the myometrium remains throughout life [12]. Endometrium consists of a variety of cell types including epithelial, stromal, endothelial and leucocyte cell populations. As most ECs are carcinomas, in the context of carcinogenesis, the critical endometrial cell type is the epithelial cells. Endometrial epithelium is also organised into distinct groups of cells (i) luminal epithelium, that lines the superficial surface of the uterine cavity, (ii) the hormone responsive, fully differentiated functionalis glands and (iii) the deeper SSEA-1 expressing basalis glandular epithelium [13]. Presumably, a new functionalis layer is generated each month from the remaining basalis after menstrual shedding, hence the basalis is proposed to harbour stem/progenitor cells (SPCs) [12]. ECs commonly occur in the PM endometrium, which is essentially the retained luminal and basalis epithelium. SPCs are thought to play a role in carcinogenesis and these epithelial layers are also their postulated location [14]. The particular endometrial epithelial cell group(s) that gives rise to ECs is not yet known. However, we can conclude that either luminal or basalis glandular compartments are likely to be the common epithelial origin of ECs.
Overview of normal endometrial endocrinology
Endometrium is the primary recipient organ for ovarian steroid hormonal signal and is intricately responsive to these hormones. The three main classical ovarian steroid hormones, oestrogens, progesterone and androgens exert their effects in the endometrial cells mainly via their cognate receptors [12, 15, 16]. Endometrium expresses both subtypes of classical nuclear oestrogen receptors (ERα and ERβ). Progesterone, acts via the progesterone receptor subtypes PRA and PRB. Androgens operate via androgen receptor (AR) and all these receptors belong to the Class I nuclear hormone receptor superfamily of ligand-inducible transcription factors that share the common, evolutionarily conserved structural and functionally distinct domains of other superfamily members [12]. These classical hormone receptors are activated upon ligand binding and may exert effects involving the classical hormone-signalling pathway [12, 15]. This involves steroid receptor dimerising, translocating to the nucleus, binding to the respective hormone responsive element located in the relevant gene promoters to initiate recruitment of co-activators, co-repressors and chromatin remodelling factors to either activate or repress transcription of target genes. Post-translational modifications of the steroid hormone receptors will also affect gene promoter targeting and subsequent target gene transcription.
The general consensus is that oestrogen induces the expression of all endometrial steroid receptor types via the action of ERα whilst progesterone may downregulate them via PR (except for ERβ) [12, 17] (Figure 1). Androgens acting via AR induce their own receptor expression [18, 19] but AR-mediated effect on the other hormone receptors in the endometrium is not yet known. The expression profile of these different steroid receptors and the levels of respective hormones at the cellular level are therefore important to ascertain the full impact of hormones on the endometrium (Figure 1).
![Figure 1: Correlation of typical hormonal changes of pituitary/ovarian axis with the consequential endometrial morphology and steroid hormone expression in women.Pituitary and ovarian hormones show typical cyclical variations during the premenopausal period and these change at menopause. The correspondent endometrial morphology and epithelial steroid receptors expression in normal [pre and postmenopausal (PM)], premalignant [endometrial hyperplasia (EH) with atypia] and malignant (low-grade endometrial cancer, LGEC and high-grade endometrial cancer, HGEC) endometrium is shown in the panels.](/document/doi/10.1515/hmbci-2016-0005/asset/graphic/j_hmbci-2016-0005_fig_001.jpg)
Correlation of typical hormonal changes of pituitary/ovarian axis with the consequential endometrial morphology and steroid hormone expression in women.
Pituitary and ovarian hormones show typical cyclical variations during the premenopausal period and these change at menopause. The correspondent endometrial morphology and epithelial steroid receptors expression in normal [pre and postmenopausal (PM)], premalignant [endometrial hyperplasia (EH) with atypia] and malignant (low-grade endometrial cancer, LGEC and high-grade endometrial cancer, HGEC) endometrium is shown in the panels.
The intra-cellular levels of steroid hormone metabolising or activating enzymes may also play a pivotal role in regulating the final effect of these hormones [20]. To add another layer of complexity, some hormone receptors make heterodimers upon hormone/ligand binding. For example, ERα dimerise with ERβ and the effect of oestrogen displayed by cells expressing equal amount of both receptors will be different to another expressing more of a particular subtype of these receptors [12]. Furthermore, most class I hormone receptors can bind to the other ovarian steroid hormones with a lower affinity due to the significant homology between their ligand binding domains [21]. Therefore, when some steroid hormones are abundant, they may also exert some effects via a different steroid receptor, as well as acting through their specific cognate receptor.
Hormonal milieu of premenopausal endometrium
The prepubertal ovary is inactive, hence, the thin endometrium grows very slowly from birth until puberty due to the lack of ovarian hormonal (mainly oestrogenic) signal [12]. In response to the increasing levels in ovarian oestrogen and adrenal androgens observed at puberty, endometrium acquires the typical adult histo-anatomical configuration, and with the oestrogenic stimuli in the proliferative phase grows a functionalis layer that differentiates with the subsequent exposure to progesterone produced by the corpus luteum [22]. At the end of an infertile cycle, with luteolysis, the drop in progesterone (and oestrogen) initiates an inflammatory cascade, vasoactive and hypoxic events result in shedding of the functionalis as menstruation, followed by epithelialisation and regrowth of the functionalis to begin the next endometrial cycle [23] (Figure 1). Ovarian production of androgens is also reported to follow a cyclical pattern, yet the exact function of androgens in the normal endometrial function and regeneration is not fully described.
Hormonal milieu of PM endometrium
Similar to the prepubertal endometrium, the main histological characteristic of the PM endometrium is the complete loss of stratum functionalis. It is composed of inactive glands lying in compact stroma that morphologically resembles the stratum basalis of premenopausal endometrium [13, 24]. The PM hormonal milieu is characterised by the absence of progesterone and oestradiol production by the ovaries, persisting levels of androgens from the adrenals and the presence of low levels of circulating oestrone produced by the extra-ovarian aromatisation of adrenal androgens [20] (Figure 1). This low level of oestrogenic signal supports the maintenance of hormone receptor expression observed in the PM endometrium hence the hormone responsiveness [25].
Hormonal aberrations associated with EC
Oestrogen and insulin in excess and lack of progesterone have been proposed as the main hormonal aberrations to affect endometrial proliferation and cell survival that can result in increasing the risk of epithelial cell transformation/carcinogenesis. This section considers these hormonal aberrations in detail in the context of EC (Figure 2).
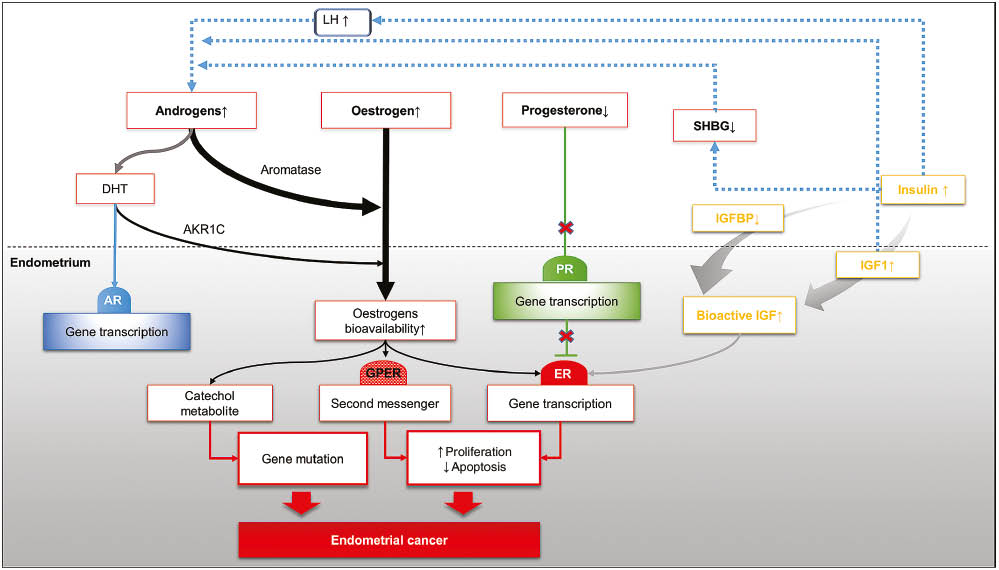
Different hormonal and metabolic pathways associated with disturbed steroid hormones homeostasis in favour of oestrogen pro-oncogenic pathway.
The figure illustrates how the aberrations associated with three major ovarian hormones, oestrogen, progesterone and androgens are involved in endometrial carcinogenesis.
AKR1C, aldoketoreductase1C; DHT, dihydrotestosterone; GPER, G protein coupled oestrogen receptor; IGF-1, insulin-like growth factor 1; IGFBP, insulin-like growth factor binding protein; LH, luteinising hormone; SHBG, sex hormone binding protein.
Unopposed oestrogens
As previously mentioned, oestrogen mediates endometrial cell growth, proliferation, apotosis inhibiton and angiogenesis in vitro and in vivo reviewed in [12]. ERβ may have opposing action on the classical ERα activity and is thought to play an antiproliferative role in endometrial proliferation [12]. Although not completely decoded, several pathways have been identified to exert oestrogenic action in endometrium, these include:
Genetic pathway: An activated ER induces gene transcription either directly by binding to oestrogen response element (ERE), hence via “classical oestrogen response ERE dependent” route or indirectly “non-classical, ERE independent” path via interacting with transcriptional factors such as SP-1 [26], insulin-like growth factor (IGF-1) receptor [27], epidermal growth factor (EGF) receptor [28], PA-1 [29], NF-κB [30]. ER gene transcription can also be induced through downstream factors such as EGF or IGF-I via the “ligand independent pathways” [31, 32].
Non-genetic pathways: Oestrogen activation of ERK1/2 signalling pathway [33] or MEK/ERK, MAPK pathway [34] has also been demonstrated via calcium influx and membrane located non-classical ER (GPR30), respectively, in the absence of nuclear ER expression.
Epigenetic: Hypermethylation of ESR1 and PRB promoters has been reported in 90% of the EC [35–37]. Furthermore, hypomethylation with subsequent activation of oestrogen downstream PAX2 has been described in EC and associated with increased tumour growth in vivo [38].
Mutagenetic: Several lines of evidence have shown that 4-hydroxylated oestrogen (catechol-oestrogens), catalysed by cytochrome P450 1B1, is able to induce DNA damage [39]. Accumulation of catechol oestrogens is observed with prolonged unopposed oestrogen exposure. Interestingly, catechol oestrogens have been associated with DNA damage at a specific DNA region (codon 130/131) on PTEN gene which is frequently mutated in type 1 ECs [40].
It is clear that there is an overlap in the pathways that derive oestrogen pro-oncogenic environment and we can conclude that unopposed oestrogenic stimuli, even with low levels of oestrogen may adversely affect the endometrium.
Progesterone insufficiency
Progesterone counteracts the above-mentioned trophic drive of oestrogen, therefore anovulation, or lack of synthesis of endogenous progesterone by the ovary that can occur without a perturbation of oestradiol production, is associated with an excessive and prolonged proliferation of the endometrial cells and thickening/hyperplasia of the glandular epithelium. For example, telomerase enzyme is pertinent for endometrial epithelial proliferation, is highly active in endometrial hyperplasia [EH] and in cancer and is induced by oestrogen and inhibited by progesterone [41].
The antiproliferative action of progesterone in the endometrial epithelial cells is exerted directly and indirectly via the stroma. Apart from the progesterone receptor, androgen and glucocorticoid receptors are also postulated to be involved in progesterone action in the endometrium [42]. The evidence for the stromal involvement in the antiproliferative action of progesterone comes from a tissue recombination study utilising PR knockout (PRKO) mice [43], and by selective inactivation of endometrial epithelial PR [44].
Thus, three possible mechanisms were proposed to explain progesterone-oestrogen antagonism (Figure 2) (i) via inhibiting ERα expression, (ii) via preventing stromal production of growth factors and subsequent transcription factor activation [45, 46] and (iii) via inducing apoptosis of endometrial epithelial cells through PR action in stromal cells [47].
Therefore, the deficiency of endogenous progesterone levels or diminished length of lifetime progesterone exposure (anovulation and lack of full-term pregnancy), are associated with the development of EH and increased risk of EC [48].
Hyperinsulinism
Hyperinsulinism, associated with either diabetes mellitus or PCOS, plays an important role in carcinogenesis as it potentiates mitotic activity in the glands and stroma by increasing the activity of IGF-1 [49, 50]. Insulin excess stimulates theca cell androgen activity, elevates serum free testosterone levels through decreased hepatic sex hormone-binding globulin (SHBG) production, amplifies LH and IGF-I-stimulated androgen production, and enhances serum IGF-I bioactivity through suppressed IGF-binding protein production [51, 52]. Insulin binding sites are also expressed in the endometrial stroma of women with EC [53] (Figure 2). Therefore, evidently, an excess in insulin signalling can result in endometrial changes with a pro-proliferative, pro-survival phenotype and inflammatory changes akin to unopposed oestrogen as mentioned above.
Hyperandrogenism
Although hyperandrogenism has been debated as a cause for the increased EC risk in women with PCOS, the in vitro and in vivo evidence to support carcinogenesis effect of androgens in the endometrium is weak [54].
Several studies have shown an increase in circulating androgen levels in EC patients nonetheless these studies have not ratified the EC risk after adjusting for the oestrogen levels. There are two androgen regulatory pathways active in the endometrium (Figure 2) (i) androgenic pathway via androgen receptor and (ii) oestrogenic pathway via ERs when androgens (androstendione and testosterone) are aromatised locally to oestrogenic compounds. Unlike in the normal endometrium, dominance of the second pathway in EC can be explained by the abundance of aromatase [55, 56] and aldo-keto reductase (AKR1C) [57] enzymes in the neoplastic endometrial cells, that increase the local availability of oestrogenic ligand. This is further exagerated by the relatively higher affinity of oestrogens to their cognate receptors, compared with that of androgens to AR [58]. Interestingly, the expression of aromatase observed to be higher in type II EC compared to type I, which will allow this subtype of EC to increase local oestrogen biosynthesis [59]. Emerging evidence also show AR to be a positive prognostic indicator and that its loss is associated with shorter disease-free survival [8, 25], therefore, androgenic pathway as a therapeutic target for EC requires further investigation.
Other hormones
GnRH
Altered or high levels of GnRH is known to have antiproliferative effect on the endometrium indirectly via its suppression of gonadotropin secretion and inhibition of ovarian oestradiol synthesis as well as working directly on the endometrium via GnRH receptor (GnRH-R). In vitro, GnRH has been shown to stimulate apoptosis in normal endometrial cells [60]. GnRH-R are expressed in breast and gynaecological tumours, wherein their activation by agonists results in antiproliferative, antimetastatic and antiangiogenic effect [9].
Luteinizing hormone/human chorionic gonadotropin
LH/hCG receptors (LH-R) are expressed in 80% of ECs [10, 61] in a grade-specific manner and may regulate the invasiveness of EC cells [62]. The overexpression of the LH-R increases the ability of EC cells to undergo local invasion and metastatic spread in animal models. Likewise, LH withdrawal strongly inhibits local and distant metastatic spread of tumors [63]. LH upregulates its own receptor, therefore it is an important target in relation to the PM period where the levels of LH remains elevated (Figure 1).
Prolactin (PRL) and thyroid stimulating hormone (TSH)
Pituitary hormones PRL and TSH may influence ovarian production of sex hormones. These hormones may have a potential to be used as serum biomarkers to detect EC patients from healthy controls with 98.3% sensitivity and 98% specificity [64]. When combined in a five marker panel (including PRL, growth hormone, eotaxin, E-selectin and TSH) it was possible to discriminate EC from ovarian and breast cancers [65]. Hyperprolactinemia is induced by the use of antipsychotics in premenopausal women, and antipsychotics have been proposed as an independent variable for risk of EC [66]. Further studies are needed to confirm these preliminary reports.
Melatonin
Melatonin (N-acetyl-5-methoxytryptamine), is a hormone produced primarily by the pineal gland, appears to protect against cancer development in general. Melatonin production is under regulation of the hypothalamus, with highest levels of melatonin secreted during the night and when sleeping. It is known to exert antioxidant, antimitotic, antiangiogenic activity in tissues, as well as being an immune modulator and a regulator of fat metabolism [67]. Melatonin interacts with membrane and nuclear receptors, and may be linked to the regulation of tumour growth [68, 69]. It is of particular relevance to EC as it may also block ERα and affect the activity of aromatase. Women with EC were found to have lower melatonin levels and a significantly increased relative risk of developing EC was reported in overweight night shift workers compared to lean women [70]. Melatonin administration in addition to HRT was associated with reduced body mass, intraperitoneal fat, reduced endometrial proliferation and prevented the appearance of histological atypia of the endometrium in an ovariectomised rat model, indicating melatonin may have a prophylactic role in preventing EC in PM women [71].
Endometrial cancer
Classification
Histological classification
The dualist model of EC proposed by several researchers has been widely accepted over the last three decades [72, 73]. Based on endocrine, clinical and histopathological characteristics, it categorises EC to type I oestrogen-dependent adenocarcinoma, associated with favourable outcome and endometrioid morphology and type II non-oestrogen-dependent EC, with worst outcome and serous papillary or clear cell morphology. Although this model was broadly supported by the reported specific molecular aberrations in each type (Table 1) [74, 75], it failed to define high grade type I endometrioid EC which showed a molecular pattern and outcome that blends between type I and type II EC [76]. Moreover, the persistent expression of steroid receptors, ER and to lesser extent PR in these so-called non-oestrogen-dependent EC [76–78] suggests hormonal regulation of Type II EC.
Common molecular alterations differentially associated with dualistic EC classification.
| Type I | Type II | ||
|---|---|---|---|
| PTEN mutation | 52%–78% | TP53 mutation | 60%–91% |
| PIK3CA mutation | 36%–52% | PIK3CA mutation | 24%–42% |
| PIK3R1 mutation | 21%–43% | PPP2R1A mutation | 15%–43% |
| KRAS mutation | 15%–43% | HER2 amplification | 27%–44% |
| ARID1A mutation | 25%–48% | ||
| CTNNB1 mutation | 23%–24% | ||
| Microsatellite instability | 28%–40% | ||
Molecular classification
The heterogeneity and overlap of the reported morphological and molecular abnormalities between different groups of ECs have restricted the traditional postsurgical risk group stratification (low, intermediate and high-risk group) and limited its prognostic and predictive value [79]. The recent emergence of a large cohort of endometrioid, serous and mixed ECs and subsequent reports of integrated proteomic and transcriptomic profile of them has added a new perspective to EC taxonomy [80]. The Cancer Genome Atlas (TCGA) study identified four genomic subgroups: POLE ultramutated (favourable outcome), microsatellite instability, copy number low (both medium risk/outcome) and copy number high (the worst outcome). This classification is in harmony with the dualistic model whereby POLE ultramutated, microsatellite instability and copy number low groups comprise 97% of type I EC while the copy number high group encompasses 94% serous EC. Nonetheless, 24% of grade 3 endometrioid were also resolved to the worst outcome, high copy number group confirming the previously predicted overlap between the subtypes [80]. This warrants integration of clinic-pathological, molecular and genomic parameters and a more holistic approach in the future to improve postsurgical stratification and tailoring personalised adjuvant therapy for women with EC to improve outcome [81].
Hormonal influence of the established risk factors of EC
The role of steroid hormones, principally oestrogen, in the aetiology of EC has been confirmed by experimental, clinical and epidemiological studies. Risk factors such as early menarche, late menopause, nulliparity (particularly when associated with ovulatory dysfunction) are directly linked to prolonged unopposed oestrogen exposure [82] yet indirect involvement of steroid hormone and imbalance that occur with other known risk factors has also been reported. This section highlights the possible contributions of steroid hormones to these widely accepted EC risk factors (Figure 3).
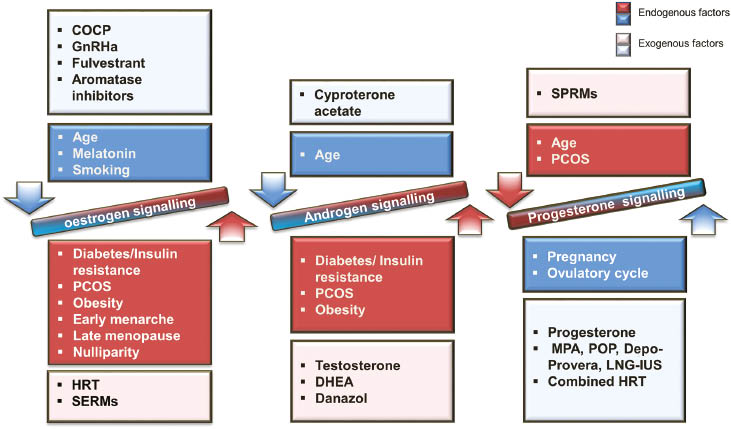
Endogenous and exogenous factors that affect steroid hormone signalling and contribute to endometrial carcinogenesis in the human endometrium.
COCP, combined oral contraceptive pills; DHEA, dehydroepiandrosterone; GnRHa, gonadotrophic-releasing hormone analogues; HRT, hormone replacement therapy; LNG-IUS, levonorgestrel-releasing intrauterine system; MPA, medroxyprogesterone acetate; PCOS, polycystic ovarian syndrome; POP, progestogen only pills; SREMs, selective oestrogen receptor modulators; SPRMS, selective progesterone receptor modulators.
Polycystic ovarian syndrome
The first report proposing a possible association between PCOS and EC appeared in 1949 [49]. Women with PCOS appear to have a three to four-fold increased risk for EC [83, 84], which translates to a lifetime risk of 9%, given the background lifetime risk of 3% in the general population [85].
PCOS is a heterogeneous hormone-imbalance disorder [86] and the commonest endocrine disease of women of reproductive age, affecting 5%–10% [87]. The pathogenesis of PCOS is multifactorial and is far from being fully understood [53]. It is a leading cause of anovulatory infertility and therefore is associated with the primary hormonal aberrations of unopposed oestrogen action, insulin resistance, hyperandrogenism and clinical features of diabetes and obesity [88, 89]. For the purpose of this review, we focused on the PCOS associated hormonal changes that have shown a causative relationship with EC, except the hormonal disturbances associated with obesity (which are independent risk factors of EC and discussed in detail in a later section).
In women with PCOS, the endometrium tends to remain in an oestrogen-mediated proliferative state due to chronic anovulation [53]. ERα and steroid receptor co-activators are overexpressed, signifying a reduced effectiveness of progesterone [90] and possibly contributing to a high sensitivity to oestrogen action [91].
The molecular mechanisms underlying endometrial progesterone resistance or sensitivity in these patients are not completely understood [86]. Impaired follicle maturation and consequent anovulation cause a chronic progesterone deficient state that affects the endometrial milieu. Alterations in the expression or function of PR co-activators, chaperones and co-chaperones that are bound to PR before activation and changes in gene expression are implicated in progesterone resistance [86, 90]. However, the classic progesterone resistance is proposed to describe the pathogenesis of endometriosis [92], and endometriosis is not associated with an increased risk of EC. Therefore, the contribution of progesterone resistance phenomenon in the pathogenesis of PCOS associated EC is debatable.
Hyperinsulinemia, a common finding in patients with PCOS regardless of body mass index (BMI) [51, 86] and anti-diabetic medications such as metformin have been used to normalise the hyperinsulinism in PCOS.
Hypersecretion of LH is also a frequent feature of PCOS and a modulator of endometrial growth, as evidenced by the ability of LH to promote growth of human EC cells in vitro. Endometrial LH receptors are overexpressed in anovulatory PCOS women with EH or EC [51].
Hyperandrogenism, as a cardinal feature of PCOS, is a common finding in EC [51]. Pituitary LH hypersecretion results in excessive theca cell stimulation and PCOS patients tend to have genetically impaired theca cell functional defect. Consequent hyperandrogenism [93] results in increased bioavailability of unopposed oestrogens due to the increased peripheral conversion [53, 94]. Peripheral aromatisation of androgens occurs preferentially in the adipose tissue and high BMI is a common feature of PCOS. This makes PCOS a self-propagating, oestrogen driven, hypermitotic condition of the endometrium. Therefore, it is obvious how all the above-mentioned hormonal changes associated with PCOS may increase the risk of EC. Normalising the hormonal aberrations with life style changes, limiting calorie intake, increasing exercise, weight loss and progesterone therapy to prevent EH may reduce the risk of EC.
Endometrial hyperplasia (EH)
EH is a pathologically heterogeneous diagnosis that ranges from histologically subtle and spontaneously reversible proliferative lesions to premalignant changes [95, 96]. It virtually always results from prolonged oestrogen stimulation, unopposed by progesterone. EH with cytological atypia is recognised as the precursor for the endometrioid type of EC, with significantly increased EC risk (RR=14, 95% CI, 5–38) [97]. Altered circulatory hormone levels are reported in EH patients, particularly in those with metabolic syndrome with elevated oestrogen, testosterone, insulin, leptin, LH and LH/FSH ratio [98, 99]. Additionally, increased oestrogen bioavailability as a consequence of increased local aromatase expression and activity, in hyperplastic endometrium has also been postulated [100]. Likewise, a significant change in AKR1C3, an enzyme catalyses progesterone, oestradiol and androstenedione, has been shown in EH compared with normal control and this is suggested to increase oestrogen signalling. However, there are conflicting reports of AKR1C3 expression profiles needing future clarification [57, 101]. Insulin resistance and carbohydrate metabolism disturbances are also common findings in EH [102], which demonstrated an increase in the mitogenic effect of oestradiol through EGF and activated IGF-1 [98] (Figure 2). Recently, reduced serum melatonin level has been described to predict EH transformation to the premalignant atypical phenotype [103].
Obesity
Excess weight has been shown to be causative for at least 40% of ECs in the UK [104, 105] and rather than BMI as a risk factor, the adult weight gain has been proposed as a better measure [106, 107]. The distribution of the excess adipose tissue within the body, specifically in visceral fat, is an independent EC risk factor [108].
There are three main theories linking obesity to EC (i) oestrogen, (ii) insulin and other growth factor production and (iii) excess production of inflammatory cytokines, adipokines in the visceral adipose tissue [109]. The most prominent out of these is the effects of unopposed oestrogen, due to aromatisation of adrenal androgens [107, 110]. Aromatase promoter in the adipose tissue is also under the influence of glucocorticoids and oestrogen-induced class 1 cytokines and TNFα [111].
Adipocytes produce adipokines such as adiponection and leptin. The serum leptin-adiponectin ratio is independently associated with increased EC development risk [112]. Adipokines may influence EC through mechanisms dependent/independent of oestrogen [113–115]. Adiponectin levels have shown to be inversely correlated with EC [116, 117] which might be due to polymorphisms in the adiponectin coding gene ADIPOQ [118]. Leptin is regulated by various hormones including insulin, glucocorticoids, TNFα and reproductive hormones [119]. Intentional weight loss has shown a decrease in several adipokines including leptin [120] and therefore it is expected to reduce EC risk.
Diabetes mellitus
There is a plethora of recent evidence suggesting that diabetes constitutes an independent risk factor for EC [121]. In addition to oestrogen-progesterone imbalance, which associates with obesity, the observed risk in type II diabetes can also be explained by the compensatory increase in the insulin production. This, consequently results in an increase in the circulating IGF-1 level [122] which acts as a mitogenic stimuli via non-classical oestrogen pathway, resulting in increasing the risk of EC [123].
Lynch syndrome (LS)
Approximately, 2%–5% of ECs may be due to inherited susceptibility, of which LS is the most common with an increased lifetime risk of EC 27%–70%. MLH1 mutations are frequent in EC but compared to colorectal cancers there is a five-fold increase in the prevalence of MSH6 mutations in EC [124–126]. Pathogenesis of LS is considered to be due to mutations of genes in the DNA mismatch repair (MMR) pathway [127]. An interesting theory proposes that MMR deficiency to be the most important abnormality in early stage EC [128]. High premenopausal level of oestradiol has shown to increase the MMR activity in vitro, which explains the lower incidence of EC in premenopausal women, whereas cells dividing in a low-oestrogen environment (such as in PM obese women) are more likely to accumulate genetic errors due to low DNA repair activity and they may be at high-risk for carcinogenesis [128]. Patients with defective MMR pathways are prone to microsatellite instability (MSI) and almost all LS-associated cancers have MSI due to MMR, but in some cases, it can also result from methylation of MLH1 promoter [129]. A later age of menarche, increased parity and the use of hormonal contraceptives may be protective against EC in LS women with MMR mutation [130]. As high levels of oestrogen may increase MMR proteins and thereby may play a protective role, combined pill in particular may be a useful chemo-preventive agent in these women at high-risk of developing EC.
Smoking
The inverse relationship between cigarette smoking and EC risk is well established [131]. According to a recent study, reduced EC risk was evident among former (RR=0.89, 95% CI 0.80, 1.00) and current (RR=0.65, 95% CI 0.55, 0.78) smokers compared with non-smokers [132]. This protective effect is independent of other EC risk factors and is fully explained via hormonal modulation affecting the function of hormone-producing organs, including adrenals and ovaries. Anti-oestrogenic mechanisms have been suggested through (i) increasing hepatic metabolism of oestrogen into minimally active 2-hydroxyoestrogens [133], (ii) reducing ovarian oestrogen production which associates with a shorter follicular phase and early menopause [134, 135] and (iii) reducing aromatisation of androgens by the adipose tissue considering the positive association between smoking and lower BMI [132]. Adrenal androgen production in smokers is higher than in non-smokers [136] and smoking is associated with higher circulating androstenedione and testosterone levels [137, 138]. PM smokers with high BMI maintained a lower risk for EC [139]. Serum oestrogen level for these women were not measured in that particular study, but it is tempting to speculate that aromatisation of adrenal androgen in the adipose tissue of women who smoke may be impaired, which needs confirmation in future studies.
Parkinson’s disease (PD)
Oestrogens are implicated in neurodegenerative diseases and PM HRT has been associated with lower risk of PD [140, 141]. Contrasting with EC, women with short menstrual span (late menarche, early menopause) seem to have higher risk of developing PD [142]. Furthermore, menstruation and pregnancy was associated with worsening of the symptoms and rapid progression of the disease. Consistent with this observation, reports from murine models have also shown that oestrogen and progesterone (but not testosterone) rescue substantia nigra from induced toxicity [143, 144]. Although this may suggest a protective effect of PD in EC, conversely, a recent report suggests that patients with PD have an increased risk in developing EC. This was accompanied by an overall reduction in risk of developing other cancers in general [145]. This association was not maintained after adjusting for multiple testing, therefore further studies will be required to confirm the causative relationship.
Role of common hormonal pharmacological agents in endometrial carcinogenesis
Pharmacological agents that operate via altering the hormonal activity in the body are considered here for their utility as preventative and treatments strategies or due to the role, they play in altering the risk of developing EC (Figure 3).
In support of that, the recent integrated genomic study [80] has identified a subset of hormonally responsive ECs that demonstrate alterations in hormonal pathways. Interestingly, the highest expression of ESR1 and PGR was found in copy number low and microsatellite instability groups rather than the favourable outcome POLE ultramutated group [80]. This further advocates personalised endocrine therapy in EC management. In this section, we discuss the therapeutic, preventative role of hormones as well as many of the commonly used hormonal agents in EC.
Oestrogen and progesterone based therapeutic modalities
As contraceptives
Since hormonal contraceptives are used by premenopausal women, yet EC is mainly a PM disease, any influence of hormonal contraceptives on endometrial carcinogenesis in general may be regarded as a delayed consequence.
Combined Oral Contraceptive pill (COCP): The protective effect in regards to EC has been well-documented [146]. The risk reduction appears to be proportionally related to the duration of use, and every 5 years of use is associated with a relative risk of 0.76 [147]. This favourable outcome may persist up to 30 years after cessation of COCPs, though it may be amplified as time after discontinuation elapses [148]. The similar protective effect observed in high and low dose oestrogen-containing pills [147] may suggest that the protective effect seen with COCP may be due to the increase in lifetime exposure to progesterone. In support of this finding, a higher progestin-potency COCP seem to be required for women with larger BMIs [149].
Progesterone only preparations: The use of progestogen only contraceptive agents such as depot medroxyprogesterone acetate (MPA) have been associated with a reduction in the risk of developing EC [150]. Likewise, the use of levonorgestrel-releasing intrauterine system (LNG-IUS) was found to be significantly protective with prolonged use up to 10 years [151].
High-dose systemic Depo-Provera preparation will induce hypoestrogenic state with profound ovarian suppression and has a direct endometrial effect with high progestogen levels in the endometrium [150]. There are no conclusive studies examining the differential risk associated with low dose systemic regimens such as progestogen only pill and implants. Furthermore, the protective effect of the other newer combined hormonal contraceptive methods remain to be investigated.
As postmenopausal hormonal replacement therapy
Menopausal HRT is required by millions of women for climacteric symptom management and for the prevention of osteoporosis throughout the world [152]. Studies consistently find a substantial increase in EC incidence with oestrogen alone use [152, 153], which is more marked with prolonged duration of use and persists for several years after treatment discontinuation [154]. As a result, combined oestrogen-progestin therapy is the standard, to counteract the proliferative effects of oestrogen on the endometrium [153]. Results from the Women’s Health Initiative study concluded that oestrogen plus progestin use was associated with a 37% reduction in type I EC incidence [152], whilst, oestrogen therapy was found to have little impact on the risk of type II EC [153]. Continuous combined oestrogen-progestin therapy was the only HRT regimen associated with an apparent decline in risk whereas long-term (10 years) sequential regimens were associated with an increased risk of EC [153]. Furthermore, the type of synthetic progestogen does not affect the risk reduction [154]. The use of topical vaginal oestrogen preparations has not shown to significantly increase the risk of EC [154].
As hormonal replacement after endometrial cancer treatment
HRT after treatment of EC remains controversial but studies have specifically looked into this group of patients and shown no significant increase in the risk of recurrence in EC survivors using HRT relative to the control group (OR=0.53; 95% CI, 0.30–0.96) [155]. However, personalising HRT to patient with ER negative EC may be considered to reduce any potential adverse effect.
Progestogens as treatment for hyperplasia
As hyperplasia is due to excess or unopposed oestrogenic effect in the endometrium, prophylactic treatment with high dose oral progestogens such as MPA and local administration with LNG-IUS is used to prevent progression of hyperplasia to EC [156]. Systemic administration of high dose progestogens are associated with an exceptionally poor side effect profile, low compliance and lower remission rates (60%) [157] compared with the LNG-IUS [158, 159]. The local endometrial LNG levels achieved with IUS exceeds that of oral administration by over 1000 folds with higher remission rates (100%) and the additional benefit of a more beneficial side effect profile [157], hence it is the preferable method of delivery. Progestogen treatment using LNG-IUS is also commonly used in women with obesity and other high-risk conditions (e.g. PCOS, tamoxifen treatment) mentioned above as part of a risk reduction strategy.
Progestogens as treatment for endometrial cancer
Synthetic progestogens have been used to treat EC for over 60 years, and they remain to be the main hormonal pharmaceutical agent used in EC. High dose progestogens, in the form of oral MPA and megestrol acetate have been licensed for fertility-sparing treatment of carefully chosen individuals with early stage, low-grade endometrial disease [160]. They are also routinely used as an adjuvant therapy in advanced, metastatic and recurrent ECs as well as in endometrial stromal cell cancers [161]. However, the efficacy of progestogens as an adjuvant treatment in high-risk ECs has been poor or modest at its best [162]. This observed inconsistent response to progestogens in clinical studies that can be explained by the fact that treatment is not tailored according to ECs with PR expression/progestogen responsive.
When considering hormonal modulators, one striking feature specific to EC is that presently, there is no consensus in routinely assessing ECs for their hormone responsiveness (assessing the expression of steroid hormone receptors like in other hormone responsive cancers e.g. breast cancer). Due to the distinct alterations of the steroid receptor expression observed in EC subtypes and with tumour progression (Figure 1), this is an important deficiency in current practise. We have recently proposed a validated scoring system to quantify PR (and other hormone receptor expression) in ECs, as an endeavour to provide means to identify potential responders [25]. Prolonged therapy with progestogens can render the endometrial cells unresponsive to hormone treatment by downregulating most (if not all) ovarian steroid hormone receptors. Considering this complex interplay of hormonal actions in the endometrium, a suitable sequential regimen of hormonal agents (progesterone, followed by anti-oestrogens) [157] and personalising/tailoring treatment according to sequential assessment of the tumour for hormone responsiveness is envisaged to increase the therapeutic potential of endocrine therapy in EC.
Selective progesterone receptor modulators (SPRMs)
Mifepristone is the first SPRM, and has partial agonist/antagonist activity with significant affinity to AR and GR. Therefore, the observed endometrial effect depends on the availability of progesterone, and at least some of the observed effects of mifepristone may be via the AR [163]. Long-term use of high-dose mifepristone thickens the endometrium, although the associated histology shows cystic glandular atrophy with reduction in glandular mitosis. Nevertheless, concerns have been raised regarding a potential trophic effect of SPRMs on the endometrium [164]. Ullipristol, a 2nd generation SPRM, is also licenced to be used for fibroid-associated symptoms and has been reported to have lower endometrial trophic effects. There are reports of hyperplasia without atypia and polyps after 13 weeks of Ullipristol treatment, yet the endometrium reverted to normal, 6 months after treatment [165]. In a small Phase II study, high-dose mifepristone as a single agent therapy in women with either advanced or recurrent EC, revealed very limited stabilisation of the disease, and no partial or complete response [166]. Therefore, until further conclusive data are available, clinicians using high dose prolonged therapy with SPRMs in women need to be aware of possible endometrial changes similar to that occur with tamoxifen [166, 167].
Selective oestrogen receptor modulators (SERMs)
SERMs represent a group of non-steroidal non-hormonal compounds that bind to the ER, each inducing a distinct set of tissue-specific effects. The agonistic or antagonistic effect of SERMs depend on the availability of oestrogen [168].
Tamoxifen, the first clinically applicable SERM is used as an adjuvant treatment and as a chemoprevention agent in breast cancer. Tamoxifen therapy, is associated with subsequent endometrial proliferation in PM women, with the development of a spectrum of endometrial lesions, ranging from hyperplasia and polyps to invasive carcinomas and sarcomas due to its ER agonist activity in the uterus [169]. The increased risk of EC with tamoxifen use seems to be proportionally related to longer duration, accumulative usage, and increased BMI. A recent opinion paper suggested that premenopausal women treated with tamoxifen require no additional monitoring beyond routine gynaecologic care due to lack of evidence of an increased risk of EC [170].
Conversely, the newer SERMs, such as Raloxifene, Bazedoxifene and Ospemifene, seem to have neutral effects on the endometrium [171–173]. Lasofoxifene, on the other hand, may increase endometrial thickness but the early studies have not confirmed a higher risk of hyperplasia or EC after 5 years of follow-up [174].
Tibolone, a synthetic steroid with oestrogenic, (and some progestogenic and androgenic) properties, is commonly used for climacteric symptoms and prevention of osteoporosis. It has also been recently reported to increase the risk of EC [153], however, a previous RCT suggested that tibolone use has no concerns on endometrial safety [175]. Therefore, further research is necessary to conclude on the effects of SERMs and tibolone on endometrial carcinogenesis.
Antioestrogens
Fulvestrant is a full ERα and ERβ antagonist used as a second line drug for the treatment of breast cancer. Prospective endometrial assessment of breast cancer patients treated with fulvestrant showed a significant decrease in endometrial growth without the development of new endometrial pathologies [176]. Phase II studies have shown modest activity in advanced or recurrent EC patients with ER and/or PR positive tumours [177, 178]. The effectiveness of fulvestrant in EH and fertility-sparing EC management has not yet been assessed.
Aromatase inhibitors (AI)
Third generation AIs (letrozole and anastrozole) are non-steroidal competitive inhibitors of the aromatase enzyme that act to decrease systemic and intra-tumoral oestrogen levels. Prospective endometrial assessment of breast cancer patients treated with AI show no effect on endometrial thickening and possible regression of baseline endometrial proliferation [179]. Further studies also confirm that EH respond to AI, and the efficacy is comparable to progestogens [180–182]. Likewise, regression in response to combined AI and MPA was reported for fertility-sparing low-grade EC, whereas AI monotherapy failed to show any benefit for advanced or recurrent EC patients [183, 184]. Collectively, these results suggest a potential role for AI in early EC stages, requiring further investigation to identify synergistic pathways.
Androgens
Testosterone transdermal patch and subcutaneous preparations are used as part of some HRT regimens, particularly in women suffering from symptoms of androgen deficiency, such as low sexual desire and well-being. Current data does not indicate any adverse endometrial, cardiovascular or breast effects with transdermal preparations, although data on long-term risks and benefits of testosterone therapy after the diagnosis of EC are lacking [185, 186]. Endometrial atrophy is the common finding in transgender women on supraphysiological doses of testosterone (Testoviron Depot), instead of hyperplasia [187].
Oral and transvaginal dehydroepiandrosterone (DHEA) has been used for controlling PM symptoms. Although significant benefit remains to be determined, reports have shown a persistent atrophic phenotype in PM endometria after 52 weeks of this treatment [188].
Local danazol therapy which is used to improve symptoms associated with endometriosis has shown to act on endometrial epithelial and stromal cells directly through AR (also binds PR) by reducing cell survival [189] and cell proliferation [190, 191]. Data from a murine model shows a protective effect of long-term danazol against oestrogen induced carcinogenesis [192]. Likewise, clinical trials have also confirmed the effectiveness of danazol in the management of EH with and without cytological atypia with very low incidence of recurrence [193, 194]. By contrast, minimal response to danazol was reported in advanced, recurrent or persistent EC [195], yet sample size and lack of data on AR expression status limits the results of this particular trial.
Cyproterone acetate, an AR antagonist, has been used in HRT combined with cyclic oestradiol. Studies have shown an increased risk of EC for patients on this regimen compared with non-users [153]. Therefore, androgens are likely to play an important role in the pathogenesis of EC and the emerging evidence suggests that pharmaceutical agents with AR modulatory activity may have a potential therapeutic effect. Further studies on the effects of androgen on endometrial cell proliferation and function are therefore needed to identify the potential therapeutic strategies.
GnRH agonists and antagonists
Due to differences in the intracellular signalling cascades, the effect observed with GnRH agonists and antagonists is tissue-specific [196–198]. The clinical effects of the GnRH analogues seen after sustained administration, leads to desensitisation of receptors, suppression of gonadotropin secretion and finally inhibits the gonadal steroid synthesis.
The GnRH-R are currently being targeted successfully in the treatment of many hormonal dependent tumours using GnRH agonists and antagonists. Preparations with pharmacological properties of GnRH agonists, antagonists, GnRH agonist-based cytotoxic hybrids, GnRH-R targeted nanoparticles delivering anti-cancer medications act through GnRH-R are being explored in this respect. GnRH agonists show promise in treating advanced, and metastatic prostatic and breast cancer treatment. Unlike the 1st and 2nd generation antagonists with limited solubility and anaphylactic reactions due to histamine release [199], the currently available 3rd and 4th generation antagonists like cetrorelix and ganirelix have a more favourable side effect profile and for example, have immensely improved prostate cancer therapy. Although some in vitro studies have suggested a possible anti-tumour effect with GnRH analogues in EC, there are no clinical studies reported to date evaluating their efficacy in EC. Therefore, further research is urgently needed to assess if GnRH has a therapeutic role in EC.
Summary/Conclusion
Hormones intricately influence the carcinogenesis process in endometrium. Whilst oestrogen is the most conspicuous driver of this, many other hormones also appear to play an important role. Most known risk factors for EC are associated with alteration of the endometrial hormonal milieu, with many routinely used pharmacological hormonal agents impinging on the endometrial carcinogenesis process. Routine assessment of ECs for their hormone responsiveness (assessing the expression of steroid hormone receptors as in other hormone responsive cancers), using a validated scoring system, personalisation of treatment according to sequential assessment of the tumours and further evaluation of interplay between all hormones in the endometrium are envisaged to unlock novel, therapeutic avenues, based on hormones, for this increasingly common malignancy.
Acknowledgements
The authors would like to acknowledge the support from Wellbeing of Women project grant RG1487 (DKH), Higher Committee for Education Development in Iraq (AK and RA), Wellbeing of Women Clinical Training Fellowship RTF510 (NT), Liverpool Women’s Hospital (CP, SM, MA) and Institute of Translational medicine, University of Liverpool (CP, AK, RA). All authors declare that there is no conflict of interest.
References
1. Husing A, Dossus L, Ferrari P, Tjonneland A, Hansen L, Fagherazzi G, Baglietto L, Schock H, Chang-Claude J, Boeing H, Steffen A, Trichopoulou A, Bamia C, Katsoulis M, Krogh V, Palli D, Panico S, Onland-Moret NC, Peeters PH, Bueno-de-Mesquita HB, Weiderpass E, Gram IT, Ardanaz E, Obon-Santacana M, Navarro C, Sanchez-Cantalejo E, Etxezarreta N, Allen NE, Khaw KT, Wareham N, Rinaldi S, Romieu I, Merritt MA, Gunter M, Riboli E, Kaaks R. An epidemiological model for prediction of endometrial cancer risk in europe. Eur J Epidemiol 2016;31:51–60.10.1007/s10654-015-0030-9Search in Google Scholar PubMed
2. Anderson AS, Key TJ, Norat T, Scoccianti C, Cecchini M, Berrino F, Boutron-Ruault MC, Espina C, Leitzmann M, Powers H, Wiseman M, Romieu I. European code against cancer 4th edition: obesity, body fatness and cancer. Cancer Epidemiol 2015;39:Suppl 1:S34–45.10.1016/j.canep.2015.01.017Search in Google Scholar PubMed
3. Lindemann K, Eskild A, Vatten LJ, Bray F. Endometrial cancer incidence trends in norway during 1953–2007 and predictions for 2008–2027. Int J Cancer 2010;127:2661–8.10.1002/ijc.25267Search in Google Scholar PubMed
4. Iyer R, Gentry-Maharaj A, Liston R, Desai R, Gornall R, Leeson S, Linder A, Lopes A, Meechan D, Nevin J, TMould I, A Olaitan d, Rufford B, Shanbhag S, Thackeray A, Nick Wood SV, Reynolds K, Nordin A, MenonUsha. Gynaecological oncology surgical outcomes and complications (ukgosoc). ESGO 18. Liverpool2013.Search in Google Scholar
5. Cancer research uk, uk cancer incidence (2009) and mortality (2010) summary july 2012 european age-standardised (as) rates 2012 [cited 2015 July].Search in Google Scholar
6. Setiawan VW, Yang HP, Pike MC, McCann SE, Yu H, Xiang YB, Wolk A, Wentzensen N, Weiss NS, Webb PM, van den Brandt PA, van de Vijver K, Thompson PJ, Australian National Endometrial Cancer Study G, Strom BL, Spurdle AB, Soslow RA, Shu XO, Schairer C, Sacerdote C, Rohan TE, Robien K, Risch HA, Ricceri F, Rebbeck TR, Rastogi R, Prescott J, Polidoro S, Park Y, Olson SH, Moysich KB, Miller AB, McCullough ML, Matsuno RK, Magliocco AM, Lurie G, Lu L, Lissowska J, Liang X, Lacey JV, Jr., Kolonel LN, Henderson BE, Hankinson SE, Hakansson N, Goodman MT, Gaudet MM, Garcia-Closas M, Friedenreich CM, Freudenheim JL, Doherty J, De Vivo I, Courneya KS, Cook LS, Chen C, Cerhan JR, Cai H, Brinton LA, Bernstein L, Anderson KE, Anton-Culver H, Schouten LJ, Horn-Ross PL. Type i and ii endometrial cancers: have they different risk factors? J Clin Oncol 2013;31:2607–18.10.1200/JCO.2012.48.2596Search in Google Scholar
7. Hanamura T, Niwa T, Gohno T, Kurosumi M, Takei H, Yamaguchi Y, Ito K-i, Hayashi S-i. Possible role of the aromatase-independent steroid metabolism pathways in hormone responsive primary breast cancers. Breast Cancer Res Treat 2014;143:69–80.10.1007/s10549-013-2788-3Search in Google Scholar PubMed
8. Tanaka S, Miki Y, Hashimoto C, Takagi K, Doe Z, Li B, Yaegashi N, Suzuki T, Ito K. The role of 5α-reductase type 1 associated with intratumoral dihydrotestosterone concentrations in human endometrial carcinoma. Mol Cell Endocrinol 2015;401:56–64.10.1016/j.mce.2014.11.022Search in Google Scholar PubMed
9. Limonta P, Montagnani Marelli M, Mai S, Motta M, Martini L, Moretti RM. Gnrh receptors in cancer: from cell biology to novel targeted therapeutic strategies. Endocr Rev 2012;33:784–811.10.1210/er.2012-1014Search in Google Scholar PubMed
10. Emons G, Gorchev G, Harter P, Wimberger P, Stahle A, Hanker L, Hilpert F, Beckmann MW, Dall P, Grundker C, Sindermann H, Sehouli J. Efficacy and safety of aezs-108 (lhrh agonist linked to doxorubicin) in women with advanced or recurrent endometrial cancer expressing lhrh receptors: a multicenter phase 2 trial (ago-gyn5). Int J Gynecol Cancer 2014;24:260–5.10.1097/IGC.0000000000000044Search in Google Scholar PubMed PubMed Central
11. MA. V-D. The development of the uterus in late fetal life, infancy, and childhood. In: Norris HJ HA, Abell, MR, editors. The uterus. Baltimore, MD: Williams & Wilkins, 1973:40–67.Search in Google Scholar
12. Hapangama DK, Kamal AM, Bulmer JN. Estrogen receptor β: the guardian of the endometrium. Hum Reprod Update 2015;21:174–93.10.1093/humupd/dmu053Search in Google Scholar PubMed
13. Valentijn AJ, Palial K, Al-Lamee H, Tempest N, Drury J, Hapangama DK, Murray P, Von Zglinicki T, Saretzki G, Gargett CE. Ssea-1 isolates human endometrial basal glandular epithelial cells: phenotypic and functional characterization and implications in the pathogenesis of endometriosis. Hum Reprod 2013;28:2695–708.10.1093/humrep/det285Search in Google Scholar PubMed
14. Chen SY, Huang YC, Liu SP, Tsai FJ, Shyu WC, Lin SZ. An overview of concepts for cancer stem cells. Cell Transplant 2011;20:113–20.10.3727/096368910X532837Search in Google Scholar PubMed
15. Hapangama D. Mifepristone: the multi-faceted anti-hormone. J Drug Eval 2003;1:149–75.Search in Google Scholar
16. Slayden D, Brenner RM. Hormonal regulation and localization of estrogen, progestin and androgen receptors in the endometrium of nonhuman primates: effects of progesterone receptor antagonists. Arch Histol Cytol 2004;67:393–409.10.1679/aohc.67.393Search in Google Scholar
17. Wada-Hiraike O, Hiraike H, Okinaga H, Imamov O, Barros RP, Morani A, Omoto Y, Warner M, Gustafsson JA. Role of estrogen receptor beta in uterine stroma and epithelium: insights from estrogen receptor beta-/- mice. Proc Natl Acad Sci USA 2006;103:18350–5.10.1073/pnas.0608861103Search in Google Scholar
18. Lovely LP, Appa Rao KB, Gui Y, Lessey BA. Characterization of androgen receptors in a well-differentiated endometrial adenocarcinoma cell line (ishikawa). J Steroid Biochem Mol Biol 2000;74:235–41.10.1016/S0960-0760(00)00127-8Search in Google Scholar
19. Apparao KB, Lovely LP, Gui Y, Lininger RA, Lessey BA. Elevated endometrial androgen receptor expression in women with polycystic ovarian syndrome. Biol Reprod 2002;66:297–304.10.1095/biolreprod66.2.297Search in Google Scholar
20. Labrie F. All sex steroids are made intracellularly in peripheral tissues by the mechanisms of intracrinology after menopause. J Steroid Biochem Mol Biol 2015;145:133–8.10.1016/j.jsbmb.2014.06.001Search in Google Scholar
21. Teulings FA, van Gilse HA, Henkelman MS, Portengen H, Alexieva-Figusch J. Estrogen, androgen, glucocorticoid, and progesterone receptors in progestin-induced regression of human breast cancer. Cancer Res 1980;40:2557–61.Search in Google Scholar
22. Kurman RJ, Ellenson LH, Ronnett BM. Pathology of the female genital tract [Online system or service]. Boston, MA: Springer Science+Business Media, LLC: Springer, 2011. Available from: http://search.ebscohost.com.ezproxy.liv.ac.uk/login.aspx?direct=true&db=cat00003a&AN=lvp.b2389878&site=eds-live&scope=site.Search in Google Scholar
23. Hapangama DK, Bulmer JN. Pathophysiology of heavy menstrual bleeding. Womens Health (Lond Engl). 2016;12:3–13.10.2217/whe.15.81Search in Google Scholar
24. Gargett CE, Chan RW, Schwab KE. Hormone and growth factor signaling in endometrial renewal: role of stem/progenitor cells. Mol Cell Endocrinol 2008;288:22–9.10.1016/j.mce.2008.02.026Search in Google Scholar
25. Kamal A, Bulmer J, DeCruze S, Stringfellow H, Martin-Hirsch P, Hapangama D. Androgen receptors are acquired by healthy postmenopausal endometrial epithelium and their subsequent loss in endometrial cancer is associated with poor survival. Br J Cancer 2016. doi: 10.1038/bjc.2016.16.10.1038/bjc.2016.16Search in Google Scholar
26. O’Lone R, Frith MC, Karlsson EK, Hansen U. Genomic targets of nuclear estrogen receptors. Mol Endocrinol 2004;18:1859–75.10.1210/me.2003-0044Search in Google Scholar
27. Umayahara Y, Kawamori R, Watada H, Imano E, Iwama N, Morishima T, Yamasaki Y, Kajimoto Y, Kamada T. Estrogen regulation of the insulin-like growth factor i gene transcription involves an ap-1 enhancer. J Biol Chem 1994;269:16433–42.10.1016/S0021-9258(17)34025-5Search in Google Scholar
28. Pietras RJ, Mrquez-Garbn DC. Membrane-associated estrogen receptor signaling pathways in human cancers. Clin Cancer Res 2007;13:4672–6.10.1158/1078-0432.CCR-07-1373Search in Google Scholar
29. Kushner PJ, Agard DA, Greene GL, Scanlan TS, Shiau AK, Uht RM, Webb P. Estrogen receptor pathways to ap-1. J Steroid Biochem Mol Biol 2000;74:311–7.10.1016/S0960-0760(00)00108-4Search in Google Scholar
30. Ray A, Prefontaine KE, Ray P. Down-modulation of interleukin-6 gene expression by 17 beta-estradiol in the absence of high affinity DNA binding by the estrogen receptor. J Biol Chem 1994;269:12940–6.10.1016/S0021-9258(18)99966-7Search in Google Scholar
31. Kato S, Endoh H, Masuhiro Y, Kitamoto T, Uchiyama S, Sasaki H, Masushige S, Gotoh Y, Nishida E, Kawashima H, Metzger D, Chambon P. Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science 1995;270:1491–4.10.1126/science.270.5241.1491Search in Google Scholar PubMed
32. Hewitt SC, Harrell JC, Korach KS. Lessons in estrogen biology from knockout and transgenic animals. Annu Rev Physiol 2005;67:285–308.10.1146/annurev.physiol.67.040403.115914Search in Google Scholar PubMed
33. Zhang L, Xiaoping L, Lijun Z, Lifang Z, Guo Z, Jianliu W, Lihui W. Nongenomic effect of estrogen on the mapk signaling pathway and calcium influx in endometrial carcinoma cells. J Cell Biochem 2009;106:553–62.10.1002/jcb.22017Search in Google Scholar PubMed
34. He YY, Cai B, Yang YX, Liu XL, Wan XP. Estrogenic g protein-coupled receptor 30 signaling is involved in regulation of endometrial carcinoma by promoting proliferation, invasion potential, and interleukin-6 secretion via the mek/erk mitogen-activated protein kinase pathway. Cancer Sci 2009;100:1051–61.10.1111/j.1349-7006.2009.01148.xSearch in Google Scholar PubMed
35. Sasaki M, Dharia A, Oh BR, Tanaka Y, Fujimoto S, Dahiya R. Progesterone receptor b gene inactivation and cpg hypermethylation in human uterine endometrial cancer. Cancer Res 2001;61:97–102.Search in Google Scholar
36. Campan M, Weisenberger D, Laird P. DNA methylation profiles of female steroid hormone deriven human malignancy. Curr Top Microbiol Immunol 2006;310:141–78.10.1007/3-540-31181-5_8Search in Google Scholar
37. Sasaki M, Kotcherguina L, Dharia A, Fujimoto S, Dahiya R. Cytosine-phosphoguanine methylation of estrogen receptors in endometrial cancer. Cancer Res 2001;61:3262–6.Search in Google Scholar
38. Wu H, Yupeng C, Jing L, Bin S, Ge W, Ying Z, Dan W, Ruifang L, Xia Y, Hua Z, Luyang S, Yongfeng S. Hypomethylation-linked activation of pax2 mediates tamoxifen-stimulated endometrial carcinogenesis. Nature 2005;438:981–7.10.1038/nature04225Search in Google Scholar PubMed
39. Hayes CL, Spink DC, Spink BC, Cao JQ, Walker NJ, Sutter TR. 17 beta-estradiol hydroxylation catalyzed by human cytochrome p450 1b1. Proc Natl Acad Sci USA 1996;93:9776–81.10.1073/pnas.93.18.9776Search in Google Scholar PubMed PubMed Central
40. Ke H, Suzuki A, Miyamoto T, Kashima H, Shiozawa T. 4-hydroxy estrogen induces DNA damage on codon 130/131 of pten in endometrial carcinoma cells. Mol Cell Endocrinol 2015;400:71–7.10.1016/j.mce.2014.10.027Search in Google Scholar PubMed
41. Valentijn AJ, Saretzki G, Tempest N, Critchley HO, Hapangama DK. Human endometrial epithelial telomerase is important for epithelial proliferation and glandular formation with potential implications in endometriosis. Hum Reprod 2015;30:2816–28.10.1093/humrep/dev267Search in Google Scholar PubMed
42. Henderson TA, Saunders PT, Moffett-King A, Groome NP, Critchley HO. Steroid receptor expression in uterine natural killer cells. J Clin Endocrinol Metab 2003;88:440–9.10.1210/jc.2002-021174Search in Google Scholar PubMed
43. Kurita T, Young P, Brody JR, Lydon JP, O’Malley BW, Cunha GR. Stromal progesterone receptors mediate the inhibitory effects of progesterone on estrogen-induced uterine epithelial cell deoxyribonucleic acid synthesis. Endocrinology 1998;139:4708–13.10.1210/endo.139.11.6317Search in Google Scholar PubMed
44. Franco HL, Rubel CA, Large MJ, Wetendorf M, Fernandez-Valdivia R, Jeong JW, Spencer TE, Behringer RR, Lydon JP, Demayo FJ. Epithelial progesterone receptor exhibits pleiotropic roles in uterine development and function. FASEB J. 2012;26:1218–27.10.1096/fj.11-193334Search in Google Scholar PubMed PubMed Central
45. Li Q, Kannan A, DeMayo FJ, Lydon JP, Cooke PS, Yamagishi H, Srivastava D, Bagchi MK, Bagchi IC. The antiproliferative action of progesterone in uterine epithelium is mediated by hand2. Science 2011;331:912–6.10.1126/science.1197454Search in Google Scholar PubMed PubMed Central
46. Mathew D, Drury JA, Valentijn AJ, Vasieva O, Hapangama DK. In silico, in vitro and in vivo analysis identifies a potential role for steroid hormone regulation of foxd3 in endometriosis-associated genes. Hum Reprod 2016;31:345–54.10.1093/humrep/dev307Search in Google Scholar
47. Kurita T, Wang YZ, Donjacour AA, Zhao C, Lydon JP, O’Malley BW, Isaacs JT, Dahiya R, Cunha GR. Paracrine regulation of apoptosis by steroid hormones in the male and female reproductive system. Cell Death Differ 2001;8:192–200.10.1038/sj.cdd.4400797Search in Google Scholar PubMed
48. Schindler AE. Progestogen deficiency and endometrial cancer risk. Maturitas 2009;62:334–7.10.1016/j.maturitas.2008.12.018Search in Google Scholar PubMed
49. Fanta M. Is polycystic ovary syndrome, a state of relative estrogen excess, a real risk factor for estrogen-dependant malignancies? Gynecol Endocrinol 2013;29:145–7.10.3109/09513590.2012.730575Search in Google Scholar PubMed
50. Park JC, Lim SY, Jang TK, Bae JG, Kim JI, Rhee JH. Endometrial histology and predictable clinical factors for endometrial disease in women with polycystic ovary syndrome. Clin Exp Reprod Med 2011;38:42–6.10.5653/cerm.2011.38.1.42Search in Google Scholar PubMed PubMed Central
51. Dumesic DA, Lobo RA. Cancer risk and pcos. Steroids 2013;78:782–5.10.1016/j.steroids.2013.04.004Search in Google Scholar PubMed
52. Holm NS, Glintborg D, Andersen MS, Schledermann D, Ravn P. The prevalence of endometrial hyperplasia and endometrial cancer in women with polycystic ovary syndrome or hyperandrogenism. Acta Obstet Gynecol Scand 2012;91:1173–6.10.1111/j.1600-0412.2012.01458.xSearch in Google Scholar PubMed
53. Shao R, Li X, Feng Y, Lin JF, Billig H. Direct effects of metformin in the endometrium: a hypothetical mechanism for the treatment of women with pcos and endometrial carcinoma. J Exp Clin Cancer Res 2014;33:41.10.1186/1756-9966-33-41Search in Google Scholar PubMed PubMed Central
54. Gibson DA, Simitsidellis I, Collins F, Saunders PT. Evidence of androgen action in endometrial and ovarian cancers. Endocr Relat Cancer 2014;21:T203–18.10.1530/ERC-13-0551Search in Google Scholar PubMed
55. Bulun SE, Lin Z, Imir G, Amin S, Demura M, Yilmaz B, Martin R, Utsunomiya H, Thung S, Gurates B, Tamura M, Langoi D, Deb S. Regulation of aromatase expression in estrogen-responsive breast and uterine disease: from bench to treatment. Pharmacol Rev 2005;57:359–83.10.1124/pr.57.3.6Search in Google Scholar PubMed
56. Gao C, Wang Y, Tian W, Zhu Y, Xue F. The therapeutic significance of aromatase inhibitors in endometrial carcinoma. Gynecol Oncol 2014;134:190–5.10.1016/j.ygyno.2014.04.060Search in Google Scholar PubMed
57. Zakharov V, Lin HK, Azzarello J, McMeekin S, Moore KN, Penning TM, Fung KM. Suppressed expression of type 2 3α/type 5 17β-hydroxysteroid dehydrogenase (akr1c3) in endometrial hyperplasia and carcinoma. Int J Clin Exp Pathol 2010;3:608–17.Search in Google Scholar
58. Pereira de Jésus-Tran K, Côté P-L, Cantin L, Blanchet J, Labrie F, Breton R. Comparison of crystal structures of human androgen receptor ligand-binding domain complexed with various agonists reveals molecular determinants responsible for binding affinity. Protein Sci 2006;15:987–99.10.1110/ps.051905906Search in Google Scholar PubMed PubMed Central
59. Berstein L, Kovalevskij A, Zimarina T, Maximov S, Gershfeld E, Vasilyev D, Baisheva S, Baymakhasheva A, Thijssen JH. Aromatase and comparative response to its inhibitors in two types of endometrial cancer. J Steroid Biochem Mol Biol 2005;95:71–4.10.1016/j.jsbmb.2005.04.008Search in Google Scholar PubMed
60. Kang JL, Wang DY, Wang XX, Yu J. Up-regulation of apoptosis by gonadotrophin-releasing hormone agonist in cultures of endometrial cells from women with symptomatic myomas. Hum Reprod 2010;25:2270–5.10.1093/humrep/deq178Search in Google Scholar PubMed
61. Engel JB, Schally AV, Buchholz S, Seitz S, Emons G, Ortmann O. Targeted chemotherapy of endometrial, ovarian and breast cancers with cytotoxic analogs of luteinizing hormone-releasing hormone (lhrh). Arch Gynecol Obstet 2012;286:437–42.10.1007/s00404-012-2335-1Search in Google Scholar PubMed
62. Noci I, Pillozzi S, Lastraioli E, Dabizzi S, Giachi M, Borrani E, Wimalasena J, Taddei GL, Scarselli G, Arcangeli A. Hlh/hcg-receptor expression correlates with in vitro invasiveness in human primary endometrial cancer. Gynecol Oncol 2008;111:496–501.10.1016/j.ygyno.2008.08.018Search in Google Scholar PubMed
63. Pillozzi S, Fortunato A, De Lorenzo E, Borrani E, Giachi M, Scarselli G, Arcangeli A, Noci I. Over-expression of the lh receptor increases distant metastases in an endometrial cancer mouse model. Front Oncol 2013;3:285.10.3389/fonc.2013.00285Search in Google Scholar
64. Yurkovetsky Z, Ta’asan S, Skates S, Rand A, Lomakin A, Linkov F, Marrangoni A, Velikokhatnaya L, Winans M, Gorelik E, Maxwell GL, Lu K, Lokshin A. Development of multimarker panel for early detection of endometrial cancer. High diagnostic power of prolactin. Gynecol Oncol 2007;107:58–65.10.1016/j.ygyno.2007.05.041Search in Google Scholar
65. Fader AN, Arriba LN, Frasure HE, von Gruenigen VE. Endometrial cancer and obesity: epidemiology, biomarkers, prevention and survivorship. Gynecol Oncol 2009;114:121–7.10.1016/j.ygyno.2009.03.039Search in Google Scholar
66. Yamazawa K, Matsui H, Seki K, Sekiya S. A case-control study of endometrial cancer after antipsychotics exposure in premenopausal women. Oncology 2003;64:116–23.10.1159/000067769Search in Google Scholar
67. Gurer-Orhan H, Suzen S. Melatonin, its metabolites and its synthetic analogs as multi-faceted compounds: antioxidant, prooxidant and inhibitor of bioactivation reactions. Curr Med Chem 2015;22:490–9.10.2174/0929867321666141215095259Search in Google Scholar
68. Zamfir Chiru AA, Popescu CR, Gheorghe DC. Melatonin and cancer. J Med Life 2014;7:373–4.Search in Google Scholar
69. Ekmekcioglu C. Expression and putative functions of melatonin receptors in malignant cells and tissues. Wien Med Wochenschr 2014;164:472–8.10.1007/s10354-014-0289-6Search in Google Scholar
70. Viswanathan AN, Hankinson SE, Schernhammer ES. Night shift work and the risk of endometrial cancer. Cancer Res 2007;67:10618–22.10.1158/0008-5472.CAN-07-2485Search in Google Scholar
71. Ciortea R, Costin N, Braicu I, Haragas D, Hudacsko A, Bondor C, Mihu D, Mihu CM. Effect of melatonin on intra-abdominal fat in correlation with endometrial proliferation in ovariectomized rats. Anticancer Res 2011;31:2637–43.Search in Google Scholar
72. Bokhman JV. Two pathogenetic types of endometrial carcinoma. Gynecol Oncol 1983;15:10–7.10.1016/0090-8258(83)90111-7Search in Google Scholar
73. Sherman ME. Theories of endometrial carcinogenesis: a multidisciplinary approach. Mod Pathol 2000;13:295–308.10.1038/modpathol.3880051Search in Google Scholar PubMed
74. McConechy MK, Ding JR, Cheang MC, Wiegand KC, Senz J, Tone AA, Yang WN, Prentice LM, Tse K, Zeng T, McDonald H, Schmidt AP, Mutch DG, McAlpine JN, Hirst M, Shah SP, Lee CH, Goodfellow PJ, Gilks CB, Huntsman DG. Use of mutation profiles to refine the classification of endometrial carcinomas. J Pathol 2012;228:20–30.10.1002/path.4056Search in Google Scholar PubMed PubMed Central
75. Lax SF, Kendall B, Tashiro H, Slebos RJ, Hedrick L. The frequency of p53, k-ras mutations, and microsatellite instability differs in uterine endometrioid and serous carcinoma: evidence of distinct molecular genetic pathways. Cancer 2000;88:814–24.10.1002/(SICI)1097-0142(20000215)88:4<814::AID-CNCR12>3.0.CO;2-USearch in Google Scholar
76. Voss MA, Ganesan R, Ludeman L, McCarthy K, Gornall R, Schaller G, Wei W, Sundar S. Should grade 3 endometrioid endometrial carcinoma be considered a type 2 cancer–a clinical and pathological evaluation. Gynecol Oncol 2012;124:15–20.10.1016/j.ygyno.2011.07.030Search in Google Scholar
77. Zannoni GF, Monterossi G, De Stefano I, Gargini A, Salerno MG, Farulla I, Travaglia D, Vellone VG, Scambia G, Gallo D. The expression ratios of estrogen receptor alpha (eralpha) to estrogen receptor beta1 (erbeta1) and eralpha to erbeta2 identify poor clinical outcome in endometrioid endometrial cancer. Hum Pathol 2013;44:1047–54.10.1016/j.humpath.2012.09.007Search in Google Scholar
78. Reid-Nicholson M, Iyengar P, Asher M, Soslow RA, Hummer AJ, Linkov I. Immunophenotypic diversity of endometrial adenocarcinomas: implications for differential diagnosis. Mod Pathol 2006;19:1091–100.10.1038/modpathol.3800620Search in Google Scholar
79. Murali R, Soslow RA, Weigelt B. Classification of endometrial carcinoma: more than two types. Lancet Oncol 2014;15:e268–e78.10.1016/S1470-2045(13)70591-6Search in Google Scholar
80. Cancer Genome Atlas Research N, Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, Shen H, Robertson AG, Pashtan I, Shen R, Benz CC, Yau C, Laird PW, Ding L, Zhang W, Mills GB, Kucherlapati R, Mardis ER, Levine DA. Integrated genomic characterization of endometrial carcinoma. Nature 2013;497:67–73.10.1038/nature12113Search in Google Scholar
81. Talhouk A, McConechy MK, Leung S, Li-Chang HH, Kwon JS, Melnyk N, Yang W, Senz J, Boyd N, Karnezis AN, Huntsman DG, Gilks CB, McAlpine JN. A clinically applicable molecular-based classification for endometrial cancers. Br J Cancer 2015;113:299–310.10.1038/bjc.2015.190Search in Google Scholar
82. McPherson CP, Sellers TA, Potter JD, Bostick RM, Folsom AR. Reproductive factors and risk of endometrial cancer. The iowa women’s health study. Am J Epidemiol 1996;143:1195–202.10.1093/oxfordjournals.aje.a008707Search in Google Scholar
83. Fearnley EJ, Marquart L, Spurdle AB, Weinstein P, Webb PM. Polycystic ovary syndrome increases the risk of endometrial cancer in women aged less than 50 years: an australian case-control study. Cancer Causes Control 2010;21:2303–8.10.1007/s10552-010-9658-7Search in Google Scholar
84. Gottschau M, Kjaer SK, Jensen A, Munk C, Mellemkjaer L. Risk of cancer among women with polycystic ovary syndrome: a danish cohort study. Gynecol Oncol 2015;136:99–103.10.1016/j.ygyno.2014.11.012Search in Google Scholar
85. Haoula Z, Salman M, Atiomo W. Evaluating the association between endometrial cancer and polycystic ovary syndrome. Hum Reprod 2012;27:1327–31.10.1093/humrep/des042Search in Google Scholar PubMed
86. Li X, Feng Y, Lin JF, Billig H, Shao R. Endometrial progesterone resistance and pcos. J Biomed Sci 2014;21:2.10.1186/1423-0127-21-2Search in Google Scholar PubMed PubMed Central
87. Thornton EC, Von Wald T, Hansen K. Polycystic ovarian syndrome: a primer. S D Med 2015;68:257–61.Search in Google Scholar
88. Aghajanova L, Velarde MC, Giudice LC. Altered gene expression profiling in endometrium: evidence for progesterone resistance. Semin Reprod Med 2010;28:51–8.10.1055/s-0029-1242994Search in Google Scholar PubMed
89. Piltonen TT, Chen JC, Khatun M, Kangasniemi M, Liakka A, Spitzer T, Tran N, Huddleston H, Irwin JC, Giudice LC. Endometrial stromal fibroblasts from women with polycystic ovary syndrome have impaired progesterone-mediated decidualization, aberrant cytokine profiles and promote enhanced immune cell migration in vitro. Hum Reprod 2015;30:1203–15.10.1093/humrep/dev055Search in Google Scholar PubMed PubMed Central
90. Savaris RF, Groll JM, Young SL, DeMayo FJ, Jeong JW, Hamilton AE, Giudice LC, Lessey BA. Progesterone resistance in pcos endometrium: a microarray analysis in clomiphene citrate-treated and artificial menstrual cycles. J Clin Endocrinol Metab 2011;96:1737–46.10.1210/jc.2010-2600Search in Google Scholar PubMed PubMed Central
91. Villavicencio A, Bacallao K, Avellaira C, Fuentes A, Vega M, Gabler F, Wu MH, Lu CW, Chang FM, Tsai SJ, Taylor AH, Al-Azzawi F. Androgen and estrogen receptors and co-regulators levels in endometria from patients with polycystic ovarian syndrome with and without endometrial hyperplasia. 2000. p. 307; 50; 145–314; 54; 155.10.1016/j.ygyno.2006.03.029Search in Google Scholar PubMed
92. Bulun SE, Chen D, Lu M, Zhao H, Cheng Y, Demura M, Yilmaz B, Martin R, Utsunomiya H, Thung S, Su E, Marsh E, Hakim A, Yin P, Ishikawa H, Amin S, Imir G, Gurates B, Attar E, Reierstad S, Innes J, Lin Z. Aromatase excess in cancers of breast, endometrium and ovary. J Steroid Biochem Mol Biol 2007;106:81–96.10.1016/j.jsbmb.2007.05.027Search in Google Scholar PubMed PubMed Central
93. Livadas S, Pappas C, Karachalios A, Marinakis E, Tolia N, Drakou M, Kaldrymides P, Panidis D, Diamanti-Kandarakis E. Prevalence and impact of hyperandrogenemia in 1,218 women with polycystic ovary syndrome. Endocrine 2014;47:631–8.10.1007/s12020-014-0200-7Search in Google Scholar PubMed
94. Kaaks R, Lukanova A, Kurzer MS. Obesity, endogenous hormones, and endometrial cancer risk: a synthetic review. Cancer Epidemiol Biomarkers Prev 2002;11:1531–43.Search in Google Scholar
95. Montgomery BE, Daum GS, Dunton CJ. Endometrial hyperplasia: a review. Obstet Gynecol Surv 2004;59:368–78.10.1097/00006254-200405000-00025Search in Google Scholar PubMed
96. Mazur MT. Endometrial hyperplasia/adenocarcinoma. A conventional approach. Ann Diagn Pathol 2005;9:174–81.10.1016/j.anndiagpath.2005.03.001Search in Google Scholar PubMed
97. Lacey JV, Jr., Ioffe OB, Ronnett BM, Rush BB, Richesson DA, Chatterjee N, Langholz B, Glass AG, Sherman ME. Endometrial carcinoma risk among women diagnosed with endometrial hyperplasia: the 34-year experience in a large health plan. Br J Cancer 2007;98:45–53.10.1038/sj.bjc.6604102Search in Google Scholar
98. Chernyshova AL, Kolomiets LA, Bochkareva NV, Kondakova IV. [Specifics of hormonal and energy balance in patients with hyperplasia and endometrial neoplasia with metabolic syndrome in the background]. Vopr Onkol 2013;59:65–71.Search in Google Scholar
99. Jnne O, Kauppila A, Kontula K, Syrjl SP, Vihko R. Female sex steroid receptors in normal, hyperplastic and carcinomatous endometrium. The relationship to serum steroid hormones and gonadotropins and changes during medroxyprogesterone acetate administration. Int J Cancer 1979;24:545–54.10.1002/ijc.2910240505Search in Google Scholar
100. Zhao P-L, Zhang Q-F, Yan L-Y, Huang S, Chen Y, Qiao J. Functional investigation on aromatase in endometrial hyperplasia in polycystic ovary syndrome cases. Asian Pac J Cancer Prev 2014;15:8975–9.10.7314/APJCP.2014.15.20.8975Search in Google Scholar
101. Šmuc T, Rižner TL. Aberrant pre-receptor regulation of estrogen and progesterone action in endometrial cancer. Mol Cell Endocrinol 2009;301:74–82.10.1016/j.mce.2008.09.019Search in Google Scholar
102. Eritja N, Mirantes C, Llobet D, Yeramian A, Bergada L, Dosil MA, Domingo M, Matias-Guiu X, Dolcet X. Long-term estradiol exposure is a direct mitogen for insulin/egf-primed endometrial cells and drives pten loss-induced hyperplasic growth. Am J Pathol 2013;183:277–87.10.1016/j.ajpath.2013.03.008Search in Google Scholar
103. Dznelashvili NO, Kasradze DG, Tavartkiladze AG, Mariamidze AG, Dzhinchveladze DN. [Expression of epidermal growth factor receptor and plasmatic level of melatonin in simple and complex endometrial hyperplasia]. Georgian Med News 2013:91–5.Search in Google Scholar
104. Bhaskaran K, Douglas I, Forbes H, dos-Santos-Silva I, Leon DA, Smeeth L. Body-mass index and risk of 22 specific cancers: a population-based cohort study of 5·24 million uk adults. Lancet 2014;384:755–65.10.1016/S0140-6736(14)60892-8Search in Google Scholar
105. Burleigh A, Talhouk A, Gilks CB, McAlpine JN. Clinical and pathological characterization of endometrial cancer in young women: identification of a cohort without classical risk factors. Gynecol Oncol 2015;138:141–6.10.1016/j.ygyno.2015.02.028Search in Google Scholar PubMed
106. Keum N, Greenwood D, Hoon Lee D, Kim R, Aune D, Ju W, Hu F, Giovannucci E. Adult weight gain and adiposity-related cancers: a dose-response meta-analysis of prospective observational studies. J Natl Cancer Inst 2015;107.10.1093/jnci/djv088Search in Google Scholar PubMed
107. Dougan MM, Hankinson SE, Vivo ID, Tworoger SS, Glynn RJ, Michels KB. Prospective study of body size throughout the life-course and the incidence of endometrial cancer among premenopausal and postmenopausal women. Int J Cancer 2015;137:625–37.10.1002/ijc.29427Search in Google Scholar PubMed PubMed Central
108. Canchola A, Chang E, Bernstein L, Largent J, Reynolds P, Deapen D, Ursin G, Horn-Ross P. Body size and the risk of endometrial cancer by hormone therapy use in postmenopausal women in the california teachers study cohort. Cancer Causes Control 2010;21:1407–16.10.1007/s10552-010-9568-8Search in Google Scholar PubMed PubMed Central
109. Benedetto C, Salvagno F, Canuto EM, Gennarelli G. Obesity and female malignancies. Best Pract Res Clin Obstet Gynaecol 2015;29:528–40.10.1016/j.bpobgyn.2015.01.003Search in Google Scholar
110. Ali AT. Risk factors for endometrial cancer. Ceska Gynekol 2013;78:448–59.Search in Google Scholar
111. Simpson ER. Sources of estrogen and their importance. J Steroid Biochem Mol Biol 2003;86:225–30.10.1016/S0960-0760(03)00360-1Search in Google Scholar
112. Ashizawa N, Yahata T, Quan J, Adachi S, Yoshihara K, Tanaka K. Serum leptin–adiponectin ratio and endometrial cancer risk in postmenopausal female subjects. Gynecol Oncol 2010;119:65–9.10.1016/j.ygyno.2010.07.007Search in Google Scholar PubMed
113. Luhn P, Dallal CM, Weiss JM, Black A, Huang W-Y, Lacey JV, Hayes RB, Stanczyk FZ, Wentzensen N, Brinton LA. Circulating adipokine levels and endometrial cancer risk in the prostate, lung, colorectal and ovarian cancer screening trial. Cancer Epidemiol Biomarkers Prev 2013;22:1304–12.10.1158/1055-9965.EPI-13-0258Search in Google Scholar PubMed PubMed Central
114. Moon H-S, Chamberland JP, Aronis K, Tseleni-Balafouta S, Mantzoros CS. Direct role of adiponectin and adiponectin receptors in endometrial cancer: in vitro and ex vivo studies in humans. Mol Cancer Ther 2011;10:2234–43.10.1158/1535-7163.MCT-11-0545Search in Google Scholar PubMed PubMed Central
115. Cong L, Gasser J, Zhao J, Yang B, Li F, Zhao AZ. Human adiponectin inhibits cell growth and induces apoptosis in human endometrial carcinoma cells, hec-1-a and rl95–2. Endocr Relat Cancer 2007;14:713–20.10.1677/ERC-07-0065Search in Google Scholar PubMed
116. Maso LD, Augustin LS, Karalis A, Talamini R, Franceschi S, Trichopoulos D, Mantzoros CS, Vecchia CL. Circulating adiponectin and endometrial cancer risk. J Clin Endocrinol Metab 2004;89:1160–3.10.1210/jc.2003-031716Search in Google Scholar PubMed
117. Kishida K, Funahashi T, Shimomura I. Adiponectin as a routine clinical biomarker. Best Pract Res Clin Endocrinol Metab 2014;28:119–30.10.1016/j.beem.2013.08.006Search in Google Scholar PubMed
118. Chen X, Xiang Y-B, Long J-R, Cai H, Cai Q, Cheng J, Wen W, Gao Y-T, Zheng W, Shu X-O. Genetic polymorphisms in obesity-related genes and endometrial cancer risk. Cancer 2012;118:3356–64.10.1002/cncr.26552Search in Google Scholar PubMed PubMed Central
119. Garofalo C, Surmacz E. Leptin and cancer. J Cell Physiol 2006;207:12–22.10.1002/jcp.20472Search in Google Scholar PubMed
120. Linkov F, Burke LE, Komaroff M, Edwards RP, Lokshin A, Styn MA, Tseytlin E, Freese KE, Bovbjerg DH. An exploratory investigation of links between changes in adipokines and quality of life in individuals undergoing weight loss interventions: possible implications for cancer research. Gynecol Oncol 2014;133:67–72.10.1016/j.ygyno.2014.01.019Search in Google Scholar PubMed PubMed Central
121. Friberg E, Orsini N, Mantzoros CS, Wolk A. Diabetes mellitus and risk of endometrial cancer: a meta-analysis. Diabetologia 2007;50:1365–74.10.1007/s00125-007-0681-5Search in Google Scholar PubMed
122. Mantzoros CS. Obesity and diabetes. [electronic book]: Totowa, NJ: Humana Press, 2006.10.1007/978-1-59259-985-1Search in Google Scholar
123. Markowska A, Pawalowska M, Filas V, Korski K, Grybos M, Sajdak S, Olejek A, Bednarek W, Spiewankiewicz B, Lubin J, Markowska J. Does metformin affect er, pr, igf-1r, beta-catenin and pax-2 expression in women with diabetes mellitus and endometrial cancer? Diabetol Metab Syndr 2013;5:76.10.1186/1758-5996-5-76Search in Google Scholar PubMed PubMed Central
124. Hendriks YM, Wagner A, Morreau H, Menko F, Stormorken A, Quehenberger F, Sandkuijl L, Moller P, Genuardi M, Van Houwelingen H, Tops C, Van Puijenbroek M, Verkuijlen P, Kenter G, Van Mil A, Meijers-Heijboer H, Tan GB, Breuning MH, Fodde R, Wijnen JT, Brocker-Vriends AH, Vasen H. Cancer risk in hereditary nonpolyposis colorectal cancer due to msh6 mutations: impact on counseling and surveillance. Gastroenterology 2004;127:17–25.10.1053/j.gastro.2004.03.068Search in Google Scholar PubMed
125. Tafe LJ. Targeted next-generation sequencing for hereditary cancer syndromes: a focus on lynch syndrome and associated endometrial cancer. J Mol Diagn 2015;17:472–82.10.1016/j.jmoldx.2015.06.001Search in Google Scholar PubMed
126. Goodfellow PJ, Billingsley CC, Lankes HA, Ali S, Cohn DE, Broaddus RJ, Ramirez N, Pritchard CC, Hampel H, Chassen AS, Simmons LV, Schmidt AP, Gao F, Brinton LA, Backes F, Landrum LM, Geller MA, DiSilvestro PA, Pearl ML, Lele SB, Powell MA, Zaino RJ, Mutch D. Combined microsatellite instability, mlh1 methylation analysis, and immunohistochemistry for lynch syndrome screening in endometrial cancers from gog210: an nrg oncology and gynecologic oncology group study. J Clin Oncol 2015;33:4301–8.10.1200/JCO.2015.63.9518Search in Google Scholar PubMed PubMed Central
127. Douglas JA, Gruber SB, Meister KA, Bonner J, Watson P, Krush AJ, Lynch HT. History and molecular genetics of lynch syndrome in family g: a century later. J Am Med Assoc 2005;294:2195–202.10.1001/jama.294.17.2195Search in Google Scholar PubMed
128. Miyamoto T, Shiozawa T, Kashima H, Feng YZ, Suzuki A, Kurai M, Nikaido T, Konishi I. Estrogen up-regulates mismatch repair activity in normal and malignant endometrial glandular cells. Endocrinology 2006;147:4863–70.10.1210/en.2006-0632Search in Google Scholar PubMed
129. Kane MF, Loda M, Gaida GM, Lipman J, Mishra R, Goldman H, Jessup JM, Kolodner R. Methylation of the hmlh1 promoter correlates with lack of expression of hmlh1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res 1997;57:808–11.Search in Google Scholar
130. Dashti SG, Chau R, Ouakrim DA, Buchanan DD, Clendenning M, Young JP, Winship IM, Arnold J, Ahnen DJ, Haile RW, Casey G, Gallinger S, Thibodeau SN, Lindor NM, Le Marchand L, Newcomb PA, Potter JD, Baron JA, Hopper JL, Jenkins MA, Win AK. Female hormonal factors and the risk of endometrial cancer in lynch syndrome. J Am Med Assoc 2015;314:61–71.10.1001/jama.2015.6789Search in Google Scholar PubMed PubMed Central
131. Cancer Research UK. Available from: http://www.cancerresearchuk.org/health-professional/uterine-cancer-risk-factors#heading-Eightteen. Accessed January 2016.Search in Google Scholar
132. Felix AS, Yang HP, Gierach GL, Park Y, Brinton LA. Cigarette smoking and endometrial carcinoma risk: the role of effect modification and tumor heterogeneity. Cancer Causes Control 2014;25:479–89.10.1007/s10552-014-0350-1Search in Google Scholar
133. Michnovicz JJ, Hershcopf RJ, Naganuma H, Bradlow HL, Fishman J. Increased 2-hydroxylation of estradiol as a possible mechanism for the anti-estrogenic effect of cigarette smoking. N Engl J Med 1986;315:1305–9.10.1056/NEJM198611203152101Search in Google Scholar
134. Mattison DR, Thorgeirsson SS. Smoking and industrial pollution, and their effects on menopause and ovarian cancer. Lancet (London, England) 1978;1:187–8.10.1016/S0140-6736(78)90617-7Search in Google Scholar
135. Spangler JG. Smoking and hormone-related disorders. Prim Care 1999;26:499–511.10.1016/S0095-4543(05)70114-7Search in Google Scholar
136. Khaw KT, Tazuke S, Barrett-Connor E. Cigarette smoking and levels of adrenal androgens in postmenopausal women. N Engl J Med 1988;318:1705–9.10.1056/NEJM198806303182601Search in Google Scholar PubMed
137. Kapoor D, Jones TH. Smoking and hormones in health and endocrine disorders. Eur J Endocrinol 2005;152:491–9.10.1530/eje.1.01867Search in Google Scholar PubMed
138. Brand JS, Chan MF, Dowsett M, Folkerd E, Wareham NJ, Luben RN, van der Schouw YT, Khaw KT. Cigarette smoking and endogenous sex hormones in postmenopausal women. J Clin Endocrinol Metab 2011;96:3184–92.10.1210/jc.2011-1165Search in Google Scholar PubMed
139. Polesel J, Serraino D, Zucchetto A, Lucenteforte E. Cigarette smoking and endometrial cancer risk: the modifying effect of obesity. Eur J Cancer Prev 2009;18:476–81.10.1097/CEJ.0b013e32832f9bc4Search in Google Scholar PubMed
140. Minami Y, Yamamoto R, Nishikouri M, Fukao A, Hisamichi S. Mortality and cancer incidence in patients with parkinson’s disease. J Neurol 2000;247:429–34.10.1007/s004150070171Search in Google Scholar PubMed
141. Currie LJ, Harrison MB, Trugman JM, Bennett JP, Wooten GF. Postmenopausal estrogen use affects risk for parkinson disease. Arch Neurol 2004;61:886–8.10.1001/archneur.61.6.886Search in Google Scholar PubMed
142. Bourque M, Dluzen DE, Di Paolo T. Neuroprotective actions of sex steroids in parkinson’s disease. Front Neuroendocrinol 2009;30:142–57.10.1016/j.yfrne.2009.04.014Search in Google Scholar PubMed
143. Ekue A, Boulanger JF, Morissette M, Di Paolo T. Lack of effect of testosterone and dihydrotestosterone compared to 17beta-oestradiol in 1-methyl-4-phenyl-1,2,3,6, tetrahydropyridine-mice. J Neuroendocrinol 2002;14:731–6.10.1046/j.1365-2826.2002.00833.xSearch in Google Scholar
144. Yu L, Liao PC. Estrogen and progesterone distinctively modulate methamphetamine-induced dopamine and serotonin depletions in C57BL/6J mice. J Neural Transm (Vienna) 2000;107:1139–47.10.1007/s007020070027Search in Google Scholar
145. Ong EL, Goldacre R, Goldacre M. Differential risks of cancer types in people with parkinson’s disease: a national record-linkage study. Eur J Cancer 2014;50:2456–62.10.1016/j.ejca.2014.06.018Search in Google Scholar
146. Mueck AO, Seeger H, Rabe T. Hormonal contraception and risk of endometrial cancer: a systematic review. Endocr Relat Cancer 2010;17:R263–71.10.1677/ERC-10-0076Search in Google Scholar
147. Endometrial cancer and oral contraceptives: an individual participant meta-analysis of 27 276 women with endometrial cancer from 36 epidemiological studies. Lancet Oncol 2015:1061–70.10.1016/S1470-2045(15)00212-0Search in Google Scholar
148. Cook LS, Dong Y, Round P, Huang X, Magliocco AM, Friedenreich CM. Hormone contraception before the first birth and endometrial cancer risk. Cancer Epidemiol Biomarkers Prev 2014;23:356–61.10.1158/1055-9965.EPI-13-0943Search in Google Scholar PubMed
149. Maxwell GL, Schildkraut JM, Calingaert B, Risinger JI, Dainty L, Marchbanks PA, Berchuck A, Barrett JC, Rodriguez GC. Progestin and estrogen potency of combination oral contraceptives and endometrial cancer risk. Gynecol Oncol 2006;103:535–40.10.1016/j.ygyno.2006.03.046Search in Google Scholar PubMed
150. Kaunitz AM. Depot medroxyprogesterone acetate contraception and the risk of breast and gynecologic cancer. J Reprod Med 1996;41:419–27.Search in Google Scholar
151. Soini T, Hurskainen R, Grénman S, Mäenpää J, Paavonen J, Pukkala E. Cancer risk in women using the levonorgestrel-releasing intrauterine system in finland. Obstet Gynecol 2014;124:292–9.10.1097/AOG.0000000000000356Search in Google Scholar PubMed
152. Chlebowski RT, Anderson GL. Menopausal hormone therapy and cancer: changing clinical observations of target site specificity. Steroids 2014;90:53–9.10.1016/j.steroids.2014.06.001Search in Google Scholar PubMed PubMed Central
153. Morch LS, Kjaer SK, Keiding N, Lokkegaard E, Lidegaard O. The influence of hormone therapies on type i and ii endometrial cancer: a nationwide cohort study. Int J Cancer 2016;138:1506–15.10.1002/ijc.29878Search in Google Scholar PubMed
154. Fournier A, Dossus L, Mesrine S, Vilier A, Boutron-Ruault MC, Clavel-Chapelon F, Chabbert-Buffet N. Risks of endometrial cancer associated with different hormone replacement therapies in the e3n cohort, 1992–2008. Am J Epidemiol 2014;180:508–17.10.1093/aje/kwu146Search in Google Scholar PubMed
155. Shim SH, Lee SJ, Kim SN. Effects of hormone replacement therapy on the rate of recurrence in endometrial cancer survivors: a meta-analysis. Eur J Cancer 2014;50:1628–37.10.1016/j.ejca.2014.03.006Search in Google Scholar PubMed
156. Behnamfar F, Ghahiri A, Tavakoli M. Levonorgestrel-releasing intrauterine system (mirena) in compare to medroxyprogesterone acetate as a therapy for endometrial hyperplasia. J Res Med Sci 2014;19:686–90.Search in Google Scholar
157. Lai CH, Huang HJ. The role of hormones for the treatment of endometrial hyperplasia and endometrial cancer. Curr Opin Obstet Gynecol 2006;18:29–34.10.1097/01.gco.0000192994.37965.c6Search in Google Scholar PubMed
158. Gallos ID, Krishan P, Shehmar M, Ganesan R, Gupta JK. Lng-ius versus oral progestogen treatment for endometrial hyperplasia: a long-term comparative cohort study. Hum Reprod 2013;28:2966–71.10.1093/humrep/det320Search in Google Scholar PubMed
159. Gallos ID, Krishan P, Shehmar M, Ganesan R, Gupta JK. Relapse of endometrial hyperplasia after conservative treatment: a cohort study with long-term follow-up. Hum Reprod 2013;28:1231–6.10.1093/humrep/det049Search in Google Scholar PubMed
160. Park JY, Nam JH. Progestins in the fertility-sparing treatment and retreatment of patients with primary and recurrent endometrial cancer. Oncologist 2015;20:270–8.10.1634/theoncologist.2013-0445Search in Google Scholar PubMed PubMed Central
161. Colombo N, Preti E, Landoni F, Carinelli S, Colombo A, Marini C, Sessa C, Group EG. Endometrial cancer: esmo clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol 2013;24 Suppl 6:vi33–8.10.1093/annonc/mdt353Search in Google Scholar PubMed
162. Martin-Hirsch PP, Bryant A, Keep SL, Kitchener HC, Lilford R. Adjuvant progestagens for endometrial cancer. Cochrane Database Syst Rev 2011:Cd001040.10.1002/14651858.CD001040.pub2Search in Google Scholar PubMed PubMed Central
163. Slayden OD. Progesterone antagonists increase androgen receptor expression in the rhesus macaque and human endometrium. J Clin Endocrinol Metab 2001;86:2668.10.1210/jc.86.6.2668Search in Google Scholar
164. Spitz IM, Grunberg SM, Chabbert-Buffet N, Lindenberg T, Gelber H, Sitruk-Ware R. Management of patients receiving long-term treatment with mifepristone. Fertil Steril 2005;84:1719–26.10.1016/j.fertnstert.2005.05.056Search in Google Scholar PubMed
165. Romero-Pérez L, Díaz-Martín J, López-García MA, Castilla MA, Palacios J, Garcia-Sanz P, Mota A, Hergueta-Redondo M, Moreno-Bueno G, Leskelä S, Martínez-Ramírez A, Soslow RA, Matias-Guiu X. A role for the transducer of the hippo pathway, taz, in the development of aggressive types of endometrial cancer. Mod Pathol 2015;28:1492–503.10.1038/modpathol.2015.102Search in Google Scholar PubMed
166. Ramondetta LM, Johnson AJ, Sun CC, Atkinson N, Smith JA, Jung MS, Broaddus R, Iyer RB, Burke T. Phase 2 trial of mifepristone (ru-486) in advanced or recurrent endometrioid adenocarcinoma or low-grade endometrial stromal sarcoma. Cancer 2009;115:1867–74.10.1002/cncr.24197Search in Google Scholar PubMed
167. Grunberg SM, Weiss MH, Russell CA, Spitz IM, Ahmadi J, Sadun A, Sitruk-Ware R. Long-term administration of mifepristone (ru486): clinical tolerance during extended treatment of meningioma. Cancer Invest 2006;24:727–33.10.1080/07357900601062339Search in Google Scholar PubMed
168. Ellis AJ, Hendrick VM, Williams R, Komm BS. Selective estrogen receptor modulators in clinical practice: a safety overview. Expert Opin Drug Saf 2015;14:921–34.10.1517/14740338.2015.1014799Search in Google Scholar PubMed
169. Cohen I. Endometrial pathologies associated with postmenopausal tamoxifen treatment. Gynecol Oncol 2004;94:256–66.10.1016/j.ygyno.2004.03.048Search in Google Scholar PubMed
170. Committee opinion no. 601. Tamoxifen and uterine cancer. Obstet Gynecol 2014;123:1394–7.10.1097/01.AOG.0000450757.18294.cfSearch in Google Scholar PubMed
171. Mirkin S, Pickar JH. Selective estrogen receptor modulators (serms): a review of clinical data. Maturitas 2015;80:52–7.10.1016/j.maturitas.2014.10.010Search in Google Scholar PubMed
172. Nakamura K, Sawada K, Sugiyama M, Mabuchi S, Hisamatsu T, Nishio Y, Ito K, Kimura T, Kamiura S, Morishige K. Efficacy of raloxifene hydrochloride for the prevention of health care problems in patients who undergo surgery for endometrial cancer: a multicenter randomized clinical trial. Int J Gynecol Cancer 2015;25:288–95.10.1097/IGC.0000000000000333Search in Google Scholar PubMed
173. Constantine GD, Goldstein SR, Archer DF. Endometrial safety of ospemifene: results of the phase 2/3 clinical development program. Menopause 2015;22:36–43.10.1097/GME.0000000000000275Search in Google Scholar PubMed PubMed Central
174. Gennari L. Lasofoxifene, a new selective estrogen receptor modulator for the treatment of osteoporosis and vaginal atrophy. Expert Opin Pharmacother 2009;10:2209–20.10.1517/14656560903127241Search in Google Scholar PubMed
175. Archer DF, Hendrix S, Ferenczy A, Felix J, Gallagher JC, Rymer J, Skouby SO, den Hollander W, Stathopoulos V, Helmond FA. Tibolone histology of the endometrium and breast endpoints study: design of the trial and endometrial histology at baseline in postmenopausal women. Fertil Steril 2007;88:866–78.10.1016/j.fertnstert.2006.12.052Search in Google Scholar PubMed
176. Morales L, Neven P, Timmerman D, Wildiers H, Konstantinovic ML, Christiaens MR, Tan PN, Paridaens R. Prospective assessment of the endometrium in postmenopausal breast cancer patients treated with fulvestrant. Breast Cancer Res Treat 2009;117:77–81.10.1007/s10549-008-0248-2Search in Google Scholar PubMed
177. Covens AL, Filiaci V, Gersell D, Lutman CV, Bonebrake A, Lee YC. Phase ii study of fulvestrant in recurrent/metastatic endometrial carcinoma: a gynecologic oncology group study. Gynecol Oncol 2011;120:185–8.10.1016/j.ygyno.2010.10.015Search in Google Scholar PubMed
178. Emons G, Günthert A, Thiel FC, Camara O, Strauss H-G, Breitbach G-P, Kölbl H, Reimer T, Finas D, Rensing K. Phase ii study of fulvestrant 250 mg/month in patients with recurrent or metastatic endometrial cancer: a study of the arbeitsgemeinschaft gynäkologische onkologie. Gynecol Oncol 2013;129:495–9.10.1016/j.ygyno.2013.02.039Search in Google Scholar PubMed
179. Garuti G, Cellani F, Centinaio G, Montanari G, Nalli G, Luerti M. Prospective endometrial assessment of breast cancer patients treated with third generation aromatase inhibitors. Gynecol Oncol 2006;103:599–603.10.1016/j.ygyno.2006.04.004Search in Google Scholar PubMed
180. Agorastos T, Vaitsi V, Pantazis K, Efstathiadis E, Vavilis D, Bontis JN. Aromatase inhibitor anastrozole for treating endometrial hyperplasia in obese postmenopausal women. Eur J Obstet Gynecol Reprod Biol 2005;118:239–40.10.1016/j.ejogrb.2004.07.002Search in Google Scholar PubMed
181. Barker LC, Brand IR, Crawford SM. Sustained effect of the aromatase inhibitors anastrozole and letrozole on endometrial thickness in patients with endometrial hyperplasia and endometrial carcinoma. Curr Med Res Opin 2009;25:1105–9.10.1185/03007990902860549Search in Google Scholar PubMed
182. Carlson MJ, Thiel KW, Leslie KK. Past, present, and future of hormonal therapy in recurrent endometrial cancer. Int J Womens Health 2014;6:429–35.10.2147/IJWH.S40942Search in Google Scholar
183. Ma BB, Oza A, Eisenhauer E, Stanimir G, Carey M, Chapman W, Latta E, Sidhu K, Powers J, Walsh W, Fyles A. The activity of letrozole in patients with advanced or recurrent endometrial cancer and correlation with biological markers – a study of the national cancer institute of canada clinical trials group. Int J Gynecol Cancer 2004;14:650–8.10.1136/ijgc-00009577-200407000-00013Search in Google Scholar
184. Rose PG, Brunetto VL, VanLe L, Bell J, Walker JL, Lee RB. Regular article: a phase ii trial of anastrozole in advanced recurrent or persistent endometrial carcinoma: a gynecologic oncology group study. Gynecol Oncol 2000;78:212–6.10.1006/gyno.2000.5865Search in Google Scholar PubMed
185. Maclaran K, Panay N. The safety of postmenopausal testosterone therapy. Womens Health (Lond Engl) 2012;8:263–75.10.2217/WHE.12.11Search in Google Scholar PubMed
186. Manley K, Edey K, Braybrooke J, Murdoch J. Hormone replacement therapy after endometrial cancer. Menopause Int 2012;18:134–8.10.1258/mi.2012.012024Search in Google Scholar PubMed
187. Perrone AM, Cerpolini S, Maria Salfi NC, Ceccarelli C, De Giorgi LB, Formelli G, Casadio P, Ghi T, Pelusi G, Pelusi C, Meriggiola MC. Effect of long-term testosterone administration on the endometrium of female-to-male (ftm) transsexuals. J Sex Med 2009;6:3193–200.10.1111/j.1743-6109.2009.01380.xSearch in Google Scholar
188. Portman DJ, Labrie F, Archer DF, Bouchard C, Cusan L, Girard G, Ayotte N, Koltun W, Blouin F, Young D, Wade A, Martel C, Dube R. Lack of effect of intravaginal dehydroepiandrosterone (dhea, prasterone) on the endometrium in postmenopausal women. Menopause 2015;22:1289–95.10.1097/GME.0000000000000470Search in Google Scholar
189. Tanaka T, Umesaki N. Danazol enhances fas-mediated apoptosis in human endometrial epithelial cells within normal physiology. Int J Mol Med 2009;23:237–43.10.3892/ijmm_00000147Search in Google Scholar
190. Terakawa N, Ikegami H, Shimizu I, Aono T, Tanizawa O, Matsumoto K. Growth inhibition by danazol in a human endometrial cancer cell line with estrogen-independent progesterone receptors. J Steroid Biochem 1987;28:571–4.10.1016/0022-4731(87)90517-6Search in Google Scholar
191. Surrey ES, Halme J. Direct effects of medroxyprogesterone acetate, danazol, and leuprolide acetate on endometrial stromal cell proliferation in vitro. Fertil Steril 1992;58:273–8.10.1016/S0015-0282(16)55199-1Search in Google Scholar
192. Niwa K, Hashimoto M, Morishita S, Yokoyama Y, Lian Z, Tagami K, Mori H, Tamaya T. Preventive effects of danazol on endometrial carcinogenesis in mice. Cancer Lett 2000;158:133–9.10.1016/S0304-3835(00)00497-3Search in Google Scholar
193. Tamaoka Y, Orikasa H, Sumi Y, Sakakura K, Kamei K, Nagatani M, Ezawa S. Treatment of endometrial hyperplasia with a danazol-releasing intrauterine device: a prospective study. Gynecol Obstet Invest 2004;58:42–8.10.1159/000077882Search in Google Scholar
194. Mariani L, Sedati A, Giovinazzi R, Sindico R, Atlante G. Postmenopausal endometrial hyperplasia: role of danazol therapy. Int J Gynaecol Obstet 1994;44:155–9.10.1016/0020-7292(94)90071-XSearch in Google Scholar
195. Covens A, Brunetto VL, Markman M, Orr JW, Lentz SS, Benda J. Phase ii trial of danazol in advanced, recurrent, or persistent endometrial cancer: a gynecologic oncology group study. Gynecol Oncol 2003;89:470–4.10.1016/S0090-8258(03)00149-5Search in Google Scholar
196. Montagnani Marelli M, Moretti RM, Januszkiewicz-Caulier J, Motta M, Limonta P. Gonadotropin-releasing hormone (gnrh) receptors in tumors: a new rationale for the therapeutical application of gnrh analogs in cancer patients? Curr Cancer Drug Targets 2006;6:257–69.10.2174/156800906776842966Search in Google Scholar PubMed
197. Grundker C, Gunthert AR, Westphalen S, Emons G. Biology of the gonadotropin-releasing hormone system in gynecological cancers. Eur J Endocrinol 2001;146:1–14.10.1530/eje.0.1460001Search in Google Scholar
198. Grundker C, Schlotawa L, Viereck V, Emons G. Protein kinase c-independent stimulation of activator protein-1 and c-jun n-terminal kinase activity in human endometrial cancer cells by the lhrh agonist triptorelin. Eur J Endocrinol 2001;145:651–8.10.1530/eje.0.1450651Search in Google Scholar PubMed
199. Herbst KL. Gonadotropin-releasing hormone antagonists. Curr Opin Pharmacol 2003;3:660–6.10.1016/j.coph.2003.06.009Search in Google Scholar PubMed
©2016 by De Gruyter
Articles in the same Issue
- Frontmatter
- Topic B: Neuroendocrine Mechanisms and Hormonal Impact on Eating Disorders, Sexuality and Reproductive System
- Review Articles
- Sexuality in eating disorders patients: etiological factors, sexual dysfunction and identity issues. A systematic review
- The physiology of functional hypothalamic amenorrhea associated with energy deficiency in exercising women and in women with anorexia nervosa
- Ghrelin action on GnRH neurons and pituitary gonadotropes might be mediated by GnIH-GPR147 system
- Hormones and endometrial carcinogenesis
- Original Article
- Neonatal exposure to estradiol-17β modulates tumour necrosis factor alpha and cyclooxygenase-2 expression in brain and also in ovaries of adult female rats
Articles in the same Issue
- Frontmatter
- Topic B: Neuroendocrine Mechanisms and Hormonal Impact on Eating Disorders, Sexuality and Reproductive System
- Review Articles
- Sexuality in eating disorders patients: etiological factors, sexual dysfunction and identity issues. A systematic review
- The physiology of functional hypothalamic amenorrhea associated with energy deficiency in exercising women and in women with anorexia nervosa
- Ghrelin action on GnRH neurons and pituitary gonadotropes might be mediated by GnIH-GPR147 system
- Hormones and endometrial carcinogenesis
- Original Article
- Neonatal exposure to estradiol-17β modulates tumour necrosis factor alpha and cyclooxygenase-2 expression in brain and also in ovaries of adult female rats